

Abstract
Alcohol abuse has been associated with increased susceptibility to pulmonary infection. It is not fully defined how alcohol contributes to the host defense compromise. Here primary human airway epithelial cells were cultured at an air-liquid interface to form a differentiated and polarized epithelium. This unique culture model allowed us to closely mimic lung infection in the context of alcohol abuse by basolateral alcohol exposure and apical live bacterial challenge. Application of clinically relevant concentrations of alcohol for 24 hours did not significantly alter epithelial integrity or barrier function. When apically challenged with viable Klebsiella pneumoniae, the cultured epithelia had an enhanced tightness which was unaffected by alcohol. Further, alcohol enhanced apical bacterial growth, but not bacterial binding to the cells. The cultured epithelium in the absence of any treatment or stimulation had a base-level IL-6 and IL-8 secretion. Apical bacterial challenge significantly elevated the basolateral secretion of inflammatory cytokines including IL-2, IL-4, IL-6, IL-8, IFN-γ, GM-CSF, and TNF-α. However, alcohol suppressed the observed cytokine burst in response to infection. Addition of adenosine receptor agonists negated the suppression of IL-6 and TNF-α. Thus, acute alcohol alters the epithelial cytokine response to infection, which can be partially mitigated by adenosine receptor agonists.
Keywords: Alcohol, Human airway epithelia, Air-liquid interface culture, Klebsiella pneumoniae bacteria, Epithelial barrier function, Cytokines, Adenosine receptor
Introduction
The respiratory tract is constantly exposed to airborne microorganisms during respiration. It has long been observed that alcohol abuse increases the risk of pulmonary infection such as bacterial pneumonia and bronchitis [1–3]. The mechanism whereby alcohol abuse impairs lung host defense is not fully defined.
Lung host defense reflects the combined activities of resident cells, including the pulmonary epithelial cells and macrophages, and recruited cells, most notably polymorphonuclear leukocytes. Multi-tiered mechanisms have been developed to effectively defend against microbial colonization and invasion. First, pulmonary epithelial cells form a tight epithelial sheet via their junctional complexes, serving as a structural barrier [4]. Second, the polarized airway epithelia have mucociliary clearance as a means to sweep away the deposited microbes [5–6]. Third, airway epithelia produce host defense factors, including antimicrobial peptides like lysozyme, defensins and the short palate, lung, nasal epithelium clone 1 (SPLUNC1) protein to directly kill the invading microorganisms [7–10]. Fourth, airways and alveoli are patrolled by phagocytic cells, most notably the alveolar macrophage, which can engulf aspirated microbes [11–13]. Moreover, airways can also recruit neutrophils, if needed, to the sites of inflammation for elimination of invading bacteria [14–17]. Fifth, the acquired immune system, including antigen-stimulated T and B lymphocytes, provides cellular and humoral defenses for the airways [13, 18]. Therefore, impairment of any of these mechanisms may compromise the host defense capacities of the lung.
Alcohol affects multiple lung host defense mechanisms. It was documented that alcohol slows ciliary beating due to desensitization of airway protein kinase A (PKA) activity [19–21]. Alcohol abusers have an increased susceptibility to lung infection by Streptococcus pneumoniae and Klebsiella pneumoniae (K. Pneumoniae) [22–24]. In response to the microbial exposure, the pulmonary epithelium produces inflammatory mediators which act in a paracrine or autocrine fashion to mobilize an effective immune defense [25]. Acute alcohol suppresses CXC chemokine production in the lung during pulmonary infection and inflammation [26–28]. In contrast, chronic alcohol has been associated with increased proinflammatory cytokine activation after infection [29]. Toll-like receptor and NF-κB signaling pathway appears involved in the cytokine responses [30–35]. These studies provide important information to define alcohol alteration of pulmonary epithelial function and response to infection. However, fully understanding of alcohol-induced impairment of lung host defense requires further studies on a physiologically relevant model.
In this study we hypothesized that air-liquid interface culture of human airway epithelia better recapitulates in vivo airways in response to infection and alcohol exposure, which is critical to delineate the molecular mechanisms of alcohol-associated airway infection. To test the hypothesis we cultured primary human airway epithelial cells at the air-liquid interface. The airway epithelia fully differentiated and polarized. We were able to expose the basolateral side to alcohol and the apical side to live bacteria. Alcohol effects on airway epithelial functions including epithelial integrity, bacterial binding and viability, and cytokine response to K. pneumoniae infection were investigated. Pharmacological mitigation of the alcohol cytokine effect was also explored.
Materials and Methods
Cell culture
Primary human bronchial airway epithelial cells (NHBE) were purchased commercially (Lonza Walkersville, MD). After expansion for 1–2 passages, 5×105 NHBE cells were trypsinized off, seeded on a collagen-coated 0.6 cm2 Millicell®-PCF membrane insert (Millipore, Billerica, MA) and cultured at the air-liquid interface using the Lonza BEGM™ bronchial epithelial cell growth medium according to the previously published protocol [36–37]. Fully differentiated airway epithelial cultures, reflected by completely dry apical surfaces and transepithelial electrical resistances (TEER) greater than 700 Ω per one cm2 surface area, were selected for experiments. TEER was measured using the “chop stick” epithelial ohm meter (World Precision Instruments, Sarasota, FL), as described previously [38–39].
Ethanol exposure and bacterial challenge
The air-liquid interface cultures of NHBE cells were basolaterally exposed to different concentrations of ethanol (200 Proof; AAPER Alcohol and Chemical Co., Shelbyville, KY), as indicated in individual experiments. Alcohol concentrations used in this study were biologically relevant levels. As calculated [40], legal driving blood alcohol content (BAC) is less than 0.08%, which equates to a concentration of 22 mM alcohol. BAC of 0.20% is equivalent to 50 mM, commonly observed in individuals presenting to emergency rooms for intoxication. The highest alcohol dose (100 mM) used in this report is consistent with blood alcohol concentrations of chronic alcohol abusers [41]. All the cultures were kept in 37°C, 5% CO2 incubators that had been pre-saturated with the specified ethanol concentrations in the bottom water pan. For bacterial challenge, overnight culture of K. pneumoniae (Strain 43816, serotype 2) (ATCC, Manassas, VA) was spun down, washed twice with PBS and resuspended in PBS to the density of 109 CFU/ml. Fifty microliters of this suspension containing 5×107 CFU bacteria were applied to the apical side of each culture insert for varied durations of time as indicated.
Confocal microscopy
CellTracker™ fluorescent probe, 5-chloromethyl-fluorescein diacetate (CMFDA; Molecular Probes-Invitrogen, Eugene, OR), was added to the basal medium at 10 μM overnight to label the NHBE epithelial cells. Then, the cultures were washed with PBS 4 times for 20 min and fixed in 4% Paraformaldehyde in PBS for 20 min. The cell-grown Millicell® filter membranes were excised, mounted on microscopic slides, and examined by confocal microscopy.
Transmission electron microscopy
Differentiated human airway epithelia were fixed in 2.5% glutaraldehyde (0.1 M sodium cacodylate buffer, pH 7.4) overnight at 4°C and then post-fixed with 1% osmium tetroxide for 1 hour. After serial alcohol dehydration, the samples were embedded in Eponate 12 (Ted Pella, Inc., Redding, CA). Ultrathin sectioning and post staining were performed using the routine procedure. Samples were examined by transmission electron microscopy.
Transepithelial permeability assay
Transepithelial permeability was estimated by diffusion of fluorescent inulin from the apical to the basolateral side. Briefly, NHBE air-liquid cultures were treated with or without alcohol for 24 hours, as indicated. Then, the culture inserts were placed in a new 24-well plate. Phosphate-buffered saline (PBS, 400 μl) was added to the apical and basolateral sides, separately. The apical PBS contained 1 mg/ml FITC-inulin (MW. 2000–5000 KDa; Sigma-Aldrich, St Louis, MO). After 1 hour, 10 μl of the basal buffer was collected and the extent of FITC-inulin permeation determined by measuring the fluorescence at 528 nm in the basal medium samples with a 96-well fluorescent plate reader (BioTEK, Winooski, VT).
Bacterial viability assay
Overnight culture of K. pneunomiae was washed and resuspended to 1×109 CFU/ml. Fifty microliters of the bacterial suspension (5×107 CFU) were added to the apical side of the NHBE cultures which were treated with and without 50 mM basolateral alcohol for 24 hours. Onto each NHBE culture, 350 μl of 0.05% saponin in water was added to lyse the cells. After three passes through a 25-gauge needle, the samples were diluted further in sterile PBS, and 25 μl were plated on LB agar plates. After overnight culture, viable colonies were counted.
Bacterial binding assay
The air-liquid interface cultures of NHBE cells were exposed to 0 and 50 mM of alcohol basolaterally for 24 hours. Then, radioactive 14C-labelled K. pneumoniae (MOI of 50) were applied to the apical side for 1 hour at 37°C. The free bacteria were washed off thrice with PBS and the cells were lysed using 0.1% Triton-X in PBS. The lysed samples (100 μl) were used for measuring the 14C radioactivity with a scintillation counter. By comparing with the scintillation counts of the standard curve established from known numbers of 14C-labelled K. pneumoniae, the numbers of bound bacteria were obtained.
Cytokine measurements
The air-liquid interface culture of NHBE cells were basolatorally exposed to varied concentrations of alcohol (0, 25, 50 and 100 mM) 3 hours prior to an apical challenge with K. pneunomiae (5×107 CFU) for 24 hours. Basal media (300 μl) from each well were collected and centrifuged at 14,000 rpm for 20 minutes at 4°C. The supernatants were assayed for multiple cytokines by Bioplex® Human Cytokine Assay (Bio-Rad, Hercules, CA) or Milliplex® (Millipore, Billerica, MA) following the manufacturers’ directions.
Statistical Analysis
Data presented represent mean of multiple repeats, as indicated in each experiment, with error bar indicating standard deviation from the mean. The data points were analyzed by Student’s t-test and a P-value of ≤0.05 was considered significant.
Results
Differentiated and Polarized human airway epithelia for alcohol studies
Human airway epithelia are polarized, which renders the apical properties of the cells distinct from the basolateral ones. We hypothesized that alcohol exposure mode might affect the responses of airway epithelial cells to infection. To establish a physiologically relevant epithelial model, we seeded primary human airway epithelial cells (NHBEs) on semi-permeable filters and cultured them at the air-liquid interface (Fig. 1A). This system produced well-differentiated and polarized airway epithelia in 2–3 weeks. As examined by confocal microscopy, the cultures that were pre-labeled with the fluorescent dye CMFDA demonstrated a tight epithelial sheet under horizontal optical scanning (Fig. 1B). Apical ciliation was discernable by vertical scanning (Fig. 1C). To appreciate the subcellular structures, the cultures were embedded, followed by ultra-thin section. Transmission electron microscopic observation showed a typical polarized airway epithelium with apical cilia, microvilli and basal pseudopodia extended to the basal filter membrane (Fig. 1D). This culture model permitted separate apical and basolateral experimental manipulations to closely mimic airway infections under the influence of alcohol.
Fig. 1. Air-liquid interface culture of airway epithelial cells.
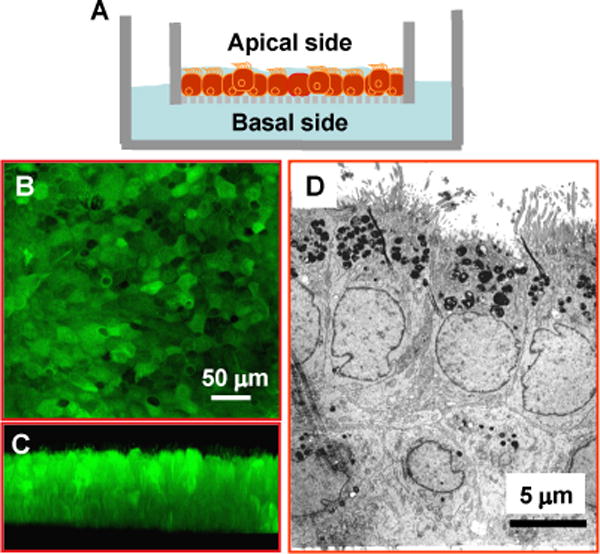
A) A drawing depicting the air-liquid interface culture. NHBE cells are seeded on a semi-permeable filter and grown at the air-liquid interface. Nutritional support comes from the basolateral side and the apical side is exposed to the air. Such a setting facilitates epithelial polarization and differentiation. B & C) Confocal micrographs demonstrate that the cultured airway epithelia form a tight epithelial sheet as seen under horizontal optical scanning with apical ciliation discernable by vertical scanning. D) The electron micrograph shows the typical characteristics of polarized epithelium with apical cilia and microvilli, and pseudopodia extending into the basal filter membrane.
Alcohol at clinically relevant concentrations does not significantly alter airway epithelial barrier function
Airway epithelium provides an intact physical barrier which is vital for blocking airborne infectious pathogens. We first investigated the effect of alcohol on the integrity of our cultured epithelia by measuring transepithelial electrical resistance (TEER). The cultured airway epithelia were exposed basolaterally to varied concentrations of alcohol (0, 25, 50 and 100 mM) for 24 hours, followed by TEER measurement. The results (Fig. 2A) show that alcohol did not significantly affect the TEER values. To employ an alternative method to validate this finding, we tested the transepithelial permeability of FITC-inulin. Similarly, the cultured epithelia were treated basolaterally with alcohol (0, 25, 50 or 100 mM) for 24 hours. Then, the culture inserts were placed in a new 24-well plate. Phosphate-buffered saline (PBS, 400 μl) was added to the apical and basolateral sides, separately. The apical PBS contained 1 mg/ml FITC-inulin. After 1 hour, 10 μl of the basal buffer was collected and measured for FITC fluorescence. The data (Fig. 2B) show that alcohol did not significantly increase FITC-inulin transepithelial diffusion, suggesting an intact epithelial barrier function.
Fig. 2. Effect of alcohol exposure on epithelial integrity and transepithelial permeability.
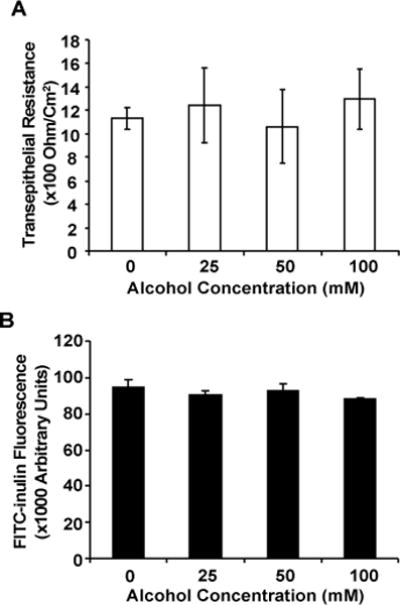
NHBE cells were grown at the air-liquid interface and were exposed to different concentrations of alcohol for 24 hours. A) The effect of alcohol on epithelial cell monolayer integrity was investigated by measuring the transepithelial electrical resistance (TEER). As displayed, alcohol did not induce any significant changes in TEER. B) The effect of alcohol on the transepithelial permeability was estimated by measuring the apical to basolateral diffusion of FITC-Inulin. The acute alcohol exposure did not markedly affect the transepithelial permeability. Data expressed is representative of 2 independent experiments carried out in quadruplicates.
Apical challenge with live K. pneumoniae enhances the airway epithelial tightness regardless of basolateral alcohol presence
We then studied if basolateral alcohol exposure and apical bacterial challenge in combination would affect airway epithelial barrier function. To address this question, we challenged the polarized NHBE cultures apically with K. pneumoniae (5×107 CFU) for 24 hours. Surprisingly, TEER measurement showed an increase in epithelial tightness when compared to baseline controls. Alcohol presence did not significantly affect the TEER readings (Fig 3A). To confirm the finding, the polarized NHBE cultures were similarly challenged with K. pneumoniae in the presence or absence of alcohol for 24 hours. FITC-inulin transepithelial diffusion was estimated as described above. FITC-inulin transepithelial flux dropped in the bacterium-challenged cells irrespective of alcohol exposure (Fig 3B). These data suggest that the epithelial cells respond to bacterial insult by augmenting their cell-cell contacts to strengthen their integrity and barrier function. Alcohol exposure at the tested concentrations did not negatively influence the barrier function of the epithelium under this experimental condition.
Fig. 3. Effect of alcohol on epithelial integrity and transepithelial permeability under apical K. pneumoniae challenge.
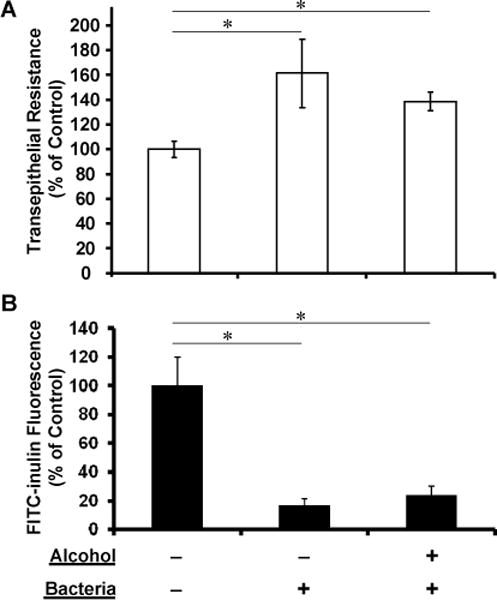
Polarized NHBE cells were challenged with ~10 MOIs of apical K. pneumoniae for 24 hours in the absence (0mM) or presence (100mM) of basolateral alcohol for 24 hours. A) TEER measurements of NHBE cells showed a marked TEER increase with K. pneumoniae challenge as compared to the no alcohol treatment and no bacterial challenge control. Further, alcohol did not have any negative effect on such bacterium-induced TEER increase. B) Similar to the TEER data, transepithelial FITC-inulin diffusion measurements indicated that K. pneumoniae challenge enhanced the epithelial barrier function by significant reduction in FITC-inulin transepithelial diffusion. Under this condition, alcohol exposure did not influence the K. pneumoniae effects on epithelial integrity. Asterisks denote statistically significant difference (p ≤0.05, n=5).
Basolateral alcohol exposure facilitates apical bacterial growth but does not affect apical bacterial binding
We further sought to determine whether basolateral alcohol exposure would affect the apical bacterial viability and binding avidity to airway epithelia. To address the viability issue, K. pneumoniae bacteria (5×107 CFU) were added to the apical surface of polarized NHBEs which had been exposed to 0 and 50 mM of alcohol basolaterally. After 24 hours, the epithelia were lysed with 0.1% saponin and the lysates were serially diluted and plated for overnight culture at 37°C. Bacterial viability was determined by colony counting. Alcohol exposure at 50 mM concentration enhanced apical K. pneumoniae growth (Fig 4A).
Fig. 4. Alcohol effect on apical bacterial viability and binding.
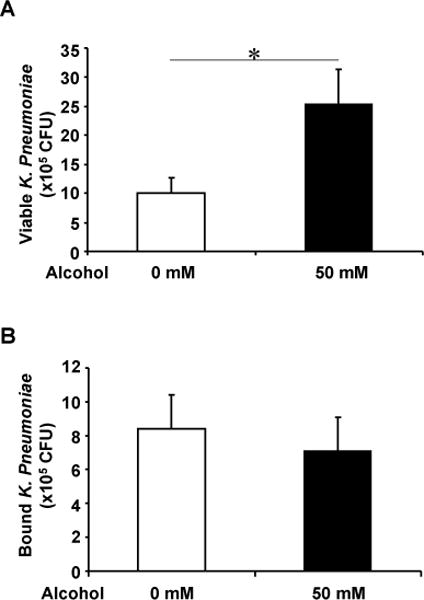
A) Polarized NHBEs were treated with 0 and 50 mM of basolateral alcohol for 24 hours and apically challenged with K. pneumoniae (MOI= ~10). The epithelia were lysed and bacterial supernatants were diluted and plated on agar plates for 24 hours. The colonies were counted to estimate the viable colonies. Alcohol presence promoted the number of colonies on airway epithelia. B) NHBEs were exposed to 0 and 50 mM of basolateral alcohol for 24 hours and then apically applied with 14C-labelled K. pneumoniae for an hour. The cells were washed and lysed to count radioactivity. Bound bacterial numbers were determined by comparing the cell-retaining radioactivity with a pre-established standard curve with various bacterial numbers. As demonstrated, alcohol did not alter the number of K. pneumoniae bound to NHBEs. Asterisk denotes statistically significant difference (p≤0.05, n=4–6).
Bacterial binding to airway epithelium is the initial step for pathogen-host interaction. To study the effect of alcohol on bacterial binding to airway epithelium, polarized NHBE cells were basolaterally exposed for 24 hours to 0 and 50 mM of alcohol and apically applied with 14C-labelled K. pneumoniae (5×107 CFU)) for 1 hour. After intense washes, these cells were lysed with 0.1% Triton-X 100. The number of bacteria that remained bound to the epithelia was calculated by measuring the 14C-label and compared with an established standard curve with a known number of bacteria. The presence or absence of alcohol did not significantly change the number of bacteria that were bound to the NHBE cell monolayer (Fig 4B).
Alcohol suppresses human airway epithelial cytokine release under apical bacterial challenge
Cytokine secretion is an important property of airway epithelia to modulate innate and adaptive immunities against infections. To investigate how alcohol affects cytokine secretion by polarized airway epithelia under apical bacterial challenge, we treated the cultured epithelia basolaterally with 0 or 50 mM alcohol and then challenged apically with live K. pneumoniae (5×107 CFU) for 24 hours. The basal media were collected for cytokine measurement by cytokine bioplex assays. As displayed in Table 1, at baseline the cultured epithelia without any treatment secreted negligible levels of IL-2, IL-4, GM-CSF, IFN-γ, TNF-α and IL-10, but did produce IL-6 (767.66 ± 294.89 pg/ml) and IL-8 (12905.17 ± 1915.45 pg/ml). Apical challenge with live bacteria significantly increased the secretion of IL-6, IL-2, GM-CSF, IL-4, IFN-γ and TNF-α by ~4.0, ~3.5, ~3.3, ~4.9, ~3.9 and ~5.9 fold, correspondingly (Fig. 5). Basolateral alcohol exposure decreased the bacterial-stimulated cytokine response. These data show that acute exposure of clinically relevant concentrations of alcohol significantly decreased the levels of inflammatory mediators.
Table 1.
Base-level Secretion of Eight Cytokines
| BY BIO-PLEX CYTOKINE ASSAYS*
| |
|---|---|
| Cytokine | Concentration in Basal Media (pg/ml) |
| IL-2 | 7.66 ± 1.52 |
| TNF-α | 9.33 ± 2.51 |
| IL-4 | 5.66 ± 1.51 |
| IL-6 | 767.66 ± 294.89 |
| IL-8 | 12905.17 ± 1915.45 |
| GM-CSF | 14.33 ± 2.30 |
| IFN-γ | 11.00 ± 2.64 |
Polarized human airway epithelia were cultured in newly replaced basal media at the air-liquid interface for 24 hours. The collected basal media were assayed for 8 different human cytokines. The detected cytokine levels represent the base level secretion by the cells. The data were averaged of triplicates.
Fig. 5. Alcohol effect on epithelial basolateral cytokine release under apical bacterial challenge.
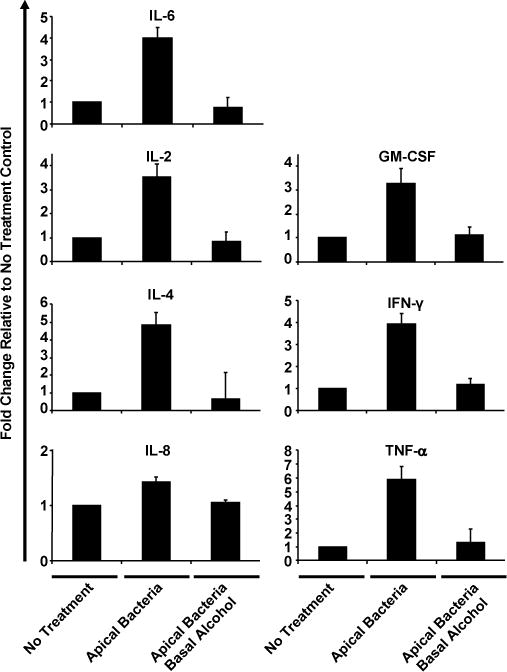
NHBE cells were grown at the air-liquid interface and challenged with live K. pneumonia apically in the presence (50 mM) or absence (0 mM) of basolateral alcohol for 24 hours. Eight different cytokines in the basolateral media were estimated by cytokine Bio-plex assay. Data are expressed as fold changes as compared to the base levels of cytokines secreted by the no treatment control. The absolute amount of the base level of each cytokine is presented in Table 1.
Adenosine receptor activation partially reverses alcohol suppressed inflammatory cytokine release
Extracellular adenosine, released from cells or converted from released adenine nucleotides, interacts with the cell surface adenosine receptors [42–43]. Such a signaling pathway plays a critical role in wound healing, matrix production and regulation of inflammation [44]. Previous work with airway epithelia has established that adenosine promotes release of IL-6 and other inflammatory cytokines [45–47]. Adenosine receptor agonists like NECA induce inflammatory cytokine release in different cell types [48]. We hypothesized that adenosine receptor activation in airway epithelia might attenuate the immune suppressive effects of alcohol during bacterial challenge. To test the hypothesis, polarized NHBEs were treated with 0 and 50 mM of basolateral alcohol for 3 hours followed by an apical challenge with live K. pneumoniae (5×107 CFU) for 6 more hours in the presence of apical adenosine (100 μM) or N-ethylcarboxamidoadenosine (NECA, 10 μM). Adenosine is a natural ligand for adenosine receptors and NECA a potent adenosine receptor agonist. The basolateral media were collected and cytokine release was estimated by multi-cytokine assay. Data presented in Figure 6 confirmed that in the absence of bacterial challenge NHBE cells produced negligible levels of IL-6, TNF-α and MCP-1. Apical bacterial application significantly stimulated production of these three cytokines. Alcohol application significantly inhibited the response on all three cytokines. The suppressive effects of alcohol on IL-6 and TNF-α were attenuated by adenosine and NECA. Neither agent had any effect on MCP-1.
Fig. 6. Adenosine receptor agonists partially restore alcohol-suppressed cytokines.
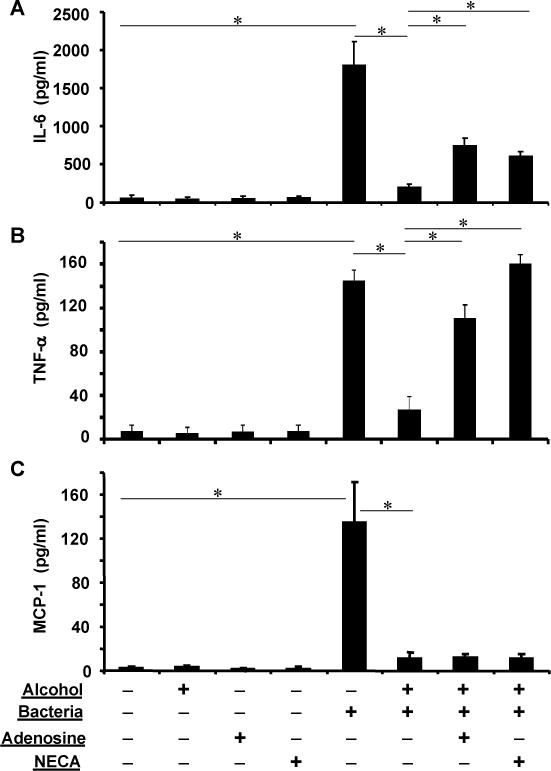
Polarized epithelia were exposed to 0 or 50 mM of alcohol basolaterally and challenged with K. pneumoniae apically in presence of adenosine (100 μM) or NECA (10 μM). Bacterial challenge significantly elevated IL-6, TNF-α and MCP-1 release. Alcohol blunted such an increase. However, adenosine and NECA partially restored the alcohol-suppressed IL-6 (A) and almost completely restored the alcohol-suppressed TNF-α (B). In contrast, the adenosine receptor agonists had no effect on the alcohol-suppressed MCP-1. Asterisks denote statistically significant difference (p≤0.05, n=4–6).
Discussion
Human lungs are exposed to a large number of pathogens daily. However, respiratory infections are not common, indicating the presence of an efficient host defense system at the mucosal surface of the lung. Airway epithelial cells are positioned to encounter any inhaled microbes first, thus playing a crucial role in sensing microbial presence, initiating and mobilizing effective host defense. Even though it has long been recognized that alcoholics are susceptible to lung infections, the mechanisms underlying the alcohol-caused immune deficiency in the lung are not fully understood. Evidence indicates that the impact of alcohol on lung airway functions is dependent on alcohol concentration, duration and route of exposure [2, 49]. The volatility of alcohol renders it freely diffusible from the bronchial circulation directly through the airway epithelia into the airway lumen. Vaporized alcohol can deposit back into the airway lining fluid to act on the cells. Thus, using a physiologically relevant experimental system to closely mimic the natural alcohol airway exposure is critical to addressing these alcohol-related questions. In the current report, we used the air-liquid interface culture model to replicate the physiological mode of alcohol exposure and live bacterial infection. This system has unique features including 1) direct basolateral contact and indirect apical vapor exposure of alcohol, and 2) apical live bacterial infection and basolateral release of cytokines. This air-liquid interface culture system has been widely used in other research fields to investigate pulmonary epithelial properties and lung disease pathogenesis [36, 50–52]. Using the culture, Gross and colleagues found β2-aganists enhance host defense against bacterial infection of the cultured normal and asthmatic airway epithelia [9]. However, to our best knowledge, no alcohol studies to date have been conducted with this model. By using this culture model, we have made several interesting observations. First, apical bacterial challenge enhanced the epithelial barrier function. Even though the molecular mechanism is not clear, we postulate that bacterial infection stimulates the epithelia to strengthen their self-defense measures including reinforcing their cell-cell junctions. Future studies are warranted to examine the mechanisms underlying this phenomenon. Second, basolateral exposure of clinically relevant levels of alcohol did not significantly change epithelial integrity and permeability. This result is different from what was published previously [40]. Simet and colleagues measured the resistance of human airway epithelial cells cultured in submersion with varied doses of alcohol. They found that alcohol-treated cells had less resistance as compared to the non-alcohol-treated cells. According to their data, the cells gained resistance over culture time, indicating the submerged cultures were not fully differentiated. We believe that the disparity between the two studies may reflect the difference of the two culture systems. Third, basolateral exposure of physiologically relevant levels of alcohol facilitated apical bacterial growth. Such an increase in bacterial viability suggests that clinically relevant levels of alcohol might enervate the antimicrobial capacity of airway epithelia or that alcohol as a carbon source facilitates bacterial growth or both.
In addition to serving as a physical barrier that defends against invasion of microbial pathogens, airway epithelia respond actively to infections by secretion of immune modulators such as cytokines/chemokines. This is a normal physiological response essential for airway damage control and functional restoration. Mounting evidence indicates that inflammatory cytokine production varies by acute or chronic alcohol exposure and by cell types [17, 49, 53–55]. Acute alcohol inhibits induction of pro-inflammatory mediators, such as TNF-α, IL-1, IL-6 and IL-8, partially through the Toll-like receptor and NF-κB signaling pathways [56–57]. Unfortunately, lungs are often a target of multiple concurrent insults, for example, alcohol and infection or alcohol and cigarette. These factors act combinatorially on the cells. It was reported that pulmonary cytokine profile differs in the setting of alcohol use disorders and cigarette smoking [58]. In the current report, we found that challenging the cultured human airway epithelia apically with the live bacteria elicited a robust cytokine/chemokine production basolaterally, including IL-6, IL-2, GM-CSF, IL-4, IFN-γ, TNF-α and IL-8. Cytokine/chemokine production was decreased by acute alcohol treatment. It appears that alcohol can negatively modulate both Th1 and Th2 responses indiscriminately. IL-8 is a major chemokine attracting neutrophils to inflammatory sites. Previous reports demonstrate that rodent IL-8 homologues such as macrophage inflammatory protein 2 (MIP-2) and cytokine-induced neutrophil chemoattractant (CINC) are significantly suppressed by alcohol [22, 26], which retards neutrophil recruitment to bacteria-challenged lungs. GM-CSF is critical to alveolar macrophage differentiation and microbicidal function [59]. Alcohol-suppression of GM-CSF would impair lung phagocytic function. Moreover, IL-6 and TNF-α are key cytokines activate NF-κB and STAT3 signaling hubs to mount an effective lung innate immunity [60]. Therefore, alcohol-depressed cytokine/chemokine response from airway epithelia to bacterial challenge would compromise mobilization and orchestration of lung host defense, contributing to lung infections.
Adenosine is a signaling molecule that is generated at sites of tissue injury and inflammation and acts as a potent modulator in wound healing and inflammatory regulation by engaging the adenosine receptors of the G protein-coupled receptor family [61]. Adenosine modulates inflammation in different cells through selective production of proinflammatory or anti-inflammatory cytokines. For instance, adenosine stimulates the release of proinflammatory IL-6 and IL-8 by mast cells and neutrophils [46, 62] and inhibits IL-12 production by monocytes [63]. Adenosine promotes IL-6 release in airway epithelial cells and in mouse lungs [45]. Attenuation of chronic pulmonary inflammation occurs in A2B adenosine receptor knockout mice [64]. Further, adenosine levels are elevated in the lungs of patients with chronic lung diseases [65–66], which correlate with the extents of inflammation. In contrast, lowering elevated adenosine levels in models of chronic lung disease leads to quenching of inflammation [67–68]. Based on the fact that adenosine can provoke lung immune reactions, we tested in the current report if adenosine could overcome alcohol suppression of cytokine/chemokine production in airway epithelial cells. The results show that adenosine and NECA restored the alcohol-suppressed TNF-α and IL-6 burst upon bacterial challenge. The data suggest that the adenosine-adenosine receptor signaling pathway is not blunted by alcohol and can be employed to potentially counteract alcohol-induced cytokine suppression. Because IL-6 and TNF-α are two key cytokines in promoting inflammation, such an action of adenosine and NECA may have clinical applications in rescuing the alcohol-suppressed airway cytokine response to infection.
In summary, acute exposure of alcohol at the clinically relevant levels does not impair airway epithelial barrier function under the air-liquid interface culture condition, but compromises the normal inflammatory cytokine response to infections. Such an alternation by alcohol may pre-condition alcohol abusers to airborne infections. Adenosine receptor agonists could be promising candidates to boost inflammatory cytokine production which is suppressed by alcohol.
Acknowledgments
This work was supported by National Institutes of Health Grants 5R21AA16118 (G.W.), P60AA09803 (S.N.), and by a pilot grant to G.W. from the Dean’s Research fund of the LSU Health Sciences Center in New Orleans.
Footnotes
Disclosures
No conflicts of interest, financial or otherwise, are declared by the authors.
References
- 1.Nelson S, Kolls JK. Alcohol, host defence and society. Nat Rev Immunol. 2002;2:205–9. doi: 10.1038/nri744. [DOI] [PubMed] [Google Scholar]
- 2.Sisson JH. Alcohol and airways function in health and disease. Alcohol. 2007;41:293–307. doi: 10.1016/j.alcohol.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang P, Bagby GJ, Happel KI, Raasch CE, Nelson S. Alcohol abuse, immunosuppression, and pulmonary infection. Curr Drug Abuse Rev. 2008;1:56–67. doi: 10.2174/1874473710801010056. [DOI] [PubMed] [Google Scholar]
- 4.Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. 2003;8:432–46. doi: 10.1046/j.1440-1843.2003.00493.x. [DOI] [PubMed] [Google Scholar]
- 5.Voynow JA, Rubin BK. Mucins, mucus, and sputum. Chest. 2009;135:505–12. doi: 10.1378/chest.08-0412. [DOI] [PubMed] [Google Scholar]
- 6.Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. 2006;354:241–50. doi: 10.1056/NEJMoa043891. [DOI] [PubMed] [Google Scholar]
- 7.Schutte BC, McCray PB., Jr [beta]-defensins in lung host defense. Annu Rev Physiol. 2002;64:709–48. doi: 10.1146/annurev.physiol.64.081501.134340. [DOI] [PubMed] [Google Scholar]
- 8.Bals R, Hiemstra PS. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur Respir J. 2004;23:327–33. doi: 10.1183/09031936.03.00098803. [DOI] [PubMed] [Google Scholar]
- 9.Gross CA, Bowler RP, Green RM, Weinberger AR, Schnell C, Chu HW. beta2-agonists promote host defense against bacterial infection in primary human bronchial epithelial cells. BMC Pulm Med. 2010;10:30. doi: 10.1186/1471-2466-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Bartlett JA, Di ME, Bomberger JM, Chan YR, Gakhar L, Mallampalli RK, McCray PB, Jr, Di YP. SPLUNC1/BPIFA1 contributes to pulmonary host defense against Klebsiella pneumoniae respiratory infection. Am J Pathol. 2013;182:1519–31. doi: 10.1016/j.ajpath.2013.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sibille Y, Reynolds HY. Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am Rev Respir Dis. 1990;141:471–501. doi: 10.1164/ajrccm/141.2.471. [DOI] [PubMed] [Google Scholar]
- 12.Nelson S, Summer WR. Innate immunity, cytokines, and pulmonary host defense. Infect Dis Clin North Am. 1998;12:555–67. vii. doi: 10.1016/s0891-5520(05)70198-7. [DOI] [PubMed] [Google Scholar]
- 13.Reynolds HY. Modulating airway defenses against microbes. Curr Opin Pulm Med. 2002;8:154–65. doi: 10.1097/00063198-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Burns AR, Smith CW, Walker DC. Unique structural features that influence neutrophil emigration into the lung. Physiol Rev. 2003;83:309–36. doi: 10.1152/physrev.00023.2002. [DOI] [PubMed] [Google Scholar]
- 15.Evans SE, Xu Y, Tuvim MJ, Dickey BF. Inducible innate resistance of lung epithelium to infection. Annu Rev Physiol. 2010;72:413–35. doi: 10.1146/annurev-physiol-021909-135909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polito AJ, Proud D. Epithelia cells as regulators of airway inflammation. J Allergy Clin Immunol. 1998;102:714–8. doi: 10.1016/s0091-6749(98)70008-9. [DOI] [PubMed] [Google Scholar]
- 17.Crews FT, Bechara R, Brown LA, Guidot DM, Mandrekar P, Oak S, Qin L, Szabo G, Wheeler M, Zou J. Cytokines and alcohol. Alcohol Clin Exp Res. 2006;30:720–30. doi: 10.1111/j.1530-0277.2006.00084.x. [DOI] [PubMed] [Google Scholar]
- 18.Twigg HL., 3rd Pulmonary host defenses. J Thorac Imaging. 1998;13:221–33. doi: 10.1097/00005382-199810000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Wyatt TA, Sisson JH. Chronic ethanol downregulates PKA activation and ciliary beating in bovine bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L575–81. doi: 10.1152/ajplung.2001.281.3.L575. [DOI] [PubMed] [Google Scholar]
- 20.Wyatt TA, Gentry-Nielsen MJ, Pavlik JA, Sisson JH. Desensitization of PKA-stimulated ciliary beat frequency in an ethanol-fed rat model of cigarette smoke exposure. Alcohol Clin Exp Res. 2004;28:998–1004. doi: 10.1097/01.ALC.0000130805.75641.F4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elliott MK, Sisson JH, Wyatt TA. Effects of cigarette smoke and alcohol on ciliated tracheal epithelium and inflammatory cell recruitment. Am J Respir Cell Mol Biol. 2007;36:452–9. doi: 10.1165/rcmb.2005-0440OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boe DM, Nelson S, Zhang P, Quinton L, Bagby GJ. Alcohol-induced suppression of lung chemokine production and the host defense response to Streptococcus pneumoniae. Alcohol Clin Exp Res. 2003;27:1838–45. doi: 10.1097/01.ALC.0000095634.82310.53. [DOI] [PubMed] [Google Scholar]
- 23.NIAAA. 10th Special Report to the US Congress on Alcohol and Health. National Institute on Alcohol Abuse and Alcoholism Publications; 2000. Jun, [Google Scholar]
- 24.Schmidt W, De Lint J. Causes of death of alcoholics. Q J Stud Alcohol. 1972;33:171–85. [PubMed] [Google Scholar]
- 25.Martin LD, Rochelle LG, Fischer BM, Krunkosky TM, Adler KB. Airway epithelium as an effector of inflammation: molecular regulation of secondary mediators. Eur Respir J. 1997;10:2139–46. doi: 10.1183/09031936.97.10092139. [DOI] [PubMed] [Google Scholar]
- 26.Boe DM, Nelson S, Zhang P, Bagby GJ. Acute ethanol intoxication suppresses lung chemokine production following infection with Streptococcus pneumoniae. J Infect Dis. 2001;184:1134–42. doi: 10.1086/323661. [DOI] [PubMed] [Google Scholar]
- 27.Happel KI, Rudner X, Quinton LJ, Movassaghi JL, Clark C, Odden AR, Zhang P, Bagby GJ, Nelson S, Shellito JE. Acute alcohol intoxication suppresses the pulmonary ELR-negative CXC chemokine response to lipopolysaccharide. Alcohol. 2007;41:325–33. doi: 10.1016/j.alcohol.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang P, Bagby GJ, Stoltz DA, Summer WR, Nelson S. Granulocyte colony-stimulating factor modulates the pulmonary host response to endotoxin in the absence and presence of acute ethanol intoxication. J Infect Dis. 1999;179:1441–8. doi: 10.1086/314763. [DOI] [PubMed] [Google Scholar]
- 29.Jerrells TR, Pavlik JA, DeVasure J, Vidlak D, Costello A, Strachota JM, Wyatt TA. Association of chronic alcohol consumption and increased susceptibility to and pathogenic effects of pulmonary infection with respiratory syncytial virus in mice. Alcohol. 2007;41:357–69. doi: 10.1016/j.alcohol.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pruett SB, Schwab C, Zheng Q, Fan R. Suppression of innate immunity by acute ethanol administration: a global perspective and a new mechanism beginning with inhibition of signaling through TLR3. J Immunol. 2004;173:2715–24. doi: 10.4049/jimmunol.173.4.2715. [DOI] [PubMed] [Google Scholar]
- 31.Pruett SB, Zheng Q, Fan R, Matthews K, Schwab C. Acute exposure to ethanol affects Toll-like receptor signaling and subsequent responses: an overview of recent studies. Alcohol. 2004;33:235–9. doi: 10.1016/j.alcohol.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Pruett SB, Zheng Q, Fan R, Matthews K, Schwab C. Ethanol suppresses cytokine responses induced through Toll-like receptors as well as innate resistance to Escherichia coli in a mouse model for binge drinking. Alcohol. 2004;33:147–55. doi: 10.1016/j.alcohol.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 33.Pruett BS, Pruett SB. An explanation for the paradoxical induction and suppression of an acute phase response by ethanol. Alcohol. 2006;39:105–10. doi: 10.1016/j.alcohol.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey KL, Wyatt TA, Romberger DJ, Sisson JH. Alcohol functionally upregulates Toll-like receptor 2 in airway epithelial cells. Alcohol Clin Exp Res. 2009;33:499–504. doi: 10.1111/j.1530-0277.2008.00862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhatty M, Jan BL, Tan W, Pruett SB, Nanduri B. Role of acute ethanol exposure and TLR4 in early events of sepsis in a mouse model. Alcohol. 2011;45:795–803. doi: 10.1016/j.alcohol.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karp PH, Moninger TO, Weber SP, Nesselhauf TS, Launspach JL, Zabner J, Welsh MJ. An in vitro model of differentiated human airway epithelia. Methods for establishing primary cultures. Methods Mol Biol. 2002;188:115–37. doi: 10.1385/1-59259-185-X:115. [DOI] [PubMed] [Google Scholar]
- 37.Gomez M, Raju SV, Viswanathan A, Painter RG, Bonvillain R, Byrne P, Nguyen DH, Bagby GJ, Kolls JK, Nelson S, Wang G. Ethanol upregulates glucocorticoid-induced leucine zipper expression and modulates cellular inflammatory responses in lung epithelial cells. J Immunol. 2010;184:5715–22. doi: 10.4049/jimmunol.0903521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang G, Davidson BL, Melchert P, Slepushkin VA, van Es HH, Bodner M, Jolly DJ, McCray PB., Jr Influence of cell polarity on retrovirus-mediated gene transfer to differentiated human airway epithelia. J Virol. 1998;72:9818–26. doi: 10.1128/jvi.72.12.9818-9826.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang G, Zabner J, Deering C, Launspach J, Shao J, Bodner M, Jolly DJ, Davidson BL, McCray PB., Jr Increasing epithelial junction permeability enhances gene transfer to airway epithelia In vivo. Am J Respir Cell Mol Biol. 2000;22:129–38. doi: 10.1165/ajrcmb.22.2.3938. [DOI] [PubMed] [Google Scholar]
- 40.Simet SM, Wyatt TA, DeVasure J, Yanov D, Allen-Gipson D, Sisson JH. Alcohol increases the permeability of airway epithelial tight junctions in Beas-2B and NHBE cells. Alcohol Clin Exp Res. 2012;36:432–42. doi: 10.1111/j.1530-0277.2011.01640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teplin LA, Abram KM, Michaels SK. Blood alcohol level among emergency room patients: a multivariate analysis. J Stud Alcohol. 1989;50:441–7. doi: 10.15288/jsa.1989.50.441. [DOI] [PubMed] [Google Scholar]
- 42.Spicuzza L, Di Maria G, Polosa R. Adenosine in the airways: implications and applications. Eur J Pharmacol. 2006;533:77–88. doi: 10.1016/j.ejphar.2005.12.056. [DOI] [PubMed] [Google Scholar]
- 43.Button B, Boucher RC. Role of mechanical stress in regulating airway surface hydration and mucus clearance rates. Respir Physiol Neurobiol. 2008;163:189–201. doi: 10.1016/j.resp.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valls MD, Cronstein BN, Montesinos MC. Adenosine receptor agonists for promotion of dermal wound healing. Biochem Pharmacol. 2009;77:1117–24. doi: 10.1016/j.bcp.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun Y, Wu F, Sun F, Huang P. Adenosine promotes IL-6 release in airway epithelia. J Immunol. 2008;180:4173–81. doi: 10.4049/jimmunol.180.6.4173. [DOI] [PubMed] [Google Scholar]
- 46.Sitaraman SV, Merlin D, Wang L, Wong M, Gewirtz AT, Si-Tahar M, Madara JL. Neutrophil-epithelial crosstalk at the intestinal lumenal surface mediated by reciprocal secretion of adenosine and IL-6. J Clin Invest. 2001;107:861–9. doi: 10.1172/JCI11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun CX, Zhong H, Mohsenin A, Morschl E, Chunn JL, Molina JG, Belardinelli L, Zeng D, Blackburn MR. Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest. 2006;116:2173–2182. doi: 10.1172/JCI27303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhong H, Belardinelli L, Maa T, Feoktistov I, Biaggioni I, Zeng D. A(2B) adenosine receptors increase cytokine release by bronchial smooth muscle cells. Am J Respir Cell Mol Biol. 2004;30:118–25. doi: 10.1165/rcmb.2003-0118OC. [DOI] [PubMed] [Google Scholar]
- 49.Goral J, Karavitis J, Kovacs EJ. Exposure-dependent effects of ethanol on the innate immune system. Alcohol. 2008;42:237–47. doi: 10.1016/j.alcohol.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adler KB, Cheng PW, Kim KC. Characterization of guinea pig tracheal epithelial cells maintained in biphasic organotypic culture: cellular composition and biochemical analysis of released glycoconjugates. Am J Respir Cell Mol Biol. 1990;2:145–54. doi: 10.1165/ajrcmb/2.2.145. [DOI] [PubMed] [Google Scholar]
- 51.Adler KB, Fischer BM, Wright DT, Cohn LA, Becker S. Interactions between respiratory epithelial cells and cytokines: relationships to lung inflammation. Ann N Y Acad Sci. 1994;725:128–45. doi: 10.1111/j.1749-6632.1994.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 52.Adler KB, Li Y. Airway epithelium and mucus: intracellular signaling pathways for gene expression and secretion. Am J Respir Cell Mol Biol. 2001;25:397–400. doi: 10.1165/ajrcmb.25.4.f214. [DOI] [PubMed] [Google Scholar]
- 53.Mandrekar P, Bala S, Catalano D, Kodys K, Szabo G. The opposite effects of acute and chronic alcohol on lipopolysaccharide-induced inflammation are linked to IRAK-M in human monocytes. J Immunol. 2009;183:1320–7. doi: 10.4049/jimmunol.0803206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown LA, Cook RT, Jerrells TR, Kolls JK, Nagy LE, Szabo G, Wands JR, Kovacs EJ. Acute and chronic alcohol abuse modulate immunity. Alcohol Clin Exp Res. 2006;30:1624–31. doi: 10.1111/j.1530-0277.2006.00195.x. [DOI] [PubMed] [Google Scholar]
- 55.Happel KI, Nelson S. Alcohol, immunosuppression, and the lung. Proc Am Thorac Soc. 2005;2:428–32. doi: 10.1513/pats.200507-065JS. [DOI] [PubMed] [Google Scholar]
- 56.Pruett SB, Fan R. Ethanol inhibits LPS-induced signaling and modulates cytokine production in peritoneal macrophages in vivo in a model for binge drinking. BMC Immunol. 2009;10:49. doi: 10.1186/1471-2172-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mandrekar P, Jeliazkova V, Catalano D, Szabo G. Acute alcohol exposure exerts anti-inflammatory effects by inhibiting IkappaB kinase activity and p65 phosphorylation in human monocytes. J Immunol. 2007;178:7686–93. doi: 10.4049/jimmunol.178.12.7686. [DOI] [PubMed] [Google Scholar]
- 58.Burnham EL, Kovacs EJ, Davis CS. Pulmonary cytokine composition differs in the setting of alcohol use disorders and cigarette smoking. Am J Physiol Lung Cell Mol Physiol. 2013;304:L873–82. doi: 10.1152/ajplung.00385.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ballinger MN, Paine R, 3rd, Serezani CH, Aronoff DM, Choi ES, Standiford TJ, Toews GB, Moore BB. Role of granulocyte macrophage colony-stimulating factor during gram-negative lung infection with Pseudomonas aeruginosa. Am J Respir Cell Mol Biol. 2006;34:766–74. doi: 10.1165/rcmb.2005-0246OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinton LJ, Mizgerd JP. NF-kappaB and STAT3 signaling hubs for lung innate immunity. Cell Tissue Res. 2011;343:153–65. doi: 10.1007/s00441-010-1044-y. [DOI] [PubMed] [Google Scholar]
- 61.Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–52. [PMC free article] [PubMed] [Google Scholar]
- 62.Feoktistov I, Biaggioni I. Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. An enprofylline-sensitive mechanism with implications for asthma. J Clin Invest. 1995;96:1979–86. doi: 10.1172/JCI118245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Link AA, Kino T, Worth JA, McGuire JL, Crane ML, Chrousos GP, Wilder RL, Elenkov IJ. Ligand-activation of the adenosine A2a receptors inhibits IL-12 production by human monocytes. J Immunol. 2000;164:436–42. doi: 10.4049/jimmunol.164.1.436. [DOI] [PubMed] [Google Scholar]
- 64.Zaynagetdinov R, Ryzhov S, Goldstein AE, Yin H, Novitskiy SV, Goleniewska K, Polosukhin VV, Newcomb DC, Mitchell D, Morschl E, Zhou Y, Blackburn MR, Peebles RS, Jr, Biaggioni I, Feoktistov I. Attenuation of chronic pulmonary inflammation in A2B adenosine receptor knockout mice. Am J Respir Cell Mol Biol. 2010;42:564–71. doi: 10.1165/rcmb.2008-0391OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Driver AG, Kukoly CA, Ali S, Mustafa SJ. Adenosine in bronchoalveolar lavage fluid in asthma. Am Rev Respir Dis. 1993;148:91–7. doi: 10.1164/ajrccm/148.1.91. [DOI] [PubMed] [Google Scholar]
- 66.Huszar E, Vass G, Vizi E, Csoma Z, Barat E, Molnar Vilagos G, Herjavecz I, Horvath I. Adenosine in exhaled breath condensate in healthy volunteers and in patients with asthma. Eur Respir J. 2002;20:1393–8. doi: 10.1183/09031936.02.00005002. [DOI] [PubMed] [Google Scholar]
- 67.Blackburn MR, Lee CG, Young HW, Zhu Z, Chunn JL, Kang MJ, Banerjee SK, Elias JA. Adenosine mediates IL-13-induced inflammation and remodeling in the lung and interacts in an IL-13-adenosine amplification pathway. J Clin Invest. 2003;112:332–44. doi: 10.1172/JCI16815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma B, Blackburn MR, Lee CG, Homer RJ, Liu W, Flavell RA, Boyden L, Lifton RP, Sun CX, Young HW, Elias JA. Adenosine metabolism and murine strain-specific IL-4-induced inflammation, emphysema, and fibrosis. J Clin Invest. 2006;116:1274–83. doi: 10.1172/JCI26372. [DOI] [PMC free article] [PubMed] [Google Scholar]