

Abstract
Differentiation therapy is an attractive treatment for osteosarcoma (OS). CD99 is a cell surface molecule expressed in mesenchymal stem cells and osteoblasts that is maintained during osteoblast differentiation while lost in OS. Herein, we show that whenever OS cells regain CD99, they become prone to reactivate the terminal differentiation program. In differentiating conditions, CD99-transfected OS cells express osteocyte markers, halt proliferation, and largely die by apoptosis, resembling the fate of mature osteoblasts. CD99 induces ERK activation, increasing its membrane-bound/cytoplasmic form rather than affecting its nuclear localization. Through cytoplasmic ERK, CD99 promotes activity of the main osteogenic transcriptional factors AP1 and RUNX2, which in turn enhance osteocalcin and p21WAF1/CIP1, leading to G0/G1 arrest. These data underscore the alternative positions of active ERK into distinct subcellular compartments as key events for determining OS fate. © 2014 The Authors. Journal of Bone and Mineral Research published by Wiley Periodicals, Inc. on behalf of the American Society for Bone and Mineral Research.
Keywords: CD99, OSTEOSARCOMA, OSTEOBLAST DIFFERENTIATION, MAPK SIGNALING, RUNX2
Introduction
Osteosarcoma (OS) is the most frequent primary malignancy arising from bone in children and young adults. Thus, in spite of the relative rarity (approximately 400 new cases per year in the United States), its social impact is particularly relevant. Although targeted therapies have revolutionized the therapeutic approach to carcinomas, translation of selective inhibitors—ranging from small molecules to monoclonal antibodies—has been slow or poorly effective in the treatment of OS.(1) The presence of complicated structural and numeric genomic rearrangements in OS cells, redundancy of autocrine loops and kinase activations,(2) as well as the rarity of the tumor that hinders extensive understanding of the genetic and biologic features of OS have precluded any effective application of targeted agents. It is thus not surprising that the only drug approved for the treatment of OS patients in the recent years is indeed the immunomodulator mifamurtide (liposomal muramyl tripeptide phosphatidyl ethanolamine [L-MTP-PE]), which activates macrophages and monocytes and presents broad, although poorly characterized, mechanisms of action.(3) Despite the considerable interest, debate about whether the addition of L-MTP-PE to standard chemotherapy regimens truly results in a proven benefit is ongoing, and patients with OS still substantially lack new effective drugs. Systemic treatments continue to be based on the use of methotrexate, doxorubicin, and cisplatin, with possible inclusion of ifosfamide or etoposide; thus, patients achieved a 65% to 75% survival rate(4) by paying the price of severe toxicity and high risk of life-threatening late side events. Survivors of sarcomas, together with those of brain tumors, have the lowest health quality-of-life scores(5) and high cure costs, owing to limb-salvage procedure, prolonged dose-dense chemotherapy (around 1 year), and development of chronic severe pathologies. Thus, even in the best conditions of localized tumors at diagnosis, we need complementary therapeutic approaches to help keep treatment-related toxicity to a minimum and to optimize systemic-disease control.
OS is characterized by proliferation of malignant cells with osteoblastic features and defined by the presence of uncalcified bone matrix (osteoid). Transformation is thought to occur in mesenchymal stem cells (MSCs) and/or osteoprogenitors in any phases of their differentiation(6,7) and generates a block in normal development coupled with unregulated proliferation processes.(8) The loss of differentiation is a widespread biological feature of OS and has a strong prognostic significance; conventional OSs are high grade and poorly differentiated lesions. However, well-differentiated variants exist, and these tumors are classified as low-grade OSs, showing a much more favorable clinical history.(9) Differentiation of high-grade OS cells can be stimulated in vitro and leads to reversal of malignancy,(10) confirming that differentiation may serve as a critical clinical and biologic transition of a fatal cancer into one more amenable to management or to treatments. Thus, as for leukemia, differentiative therapy might also be a valid option for sarcomas, if essential recruited molecular pathways were understood. In this article, we focus on CD99, a cell surface molecule that we have demonstrated to serve as a new crucial modulator of OS malignancy.(11,12) CD99 is a 32-kDa transmembrane glycoprotein encoded by the pseudoautosomal MIC2 gene,(13–16) which contains an extracellular domain, followed by a transmembrane domain and a short 36-amino acid intracytoplasmic domain. Despite CD99 having a key role in several biological processes (including cell adhesion, migration, and apoptosis, differentiation of T cells and thymocytes, diapedesis of lymphocytes to inflamed vascular endothelium, maintenance of cellular morphology, and regulation of intracellular membrane protein trafficking),(17–19) its functions appeared to be cell-context dependent, and we still know little about how the molecule works. Although the mouse homologue of CD99 (designated D4) seems to act through its ligand, the paired immunoglobulin-like type 2 receptor (PILR),(20) homophilic interactions among CD99 molecules seem to be required to trigger signaling of human CD99.(21) Broadly expressed in normal tissues, CD99 is differentially expressed in neoplasms, with a strong expression in some pediatric tumors, such as Ewing's sarcoma and acute lymphoblastic leukemia cells,(22,23) and a low, but broad, distribution in many other neoplastic cell types, including osteosarcoma.(11,12) Our previous work has shown that forced expression of CD99 in OS manages to increase cell adhesion and reactivate stop-migration signals that result in significant reduction of metastasis.(11,12,24) In this article, we analyzed the possible relationship between CD99 and osteoblastic differentiation. Sporadic information suggested a possible involvement of CD99 in bone pathophysiology; Bertaux and colleagues(25) pointed out a link between CD99 and the transcription factor RUNX2, which is the master gene of osteoblastogenesis. In addition, Hamilton and colleagues(26) found that supernatants from several cancer cell lines specifically downregulated CD99 on trabecular osteoblasts (AHTO-7 cells), while inducing osteoblast activation and proliferation. In this article, we provide evidence that CD99 is expressed in human MSCs and mature human osteoblasts and its expression increases during normal osteoblastogenesis and osteoblast maturation. When CD99 is restored in OS cells, the molecule switches cells from proliferation status to cell cycle withdrawal, favoring the achievement of a terminally osteoblast-differentiated phenotype. Cell death and expression of osteocyte markers accomplish the process. From a mechanistic point of view, we propose that CD99 increases MAPK/ERK signaling and recruits activated ERK to cell membrane/cytoplasm, where it may phosphorylate RUNX2, thus leading to increased activity of the master gene of osteoblastogenesis.
Materials and Methods
Cell lines
The OS cell lines U-2 OS and Saos-2 were obtained from American Type Culture Collection (Manassas, VA, USA). The IOR/OS7, IOR/OS9, IOR/OS14, IOR/OS15, IOR/OS17, and SARG cell lines were established at the Laboratory of Experimental Oncology, Rizzoli Orthopedic Institute (Bologna, Italy) and previously characterized.(27) All cell lines were tested for mycoplasma contamination every 3 months (last control June 2013) by PCR Mycoplasma detection Set (Takara Bio Inc., Shiga, Japan). Stable transfectants expressing CD99 were obtained from the Saos-2, IOR/OS7, or U-2 OS cell lines (Sa/CD99wt22, Sa/CD99wt36, OS7/CD99wt8, OS7/CD99wt89, and U2/CD99wt136) by using calcium-phosphate transfection method. Cell lines and transfectants were grown as previously described.(11) Human primary bone marrow–derived mesenchymal stem cells (hBM-MSCs) (Table 1) were isolated, cultured, and characterized as described.(27) Primary cultures of human osteoblasts were obtained from fragments of cortical and spongy trabecular bone and maintained in 10% FBS α-MEM for 2 weeks before analysis.(28) Human bone-derived cells converted into OS cells by overexpression of the MET oncogene(29) were also used.
Table 1.
Evaluation of CD99 Expression by Flow Cytometry in hBM-MSCs and Their Origin
| hBM-MSC | % CD99 | Log mean | Origin |
|---|---|---|---|
| hBM-MSC 85 | 99% | 77 | GCT of proximal femur |
| hBM-MSC 87 | 99% | 61 | Pigmented villonodular synovitis in right hip |
| hBM-MSC 91 | 74% | 78 | Isthmic spondylolisthesis |
| hBM-MSC 93 | 77% | 65 | Low-grade osteosarcoma in right distal femur |
| hBM-MSC 95 | 78% | 83 | Right buttock synovial sarcoma |
| hBM-MSC 96 | 99% | 107 | Chondromatosis in multiple exostosis |
| hBM-MSC 98 | 70% | 10 | Ewing's sarcoma in left iliac wing |
| hBM-MSC 102 | 99% | 166 | Delay of graft joining in left humerus |
| hBM-MSC 110 | 83% | 82 | Right proximal humerus bone cyst |
| hBM-MSC 111 | Negative | Left proximal humerus bone cyst | |
| hBM-MSC 112 | 74% | 75 | Heterotopic ossification in concussion |
| hBM-MSC 113 | 28% | 30 | Right proximal femur bone cyst |
| hBM-MSC 114 | 34% | 32 | Right proximal humerus bone cyst |
| hBM-MSC 115 | 75% | 54 | Left humerus diaphysis bone cyst |
| hBM-MSC 118 | Negative | Right humerus diaphysis bone cyst | |
| hBM-MSC 119 | Negative | Right proximal humerus bone cyst | |
| hBM-MSC 121 | 40% | 45 | Left acetabular chondrosarcoma |
| hBM-MSC1 24 | 46% | 25 | Revision of intercalary prosthesis of femur |
| hBM-MSC 128 | 84% | 30 | Right proximal humerus bone cyst |
| hBM-MSC 140 | 6% | 10 | Left proximal humerus bone cyst |
| hBM-MSC 141 | 18% | 16 | Left proximal humerus bone cyst |
| hBM-MSC 143 | Negative | Right L4 paravertebral neurinoma | |
| hBM-MSC 144 | 20% | 40 | Femur osteosarcoma |
| hBM-MSC 145 | 24% | 12 | Left gluteal leiomyosarcoma |
| hBM-MSC 196 | 97% | 79 | Right hip exostoses |
| hBM-MSC 194 | 99% | 58 | Left proximal humerus exostoses |
| hBM-MSC 198 | 99% | 57 | Right femur consolidation delay |
| hBM-MSC 204 | 98% | 61 | Left proximal humerus bone cyst |
| hBM-MSC 223 | 100 | 21 | Left knee osteochondritis |
| hBM-MSC 168 | 94% | 35 | Left proximal femur angiosarcoma |
| hBM-MSC 163 | 98% | 41 | Left proximal femur fibrous dysplasia |
| hBM-MSC 205 | 70% | 23 | Left proximal femur homoplastic graft consolidation delay |
| hBM-MSC 244 | 99% | 54 | Left tibia consolidation delay |
| hBM-MSC 249 | 99% | 61 | Left distal femur consolidation delay |
All the cultures were used within 5 in vitro passages.
Osteoblast differentiation
Four days after cell seeding, cells were exposed to specific osteogenic medium (IMDM for OS cells, or α-MEM for primary human osteoblasts and hBM-MSCs), supplemented with 2% fetal bovine serum (FBS), 5 mM β-glycerophosphate, and 50 µg/mL ascorbic acid (Sigma-Aldrich, St. Louis, MO, USA) and maintained in differentiative conditions for 21 to 28 days. Medium was renewed every 4 days. Cultures were harvested at various time points to collect total RNA or proteins. Osteoblast differentiation was verified by evaluation of specific markers by quantitative real-time PCR (qRT-PCR), by liver/bone/kidney alkaline phosphatase (ALP) activity, as well as bone mineralization. Total RNA from differentiated OS cell lines was extracted using the Trizol extraction kit (Life Technologies, Grand Island, NY, USA). qRT-PCR was performed as described.(11) All primers used are indicated in Supplemental Table S1. For a more general view on osteogenic differentiation of the OS model, the Human Osteogenesis RT2 Profiler PCR Array (Qiagen-SABiosciences, Valencia, CA, USA) was also used. ALP activity was assessed using p-nytrophenyl phosphate as a substrate (Boehringer Mannheim, Mannheim, Germany) as previously described.(30) To visualize bone mineralization in primary cultures as well as in OS cells, duplicate plates were stained with 2% AgNO3 (von Kossa staining) or with 40 mM Alizarin Red S (ARS staininig) (all from Sigma-Aldrich). To quantify bone mineralization assessed for CD99-overexpressing transfectants and the respective parental cell lines, ARS was extracted from the monolayer by incubation with 10% cetylpyridinium chloride (CPC; Sigma-Aldrich). The dye was then removed, and 200 µL was transferred to a 96-well plate for reading at 550 nm.
Cell growth and proliferation
Growth was monitored in differentiating conditions by methylene blue staining (Sigma-Aldrich), which exploits the ability of the dye to intercalate to DNA. Cells were fixed in 10% formalin and then exposed to a solution of 1% methylene blue in 0.01 M borate buffer. The extracted dye with 0.1 M HCl–absolute EtOH (1:1) was quantified by spectrophotometer reading (655 nm). Vital count was performed by using the vital dye Trypan Blue. Bromodeoxyuridine (BrdU) labeling was used to assess cell proliferation. Briefly, cells were exposed to 10 µM BrdU for 1 h before being fixed in 70% ethanol. After DNA denaturation and washing, cells were processed for indirect immunofluorescence staining using anti-BrdU MAb (1:4; Becton Dickinson, Milan, Italy) as a primary antibody. For immunohistochemistry, avidin–biotin–peroxidase procedure was used (Vector Laboratories, Burlingame, CA, USA). For cell cycle analysis, cells were stained with the anti-mouse FITC secondary antibody (1:100; Thermo Scientific, South Logan, OH, USA) and 20 µg/mL propidium iodide before flow cytometric analysis (FACSCalibur; Becton Dickinson).
Western blotting
Western blotting experiments were performed as previously described using total protein lysates or fractionated protein.(11,31) The following antibodies were used: anti-CD99 12E7 (1:4000; Dako, Carpinteria, CA, USA), anti-p21 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-ERK 1/2 (1:3000; Cell Signaling Technology, Beverly, MA, USA), anti-phospho-ERK 1/2 Thr202/Tyr204 (1:1000; Covance, Princeton, NJ, USA). Membranes were incubated with secondary anti-mouse or anti-rabbit antibodies conjugated to horseradish peroxidase (GE Healthcare, Piscataway, NJ, USA) and revealed by ECL Western blotting detection reagents (Euroclone, Milan, Italy). Loading control and purity of fractionated lysates were assessed by anti-GAPDH (1:3000; Santa Cruz Biotechnology) or anti-actin (1:200,000; Merck Millipore, Billerica, MA, USA), and anti-Lamin B (1:3000; Santa Cruz Biotechnology) after stripping.
CD99 expression
CD99 expression was evaluated at mRNA by gene expression profiling by using Illumina DNA chips and Illumina BeadArray Reader (Illumina Inc., San Diego, CA) as described.(6) CD99 expression was analyzed at protein level also by indirect immunofluorescence on a FACSCalibur flow cytometer. The O13 MAb (1:80; Covance) and anti-mouse FITC (1:100; Thermo Scientific) were used as primary and secondary antibodies, respectively.
Apoptosis analysis
The Tunel assay (ApoTag Plus Peroxidase In Situ Apoptosis Detection Kit, Millipore Corporation, Bedford, MA, USA) was used to evaluate apoptosis in adherent cells during differentiation according to the manufacturer's instructions.
Chromatin immunoprecipitation (ChIP) assay
Cells were collected in basal and differentiating conditions (at day 7 of differentiation). ChIP assay was performed as previously described.(19) The immuno-cleared chromatin was precipitated with anti-RUNX2 (M-70; Santa Cruz Biotechnology) antibody. Two microliters of initial preparations of soluble chromatin were amplified to control input DNA. The following pairs of primers were used to amplify RUNX2-containing promoters of target genes: OCN promoter (221 bp) 5'-GGGGGTCTCTGAGGAAGAGT-3' (forward) and 5'-GAATCTGCCAGGGCTATTTG-3' (reverse); p21WAF/CIP1 promoter (215 bp): 5'-GGTCAGGGGTGTGAGGTAGA-3' (forward) and 5'-CACAGGACTTTTGCCTCCTG-3' (reverse). Amplification products were analyzed in 1.5% agarose gel and visualized by ethidium bromide staining. The signal was quantified by using GS-800 imaging densitometer (Bio-Rad, Hercules, CA, USA) and the Quantity One 4.6.9 software (Bio-Rad). ChIP products were also quantified by qRT-PCR using the same amount (100 ng) of IP and total DNA in a 25 µL reaction containing Sybr Green master mix and 300 nM of forward and reverse primers. Data were represented as recovery, in percent, of each DNA fragment relative to total input DNA, and it was calculated as previously described.(32)
Luciferase reporter gene assay
Cells were seeded in standard medium in 24-well plates (5 × 104 per well). At 70% confluence, the cultures were transfected with 250 ng of the SRE and AP1 reporter assays (Cignal SRE and AP1 Reporter kit; Quiagen-SA Biosciences) using Lipofectamine 2000 (Life Technologies). The mixture also contains a constitutively expressing Renilla luciferase construct to assess transfection efficacy. Forty-eight hours after transfection, cells were lysed, and luciferase activity was measured according to the manufacturer's protocol using the Dual-Glo Luciferase Assay System (Promega, Madison, WI, USA) with a GloMax Luminometer (Promega). To evaluate growth stimulatory effects, cells were starved overnight and then exposed to IGF1 (50 ng/mL; Merck Millipore) for 1 hour before luciferase activity was assessed, 24 hours after transfection.
Immunofluorescence on adherent cells
Cells were grown on coverslips and fixed in 4% paraformaldehyde (for RUNX2) or 0.5% formalin and 90% methanol (for phospho-ERK 1/2). After permeabilization with 0.15% Triton X-100 in PBS, cells were incubated with the following antibodies: anti-phospho ERK 1/2 (phospho Thr202/Tyr204) (1:10; Covance) and anti-RUNX2 (1:10; 27-k, Santa Cruz Biotechnology). Nuclei were counterstained with Hoechst 33258. The stained sections were visualized using the microscope Nikon (Tokyo, Japan) ECLIPSE 90i equipped with a Plan Apo VC 60× oil, NA 1.4, at RT. Images were captured under identical conditions using a digital color camera (Nikon DS5MC) and the software NIS-Elements AR 3.10 (Nikon). The same software was used to merge all images with double labeling.
Treatment with ERK 1/2 inhibitors
The ERK inhibitor PD98059 (50 µM; Merck Millipore) was added to differentiation medium and renewed with osteogenic medium every 3 to 4 days. Cell growth and bone mineralization were assessed by methylene blue or ARS staining, respectively. In basal conditions, inhibition of ERK 1/2 activation was checked by immunofluorescence on adherent cells grown on coverslips. Standard medium was replaced by IMDM plus 10% FBS with or without (control) PD98059 (50 µM), U0126 (10 µM; Cell Signaling), and its inactive analog U0124 (10 µM; Merck Millipore). The phosphorylation of ERK was assessed after 24 hours of treatment. To evaluate the effects on the transcriptional activities of ELK-1 and AP1 after ERK inhibition, cells were starved overnight and treated with PD98059 (100 µM) for 2 hours. Luciferase activity was checked 24 hours after transfection.
Treatment with siRNA against CD99
Transient CD99-silencing was assessed by siRNA (5'-GGCUGGCCAUUAUUAAGUCTT-3') (IDT, Coralville, IA, USA). Scrambled (ON-TARGETplus siCONTROL, Thermo Scientific) and a Validated STEALTH siRNA (Life Technologies) against p53 were used as controls. All were administered at a final concentration of 40 nM. Cells were seeded on coverslips in 60-mm plates in standard medium (3 × 105 cells per plate) and transfected after 24 hours by using Lipofectamine 2000 (Life Technologies). The expression of CD99, phospho-ERK 1/2, or RUNX2 was checked by Western blotting or by immunofluorescence on fixed cells, treated for 48 hours with siRNA sequences.
Metastatic ability in athymic mice
Female athymic 4- to 5-week-old Crl:CD-1-nu/nu BR mice (Charles River Italia, Como, Italy) were used. To obtain natural killer depressed animals, mice were injected iv with anti-asialo GM1 antiserum 24 hours before cell inoculation. Metastases were determined by injection of 2 × 106 viable cells in a tail lateral vein, and the number of lung colonies were obtained by counting with a steromicroscope after staining with black India ink. All animal experiments were performed according to Italian law 116/92 and European directive 2010/63/UE. Experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee (“Comitato Etico Scientifico per la Sperimentazione Animale”) of the University of Bologna, and forwarded to the Italian Ministry of Health.
Statistical analysis
Differences among means were analyzed using Student's t test.
Results
CD99 is expressed in both hBM-MSCs and osteoblasts as part of normal osteoblast differentiation
Protein expression of CD99 was observed at high levels in 24/34 (70%) and at medium/low levels in 6/34 (18%) of hBM-MSCs. Only 4/34 primary cultures were found to be negative (Table 1). Double-labeling staining with CD99 and CD166, CD105, or CD29 confirmed that cells expressing mesenchymal markers actually correspond to those expressing CD99 (Supplemental Fig. S1A). The latter was also expressed in human osteoblasts,(11) but it was downregulated when normal bone-derived cells were transformed by the MET oncogene (Supplemental Fig. S1B). Accordingly, patient-derived OS cell lines exhibited lower levels of the antigen compared with normal osteoblasts (Supplemental Fig. S1C). OS cells transduced with CD99 showed reduced aggressiveness in nude mice(11) (Table 2), according to its putative oncosuppressive role.
Table 2.
Metastatic Ability of Saos-2 Parental Cells and Sa/CD99 Transfected Clones
| Cell line | Incidence(%) | Median (range) |
|---|---|---|
| Saos-2 | 5/10 (50) | 0.5 (0–15) |
| Sa/CD99wt22 | 3/9 (33) | 0 (0–4) |
| Sa/CD99wt36 | 0/10 (0) | 0 |
To sustain the involvement of CD99 in osteoblast physiology, representative cultures of hBM-MSCs (Fig. 1A) or of human bone fragment–derived osteoblasts (Fig. 1B) were driven to differentiate toward the osteoblastic lineage. Expression of some specific osteoblast markers, such as ALP and osteocalcin (OCN),(33) as well as the production of bone matrix were investigated to verify the acquisition of the osteoblastic phenotype. CD99 expression increased during differentiation and was particularly high at the end of osteoblastic induction when cells became mature.
Fig. 1.
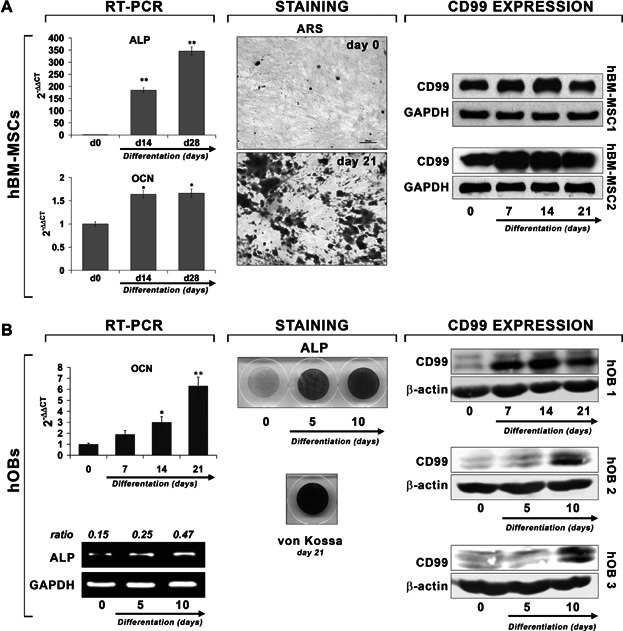
CD99 expression during osteoblast differentiation in hBM-MSCs (A) and human osteoblasts (hOBs) (B). For both cultures, expression of specific markers of differentiation in RT-PCR are shown. Relative expression of alkaline phosphatase (ALP) or osteocalcin (OCN) mRNA was calibrated on undifferentiated cells at day 0 (2-ΔΔCT = 1). Data are presented as mean ± SEM of experiments performed in triplicate. Student's t test was used (*p < 0.05; **p < 0.01). Alizarin Red S (ARS), ALP, and von Kossa stainings were performed as described in Materials and Methods to evaluate cellular differentiation and matrix mineralization. CD99 expression was detected by Western blotting.
CD99 expression favors terminal differentiation of OS cells
To investigate the role of CD99 in OS cell differentiation, we took advantage of the Saos-2 and IOR/OS7 cell lines, both displaying osteoblast-like features and ability to differentiate toward the osteogenic lineage in low-serum medium containing ascorbic acid (50 µg/mL) and β-glycerophosphate (5 mM)(27) (Fig. 2A–C). Osteoblasts produce and secrete proteins that constitute the bone matrix, such as collagens and OCN, and are essential for osteoid matrix mineralization, a process mediated by ALP. OS cells overexpressing CD99 (Supplemental Fig. S2) had higher expression of type 1 collagen, ALP, and OCN, early and late markers of osteoblastic differentiation, respectively (Fig. 2A), and displayed higher ALP expression and activity (Fig. 2A, B). Such cells produced more mineralized matrix compared with parental cells, a difference that became clearly evident whenever mineralization was normalized by cell number (Fig. 2C). In addition, consistent with the achievement of a terminal differentiation, OS cells overexpressing CD99 showed increased mRNA levels of dentin matrix protein 1 (DMP1) and matrix extracellular phosphoglycoprotein (MEPE) (Fig. 2D; Supplemental Fig. S3), two proteins highly expressed by osteocytes, which are the post-mitotic cells essential for the physiologic maintenance of bone homeostasis and integrity.(34) OS cells overexpressing CD99 were also characterized by decreased expression of the master regulators Nanog and OCT3/4 mRNA in basal conditions (Fig. 2D; Supplemental Fig. S3). Both Nanog and OCT3/4 play a pivotal role in maintaining stemness cell properties,(35) and their decrease in CD99-overexpressing cells is in line with the acquisition of a phenotype more predisposed to differentiation.
Fig. 2.
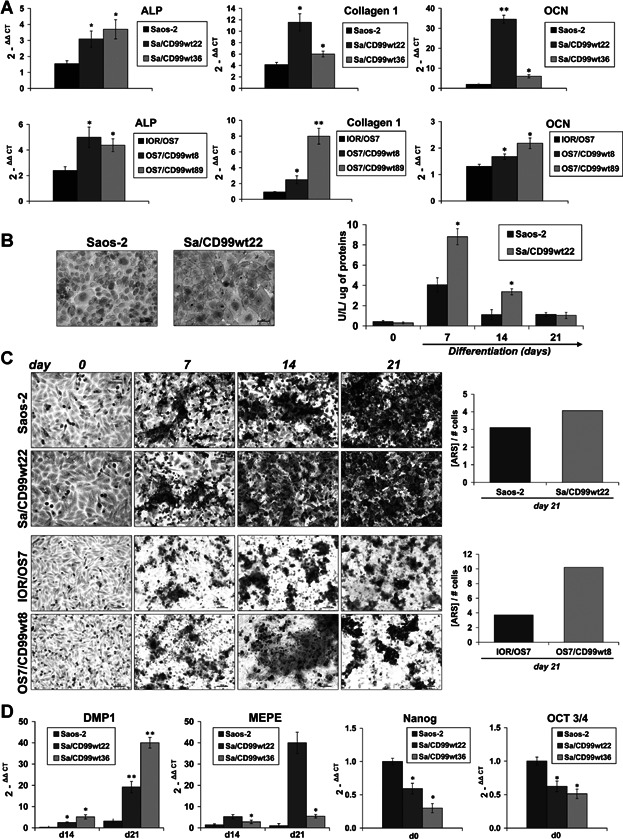
CD99 drives OS cells toward a terminally osteoblast differentiated phenotype. (A) qRT-PCR expression of representative osteoblast differentiation markers in CD99-overexpressing clones and parental cell lines: collagen 1, ALP, and OCN are displayed to sign early and late stages of the differentiation process. The relative expression of target mRNA was calibrated on undifferentiated cells at day 0 (2-ΔΔCT = 1). Data are presented as mean ± SEM of experiments performed in triplicate. Student's t test was used (*p < 0.05; **p < 0.01). (B) Evaluation of ALP expression by histochemical analysis (left panel) and activity in supernatants (right panel) data are presented as mean ± SEM of two separate experiments. Student's t test was used (*p < 0.05). (C) ARS staining of mineralized cultures over 0 to 21 days in differentiation medium. On the right of the panel, production of bone matrix corrected by cell number is shown. (D) qRT-PCR expression of the osteocyte markers DMP1 and MEPE during differentiation (days 14 and 21) and of Nanog and OCT3/4 in basal conditions (day 0). The relative expression of target mRNA was calibrated on the parental cell line (2-ΔΔCT = 1). Data are presented as mean ± SEM of experiments performed in triplicate. Student's t test was used (*p < 0.05; **p < 0.01).
CD99 promotes the transition from proliferation to cell cycle exit for terminal differentiation of OS cells
Osteoblast differentiation is associated with progressive loss of proliferative capacities, culminating in their complete block when mature osteoblasts are embedded in the bone matrix to become osteocytes.(33) The physiologic coupling of osteoblast differentiation to inhibition of proliferation is disrupted in OS cells. In fact, although Saos-2 and IOR/OS7 OS cells were capable of osteoblastic differentiation with generation of bone matrix and expression of mature osteoblast markers such as OCN, they continued to grow with an exponential trend despite being kept in differentiating conditions (Fig. 3A). This behavior was corrected by transfecting OS cells with CD99 that indeed provided growth restriction and progressive decrease of OS cell viability along with differentiation (Fig. 3A). The reduction of tumor cell growth was attributable both to inhibition of cell cycle progression and to induction of apoptosis. CD99-transfected cells showed a higher percentage of cells in the G1 phase of the cell cycle at the expense of the S phase (Fig. 3B, C). Cell growth arrest in CD99-overexpressing cells was supported by increased expression of p21WAF1/CIP1 (Fig. 3D), a key regulator of G0/G1 proliferative arrest.(36) Induction of p21WAF1/CIP1 was also observed in normal hBM-MSCs driven to osteoblast differentiation (Fig. 3D), marking the proliferative block that preludes terminal differentiation.
Fig. 3.
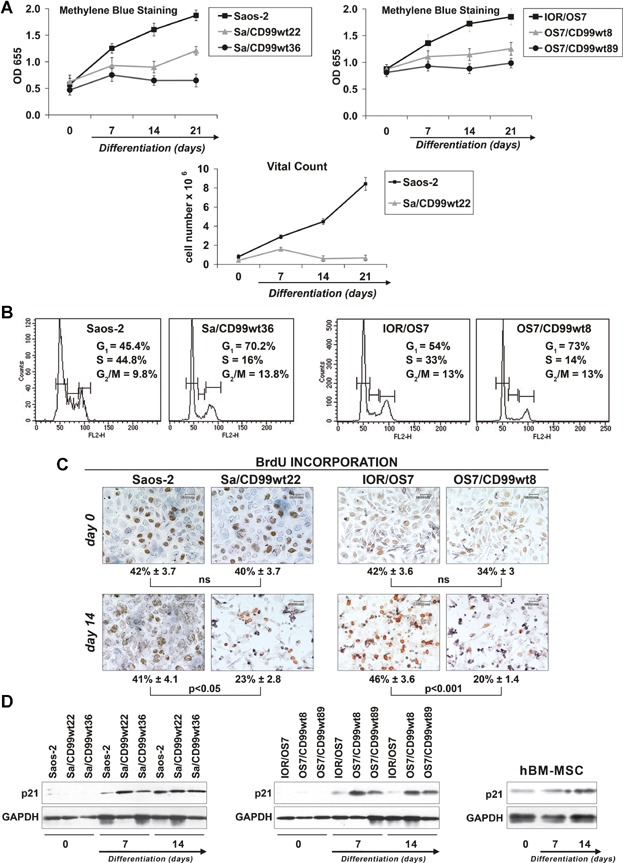
CD99 inhibits proliferation of OS cells in differentiating conditions. (A) Evaluation of cell growth by methylene blue staining in differentiating conditions. Data are presented as mean ± SEM of three separate experiments. Analysis of cell growth was also performed by vital count with Trypan blue. (B) Cell proliferation evaluated by cell cycle analysis at day 7 of differentiation using flow cytometry. (C) BrdU labeling index evaluated by immunohistochemistry at days 0 and 14. Values are expressed as mean % ± SEM. Student's t test was used. Ns = not significant. (D) Modulation of p21WAF1/CIP1 during osteoblast differentiation. Expression of the same protein in hBM-MSCs induced to differentiate toward osteogenic program is also shown.
Permanent cell cycle withdrawal is a feature of senescence other than terminal differentiation.(37) CD99-overexpressing cells did not stain for senescence-associated β-galactosidase activity(38) (data not shown). Rather, TUNEL assay clearly showed massive induction of apoptosis in cells overexpressing CD99 during differentiation (Fig. 4). Starting from day 7, clustered apoptotic nuclei, in brown, were evident in CD99-transfected cells but not in parental cells, which showed moderate presence of apoptotic cells only at day 21.
Fig. 4.
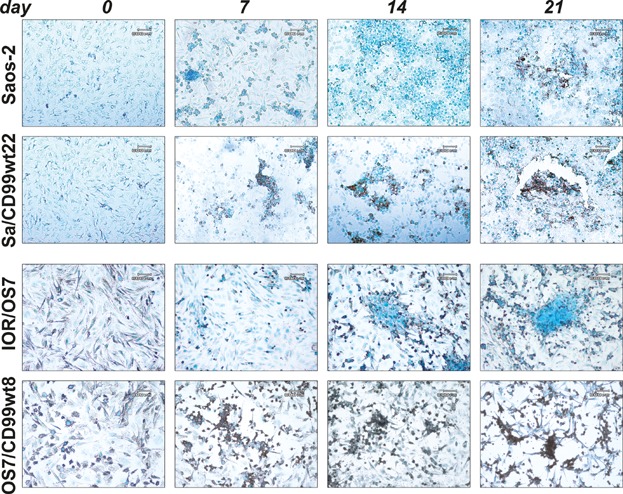
CD99 induces apoptotic cell death of OS cells during differentiation. Apoptosis was evaluated by TUNEL assay. The nuclei were counterstained with methyl green.
CD99 effects are mediated by modulation of MAPK/ERK signaling and RUNX2 activity
It has been reported that CD99 can induce a differential activation of the three MAPK members, ERK, JNK, and p38 MAPK.(39–41) We explored the possible involvement of ERK/MAPK in CD99-mediated osteogenic effects. This pathway has been shown to be activated in the osteogenic program;(42) accordingly, we found activation of ERK 1/2 in normal hBM-MSCs during osteoblast differentiation (Fig. 5A). In OS, CD99-restored expression resulted in the activation of ERK 1/2 either in basal and differentiating conditions (Fig. 5A, B). Weaker or no activation was reported for parental cell lines. Besides being expressed in the nucleus, where it may activate transcriptional factors and nuclear phosphatases,(43) phosphorylated ERK 1/2 was particularly evident at the plasma membrane/cytoplasm of CD99-expressing cells (Fig. 5B, C), where it may target proteins that regulate cell adhesion, cell–cell communication, and cell survival rather than impact on proliferative signals.(44) We analyzed some of the transcription factors known to be phosphorylated upon ERK 1/2 translocation into the nucleus by luciferase assay. In basal conditions, AP1 (Fos/Jun), a key factor in bone formation,(45) but not ELK-1 transcriptional activity, was significantly increased by CD99 transfection in comparison to parental cells (Fig. 5D). Treatments with PD98059, a selective inhibitor of the MAPK-activating enzyme MEK, and, consequently, of ERK 1/2 translocation into the nucleus, decreased ELK-1 transcriptional activity when cells were exposed to insulin-like growth factor-1 (IGF1) and increased AP1 transcriptional activity either in basal conditions or after stimulation with IGF1 (Supplemental Fig. S4A). On one side, this confirmed that increased AP1 transcriptional activity in CD99-overexpressing cells was not related to a proliferation stimulus, whereas on the other side suggested that AP1 was not a direct target of nuclear ERK 1/2 in this cellular context. Consistently with luciferase evidence, PD98059 given in differentiating conditions reduced cell proliferation of Saos-2 more effectively than the CD99-transfected counterpart (43% versus 12% of growth inhibition at day 7) but did not modify or even slightly increase levels of differentiation both in parental and in Sa/CD99 cells (Supplemental Fig. S4B, C). In keeping with its described activity,(46) PD98059 inhibited active nuclear ERK but poorly affected plasma membrane/cytoplasmic activity of ERK 1/2 in Sa/CD99 cells (Supplemental Fig. S5), which may still impact OS cell differentiation. In fact, cytoplasmic ERK 1/2 may phosphorylate either RUNX2, the master gene of osteoblastogenesis, and/or the BMP2 signaling mediators SMADs,(42) which upon translocation to the nucleus may activate the transcriptional factor AP1 described as a co-activator of RUNX2 during osteoblast differentiation.(42) We did not find significant differences in the expression of RUNX2 at the mRNA level between parental and CD99-overexpressing cells either in basal or in differentiating conditions (Supplemental Fig. S6). In contrast, at the protein level, RUNX2 was found to be increased (Fig. 6A) in OS cells ectopically expressing CD99, consistent with its stabilization owing to ERK-mediated post-translational modifications.(42) RUNX2 expression and localization in a representative culture of hBM-MSCs was also shown (Fig. 6A) for comparison, confirming that whenever MSCs were differentiated in osteoblasts, RUNX2 was increasingly expressed and localized in the nucleus. Moreover, chromatin immunoprecipitation (ChIP) indicated that the amount of RUNX2 bound to the promoter of the target genes OCN and p21WAF/CIP1 was augmented in CD99-overexpressing compared with parental cells (Fig. 6B, C). This increase was indeed confirmed also after treatments with PD98059 (+59% for OCN and +200% for p21WAF/CIP1 promoters in CD99 cells), which only partially inhibited cytoplasmic ERK. To verify that the increase of ERK 1/2 and RUNX2 activity was dependent on increased expression of CD99 and to exclude other nonspecific effects, we performed rescue knockdown experiments. OS cells expressing the CD99 construct were transiently transfected with a CD99-targeting siRNA,(19) with scrambled siRNA or p53 siRNA, which was used as additional control considering that both Saos-2 and IOR/OS7 have p53 deleted. When CD99 expression was inhibited (Fig. 7A), cytoplasmic and plasma membrane expression of phospho-ERK 1/2 was also remarkably decreased (Fig. 7A–C) and the RUNX2 level was back to that of parental cells (Fig. 7B, C). Accordingly, cells silenced for CD99 expression showed lower levels of collagen 1 and ALP (Fig. 7D), two early markers of osteoblastic differentiation, further confirming the functional connections between CD99-pERK-RUNX2 axis and differentiation in OS cells. To further support the specificity of the relationship between sustained cytoplasmic ERK 1/2 activation and RUNX2, cells were also exposed to the MEK inhibitor U0126 and its inactive analog U0124 (Supplemental Fig. S5). According to the higher affinity and selectivity for MEK of U0126 over PD98059, we observed a complete inhibition of nuclear and cytoplasmic ERK 1/2 that went in parallel with decreased RUNX2 expression in the nucleus.
Fig. 5.
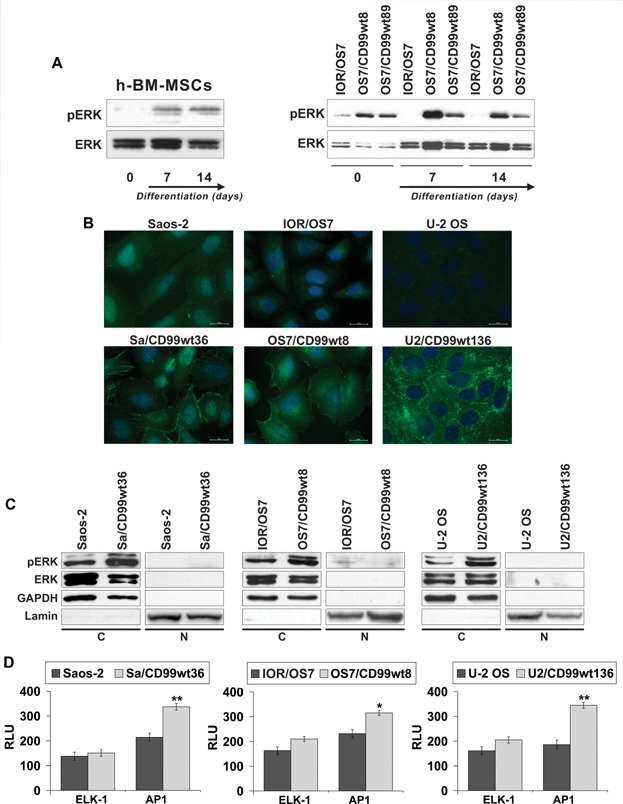
CD99 favors ERK 1/2 activity inducing its localization mainly at plasma membrane and/or in the cytoplasm rather than in the nucleus. (A) Western blotting analysis of phospho-ERK (pERK) 1/2 and ERK 1/2 confirmed the higher activation levels during osteoblast differentiation either in hBM-MSCs or in OS cells overexpressing CD99. (B, C) Immunofluorescence and Western blotting of cytoplasmic (C) and nuclear (N) fractions further supported higher expression of the activated ERK 1/2 in CD99-overexpressing cells compared with the parental cell lines. (D) ELK-1 and AP1 transcriptional activity evaluated by luciferase reporter gene assay in parental cell lines and CD99 clones. Data are presented as mean ± SEM of experiments performed in triplicate and expressed as relative light units (RLU). Values were normalized to Renilla luciferase activity and expressed as fold induction over the negative control. Student's t test was used (*p < 0.05; **p < 0.01).
Fig. 6.
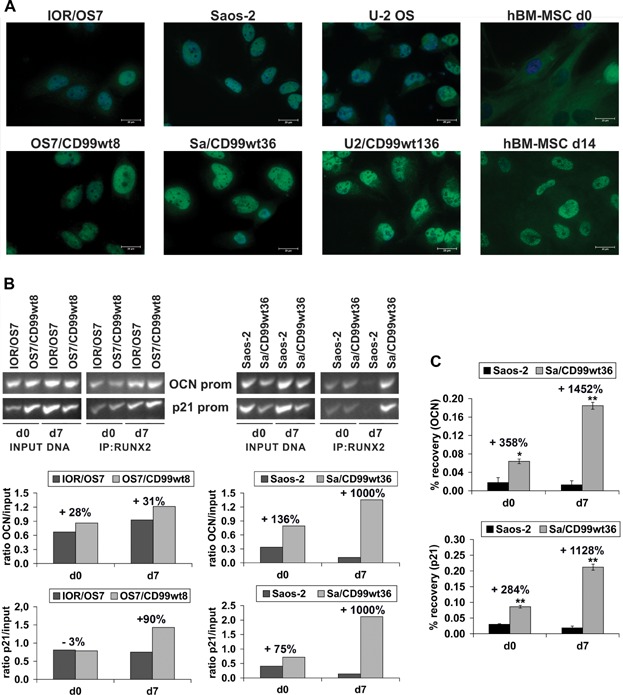
CD99 increases osteogenic stimuli and enhances RUNX2 activity. (A) Immunofluorescence confirmed higher expression of RUNX2 in CD99-overexpressing cells compared with controls. For OS cells, analysis was performed in basal conditions (day 0); for hBM-MSCs, the protein expression was analyzed during the osteoblast differentiation (day 0 and day 14). Scale bar = 20 µm. (B) The presence of CD99 in OS cells increased the recruitment of RUNX2 on OCN and p21WAF1/CIP1 promoters either in basal (day 0) or in differentiating conditions (day 7). Results were quantified and reported as OD ratio of RUNX2 occupancy on the specific promoter versus input DNA. (C) For Saos-2 model, enriched DNA was also quantified by qRT-PCR. Data represent recovery, in percent, of each DNA fragment relative to total input DNA. ▵CT = CT (input)- CT (IP) where CT (cycle threshold) is the cycle number at which each PCR reaction reaches a predetermined fluorescence threshold, set within the linear range of all reactions. Percent recovery = 2▵CT × 2 considering that the input chromatin was 2% of the total.
Fig. 7.
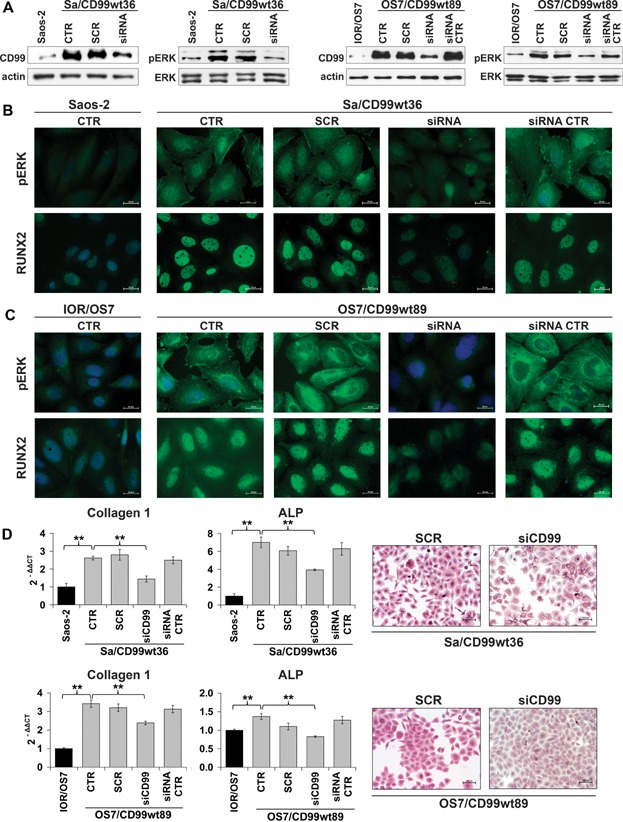
CD99 regulates RUNX2 activity modulating ERK 1/2 signaling. (A) Expression of CD99 and pERK 1/2 was analyzed by Westen blotting after CD99 silencing for 48 hours by siRNA (40 nM). Cells were transfected by a scrambled (SCR) and a siRNA against p53 (siRNA CTR) as controls at the same concentration. (B, C) Expression of active ERK 1/2 and RUNX2 was evaluated by immunofluorescence in CD99-expressing cells in the same conditions described before. Scale bar = 20 µm. (D) After CD99 silencing in OS cells for 48 hours, the expression of osteoblast differentiation markers was analyzed: collagen 1 and ALP decreased, as shown by qRT-PCR and by histochemical analysis.
Discussion
Differentiation therapy aims at reactivating endogenous differentiation program in cancer cells toward tumor cellular maturation and concurrent reversal of a fatal cancer into one more amenable to management. So far, positive examples of agents able to reverse the tumorigenicity of cancer cells and place them back on the road to normal differentiation are limited to acute promyelocytic leukemia.(47) However, unlike other solid tumors, sarcomas could be reprogrammed to resume normal differentiation, as recently described in high-grade undifferentiated pleomorphic sarcoma and liposarcoma by using PPARgamma agonists.(48) A serious limit in the identification of new therapies is that OS has not been studied extensively as other cancers with multiple rearrangements that have been proven responsive to novel strategies. In this article, we sought to ameliorate the knowledge in the field, shedding new light on a still poorly understood molecule that is lost in the majority of OS or after MET-driven osteoblast transformation but present in normal osteoblasts and in hBM-MSCs. During differentiation, osteoblasts and hBM-MSCs maintained and even increased the expression of CD99, suggesting a role of this molecule in osteoblastogenesis. We demonstrated that by restoring CD99 expression to physiologic levels, OS cells differentiate into mature osteoblasts with concomitant minimization of the tumor phenotype. The relationship between CD99 and differentiation is complex but clearly documented at least in hematopoietic elements with the highest expression among immature lymphoid and myeloid precursors, particularly in immature thymic T-cell precursors and gradual loss along differentiation.(49) Similarly, we have shown progressive loss of CD99 expression when hBM-MSCs are induced to neural differentiation.(19) In these contexts, CD99 appeared as a marker of stemness. Interestingly, in related tumors, such as T-cell acute lymphoblastic leukemia (ALL), which originates from T cells arrested at any of the immature stages of development, or in Ewing's sarcoma, which derives from undifferentiated pluripotential MSCs, CD99 is highly expressed and acts as an oncogene.(19,23) In contrast, the protein is maintained during osteoblast differentiation and increased in lining cells(11) or in mature osteoblasts. Here, we show that CD99 is pivotal in controlling the dichotomy between proliferation and differentiation. Although most OS cells can be induced to osteogenic differentiation program and to production of bone matrix if cultured in differentiating conditions, they cannot complete the terminal stages of differentiation, preserving their proliferative abilities. Conversely, OS cells transfected with CD99 can be induced to reproduce the physiologic osteogenic program because they undergo not only expression of typical differentiation markers but also block of proliferation and induction of apoptosis. One of the possible osteoblast behaviors besides becoming osteocytes or lining cells is the apoptotic death that seems required to generate a physiologic signal for bone resorption and regulation of bone turnover. Thus, it seems that CD99 can program cell fate in OS toward terminal osteoblast differentiation to osteocytes or lining cells and that apoptosis could either be a true end product of terminal differentiation or with identical death fate, the result of abortive differentiation once growth arrested.
With a short cytoplasmic tail devoid of signaling motifs except a site for PKCα phosphorylation,(17) it is unlikely that CD99 could provide direct signaling. Interactions with other cell surface molecules are, therefore, assumed to be necessary to regulate its multiple functions. In OS, CD99 was found to interact with caveolin-1 to form a complex that maintains c-Src in its inactive conformation.(11,12) Caveolin-1 has a dual role in sarcomas, thus mirroring CD99; it is required for osteoblast differentiation but is downregulated during osteoblast transformation and osteosarcoma progression.(50) When CD99 is re-expressed in OS cells, caveolin-1 is increased and c-Src signaling inhibited. Accordingly, the requirement of c-Src inhibition for osteoblast differentiation may partly explain CD99-induced phenotype. In this article, however, we provide new evidence that CD99 restoration into OS cells modulates other important signaling pathways converging on MAPK/ERK 1/2 and RUNX2 activation. In cells with high expression of CD99, ERK 1/2 phosphorylation was constitutively higher compared with parental OS cells, and active ERK was found to be located at cell membrane and cytoplasm other than the nucleus. Space-time control of the MAPK/ERK signaling pathway is a key factor to determine the specificity of response in the cell. Although phosphorylation of ERK 1/2 by growth factors leads to dissociation of ERK from MEK and rapid translocation into the nucleus where it activates cell proliferation, in quiescent cells ERK 1/2 is located in the cytoplasm or anchored at cell membrane, where it targets proteins, such as Myosin Light Chain Kinase, paxillin, FAK, and connexin43, all involved in cell migration, cell adhesion, and cell survival.(51) Localization of active ERK into distinct subcellular compartments has a role in determining cell fate decisions(44) including thymocytes selection in the thymus, a tissue characterized by high expression of CD99.(49) In OS cells, we show that sustained activation of plasma membrane/cytoplasmic ERK in CD99-overexpressing cells may increase activity of RUNX2 and AP1, the latter being another critical controller of osteogenic genes that acts as a co-regulator of RUNX2 itself. In OS cells ectopically expressing CD99, we demonstrated higher RUNX2 affinity on the promoter of p21WAF1/CIP1 (a crucial inhibitor of cell proliferation) and of OCN (a marker of mature osteoblasts) consistently with the terminal differentiated phenotype observed. The RUNX2 transcription factor has multiple regulatory activities, not only gene expression activation or repression but also scaffolding of co-regulators that establish a hub for the coordination of functions essential to the expansion and differentiation to the osteogenic lineage. Toward osteoblast maturation, RUNX2 promotes osteoblastogenesis and has antiproliferative effects.(52) However, RUNX2 is also aberrantly expressed in OS owing to amplification of the chromosomal region 6p12-21(53) and/or loss of p53 functions, which is permissive for or even contributes to elevated RUNX2 protein level.(2) Previous studies have shown that RUNX2 interacts specifically with hypophosphorylated pRB during the early stages of cell cycle withdrawal in terminal osteoblast differentiation.(9) RUNX2-pRB-induced cell cycle exit would not be possible because of pRb inactivation, a condition common to 50% to 70% of OS,(54) which could lead to uninhibited proliferation of tumor cells. Re-expression of CD99 is sufficient to resume the block of cell proliferation even in the pRb loss of function. This may again be favored by sustained activation of ERK 1/2, which likely through BMP-SMAD-AP1 signaling and RUNX2 activity(36) may increase transcriptional activation of p21WAF1/CIP1, therefore favoring G0/G1 arrest. CD99 did not immunoprecipitate with ERK 1/2 (data not shown), but evidence so far collected suggests that CD99 is able to activate multiple events at the cell membrane, such as modulation of caveolin-1,(12) cadherins,(24) increased BMP2 or FGF2 osteogenic signaling (Supplemental Fig. S7), as well as ECM-mediated mechanoresponse (collagen 1 production is increased in CD99 OS cells). These varied stimuli inhibit c-Src(12) while activating MAPK/ERK. The role of MAPK/ERK in physiologic bone remodeling and osteoblast differentiation is well known.(42,55) We demonstrated that unabated MAPK/ERK signaling and its location to cell membrane/cytoplasm rather that in the nucleus may drive a differentiated phenotype under specific circumstances also in tumor cells, challenging the classic paradigm according to which RAS/MAPK is invariably a primary driver of oncogenic transformation. It is intriguing that in Ewing's sarcoma, where the expression of CD99 is high and the molecule acts as an oncogene, the achievement of a terminally differentiated phenotype requires the loss of CD99 but still the persistent activation of ERK,(19) further confirming the crucial role of this pathway in the differentiation of sarcomas. The fact that CD99 activates pathways favoring normal cell differentiation may be considered a further confirmation of its importance in the reversal of malignancy of sarcoma cells.
In summary, we have shown that CD99 deficiency is required for OS oncogenic phenotype. Restoration of CD99 expression at the physiologic levels in OS cells affects their malignancy potential(11,12) and results in the achievement of a terminally differentiated phenotype also in the genetically compromised OS cells. CD99 activates pathways that target ERK 1/2 to the cell membrane/cytoplasm, connecting it with increased activity of major osteogenic transcriptional factors, such as AP1 and RUNX2.
Acknowledgments
We are in debt to Dr Pier-Luigi Tazzari, Immunohematology and Transfusion Unit, S. Orsola-Malpighi Polyclinic, Bologna, Italy, for immunophenotyping hBM-MSCs; Cristina Ghinelli for figures editing and Alba Balladelli for editing the manuscript. This work was supported by AIRC (IG10452 to KS), Italian Ministry of Health (MinSAL 1628-2010), and by Associazione Onlus “il Pensatore: Matteo Amitrano” and “Liberi di Vivere Luca Righi”, and Liddy Shriver Foundation.
Authors' roles: Study design: KS. Study conduct: MS, MTM, MCM, CG, MG, AO, PLL, BD, LP, and MFDR. Data collection: MS, MTM, and MCM. Data analysis: MS, MTM, MCM, and CG. Data interpretation: KS and MS. Drafting manuscript: KS. Revising manuscript content: EL, MFD, MPC, and PP. Approving final version of manuscript: MS, MTM, MCM, CG, MG, AO, EL, PLL, BD, LP, MFDR, MPC, PP, and KS. MS and KS take responsibility for the integrity of the data analysis.
Disclosures
All authors state that they have no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Figure S1.
Supplementary Figure S2.
Supplementary Figure S3.
Supplementary Figure S4.
Supplementary Figure S5.
Supplementary Figure S6.
Supplementary Figure S7.
Supplementary Table S1.
References
- 1.Gill J, Ahluwalia MK, Geller D, Gorlick R. New targets and approaches in osteosarcoma. Pharmacol Ther. 2013;137(1):89–99. doi: 10.1016/j.pharmthera.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Martin JW, Squire JA, Zielenska M. The genetics of osteosarcoma. Sarcoma. 2012;2012:627254. doi: 10.1155/2012/627254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyers PA. Muramyl tripeptide (mifamurtide) for the treatment of osteosarcoma. Expert Rev Anticancer Ther. 2009;9(8):1035–49. doi: 10.1586/era.09.69. [DOI] [PubMed] [Google Scholar]
- 4.Ferrari S, Smeland S, Mercuri M, et al. Neoadjuvant chemotherapy with high-dose ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol. 2005;23(34):8845–52. doi: 10.1200/JCO.2004.00.5785. [DOI] [PubMed] [Google Scholar]
- 5.Barr RD, Wunder JS. Bone and soft tissue sarcomas are often curable—but at what cost?: a call to arms (and legs) Cancer. 2009;115(18):4046–54. doi: 10.1002/cncr.24458. [DOI] [PubMed] [Google Scholar]
- 6.Dani N, Olivero M, Mareschi K, et al. The MET oncogene transforms human primary bone-derived cells into osteosarcomas by targeting committed osteo-progenitors. J Bone Miner Res. 2012;27(6):1322–34. doi: 10.1002/jbmr.1578. [DOI] [PubMed] [Google Scholar]
- 7.Matushansky I, Hernando E, Socci ND, et al. Derivation of sarcomas from mesenchymal stem cells via inactivation of the Wnt pathway. J Clin Invest. 2007;117(11):3248–57. doi: 10.1172/JCI31377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang N, Song WX, Luo J, Haydon RC. He TC. Osteosarcoma development and stem cell differentiation. Clin Orthop Relat Res. 2008;466(9):2114–30. doi: 10.1007/s11999-008-0335-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas DM, Johnson SA, Sims NA, et al. Terminal osteoblast differentiation, mediated by runx2 and p27KIP1, is disrupted in osteosarcoma. J Cell Biol. 2004;167(5):925–34. doi: 10.1083/jcb.200409187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo P, Yang X, Ying M, et al. Retinoid-suppressed phosphorylation of RARalpha mediates the differentiation pathway of osteosarcoma cells. Oncogene. 2010;29(19):2772–83. doi: 10.1038/onc.2010.50. [DOI] [PubMed] [Google Scholar]
- 11.Manara MC, Bernard G, Lollini PL, et al. CD99 acts as an oncosuppressor in osteosarcoma. Mol Biol Cell. 2006;17(4):1910–21. doi: 10.1091/mbc.E05-10-0971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scotlandi K, Zuntini M, Manara MC, et al. CD99 isoforms dictate opposite functions in tumour malignancy and metastases by activating or repressing c-Src kinase activity. Oncogene. 2007;26(46):6604–18. doi: 10.1038/sj.onc.1210481. [DOI] [PubMed] [Google Scholar]
- 13.Ellis NA, Ye TZ, Patton S, German J, Goodfellow PN, Weller P. Cloning of PBDX, an MIC2-related gene that spans the pseudoautosomal boundary on chromosome Xp. Nat Genet. 1994;6(4):394–400. doi: 10.1038/ng0494-394. [DOI] [PubMed] [Google Scholar]
- 14.Gelin C, Aubrit F, Phalipon A, et al. The E2 antigen,a 32 kd glycoprotein involved in T-cell adhesion processes,is the MIC2 gene product. EMBO.J. 1989;8(11):3253–9. doi: 10.1002/j.1460-2075.1989.tb08485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodfellow PJ, Darling SM, Thomas NS, Goodfellow PN. A pseudoautosomal gene in man. Science. 1986;234(4777):740–3. doi: 10.1126/science.2877492. [DOI] [PubMed] [Google Scholar]
- 16.Levy R, Dilley J, Fox RI, Warnke R. A human thymus-leukemia antigen defined by hybridoma monoclonal antibodies. Proc Natl Acad Sci USA. 1979;76(12):6552–6. doi: 10.1073/pnas.76.12.6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alberti I, Bernard G, Rouquette-Jazdanian AK, et al. CD99 isoforms expression dictates T cell functional outcomes. FASEB.J. 2002;16(14):1946–8. doi: 10.1096/fj.02-0049fje. [DOI] [PubMed] [Google Scholar]
- 18.Dufour EM, Deroche A, Bae Y, Muller WA. CD99 is essential for leukocyte diapedesis in vivo. Cell Commun Adhes. 2008;15(4):351–63. doi: 10.1080/15419060802442191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rocchi A, Manara MC, Sciandra M, et al. CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. J Clin Invest. 2010;120(3):668–80. doi: 10.1172/JCI36667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shiratori I, Ogasawara K, Saito T, Lanier LL, Arase H. Activation of natural killer cells and dendritic cells upon recognition of a novel CD99-like ligand by paired immunoglobulin-like type 2 receptor. J Exp Med. 2004;199(4):525–33. doi: 10.1084/jem.20031885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol. 2002;3(2):143–50. doi: 10.1038/ni749. [DOI] [PubMed] [Google Scholar]
- 22.Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M. MIC2 is a specific marker for Ewing's sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing's sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer. 1991;67(7):1886–93. doi: 10.1002/1097-0142(19910401)67:7<1886::aid-cncr2820670712>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 23.Dworzak MN, Froschl G, Printz D, et al. CD99 expression in T-lineage ALL: implications for flow cytometric detection of minimal residual disease. Leukemia. 2004;18(4):703–8. doi: 10.1038/sj.leu.2403303. [DOI] [PubMed] [Google Scholar]
- 24.Zucchini C, Manara MC, Pinca RS, et al. CD99 suppresses osteosarcoma cell migration through inhibition of ROCK2 activity. Oncogene. 2013 doi: 10.1038/onc.2013.152. May 6. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 25.Bertaux K, Broux O, Chauveau C, Jeanfils J, Devedjian JC. Identification of CBFA1-regulated genes on SaOs-2 cells. J Bone Miner Metab. 2005;23(2):114–22. doi: 10.1007/s00774-004-0549-4. [DOI] [PubMed] [Google Scholar]
- 26.Hamilton D, Mallinger R, Millesi H, Engel A, Baumgartner G, Raderer M. Modulation of CD99/MIC2 expression of human AHTO-7 osteoblasts by carcinoma cell line-conditioned media. Anticancer Res. 2001;21(6A):3909–13. [PubMed] [Google Scholar]
- 27.Perbal B, Zuntini M, Zambelli D, et al. Prognostic value of CCN3 in osteosarcoma. Clin Cancer Res. 2008;14(3):701–9. doi: 10.1158/1078-0432.CCR-07-0806. [DOI] [PubMed] [Google Scholar]
- 28.Colucci S, Mori G, Vaira S, et al. L-carnitine and isovaleryl L-carnitine fumarate positively affect human osteoblast proliferation and differentiation in vitro. Calcif Tissue Int. 2005;76(6):458–65. doi: 10.1007/s00223-004-0147-4. [DOI] [PubMed] [Google Scholar]
- 29.Patane S, Avnet S, Coltella N, et al. MET overexpression turns human primary osteoblasts into osteosarcomas. Cancer Res. 2006;66(9):4750–7. doi: 10.1158/0008-5472.CAN-05-4422. [DOI] [PubMed] [Google Scholar]
- 30.Manara MC, Baldini N, Serra M, et al. Reversal of malignant phenotype in human osteosarcoma cells transduced with the alkaline phosphatase gene. Bone. 2000;26(3):215–20. doi: 10.1016/s8756-3282(99)00266-5. [DOI] [PubMed] [Google Scholar]
- 31.Garofalo C, Sisci D, Surmacz E. Leptin interferes with the effects of the antiestrogen ICI 182,780 in MCF-7 breast cancer cells. Clin Cancer Res. 2004;10(19):6466–75. doi: 10.1158/1078-0432.CCR-04-0203. [DOI] [PubMed] [Google Scholar]
- 32.Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 2001;15(16):2069–82. doi: 10.1101/gad.906601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lian JB, Javed A, Zaidi SK, et al. Regulatory controls for osteoblast growth and differentiation: role of Runx/Cbfa/AML factors. Crit Rev Eukaryot Gene Expr. 2004;14(1–2):1–41. [PubMed] [Google Scholar]
- 34.Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26(2):229–38. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JB, Zaehres H, Wu G, et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature. 2008;454(7204):646–50. doi: 10.1038/nature07061. [DOI] [PubMed] [Google Scholar]
- 36.Chang SF, Chang TK, Peng HH, et al. BMP-4 induction of arrest and differentiation of osteoblast-like cells via p21 CIP1 and p27 KIP1 regulation. Mol Endocrinol. 2009;23(11):1827–38. doi: 10.1210/me.2009-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 38.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture,in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92(20):9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Byun HJ, Hong IK, Kim E, et al. A splice variant of CD99 increases motility and MMP-9 expression of human breast cancer cells through the AKT-, ERK-, and JNK-dependent AP-1 activation signaling pathways. J Biol Chem. 2006;281(46):34833–47. doi: 10.1074/jbc.M605483200. [DOI] [PubMed] [Google Scholar]
- 40.Hahn MJ, Yoon SS, Sohn HW, Song HG, Park SH, Kim TJ. Differential activation of MAP kinase family members triggered by CD99 engagement. FEBS Lett. 2000;470(3):350–4. doi: 10.1016/s0014-5793(00)01330-2. [DOI] [PubMed] [Google Scholar]
- 41.Kasinrerk W, Tokrasinwit N, Moonsom S, Stockinger H. CD99 monoclonal antibody induce homotypic adhesion of Jurkat cells through protein tyrosine kinase and protein kinase C-dependent pathway. Immunol Lett. 2000;71(1):33–41. doi: 10.1016/s0165-2478(99)00165-0. [DOI] [PubMed] [Google Scholar]
- 42.Franceschi RT, Ge C, Xiao G, Roca H, Jiang D. Transcriptional regulation of osteoblasts. Ann NY Acad Sci. 2007;1116:196–207. doi: 10.1196/annals.1402.081. [DOI] [PubMed] [Google Scholar]
- 43.Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer. Cell Cycle. 2009;8(8):1168–75. doi: 10.4161/cc.8.8.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mor A, Philips MR. Compartmentalized Ras/MAPK signaling. Annu Rev Immunol. 2006;24:771–800. doi: 10.1146/annurev.immunol.24.021605.090723. [DOI] [PubMed] [Google Scholar]
- 45.Marie PJ. Transcription factors controlling osteoblastogenesis. Arch Biochem Biophys. 2008;473(2):98–105. doi: 10.1016/j.abb.2008.02.030. [DOI] [PubMed] [Google Scholar]
- 46.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270(46):27489–94. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 47.Grimwade D, Mistry AR, Solomon E, Guidez F. Acute promyelocytic leukemia: a paradigm for differentiation therapy. Cancer Treat Res. 2010;145:219–35. doi: 10.1007/978-0-387-69259-3_13. [DOI] [PubMed] [Google Scholar]
- 48.Charytonowicz E, Terry M, Coakley K, et al. PPARgamma agonists enhance ET-743-induced adipogenic differentiation in a transgenic mouse model of myxoid round cell liposarcoma. J Clin Invest. 2012;122(3):886–98. doi: 10.1172/JCI60015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dworzak MN, Fritsch G, Buchinger P, et al. Flow cytometric assessment of human MIC2 expression in bone marrow, thymus, and peripheral blood. Blood. 1994;83(2):415–25. [PubMed] [Google Scholar]
- 50.Cantiani L, Manara MC, Zucchini C, et al. Caveolin-1 reduces osteosarcoma metastases by inhibiting c-Src activity and met signaling. Cancer Res. 2007;67(16):7675–85. doi: 10.1158/0008-5472.CAN-06-4697. [DOI] [PubMed] [Google Scholar]
- 51.Ramos JW. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol. 2008;40(12):2707–19. doi: 10.1016/j.biocel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 52.Pratap J, Galindo M, Zaidi SK, et al. Cell growth regulatory role of Runx2 during proliferative expansion of preosteoblasts. Cancer Res. 2003;63(17):5357–62. [PubMed] [Google Scholar]
- 53.Lau CC, Harris CP, Lu XY, et al. Frequent amplification and rearrangement of chromosomal bands 6p12-p21 and 17p11.2 in osteosarcoma. Genes Chromosomes Cancer. 2004;39(1):11–21. doi: 10.1002/gcc.10291. [DOI] [PubMed] [Google Scholar]
- 54.Berman SD, Calo E, Landman AS, et al. Metastatic osteosarcoma induced by inactivation of Rb,p53 in the osteoblast lineage. Proc Natl Acad Sci USA. 2008;105(33):11851–6. doi: 10.1073/pnas.0805462105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson WR, Rubin CT, Rubin J. Mechanical regulation of signaling pathways in bone. Gene. 2012;503(2):179–93. doi: 10.1016/j.gene.2012.04.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1.
Supplementary Figure S2.
Supplementary Figure S3.
Supplementary Figure S4.
Supplementary Figure S5.
Supplementary Figure S6.
Supplementary Figure S7.
Supplementary Table S1.