

Abstract
During the last decade the use of transaminases for the production of pharmaceutical and fine chemical intermediates has attracted a great deal of attention. Transaminases are versatile biocatalysts for the efficient production of amine intermediates and many have (S)-enantiospecificity. Transaminases with (R)-specificity are needed to expand the applications of these enzymes in biocatalysis. In this work we have identified a fungal putative (R)-specific transaminase from the Eurotiomycetes Nectria haematococca, cloned a synthetic version of this gene, demonstrated (R)-selective deamination of several substrates including (R)-α-methylbenzylamine, as well as production of (R)-amines, and determined its crystal structure. The crystal structures of the holoenzyme and the complex with an inhibitor gabaculine offer the first detailed insight into the structural basis for substrate specificity and enantioselectivity of the industrially important class of (R)-selective amine : pyruvate transaminases.
Database
The atomic coordinates and structure factors for the Nectria TAm in holoenzyme and gabaculine-bound forms have been deposited in the PDB as entries 4cmd and 4cmf respectively.
Structured digital abstract
• TAm and TAm bind by x-ray crystallography (View interaction)
Keywords: (R)-selective, aminotransferase, industrial biocatalysis, protein structure, substrate specificity, transaminase
Introduction
Transaminase (TAm; aminotransferase; EC 2.6.1.) enzymes catalyse the reversible transfer of an amino group from an amino substrate onto a ketone or aldehyde, producing a chiral amine in the case of a keto-acceptor 1–4. TAms are involved in the metabolism of most of the 20 naturally occurring proteinogenic amino acids and many other amine-containing compounds. TAm enzymes with sufficiently relaxed substrate range are useful tools in the synthesis of chiral amino acid analogues 5–7, e.g. tert-leucine 8, homophenylalanine 9, and glutamate analogues for the study of neuronal receptors 10.
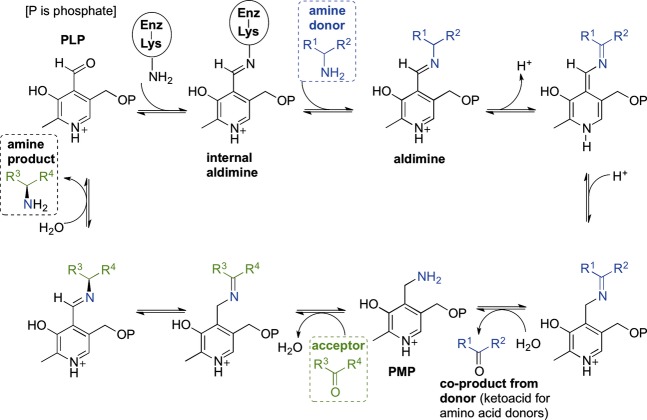
TAms use the cofactor pyridoxal 5′-phosphate (PLP), the biologically active form of vitamin B6 11,12 which normally covalently binds to an active site lysine via a Schiff base (internal aldimine; Scheme6). The TAm mechanism is made up of two half-reactions. In the first half-reaction the donor substrate forms a Schiff base with the cofactor and after further intermediate steps, including an α-proton abstraction step, transfers its amino group to the cofactor (external aldimine), resulting in a keto acid and enzyme-bound pyridoxamine 5′-phosphate (PMP). In the second half-reaction the amino group is transferred from PMP to an acceptor ketone or aldehyde restoring the PLP internal aldimine and producing a chiral amino group if the acceptor was a ketone or a primary amine if the acceptor was an aldehyde. α-ketoglutarate is a common amino acceptor substrate of TAms, but some use pyruvate as an amino group acceptor and are called in such cases substrate : pyruvate TAms, e.g. serine : pyruvate TAm from Sulfolobus solfataricus from TAm class V 13,14.
Figure 6. Scheme.

A representation of the transaminase enzyme mechanism showing key intermediates.
TAms are currently of key interest to biotechnology for both the asymmetric synthesis and resolution of chiral amines. Within the many types of TAm enzymes, specifically interesting for industry are the (S)- and (R)-selective TAms which can catalyse the transfer of an amino group from substrates containing an amino group distal to the carboxyl group (ωATms 15) or no carboxyl group (amine transaminases 16). Several of these enzymes have been biochemically characterized. In particular the ωTAms from Vibrio fluvialis 16 and Chromobacterium violaceum 17 have been studied as potential biocatalysts in the production of chiral amines in a stereoselective fashion. These enzymes all belong to the Pfam class III of transaminases 18 and have a PLP type I protein structure fold. They show activity towards (S)-α-methylbenzylamine [(S)-MBA] and have varying activities towards other amines such as β-alanine and other aliphatic amines. They also use pyruvate and many other ketones and aldehydes as the amino group acceptor 17.
To date enzymes capable of catalysing the (R)-selective transamination of ω-substituted amines have not been widely studied but the production of such chiral amines remains very challenging synthetically. Recently a collaboration between Codexis and Merck resulted in the production of a mutant (R)-specific transaminase from Arthrobacter sp. (ArRMut11) to aminate sterically demanding 1,3-ketoamides and generate an (R)-chiral amine used in the manufacture of sitagliptin, which is used in the treatment of type II diabetes 19. The mutant ArRMut11 has also been reported to transaminate bicyclic ketones such as tetralone 20,21. Most transaminases (such as the (S)-ωTAms) belong to PLP type I fold and are all specific towards the (S)-enantiomer of their substrates. Interestingly, enzymes which belong to Pfam TAm class IV and have PLP type IV fold can catalyse transamination of either d-amino acids or branched chain l-amino acids. Unlike most of the other PLP enzymes the catalysis in these enzymes occurs on the re side of the cofactor. Since both (S)- and (R)-enantiomers can be accommodated within one architectural fold depending on the specific sequences of the enzymes, this class of TAms potentially contains enzymes capable of transamination to give ω-substituted amines. Höhne et al. 22 analysed sequences of a large group of TAms of this Pfam class and found a fingerprint sequence for enzymes potentially capable of (R)- ωTAm activity. Several of these enzymes demonstrated high enantioselective activities towards R-α-methylbenzylamine [(R)-MBA], (2R)-aminohexane and (2R)-amino-4-phenylbutane. Several three-dimensional structures of (S)-specific TAms have been elucidated 23,24. Recently the crystallization of an (R)-TAm has been reported 25; however, to date no structure is available.
Gabaculine (5-amino-1,3-cyclohexadienylcarboxylic acid) is a common suicide inhibitor of both α- and ω-transaminases. Gabaculine binds to the TAm enzyme to form a Schiff base with the PLP cofactor. The complex then undergoes a number of bond rearrangements to form an unstable intermediate, which is spontaneously converted to m-carboxyphenylpyridoxamine phosphate (mCPP). This compound results in an irreversible aromatic modification of the cofactor where the Schiff base formed between gabaculine and PLP becomes a non-hydrolysable single bond 26, which binds tightly to the active sites of TAms. The formation of this complex can aid our understanding of the active site conformation adopted for determining substrate specificity and enzymatic turnover 13,24.
In this paper we describe the substrate specificity for an (R)-specific ωTAm from Nectria haematococca and its three-dimensional structures in both the PLP and gabaculine-bound forms.
Results
(R)-selective TAm design, cloning and initial assays
Previous sequence structure studies on both d-amino acid TAms (DATAs) and branched chain TAms (BCATs; class IV 14) indicated that there was a possibility that these types of TAms might be able to perform (R)-selective transamination. A blast search for class IV TAms was performed and several fungal branched chain enzymes were identified and aligned using clustalw (http://embnet.vital-it.ch/software/ClustalW.html) followed by the creation of an unrooted tree. From one of the subgroups of this unrooted tree, four amino acid sequences were selected and these were later shown to match TAms reported to have (R)-selective activity [i.e. accepting (R)-compounds such as (R)-MBA or d-alanine] 22. A detailed analysis of the retrieved sequences led us in 2009 to the identification of a TAm belonging to N. haematococca, also known by its asexual name Fusarium solani 27,28, as a new TAm whose properties had not been previously reported.
The Nectria TAm protein sequence was used to design a synthetic gene using optimal codons for Escherichia coli. The synthetic genes were designed to contain three flanking restriction sites, NdeI, HindIII and XhoI, which were used to create a wild-type (wt) and a His-tagged version of the Nectria TAm synthetic gene respectively (Fig. S1). The gene was cloned into the pET29a vector and overexpressed in E. coli in a soluble form.
The amino acid sequence of the Nectria TAm is shown in Fig.1 highlighting the two motifs identified by Höhne et al. 22 as being responsible for (R)-selective transamination.
Figure 1.
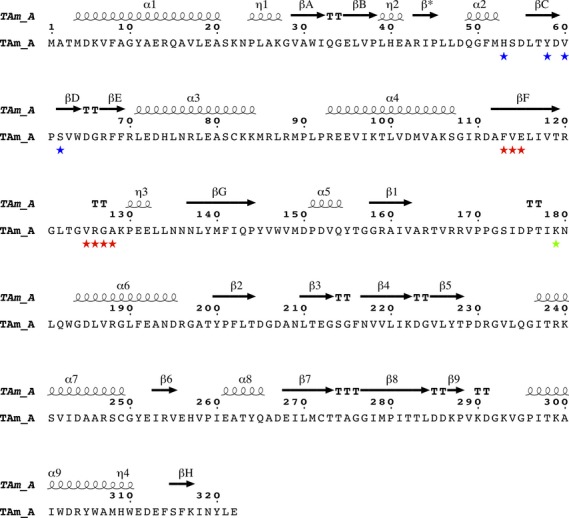
The amino acid sequence and secondary structure of the Nectria TAm. The residues proposed by consensus analysis to be important for (R)-selective amine activity are marked 22. Motif 1 (blue stars): H(53); Y(58); V(60) and S(62). Motif 2 (red stars): F(113); V(114); E(115); V(125); R(126); G(127); A(128). The active site Lys179 is highlighted by a green star. The secondary structural elements are indicated above the sequence, respectively, as α-helices, η-310 helices and β-strands. The N-terminal domain β-sheet is shown as βA–βH, the PLP binding domain β-sheet as β1–β9. The β-strand forming the inter-subunit β-strand with its symmetry equivalent is marked as β*. The secondary structure assignment and the figure were produced using espript 45.
Preliminary enzyme activity from small scale cultures indicated that crude lysate from constructs pQR1022 (wt) and pQR1023 (His-tagged) produced acetophenone when using (R)-MBA plus pyruvate as substrate pairs (Fig. S2). However, no significant activity was noted when using (S)-MBA (crude lysate or resuspended pellets).
Nectria TAm substrate specificity and the production of (R)-amines
Based on the analysis of the primary amino acid sequence and the preliminary assay data the Nectria TAm was believed to be (R)-selective. The substrate specificity and the enantioselectivity of the transaminase were then investigated in detail using a colorimetric assay previously described by Hopwood et al. 29 (Scheme7). The specific and relative activities of the Nectria TAm in the deamination of various racemic (rac) and single isomer amines (1–7) are summarized in Table1. In general, the Nectria TAm accepted a number of aromatic and cyclic amines with different activities. The highest specific activity was determined to be in the oxidative deamination of (R)-MBA (R)-1, while (R)-1-cyclohexylethylamine (R)-2, (R)-4-chloro-α-methylbenzylamine (R)-3 and (rac)-1-methyl-3-phenylpropylamine 6 were converted with good specific activities as well (25%–62% relative activity). With the amines 1,2,3,4-tetrahydro-1-naphthylamine 4 and α-ethylbenzylamine 5 only a low activity could be detected, and no conversion of cyclohexylamine 7 to cyclohexanone was detected at all.
Figure 7. Scheme.
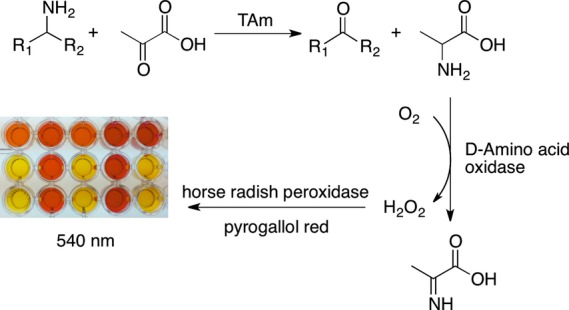
Spectophotometric assay used to screen the Nectria TAm with several chiral amines.
Table 1.
Spectophotometric assay used to screen the Nectria TAm against several chiral amines 1–6 and amine 7 and the substrate acceptance and relative activities.
| Substrate | Specific activity (U·mg−1) | Relative activity (%) | |
|---|---|---|---|
|
1
|
(R) | 1.83 | 100 |
| (S) | 0.02 | 0.90 | |
 2 2
|
(R) | 1.14 | 62 |
| (S) | 0 | 0 | |
 3 3
|
rac | 0.63 | 35 |
| (R) | 0.85 | 46 | |
 4 4
|
rac | 0.02 | 1.1 |
| (R) | 0.08 | 4.5 | |
 5 5
|
rac | 0.16 | 8.6 |
| (S) | 0 | 0.13 | |
 6 6
|
rac | 0.46 | 25 |
|
7
|
– | 0 | 0.20 |
Interestingly, when considering the structures of the substrates and the specific activities obtained it seems that the position of the amino group relative to the ring is a crucial parameter. While (R)-1 was converted with excellent activity, the use of (rac)-α-ethylbenzylamine 5 incorporating one further carbon in the side chain led to a significant decrease in activity (9% relative activity). However, a longer carbon chain between the ring and the amine, with the amine still in the α-position to a methyl group as in (rac)-6, led to decreased activity as well, but this was still approximately threefold higher than for (rac)-5. These results indicate that a methyl group α to the amine seems to be beneficial with respect to the specific activity of the Nectria TAm.
The results from the colorimetric assay (Scheme7, Table1) using enantiopure and racemic amines also confirmed the (R)-selectivity of the Nectria TAm. In particular, the activities observed in the conversions of 1 and 2 to the corresponding amines, where both enantiomers were available, clearly demonstrated the enzyme preference for the (R)-enantiomer.
To further confirm the enantiopreference of the enzyme, its selectivity in the reverse reaction (reductive amination) was also carried out, as shown in Scheme8, using lactate dehydrogenase (LDH) for pyruvate co-product removal as previously reported 21,30.
Figure 8. Scheme.
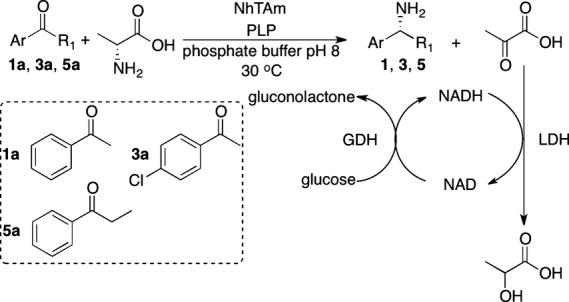
Asymmetric reductive amination of 1a, 3a, 5a catalysed by Nectria TAm with simultaneous co-product removal using lactate dehydrogenase (LDH) and glucose dehydrogenase (GDH) and NAD+ as cofactor.
The reaction products in the reductive amination of acetophenone 1a, 4-chloracetophenone 3a and propiophenone 5a to 1,3 and 5, respectively, were analysed by chiral HPLC. For all three ketones the (R)-amine was the sole reaction product indicating an enantiomeric excess > 98%, by comparison with HPLC data for the racemic and single isomer standards available (see Table S1 for chiral HPLC details). This provided additional support for the (R)-selectivity of the Nectria TAm.
Structure of the Nectria TAm
The native (internal aldimine form) Nectria TAm structure and its complex with the inhibitor mCPP (gabaculine-PLP adduct) were solved and refined to resolutions of 1.68 and 1.50 Å respectively. The resulting R factors for the native and complex structures were 15.8% and 15.3% with Rfree values of 18.8% and 18.0%, respectively (Table2). The G factors calculated for each model confirmed that the structures have acceptable stereochemical properties 31. There are no residues in the cis configuration.
Table 2.
Summary of data processing and refinement statistics. Values in parentheses are given for the outer resolution shell. Rmerge = Σh ΣJ|〈Ih〉 − IJ(h)|/Σh ΣJI(h), where I(h) is the intensity of reflection h, Σh is the sum over all reflections and ΣJ is the sum over J measurements of the reflection. Rcryst = Σ||Fo| − |Fc||/Σ|Fo|. Target values are given in square brackets. Wilson B factor was estimated by sfcheck 46. Ramachandran plot analysis was performed by procheck.
| Native | Gabaculine complex | |
|---|---|---|
| Space group | P21212 | I222 |
| Wavelength (Å) | 0.92 | 0.98 |
| Unit cell (Å) | a = 106.1, b = 133.9, c = 61.7 | a = 61.7, b = 100.3, c = 130.3 |
| Solvent content (%)VM (Å3·Da−1) | 59.3% (3.0) | 55.8% (2.8) |
| Number of chains per asymmetric unit | 2 | 1 |
| Resolution range (Å) | 29.9–1.68 (1.73–1.68) | 39.85–1.5 (1.54–1.50) |
| Multiplicity | 5.6 (5.2) | 6.4 (7.3) |
| Unique reflections | 93 564 | 64 789 |
| Completeness (%) | 98.8 (94.3) | 99.9 (99.9) |
| Rsym (%) | 9.6 (84.8) | 9.6 (102.7) |
| I/σ(I) | 13.9 (2.2) | 16 (3.2) |
| Wilson B factor (Å2) | 25.1 | 12.0 |
| REFMAC RMS error (Å) | 0.09 | 0.06 |
| No. water molecules | 1085 | 508 |
| Rcryst% | 15.8 | 15.3 |
| Rfree% (5% of total data) | 18.8 | 18.0 |
| Rmsd bond lengths (Å) | 0.010 [0.019] | 0.010 [0.019] |
| Rmsd bond angles (°) | 1.4 [1.99] | 1.4 [1.99] |
| Occupancy of inhibitor | N/A | 1 |
| Average B factor (Å2) | ||
| Protein | 18.2 | 16.9 |
| Solvent | 33.7 | 33.6 |
| Gabaculine | N/A | 21.9 |
| Ramachandran plot analysis, residues (%) in | ||
| Most favoured regions | 91.8 | 90.9 |
| Additional allowed regions | 7.8 | 8.0 |
| Generously allowed regions | 0 | 0.7 |
| Disallowed regions | 0.4 | 0.4 |
All amino acid residues of the Nectria TAm were modelled into the electron density of both structures along with the first two amino acids of the C-terminal His-tag. An MES buffer molecule was modelled with occupancy 0.8 in the active site of the native enzyme.
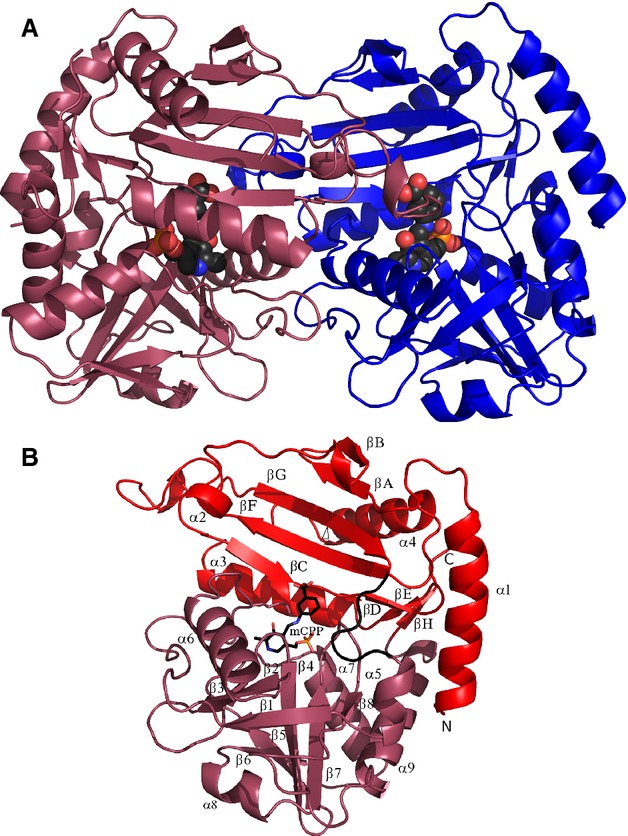
The protein eluted from the gel filtration column at a volume consistent with it forming a dimer in solution. The asymmetric unit of the holoenzyme contains the catalytic dimeric molecule of the Nectria Tam (Fig.2A). The gabaculine complex crystals contain only one subunit of TAm, with two subunits of the molecular dimer related by the crystallographic dyad. Most of the related DATAs and BCATs are dimeric, although a significant number of BCATs are hexameric, being built as a trimer of catalytic dimers.
Figure 2.
(A) A cartoon diagram of the Nectria TAm dimer viewed normal to its molecular dyad; the individual subunits are shown in blue and purple. The mCPP adduct is shown as a space-filling model. (B) A cartoon diagram of the Nectria TAm subunit with the PLP binding domain and the N-terminal domain coloured purple and red respectively. The interdomain loop region 146–151 is shown in black. The secondary structure elements are numbered, the N-terminal domain strands are labelled βA–βH, and the PLP binding domain strands β1–β8. The mCPP adduct is shown as sticks indicating the location of the active site cavity.
The Nectria TAm belongs to the fold-type IV class of PLP-dependent enzymes and is built up of two domains of α/β type (Fig.2B). It is similar to known structures of BCATs and DATAs, with the closest structural homologue being Thermus thermophilus BCAT (PDB code 1WRV; 26% sequence identity). Domain 1 consists of residues 1–145 and 315–320 in an antiparallel seven-stranded β-sheet with topology −1, +4x, +1, −2x, −1, +4x. The second PLP binding domain consisting of residues 151–310-folds into a single eight-stranded β-sheet of mixed type with topology −1x, −1, +5, +1, +1, −3, −1 and direction − + + + − + −. Residues 43–45 from each subunit of the dimer form a two-stranded antiparallel β-sheet. A long N-terminal α-helix appears to be a feature of fungal (R)-specific amine : pyruvate TAms.
The conformations of the two subunits in the native structure are very close with rmsd of their Cα positions of only 0.13 Å over all residues. The rmsd of Cα positions between the mCPP complex structure and the native structure over all residues is only 0.21 Å. This is smaller than the rmsd between subunits within a single structure of many other PLP enzymes. This suggests that the high rigidity of this enzyme may be a consequence of the requirement to maintain a high enantioselectivity within the protein scaffold for both (R) and (S) stereoselective activities.
Active site
The active site is formed by residues from both the N-terminal and the PLP binding domains of one subunit of the Nectria TAm and the N-terminal domain from the adjacent subunit. The PLP cofactor in the holoenzyme structure is bound at the bottom of the active site pocket with the re side of the pyridine ring exposed to solvent and the si side resting on the side chain of Leu234. It is covalently bound to Lys179 via a Schiff base. The PLP phosphate group is anchored by a salt bridge to the side chain of Arg77 and H-bonds with the main chain nitrogen atoms of Ile237 and Thr238 and the side chain oxygens of Thr238 and Thr274. The PLP pyridine nitrogen is coordinated by the carboxyl group of Glu212. Höhne et al. 22 describe two pockets for the active sites of Pfam class IV TAms with two binding sites for groups of the incoming substrates (methyl and phenyl groups in MBA) referred to as S (small) and L (large) pockets.
mCPP adduct binding
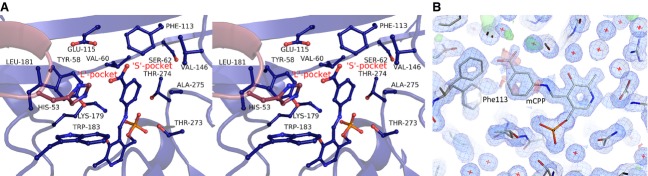
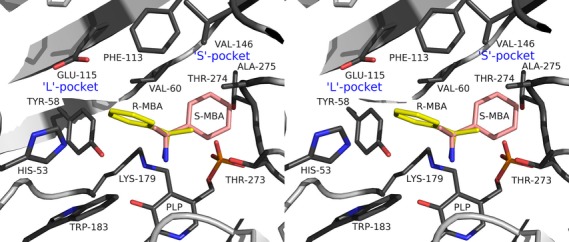
In order to analyse the active site binding pockets adjacent to the cofactor the Nectria TAm was co-crystallized with the inhibitor gabaculine, which forms the mCPP adduct with PLP in the enzyme active site. The mCPP adduct is positioned centrally in the active site in close proximity (< 4.5 Å) to the S pocket outlined by the side chains of residues Phe113, Val60 and Glu115 but does not fit into it (Fig.3A). This site is responsible for binding of the methyl group of MBA. On the opposite side of the active site the L pocket outlined by residues Tyr58, Leu181, Trp183 and His53 from the adjacent subunit forms a much larger cavity which can bind the phenyl group of substrates such as (R)-MBA. Thus the relative positioning of the small restrictive pocket and the large pocket gives the enzyme its (R)-enantioselectivity towards ω-amines. The formation of the mCPP adduct causes the rotation of the PLP pyridine ring by ˜ 35° in relation to its position in the holoenzyme structure; the positions of the PLP phosphate and pyridine nitrogen remain fixed. This angle of rotation matches those observed between the holoenzyme structure and complexes in both Bacillus sp. DATA 32 and E. coli BCAT 33. The carboxyl group of the mCPP adduct is disordered due to its clash with Phe113, which is also partially displaced to a different, strained conformer not observed in the native Nectria TAm structure (Fig.3B).
Figure 3.
(A) A stereo diagram showing the Nectria TAm active site with the bound mCPP adduct. Residues forming the L and S binding pockets are shown as sticks. (B) The 2Fo − Fc electron density map (blue) was contoured at 1 σ for the gabaculine-PLP bound adduct and neighbouring residues. The positive Fo − Fc electron density map is shown in green and negative in red with both contoured at 3.1 σ. The two conformations of the Phe113 induced by the mCPP binding are shown with the disorder of the carboxyl group of mCPP highlighted by the negative Fo − Fc density.
Determinants of the enzyme stereoselectivity
To explain this enantioselectivity of the Nectria TAm the mCPP complex was used as a template to manually position the (R)-MBA-PLP external aldimine adduct in the active site for catalysis. The adduct was modelled so that orientation of the PLP pyridine ring was the same as in the mCPP complex. The (R)-MBA part of the adduct was positioned so that the hydrogen on the chiral carbon was normal to the plane of the PLP pyridine ring and pointing towards the NZ of the catalytic Lys179, according to the Dunathan hypothesis 34 requirement for catalysis. This positions the methyl group of MBA in the S pocket as described by Höhne et al. 22 and the phenyl ring in the opposite L pocket (Fig.4), where both groups fit nicely. If the (S)-MBA-PLP adduct is positioned for catalysis in the same manner for potential amine transfer, its phenyl group points towards the S pocket. Manual modelling of this enantiomer into the Nectria TAm structure revealed that the phenyl group would not fit in this S pocket due to clashes with both the side chains and main chain atoms of residues Thr273 and Thr274 and the side chain of the Ala275 (Fig.4). The limitation in space within the S pocket ensures that the Nectria TAm has (R)-amine substrate specificity.
Figure 4.
A stereo diagram showing the structural basis of the (R)-enantioselectivity of the Nectria TAm. Both enantiomers of MBA were positioned for catalysis based on the structure of the mCPP complex and the requirement for the scissile Cα-H bond to be normal to the plane of the pyridine ring of PLP. The (S)-MBA enantiomer in this position is pointing into the S pocket where it is sterically hindered by residues Thr273-Ala275.
Substrate specificity
In the same manner in which this S pocket restricts the stereospecificity of the enzyme it also regulates the size and regioselectivity of the substrates. The enzyme shows optimal activity for (R)-MBA (relative activity 100%, see Table1); however, in the case of α-ethylbenzylamine when the ethyl group is positioned in the S pocket the relative activity is reduced to 9%. Modelling of this substrate into the active site reveals that when the ethyl group of α-ethylbenzylamine goes into the S pocket it is restricted by the side chain of Thr273. This would put the substrate-PLP external aldimine adduct in a less favourable position for proton abstraction by Lys179, accounting for a 10-fold reduction in activity.
In the case of the substrate (rac)-1-methyl-3-phenylpropylamine 6, which has an additional carbon atom between the phenyl and amine groups compared with (R)-MBA, a fourfold reduction in activity was observed. Positioning this substrate (R-isomer) into the active site reveals that the L pocket can accommodate this larger group while the methyl group neatly fits into the S pocket as observed for MBA. Whilst this larger substrate fits in the L pocket, its movement is significantly more restricted than that observed for (R)-MBA with the phenyl group being positioned close to the edge of the pocket and sandwiched between the side chains of Phe113, Glu115, Tyr58, Val60 and His53. This steric hindrance could slow down the binding of (R)-1-methyl-3-phenylpropylamine to the active site and release of the subsequent product. It could also affect the attack of Lys179 onto the external aldimine adduct which would account for the fourfold reduction in activity. Generally, it appears that the S pocket is more limiting in controlling the substrate specificity compared with the L pocket.
Most (R)-specific amine TAms listed in Höhne et al. 22, some of which have sequence similarity to the Nectria TAm, have high activity towards pyruvate as an acceptor but are inert towards the acceptor α-ketoglutarate or the amino donor glutamate. We infer from this sequence similarity that the Nectria TAm does not have this activity. It would appear that the methyl group of pyruvate or of the reaction product d-alanine fits into the S pocket, with the carboxyl group going into the L pocket. For the α-ketoglutarate to bind in a catalytic position its α-carboxyl group should fit into the S pocket, which is not feasible due to steric restrictions.
Relationship between the (R)-specific amine TAm signature motifs and its structure
While the main scaffold of Pfam class IV TAms is similar between BCATs, DATAs and (R)-selective amine : pyruvate TAms, different residues in specific positions enable the required substrate activities. The BCATs transfer the amino group of branched chain l-amino acids onto the α-ketoglutarate acceptor to produce the corresponding ketoacid and l-glutamate. The DATAs transfer the amino group from d-amino acids onto the α-ketoglutarate to produce the ketoacid and d-glutamate. The (R)-selective amine : pyruvate TAms transfer the substrate amino group onto pyruvate to produce the ketone/aldehyde and d-alanine. Höhne et al. 22 identified two signature motifs which allow the (R)-specific amine : pyruvate TAms to be distinguished from a multitude of sequence related DATAs and BCATs. These motifs are highlighted in Fig.1 for the Nectria TAm sequence.
The enzymatic mechanisms of both the Bacillus DATA 32 and the E. coli BCAT 33 have been thoroughly investigated and many structures of enzyme complexes containing reaction intermediates are available. The comparison of the Nectria TAm with structures of these other enzymes allows a greater understanding of the relationship between their sequence and enzyme activities. The conserved residues His53, Tyr58 and Val60 from the Nectria TAm from the first sequence motif and Phe113 from the second motif contribute to the formation of the L and S substrate pockets conferring specificity for the (R)-amine binding. The pair of residues, Ser62 from the first signature motif and Glu115 from the second motif, rule out the BCAT activity for the Nectria TAm enzyme. In the BCATs the equivalent residues Arg40 and Arg97 are involved in binding the substrates α-carboxyl and the γ-carboxyl groups respectively.
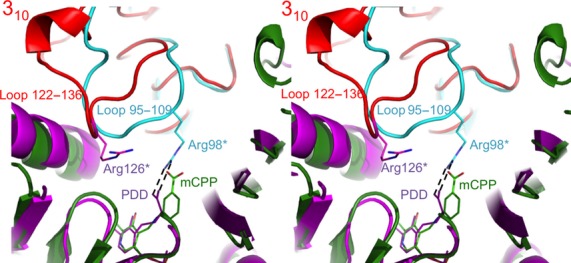
The loop 122–135 in Nectria TAm which contains the four residues Val125, Arg126, Gly127 and Ala128 of the second signature motif folds into a 310 helix. The conformation of the loop is significantly different from that observed for the same regions of BCAT and DATA (Fig.5). This results in a displacement of Arg126 away from the active site in relation to the equivalent Arg98 of DATA which binds the α-carboxyl group of d-alanine. Inability of the Arg126 of Nectria TAm to bind the substrate carboxyl group is one of the reasons for its negligible DATA activity 22.
Figure 5.
A stereo diagram showing the superposition of the dimeric Nectria TAm structure (subunits are shown as green and red cartoons) with the Bacillus sp. DATA structure (magenta and cyan cartoons). This is viewed from the solvent region into the active site cavity. The Arg98 binds the α-carboxyl group of the substrate in DATA as shown for the complex with the reaction intermediate analogue pyridoxyl-d-alanine (PDD; PDB code 3DAA). In BCAT this arginine is not conserved, although the equivalent loop retains similar conformation (not shown). In the Nectria TAm the corresponding loop 122–136, which contains four residues of the second fingerprint sequence motif, accepts a distinct conformation with part of it folding into 310 helix. This results in significant displacement of conserved Arg126, which is unable to bind the substrate analogue mCPP carboxyl group since it is too remote. The ligands and the residues are shown as stick models.
Amongst TAm enzymes, carboxylated substrates are typically coordinated by one or two arginine residues. In the PLP fold I type TAms these arginines are highly conserved. However, in the PLP fold type IV enzymes containing BCATs, DATAs and (R)-amine : pyruvate TAms, the arginine residues which could bind the substrate carboxyl groups are not conserved between the different enzyme activities, with the Nectria TAm enzyme having no arginines in the active site. It appears that the N. haemotococca enzyme has no fixed carboxyl binding position for amino acids, as was earlier predicted by Höhne et al. 22. Thus the signature sequences for the (R)-selective amine TAms appear to specify a narrow S pocket, a hydrophobic L pocket and the absence of charged carboxyl binding residues thereby favouring binding of aliphatic and aromatic amines and ketones.
Conclusion
In this work we have identified a fungal (R)-specific TAm from the Eurotiomycetes N. haematococca, and the protein sequence has been used to design a wild-type and a His-tagged version of the synthetic gene using optimal codons for E. coli. Cloning and expression gave an ωTAm that demonstrated (R)-enantioselectivity towards (R)-MBA (R)-1. The use of other chiral amines (2–6) confirmed the (R)-enantioselective behaviour of the Nectria TAm. Three ketone substrates (1a,3a,5a) were selected and the (R)-amines were formed by the Nectria TAm with simultaneous co-product removal using LDH and glucose dehydrogenase and NAD+ as a cofactor. The crystal structures of the holoenzyme of this (R)-selective TAm from N. haematococca and the complex with an inhibitor gabaculine were determined, which offers a detailed insight into the structural basis for substrate specificity and enantioselectivity. Notably, the side chains of Phe113, Val60 and Glu115 line an S pocket, and on the opposite side of the active site the L pocket is lined by residues Tyr58, Leu181, His53 and Trp183. Furthermore modelling of substrates 1,5 and 6 in the active site of the TAm was able to rationalise the relative specific activities and enantioselectivities that have been observed for this enzyme. This work highlights how the use of detailed structural information can help to understand the enantioselectivity and substrate specificity of this Nectria TAm enzyme. The information obtained will aid the use of rationally directed mutagenesis of this and other industrially important transaminases.
Materials and methods
Gene design, cloning and expression of Nectria TAm
A blast search 35 was used to screen for class IV TAms and the Nectria TAm protein was selected. The protein sequence was used to design a synthetic gene using the dna 2.0 software optimized for expression in E. coli and synthesized by dna 2.0 (Menlo Park, CA, USA). The two restriction digested versions of the gene (NdeI–HindIII and NdeI–XhoI) were then ligated into the expression vector pET29a+ and transformed into BL21(DE3) pLysS, creating the pQR1022 (wt) and pQR1023 (6x-His-tag) constructs respectively (Fig. S1).
Preliminary expression of the Nectria TAm for biochemical assays
A 5 mL overnight culture of the BL21(DE3) pLysS containing pQR1022 (no His tag) was grown in tryptone-yeast (TY) medium for 16 h at 37 °C, 220 r.p.m. The non-His-tagged version was used initially to assess enzyme activity in case the His-tag inhibited activity. The His-tag was found to have no effect on activity and later purification and work was carried out with the His-tagged enzyme. To initiate the overexpression, the overnight cultures were diluted 1 : 100 into 20 mL of TY fresh medium containing both kanamycin (0.05 mg·mL−1) and chloramphenicol (0.03 mg·mL−1) and incubated until an A600 nm of 0.7–0.8 was reached at 37 °C, 220 r.p.m. Then isopropyl thio-β-d-galactoside (IPTG; 0.5 mm) was added and the culture was left for a further 5 h at 30 °C, 220 r.p.m. Cells were harvested after 5 h and centrifuged for 10 min at 14 000 g; the cell pellet was then re-suspended in lysis buffer (100 mm HEPES and 0.4 mm PLP, pH 8) and sonicated using a 10 s per 10 s (on/off) protocol at 12 μm. The resulting material was centrifuged and separated into crude lysate or resuspended pellet and stored at −20 °C.
Optimized expression for enzyme characterization
A 5 mL overnight culture of the BL21(DE3) pLysS containing pQR1023 (6x-His-tag) was grown in LB medium containing kanamycin (0.05 mg·mL−1) for 16 h at 37 °C, 220 r.p.m. The overnight cultures were used to inoculate fresh LB medium (20 mL) containing kanamycin (0.05 mg·mL−1) to give a final 0.05 A600 nm of 0.05. Cultures were grown at 37 °C and 120 r.p.m. until an A600 nm of 0.7–0.8 was reached, IPTG (0.5 mm) was added and the temperature was reduced to 25 °C and the cultures incubated for 20 h. The cells were harvested by centrifugation for 10 min at 6000 g and stored at −20 °C until further use. For the disruption of the cells a 10% (w/v) solution was prepared using an equilibration buffer (100 mm HEPES pH8, containing 0.5 mm PLP and 20 mm imidazole). The cells were disrupted by sonication (10 × 15 s on ice, 0.10 output), and after each cycle the cells were kept on ice for 1 min. The resulting material was centrifuged at 13 000 r.p.m., and the supernatant was used in the purification procedure. For purification the crude extract was loaded onto a column (PD10; GE Healthcare, Little Chalfont, UK) containing 2 mL of Ni-nitrilotriacetic acid material. After binding, the column was washed with three volumes of equilibration buffer, and eluted with the same buffer but containing 200 mm imidazole. For desalting a PD10 desalting column (GE Healthcare) was used and 10 mm HEPES buffer at pH 8. This purification protocol provides enzyme preparations that are optimal for biocatalysis.
Overexpression and purification of the Nectria TAm for crystallization
Plasmid pQR1023 was transformed into E. coli BL21(DE3) pLysS and cultured overnight in LB medium containing kanamycin (0.05 mg·mL−1). ZYM-5052 36 medium (500 mL) was inoculated using the overnight cultures and incubated at 20 °C for 2 days with shaking at 250 r.p.m. The cells were then harvested at 4700 g and 4 °C for 20 min. The pellet was re-suspended in 50 mm Tris/HCl pH 7.5. Cells were disrupted by sonication at 10 μm (Soniprep 150; MSE (UK) Ltd, Lower Sydenham, UK) on ice for 2 min and the cell debris was removed by centrifugation at 15 000 g and 4 °C for 30 min.
The protein was purified using the 6x-His-tag using a 1 mL His-Trap FF crude column (GE Healthcare) using a gradient up to 500 mm imidazole in 50 mm Tris/HCl pH 7.5. The enzyme was then applied to a Superdex 200 HiLoad 16/60 gel filtration column (column volume 120 mL) and was eluted with 1 column volume in 50 mm Tris/HCl, 0.1 m NaCl pH 7.5 at 1.0 mL·min−1. This purification protocol gives enzyme material that can readily crystallize (see section on Crystallization).
Determination of the Nectria TAm stereoselectivity using the acetophenone production assay
(R)- or (S)-selectivity of crude lysates/resuspended pellets from pQR1022 and pQR1023 were tested using 5 mm of the substrate pairs (R)- or (S)-MBA and pyruvate. Reactions were carried out in 1 mL reaction buffer (100 mm HEPES buffer, 0.4 mm PLP, pH 8), where 20% of that final volume corresponded to either crude lysate or resuspended pellet from pQR1022 or pQR1023, respectively. Sample reactions were taken and quenched after 1, 2 and 17 h by mixing 1 : 1 (v/v) of reaction mix with 0.2% trifluoroacetic acid solution. Production of acetophenone was followed using an HPLC C18 column at 210 nm. (R)- or (S)-selectivity of the enzyme was assessed by the production of acetophenone from (R)- or (S)-MBA.
Determination of TAm substrate specificity
The transaminase activity of the Nectria TAm against selected substrates 1–7 was determined using the spectrophotometric assay previously described by Hopwood et al. 29. A volume of 10 μL of the purified Nectria TAm (pQR1023) was added to 190 μL of assay solution in a microtitre plate. The assay solution contained substrate (5 mm), sodium pyruvate (0.2 mg·mL−1), pyrogallol red (0.1 mg·mL−1), PLP (0.05 mg·mL−1), amino acid oxidase (1.2 U·mL−1) and horseradish peroxidise (12.5 U·mL−1) in sodium phosphate buffer (100 mm, pH 8). Absorbance at 510 nm was followed over a period of 30 min, and the linear slope was used to determine initial rates of the enzyme activity using the extinction coefficient (19.533 mm).
Stereoselective formation of chiral amines from acetophenone
The stereoselectivity of the Nectria TAm in the asymmetric amination of different ketones was performed with simultaneous co-product removal to shift the equilibrium of the TAm reaction towards the desired amine product. Purified TAm (0.2 mg) was added to a sodium phosphate buffer solution (1 mL, 100 mm, pH 8) containing the substrate (20 mm), the co-substrates d-alanine (100 mm), β-d-glucose (60 mm), the cofactors PLP (1 mm), NAD+ (0.5 mm) and the co-product removal system (lactate dehydrogenase and glucose dehydrogenase, both 5 U·mL−1). The reaction was incubated at 30 °C and 800 r.p.m in an Eppendorf Thermomixer (Hamburg, Germany). After 72 h the reaction was stopped by the addition of 1 mL of acetonitrile containing 0.1% trifluoroacetic acid and the protein was removed by centrifugation (5 min, 13 000 g). The enantiomeric excesses were determined (within the limits of detection) by analysis of the supernatants by HPLC (Agilent, Santa Clara, CA, USA) equipped with a CROWNPAK® CR(+) column (150 × 4 mm × 5 μm; Daicel, Illkirch, France) with UV detection at 254 nm. The conditions used for the detection of the different amines are given in Table S1.
Crystallization
The Nectria TAm was concentrated to 10 mg·mL−1 using a 10 kDa membrane Vivaspin (Vivascience, MA, USA) and microbatch crystallization trials were set up using an Oryx 6 crystallization robot (Douglas Instruments, Hungerford, Berks, UK) using the JCSG+ and Morpheus screens (Molecular Dimensions, Newmarket, Suffolk, UK). The droplet contained a 50 : 50 ratio of protein solution to precipitant ratio and was covered with a 50 : 50 mix of silicon and paraffin oils.
Crystals of the holoenzyme were grown in condition D1 from the Morpheus screen containing 30% PEG 550MME and PEG 20K. Protein prepared in the presence of 5 mm gabaculine was co-crystallized in 0.1 m MgCl2, 50 mm Na HEPES pH 7.5 and 15% v/v PEG 400.
Data were collected on beamlines I04 and I04-1 at the Diamond Synchrotron light source (Oxford, UK) under cryo conditions (100 K in a stream of gaseous nitrogen). Data were processed using xds 37 in the Xia2 38 pipeline. All further data and model manipulation was carried out using the ccp4 suite of programs 39.
Data collection and structure determination
The structure of holoenzyme was solved by molecular replacement (MR) using the program molrep 40. The functional dimer of T. thermophilus BCAT (PDB code 1WRV) was modified to match the Nectria TAm sequence and was used as a search model. The rotation function was calculated at 3 Å resolution and the translation search was conducted at 4 Å resolution. The rotation solution peak height was 6.6 σ and the correlation of translational solution was 0.183. The resulting MR solution was subjected to rigid body and subsequent restrained refinement in refmac5 41. The refined model was submitted to automated refinement using the arp/warp version 7.0.1 42. The model was manually rebuilt in coot 43 and refined with refmac5. The gabaculine complex, which crystallized in a different space group, was solved by MR using the coordinates of the refined holoenzyme structure. The statistics of the data processing and refinement are given in Table2. After refinement the quality of the model was checked using the program procheck 31. Images were created using the molecular graphics program pymol 44.
Acknowledgments
The authors thank Diamond Light Source for access to beamline I04 and I04-1 (proposal No. MX8889) and the beamline staff scientists. The work was supported by an EPSRC Follow on Fund grant EP/G005834/1 to J. M. Ward and H. C. Hailes. N. Richter was supported by a postdoctoral fellowship of the German academic exchange service (DAAD). C. Sayer was supported by a University of Exeter PhD studentship. M. Isupov is grateful to University of Exeter and BBSRC funded ERA-IB grant BB/L002035/1. Funding in JALs laboratory has been supported by the EU, Wellcome Trust, BBSRC and EPSRC.
Glossary
- BCAT
branched chain transaminase
- DATA
d-amino acid transaminase
- L
large
- LDH
lactate dehydrogenase
- (R)-MBA
(R)-α-methylbenzylamine
- (S)-MBA
(S)-α-methylbenzylamine
- mCPP
m-carboxyphenylpyridoxamine phosphate
- MR
molecular replacement
- PEG
poly(ethylene glycol)
- PLP
pyridoxal 5′-phosphate
- PMP
pyridoxamine 5′-phosphate
- S
small
- TAm
transaminase
- ωTAm
ω-transaminase
- TY
tryptone-yeast extract
- wt
wild-type
Author contributions
R.J. Martinez-Torres conceived the cloning strategy, designed, cloned and expressed the gene and carried out assays. C. Sayer crystallized the protein and determined the structure, N. Richter carried out substrate specificity assays, M.N. Isupov assisted in the structure determination, H.C. Hailes helped plan and oversee and supervise the cloning, expression and assays, J.A. Littlechild planned and oversaw the structure determination, J.M. Ward conceived the project and assisted in planning and overseeing the cloning strategy, expression and assays. All authors wrote the manuscript. CS and RJM-T are equal first authors.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher’s web site
Representation of synthetic gene design.
Enzyme activity of the Nectria TAm synthetic genes.
Chiral HPLC details for chiral amines prepared using Nectria TAm.
References
- 1.Mehta PK, Hale TI. Christen P. Aminotransferases: demonstration of homology and division into evolutionary subgroups. Eur J Biochem. 1993;214:549–561. doi: 10.1111/j.1432-1033.1993.tb17953.x. [DOI] [PubMed] [Google Scholar]
- 2.Hayashi H. Pyridoxal enzymes mechanistic diversity and uniformity. J Biochem. 1995;118:463–473. doi: 10.1093/oxfordjournals.jbchem.a124931. [DOI] [PubMed] [Google Scholar]
- 3.John RA. Pyridoxal phosphate-dependent enzymes. Biochim Biophys Acta. 1995;1248:81–96. doi: 10.1016/0167-4838(95)00025-p. [DOI] [PubMed] [Google Scholar]
- 4.Jansonius JN. Structure evolution and action of vitamin B6-dependent enzymes. Curr Opin Struct Biol. 1998;8:759–769. doi: 10.1016/s0959-440x(98)80096-1. [DOI] [PubMed] [Google Scholar]
- 5.Stewart JD. Dehydrogenases and transaminases in asymmetric synthesis. Curr Opin Chem Biol. 2001;5:120–129. doi: 10.1016/s1367-5931(00)00180-0. [DOI] [PubMed] [Google Scholar]
- 6.Leuchtenberger W, Huthmacher K. Drauz K. Biotechnological production of amino acids and derivatives: current status and prospects. Appl Microbiol Biotechnol. 2005;69:1–8. doi: 10.1007/s00253-005-0155-y. [DOI] [PubMed] [Google Scholar]
- 7.Tufvesson P, Lima-Ramos J, Jensen JS, Al-Haque N, Neto W. Woodley JM. Process considerations for the asymmetric synthesis of chiral amines using transaminases. Biotechnol Bioeng. 2011;108:1479–1493. doi: 10.1002/bit.23154. [DOI] [PubMed] [Google Scholar]
- 8.Taylor PP, Pantaleone DP, Senkpeil RF. Fotheringham IG. Novel biosynthetic approaches to the production of unnatural amino acids using transaminases. Trends Biotechnol. 1998;16:412–418. doi: 10.1016/s0167-7799(98)01240-2. [DOI] [PubMed] [Google Scholar]
- 9.Cho BK, Seo JH, Kang TW. Kim BG. Asymmetric synthesis of l-homophenylalanine by equilibrium-shift using recombinant aromatic L-amino acid transaminase. Biotechnol Bioeng. 2003;83:226–234. doi: 10.1002/bit.10661. [DOI] [PubMed] [Google Scholar]
- 10.Gefflaut T, Assaf Z. Sancelme M. Preparation of glutamate analogues by enzymatic transamination. Methods Mol Biol. 2012;794:55–72. doi: 10.1007/978-1-61779-331-8_4. [DOI] [PubMed] [Google Scholar]
- 11.Braunstein AE. Shemyakin MM. The theory of processes of amino acid metabolism catalysed by pyridoxal 5′-phosphate-dependent enzymes. Biokhimia. 1953;18:393–411. [Google Scholar]
- 12.Metzler DE, Ikawa M. Snell EE. A general mechanism for vitamin B6-catalyzed reactions. J Am Chem Soc. 1954;76:648–652. [Google Scholar]
- 13.Sayer C, Bommer M, Isupov M, Ward J. Littlechild J. Crystal structure and substrate specificity of the thermophilic serine:pyruvate aminotransferase from Sulfolobus solfataricus. Acta Crystallogr D Biol Crystallogr. 2012;68:763–772. doi: 10.1107/S0907444912011274. [DOI] [PubMed] [Google Scholar]
- 14.Ward J. Wohlgemuth R. High-yield biocatalytic amination reactions in organic synthesis. Curr Org Chem. 2010;14:1914–1927. [Google Scholar]
- 15.Malik MS, Park ES. Shin JS. Features and technical applications of ω-transaminases. Appl Microbiol Biotechnol. 2012;94:1163–1171. doi: 10.1007/s00253-012-4103-3. [DOI] [PubMed] [Google Scholar]
- 16.Shin JS, Yun H, Jang JW, Park I. Kim BG. Purification, characterization, and molecular cloning of a novel amine:pyruvate transaminase from Vibrio fluvialis JS17. Appl Microbiol Biotechnol. 2003;61:463–471. doi: 10.1007/s00253-003-1250-6. [DOI] [PubMed] [Google Scholar]
- 17.Kaulmann U, Smithies K, Smith MEB, Hailes HC. Ward JM. Substrate spectrum of ω-transaminase from Chromobacterium violaceum DSM30191 and its potential for biocatalysis. Enzyme Microb Technol. 2007;41:628–637. [Google Scholar]
- 18.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic Acids Res. 2012;14:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science. 2010;329:305–309. doi: 10.1126/science.1188934. [DOI] [PubMed] [Google Scholar]
- 20.Mutti FG, Fuchs CS, Pressnitz D, Sattler JH. Kroutil W. Stereoselectivity of four (R)-selective transaminases for the asymmetric amination of ketones. Adv Synth Catal. 2011;353:3227–3233. [Google Scholar]
- 21.Pressnitz D, Fuchs CS, Sattler JH, Knaus T, Macheroux P, Mutti FG. Kroutil W. Asymmetric amination of tetralone and chromanone derivatives employing w-transaminases. ACS Catal. 2013;3:555–559. [Google Scholar]
- 22.Höhne M, Schatzle S, Jochens H, Robins K. Bornscheuer UT. Rational assignment of key motifs for function guides in silico enzyme identification. Nat Chem Biol. 2010;6:807–813. doi: 10.1038/nchembio.447. [DOI] [PubMed] [Google Scholar]
- 23.Humble MS, Cassimjee KE, Håkansson M, Kimbung YR, Walse B, Abedi V, Federsel HJ, Berglund P. Logan DT. Crystal structures of the Chromobacterium violaceum w-transaminase reveal major structural rearrangements upon binding of coenzyme PLP. FEBS J. 2012;279:779–792. doi: 10.1111/j.1742-4658.2012.08468.x. [DOI] [PubMed] [Google Scholar]
- 24.Sayer C, Isupov MN, Westlake A. Littlechild JA. Structural studies of Pseudomonas and Chromobacterium w-aminotransferases provide insights into their differing substrate specificity. Acta Crystallogr D Biol Crystallogr. 2013;69:564–576. doi: 10.1107/S0907444912051670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomsen M, Skalden L, Palm GJ, Höhne M, Bornscheuer UT. Hinrichs W. Crystallization and preliminary X-ray diffraction studies of the (R)-selective amine transaminase from Aspergillus fumigatus. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2013;69:1415–1417. doi: 10.1107/S1744309113030923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rando RR. Mechanism of the irreversible inhibition of GABA transaminase by the neurotoxin gabaculine. Biochemistry. 1977;16:4604–4610. doi: 10.1021/bi00640a012. [DOI] [PubMed] [Google Scholar]
- 27.VanEtten HD. Identification of additional habitats of Nectria haematococca mating population VI. Phytopathology. 1978;68:1552–1556. [Google Scholar]
- 28.Coleman JJ. The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 2009;5:e1000618. doi: 10.1371/journal.pgen.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hopwood J, Truppo MD, Turner NJ. Lloyd RC. A fast and sensitive assay for measuring the activity and enantioselectivity of transaminases. Chem Commun. 2011;47:773–775. doi: 10.1039/c0cc02919j. [DOI] [PubMed] [Google Scholar]
- 30.Fuchs M, Koszelewski D, Tauber K, Kroutil W. Faber K. Chemoenzymatic asymmetric total synthesis of (S)-rivastigmine using omega-transaminases. Chem Commun. 2010;46:5500–5502. doi: 10.1039/c0cc00585a. [DOI] [PubMed] [Google Scholar]
- 31.Laskowski RA, MacArthur MW, Moss DS. Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 32.Peisach D, Chipman DM, Van Ophem PW, Manning JM. Ringe D. Crystallographic study of steps along the reaction pathway of D-amino acid aminotransferase. Biochemistry. 1998;37:4958–4967. doi: 10.1021/bi972884d. [DOI] [PubMed] [Google Scholar]
- 33.Okada K, Hirotsu K, Hayashi H. Kagamiyama H. Structures of Escherichia coli branched-chain amino acid aminotransferase and its complexes with 4-methylvalerate and 2-methylleucine: induced fit and substrate recognition of the enzyme. Biochemistry. 2001;40:7453–7463. doi: 10.1021/bi010384l. [DOI] [PubMed] [Google Scholar]
- 34.Dunathan HC. Conformation and reaction specificity in pyridoxal phosphate enzymes. Proc Natl Acad Sci USA. 1966;55:712–716. doi: 10.1073/pnas.55.4.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altschul S, Gish W, Miller W, Myers E. Lipman D. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 36.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 37.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winter G, Lobley CMC. Prince SM. Decision making in xia2. Acta Crystallogr D Biol Crystallogr. 2013;69:1260–1273. doi: 10.1107/S0907444913015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vagin A. Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 41.Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F. Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langer GG, Hazledine S, Wiegels T, Carolan C. Lamzin VS. Visual automated macromolecular model building. Acta Crystallogr D Biol Crystallogr. 2013;69:635–641. doi: 10.1107/S0907444913000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P, Lohkamp B, Scott WG. Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific; 2002. http://www.pymol.org. [Google Scholar]
- 45.Gouet P, Courcelle E, Stuart DI. Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 46.Vaguine AA, Richelle J. Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D Biol Crystallogr. 1999;55:191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representation of synthetic gene design.
Enzyme activity of the Nectria TAm synthetic genes.
Chiral HPLC details for chiral amines prepared using Nectria TAm.