

Abstract
The aim of this study was to clarify the significance of DNA methylation alterations during lung carcinogenesis. Infinium assay was performed using 139 paired samples of non-cancerous lung tissue (N) and tumorous tissue (T) from a learning cohort of patients with lung adenocarcinomas (LADCs). Fifty paired N and T samples from a validation cohort were also analyzed. DNA methylation alterations on 1,928 probes occurred in N samples relative to normal lung tissue from patients without primary lung tumors, and were inherited by, or strengthened in, T samples. Unsupervised hierarchical clustering using DNA methylation levels in N samples on all 26,447 probes subclustered patients into Cluster I (n = 32), Cluster II (n = 35) and Cluster III (n = 72). LADCs in Cluster I developed from the inflammatory background in chronic obstructive pulmonary disease (COPD) in heavy smokers and were locally invasive. Most patients in Cluster II were non-smokers and had a favorable outcome. LADCs in Cluster III developed in light smokers were most aggressive (frequently showing lymphatic and blood vessel invasion, lymph node metastasis and an advanced pathological stage), and had a poor outcome. DNA methylation levels of hallmark genes for each cluster, such as IRX2, HOXD8, SPARCL1, RGS5 and EI24, were again correlated with clinicopathological characteristics in the validation cohort. DNA methylation profiles reflecting carcinogenetic factors such as smoking and COPD appear to be established in non-cancerous lung tissue from patients with LADCs and may determine the aggressiveness of tumors developing in individual patients, and thus patient outcome.
Keywords: DNA methylation, infinium assay, lung adenocarcinoma, cigarette smoking, chronic obstructive pulmonary disease
What's new?
While genetic abnormalities are well studied in human cancers, epigenetic changes, especially in the early stages of carcinogenesis, remain largely unknown. Here, the authors perform a genome-wide analysis focusing on DNA methylation profiles in “normal” lung tissue adjacent to lung adenocarcinomas. Using single-CpG-resolution Infinium assays, they identify distinct DNA methylation profiles clustering with specific risk factors such as cigarette smoking, inflammation and chronic obstructive pulmonary disease. The authors speculate that these epigenetic profiles detected in the neighboring cells may influence the aggressiveness of tumors developing in individual patients and may thus help predict disease outcome.
Lung cancer is the leading cause of cancer-related death worldwide,1 and adenocarcinoma is the most common histological subtype, both in smokers and non-smokers. Differences in the genetic features of lung adenocarcinomas (LADCs) between smokers and non-smokers have been described.2 LADCs arising in individuals who have never smoked, especially women and those of East Asian ethnicity, have been reported to have EGFR mutation and are thus responsive to tyrosine kinase inhibitors, whereas those arising in smokers frequently show oncogenic missense mutations in KRAS. EGFR and KRAS mutations in LADCs are almost entirely mutually exclusive. With regard to TP53 mutations, G:C to T:A transversions and A:T to G:C transitions at CpG sites are characteristic of smoking-related lung cancers, whereas G:C to A:T transitions at non-CpG sites are associated with lung cancers in individuals who have never smoked. However, the molecular changes responsible for the development of LADCs in both smokers and non-smokers, especially at the very early stages, are not yet fully understood.
As well as genetic abnormalities, epigenetic changes have been described in human cancers,3 one of the most consistent being DNA methylation alterations. In LADCs, silencing of the RASSF1A, CDKN2A, RARβ, MGMT, APC, DAPK, FHIT and CDH13 genes due to DNA hypermethylation around their promoter regions has been frequently reported.4 Moreover, in various organs, DNA methylation alterations are characteristically observed even at the precancerous stage5–7: we and other groups have reported aberrant DNA methylation of specific genes or chromosomal loci in non-cancerous lung tissue from LADC patients, or in lung tissue from cancer-free smokers.4,8,9 DNA methylation alterations of tumor-related genes have been reported in airway epithelial cells from smokers. 8,10,11 Recently, methylome analysis using single-CpG-resolution Infinium assay has been introduced.12 Although studies of lung cancers using the Infinium assay by Selamat et al.13 and Lockwood et al.14 did not focus on non-cancerous lung tissue obtained from the same patients, our previous study revealed that alterations of DNA methylation status in adjacent lung tissue are not nonsensical, but in fact create alterations in the expression of mRNAs for specific genes in cancerous tissue developing in the same individual patients.15
It is known that DNA methylation profiles at the precancerous stage are determined by carcinogenetic factors. For examples, distinct DNA methylation profiles at the chronic hepatitis or liver cirrhosis stage as a precancerous condition for hepatocellular carcinoma16,17 or those in the stomach mucosa harboring Helicobacter pylori infection as a precancerous condition for stomach adenocarcinoma have been reported.18 In this study, to further understand the significance of DNA methylation alterations during lung carcinogenesis, we examined correlations between epigenetic clustering of patients with LADCs based on DNA methylation profiles in adjacent lung tissue and carcinogenetic factors such as cigarette smoking and chronic obstructive lung disease (COPD).
Material and Methods
Patients and tissue samples
As a learning cohort, 139 paired samples of non-cancerous lung tissue (N) and the corresponding tumorous tissue (T) were obtained from patients with primary LADCs who underwent lung resection at the National Cancer Center Hospital, Japan, between December 2000 and May 2008. None of these patients had received any preoperative treatment. Sixty-nine patients were males and seventy were females with a median age of 60 years (range, 30–76 years). Clinicopathological parameters in the learning cohort are summarized in Supporting Information Table S1. Pleural anthracosis, which mainly reflects the cumulative effects of smoking history, was evaluated macroscopically according to the criteria described previously.19 Presence or absence of emphysematous change, respiratory bronchiolitis, interstitial fibrosis20,21 and atypical adenomatous hyperplasia (AAH, a precancerous lesion for LADC)22,23 was evaluated microscopically on the basis of the criteria described previously. Histological diagnosis and grading were based on the 2004 World Health Organization classification.24 When, within a tumor, black dusty material25 is seen to have accumulated in foci of active fibroblast proliferation, reflecting active cancer–stromal interaction associated with a poorer outcome in LADC patients,26 the tumor is considered to be tumor anthracosis-positive (Supporting Information Fig. S1). All the tumors were classified according to the pathological tumor-node-metastasis (TNM) classification.27 Recurrence was diagnosed by clinicians on the basis of physical examination and imaging modalities such as computed tomography, magnetic resonance imaging, scintigraphy or positron-emission tomography, and sometimes confirmed histopathologically by biopsy. A proportion of this cohort had also been included in our previous study focusing on recurrence-related genes.15
DNA methylation profiles of the 139 N samples and 139 T samples were compared with previously reported DNA methylation profiles of 36 samples of normal lung tissue (C) obtained from specimens surgically resected from 36 patients without any primary lung tumors.15 Briefly, 22 of these patients were males and 14 were females, with a median age of 63 years (range, 27–83 years). Thirty-five had undergone lung resection for metastatic lesions from primary cancers of the colon, rectum, kidney, urinary bladder, thyroid, breast, pancreas, ampulla of Vater and salivary gland, osteosarcoma, synovial sarcoma, leiomyosarcoma, rhabdomyosarcoma, liposarcoma, dermatofibrosarcoma and myxofibrosarcoma. The remaining one patient had undergone chest wall resection for lipoma with removal of adjacent lung tissue.
As a validation cohort, 50 paired samples of N and the corresponding T were obtained from patients with primary LADCs who underwent lung resection at the National Cancer Center Hospital, Japan, between December 1997 and May 2000. None of these patients had received any preoperative treatment. Thirty-three patients were males and seventeen were females with a median age of 63 years (range, 40–81 years). Clinicopathological parameters in the validation cohort are summarized in Supporting Information Table S1.
Tissue specimens were provided by the National Cancer Center Biobank, Japan. This study was approved by the Ethics Committee of the National Cancer Center, Japan, and was performed in accordance with the Declaration of Helsinki. All patients included in this study provided written informed consent.
Infinium assay
Genomic DNA was extracted from all tissue samples using a QIAamp DNA Mini kit (Qiagen, Valencia, CA). Five-hundred-nanogram aliquots of DNA were subjected to bisulfite conversion using an EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA). Subsequently, DNA methylation status at 27,578 CpG loci was examined at single-CpG resolution using the Infinium HumanMethylation27 Bead Array (Illumina, San Diego, CA). This array contains CpG sites located mainly within the proximal promoter regions of the transcription start sites of 14,475 consensus coding sequences in the National Center for Biotechnology Information Database. An Evo robot (Tecan, Männedorf, Switzerland) was used for automated sample processing. After whole-genome amplification and hybridization, the specifically hybridized DNA was fluorescence-labeled by a single-base extension reaction and detected using a BeadScan reader (Illumina) in accordance with the manufacturer's protocols. The data were then assembled using GenomeStudio methylation software (Illumina). At each CpG site, the ratio of the fluorescence signal was measured using a methylated probe relative to the sum of the methylated and unmethylated probes, that is, the so-called β-value, which ranges from 0.00 to 1.00, reflecting the methylation level of an individual CpG site.
The reliability of DNA methylation levels (β-values) determined by Infinium assay has been verified in our previous studies.7,15 In addition, DNA methylation levels of the representative genes (NUPR1, EVI2B, CASP8 and KRTAP11-1 genes) based on the Infinium assay in representative samples included in this study were verified using the quantitative pyrosequencing method (Supporting Information Fig. S2), thus confirming the reliability of the Infinium assay. Moreover, we compared the DNA methylation levels of 545 representative Infinium probes, whose β values were unrelated to the clinicopathological parameters of the tumors or patient outcome (recurrence or death), between all samples in the learning cohort (obtained between December 2000 and May 2008) and the validation cohort (obtained between December 1997 and May 2000). No significant differences in DNA methylation levels between the learning and validation cohorts were observed in any of the 545 probes examined (Supporting Information Fig. S3). Supporting Information Figure S3 clearly indicates the excellent concordance of DNA methylation status between the two cohorts (r = 1.000, p < 2.20 × 10−16), confirming that the epigenetic changes did not degrade over time.
Statistics
In the Infinium assay, all CpG sites on chromosomes X and Y were excluded, to avoid any gender-specific methylation bias. In addition, the call proportions (p-value of <0.01 for detection of signals above the background) for 39 probes (shown in Supporting Information Table S2) in 36 C samples, 139 N samples and 139 corresponding T samples in the learning cohort were less than 90%. As such a low proportion may be attributable to polymorphism at the probe CpG sites, these 39 probes were excluded from the present assay, leaving a final total of 26,447 autosomal CpG sites.
Infinium probes showing significant differences in DNA methylation levels between the 36 C samples and 139 N samples in the learning cohort were identified by the Welch's t-test. Ordered differences from 36 C to 139 N, and then to 139 T samples themselves in the learning cohort were examined by the Jonckheere–Terpstra trend test. A false discovery rate (FDR) of q = 0.01 was considered significant. Unsupervised hierarchical clustering (Euclidean distance, Ward method) based on DNA methylation levels of the 139 N samples in the learning cohort was performed. Correlations between clusters of patients and clinicopathological parameters were examined using Kruskal–Wallis test, Fisher's exact test and Kruskal–Wallis exact test at a significance level of p < 0.05. Survival curves of patients belonging to each cluster were calculated by the Kaplan–Meier method, and the differences were compared by the Log-rank test. The hallmark genes discriminating the clusters were identified by Welch's t-test. Correlations between DNA methylation levels of such hallmark genes in N samples and clinicopathological parameters of patients in the validation cohort were examined using Welch's t-test and ANOVA test at a significance level of p < 0.05. All statistical analyses were performed using programming language R.
Results
DNA methylation alterations during lung carcinogenesis
(i) Welch's t-test revealed that DNA methylation levels on the 3,778 probes were already altered in N samples in the learning cohort relative to those in C samples (FDR, q = 0.01, Table 1A). (ii) The Jonckheere–Terpstra trend test revealed ordered differences in the DNA methylation level from the 39 C samples to the 139 N samples, and then to the 139 T samples themselves in the learning cohort on the 12,368 probes (FDR, q = 0.01, Table 1B). (iii) Among the probes, 1,928 satisfied the above criteria (i) and (ii): DNA methylation alterations on the 1,928 probes occurred even in N samples relative to C samples, and such DNA methylation alterations were inherited by, or strengthened in, the T samples (Table 1C).
Table 1.
DNA methylation alterations during lung carcinogenesis
| The number of probes showing DNA hypermethylation and DNA hypomethylation | |
|---|---|
| (A) The probes on which DNA methylation levels were altered in 139 samples of non-cancerous lung tissue (N) obtained from patients with lung adenocarcinomas (LADCs) in the learning cohort relative to those in 39 samples of normal lung tissue (C) obtained from patients without any primary lung tumors. (Welch's t-test, False discovery rate [FDR] q = 0.01) | |
| DNA hypermethylation (βC < βN) | 1,526 |
| DNA hypomethylation (βC > βN) | 2,252 |
| Total | 3,778 |
| (B) The probes on which DNA methylation levels showed ordered differences from 39 C samples to 139 N samples, and then to 139 tumorous tissue (T) samples in the learning cohort. (Jonckheere–Terpstra trend test, FDR q = 0.01) | |
| DNA hypermethylation (βC < βN < βT, βC < βN ≒ βT or βC≒ βN < βT) | 6,460 |
| DNA hypomethylation (βC > βN > βT, βC > βN ≒ βT or βC ≒ βN > βT) | 5,908 |
| Total | 12,368 |
| (C) The probes satisfying both of the above criteria (A) and (B): DNA methylation alterations on these probes occurred even in N samples relative to C samples, and such DNA methylation alterations were inherited by, or strengthened in, T samples. | |
| DNA hypermethylation (βC < βN < βT or βC < βN ≒ βT) | 484 |
| DNA hypomethylation (βC > βN > βT or βC > βN ≒ βT) | 1,444 |
| Total | 1,928 |
Epigenetic clustering of LADCs based on DNA methylation profiles in N samples
As DNA methylation alterations already occurred in Ns, unsupervised hierarchical clustering using DNA methylation levels in N samples (βN) on all 26,447 probes was performed in 139 patients with LADCs in the learning cohort. Such clustering based on DNA methylation profiles in N samples subclustered 139 patients in the learning cohort into Cluster I (n = 32), Cluster II (n = 35) and Cluster III (n = 72, Fig. 1a). The clinicopathological parameters of the patients in these clusters are summarized in Table 2.
Figure 1.

(a) Unsupervised hierarchical clustering (Euclidean distance, Ward method) using DNA methylation levels on all 26,447 probes in samples of non-cancerous lung tissue (N) from 139 patients with lung adenocarcinomas in the learning cohort. Based on DNA methylation status in adjacent lung tissue, 139 patients were subclustered into Cluster I (n = 32), Cluster II (n = 35) and Cluster III (n = 72). Correlations between this epigenetic clustering and clinicopathological parameters of the patients are summarized in Table 2. (b) Kaplan–Meier survival curves of patients belonging to Clusters I, II and III. The period covered ranged from 196 to 3,957 days (mean, 1,634 days). The cancer-free (p = 1.24 × 10−4) and overall (p = 1.58 × 10−2) survival rates of patients in Cluster III were significantly lower than those of patients in Cluster II (log-rank test).
Table 2.
Correlation between epigenetic clustering of patients with lung adenocarcinomas based on DNA methylation profiles in adjacent lung tissue and clinicopathological parameters
| Clinicopathological parameters | Cluster I (n = 32) | Cluster II (n = 35) | Cluster III (n = 72) | P1 | |
|---|---|---|---|---|---|
| Patients | Age (year) | ||||
| Median | 64 | 57 | 60 | 2.03 × 10−22 | |
| Interquartile range | 59–68 | 54–62 | 53–64 | ||
| Sex | |||||
| Male | 24 | 11 | 34 | 1.35 × 10−33 | |
| Female | 8 | 24 | 38 | ||
| Smoking history (number of cigarettes smoked per day × year index) | |||||
| Median | 810 | 0 | 0 | 8.80 × 10−62 | |
| Interquartile range | 195–1,113 | 0–140 | 0–635 | ||
| Adjacent lung tissue | |||||
| Pleural anthracosis | |||||
| G1 | 13 | 24 | 48 | 2.46 × 10−24 | |
| G2-3 | 19 | 11 | 24 | ||
| Emphysematic change | |||||
| Negative | 8 | 24 | 46 | 2.50 × 10−44 | |
| Positive | 24 | 11 | 26 | ||
| Respiratory bronchiolitis | |||||
| Negative | 2 | 14 | 10 | 2.80 × 10−34 | |
| Positive | 22 | 21 | 58 | ||
| Interstitial fibrosis | |||||
| Negative | 24 | 35 | 68 | 5.72 × 10−44 | |
| Positive | 8 | 0 | 4 | ||
| Obstructive ventilation impairment | |||||
| Forced expiratory volume in 1 sec (FEV1): forced vital capacity (FVC) ≥0.70 | 24 | 34 | 65 | 9.86 × 10−34 | |
| FEV1:FVC <0.70 | |||||
| FEV1 ≥80% of predicted value | 4 | 1 | 6 | ||
| FEV1 <80% but ≥50% of predicted value | 4 | 0 | 1 | ||
| Atypical adenomatous hyperplasia | |||||
| Absence | 30 | 30 | 65 | 5.72 × 10−1 4 | |
| Presence | 2 | 5 | 7 | ||
| Lung adenocarcinomas | |||||
| Tumor diameter (cm) | |||||
| Median | 3.4 | 2.3 | 3.1 | 1.64 × 10−44 | |
| Interquartile range | 2.5–4.9 | 2.1–2.9 | 2.5–4.5 | ||
| Tumor stage | |||||
| T1a-T1b | 6 | 19 | 19 | 1.60 × 10−44 | |
| T2a-T2b | 12 | 14 | 39 | ||
| T3-4 | 14 | 2 | 14 | ||
| Histological grades | |||||
| G1 | 8 | 20 | 26 | 2.37 × 10−34 | |
| G2 | 11 | 12 | 34 | ||
| G3 | 13 | 3 | 12 | ||
| Tumor anthracosis | |||||
| Negative | 6 | 20 | 39 | 1.70 × 10−34 | |
| Positive | 25 | 15 | 33 | ||
| Pleural invasion | |||||
| Negative | 12 | 22 | 35 | 9.62 × 10−34 | |
| Invasion to the visceral pleura beyond the elastic fiber | 6 | 9 | 17 | ||
| Invasion to the surface of the visceral pleura | 4 | 4 | 15 | ||
| Invation to the parietal pleura | 10 | 0 | 5 | ||
| Lymphatic vessel invasion | |||||
| Negative | 9 | 18 | 16 | 8.54 × 10−34 | |
| Positive | 23 | 17 | 56 | ||
| Blood vessel invasion | |||||
| Negative | 7 | 18 | 15 | 3.02 × 10−34 | |
| Positive | 25 | 17 | 57 | ||
| Nodal status | |||||
| N0 | 17 | 26 | 25 | 8.72 × 10−54 | |
| N1 | 10 | 6 | 18 | ||
| N2-3 | 5 | 3 | 29 | ||
| Metastatic status | |||||
| M0 | 31 | 34 | 66 | 4.40 × 10−1 4 | |
| M1a-1b | 1 | 1 | 1 | ||
| Pathological Tumor-Node-Metastasis stage | |||||
| IA-IB | 5 | 24 | 18 | 4.36 × 10−64 | |
| IIA-IIB | 21 | 7 | 19 | ||
| IIIA-IV | 6 | 4 | 35 | ||
Pvalues of <0.05 are underlined.
Kruskal-Wallis test.
Fisher's exact test.
Kruskal-Wallis exact test.
Most of the patients in Cluster I were heavy smokers (median number of cigarettes smoked per day × year index: 810) and frequently showed severe pleural anthracosis, which mainly reflects the cumulative effects of smoking.19 With regard to the non-cancerous lung tissue, patients belonging to Cluster I frequently showed histological findings compatible with emphysema, respiratory bronchiolitis and interstitial fibrosis, and they frequently suffered from obstructive ventilation impairment (Table 2). In Cluster I, LADCs with a large diameter, a progressed T stage, a high histological grade and frequent pleural invasion were accumulated (Table 2). In addition, tumor anthracosis reflecting active cancer–stromal interaction26 was frequent in Cluster I (Table 2). These data indicated that LADCs in Cluster I were locally invasive tumors.
Most of the patients in Cluster II were non-smokers (median number of cigarettes smoked per day × year index: 0) and less frequently showed emphysematous changes in their adjacent lung tissue (Table 2). The correlation between epigenetic clustering of LADCs and patient age and sex may be attributable to the fact that younger female non-smokers28 were accumulated in Cluster II. LADCs in Cluster II showed less aggressive clinicopathological features (Table 2).
Most of the patients in Cluster III were light smokers and tended to have a lower incidence of emphysematous changes in their adjacent lung tissue (Table 2). LADCs in Cluster III frequently showed lymphatic vessel invasion, blood vessel invasion, high N stage and high TNM stage (Table 2), indicating that they were the most aggressive tumors.
Figure 1b shows the Kaplan–Meier survival curves of patients belonging to Clusters I, II and III. The period covered ranged from 196 to 3,957 days (mean, 1,634 days). The cancer-free and overall survival rates of patients in Cluster III were significantly lower than those of patients in Cluster II (p = 1.24 × 10−4 and p = 1.58 × 10−2, respectively, Fig. 1b).
DNA methylation profiles of N samples belonging to each cluster in the learning cohort
Scattergrams of average DNA methylation levels in N samples (averageβN) of patients belonging to Clusters I, II and III and average DNA methylation levels in C samples (averageβC) for all 26,447 probes are shown in Figure 2. In Cluster I, DNA methylation levels on probes normally showing a low or medium degree of DNA methylation (average βC < 0.6) were elevated in N samples relative to C samples, and DNA methylation levels on probes normally showing a high or medium degree of DNA methylation (average βC > 0.3) were reduced in N samples relative to C samples (Fig. 2a). In Cluster II, DNA methylation levels on probes normally showing a low degree of DNA methylation (average βC < 0.2) were elevated in N samples relative to C samples, and DNA methylation levels on probes normally showing a high degree of DNA methylation (average βC > 0.7) were reduced in N samples relative to C samples (Fig. 2b). In Cluster III, DNA methylation levels on probes normally showing a high or medium degree of DNA methylation (average βC > 0.3) were reduced in N samples relative to C samples (Fig. 2c).
Figure 2.
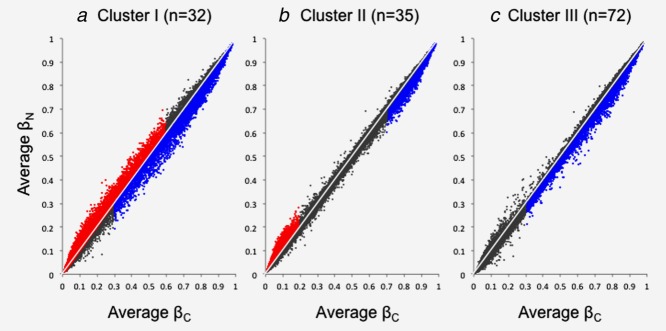
Distribution of average DNA methylation levels on all 26,447 probes of non-cancerous lung tissue (N) samples obtained from patients with lung adenocarcinomas belonging to Clusters I (a), II (b) and III (c) and 36 samples of normal lung tissue (C) obtained from patients without any primary lung tumors. (a) In Cluster I, DNA methylation levels on probes normally showing a lower or medium degree of DNA methylation (averageβC < 0.6, red) were elevated in N samples relative to C samples, and DNA methylation levels on probes normally showing a higher or medium degree of DNA methylation (average βC > 0.3, blue) were reduced in N samples relative to C samples. (b) In Cluster II, DNA methylation levels on probes normally showing a lower degree of DNA methylation (average βC <0.2,red) were elevated in N samples relative to C samples, and DNA methylation levels on probes normally showing a higher degree of DNA methylation (average βC >0.7,blue) were reduced in N samples relative to C samples. (c) In Cluster III, DNA methylation levels on probes normally showing a higher or medium degree of DNA methylation (average βC >0.3,blue) were reduced in N samples relative to C samples.
Hallmark CpG sites for each cluster in the learning cohort
One hundred sixteen CpG sites were identified as hallmarks of the DNA methylation profile (Fig. 2a) of N samples belonging to Cluster I: on these 116 CpG sites, the average βN–C values in Cluster I were significantly different from those in Clusters II and III (Welch's t-test, p < 1 × 10−3) and the average βN–C value in Cluster I was 0.1 or more higher or lower than those in Clusters II and III (Table 3A and Supporting Information Table S3). One CpG site was identified as a hallmark for the DNA methylation profile (Fig. 2b) of N samples belonging to Cluster II: on the CpG site, the average βN–C value in Cluster II was significantly different from that in Clusters I and III (Welch's t-test, p < 1 × 10−3) and the average βN–C value in Cluster II was 0.1 or more higher than those in Clusters I and III (Table 3B). Four CpG sites were identified as a hallmark for the DNA methylation profile (Fig. 2c) of N samples belonging to Cluster III: on the four CpG sites, average βN–C values in Cluster III were significantly different from those in Clusters I and II (Welch's t-test, p < 1 × 10−3) and average βN–C values in Cluster III were 0.1 or more higher or lower than those in Clusters I and II (Table 3C). In 119 of the 120 CpG sites in Table 3 or Supporting Information Table S3, which were identified based on the DNA methylation profiles in N samples, stepwise DNA methylation alterations from C to N, and then to T samples were revealed by Jonckheere–Terpstra trend test (Table 3 and Supporting Information Table S3).
Table 3.
Genes for which DNA methylation levels were hallmarks for Clusters I, II and III in the learning cohort
| (A) Hallmark genes for Cluster I | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Target ID 1 | Chrom2 | Position3 | Gene symbol | DNA methylation level in non-cancerous lung tissue (N) samples4 (mean ± SD) | p-Value of Welch's t-test (I vs. II and III)5 | Δβ (I-II and III)6 | p-Value of Jonckheere–Terpstra trend test in I7 | ||
| Cluster I | Cluster II | Cluster III | |||||||
| cg20249919 | 15 | 102,029,706 | PCSK6 | 0.091 ± 0.188 | −0.047 ± 0.109 | −0.070 ± 0.125 | 9.28 × 10−5 | 0.153 | 6.51 × 10−4 (Hyper) |
| cg23349790 | 1 | 18,434,576 | IGSF21 | 0.114 ± 0.133 | −0.011 ± 0.111 | −0.032 ± 0.108 | 2.41 × 10−6 | 0.139 | 4.43 × 10−9 (Hyper) |
| cg22285621 | 11 | 67,071,322 | SSH3 | 0.103 ± 0.116 | −0.031 ± 0.075 | −0.033 ± 0.082 | 2.32 × 10−7 | 0.136 | 3.69 × 10−7 (Hyper) |
| cg15433631 | 5 | 2,751,541 | IRX2 | 0.123 ± 0.083 | −0.007 ± 0.073 | 0.000 ± 0.070 | 8.88 × 10−10 | 0.125 | 6.60 × 10−8 (Hyper) |
| cg21949305 | 22 | 24,828,655 | ADORA2A,CYTSA | 0.109 ± 0.053 | −0.015 ± 0.040 | −0.010 ± 0.052 | 2.91 × 10−15 | 0.121 | 0 (Hyper) |
| cg10942056 | 1 | 223,101,848 | DISP1 | 0.095 ± 0.059 | −0.027 ± 0.039 | −0.026 ± 0.048 | 1.59 × 10−13 | 0.121 | 4.05 × 10−13 (Hyper) |
| cg15149645 | 16 | 28,550,619 | NUPR1 | 0.090 ± 0.067 | −0.023 ± 0.044 | −0.033 ± 0.058 | 7.39 × 10−12 | 0.12 | 1.36 × 10−12 (Hyper) |
| cg06954481 | 2 | 237,076,497 | GBX2 | 0.096 ± 0.111 | −0.012 ± 0.051 | −0.029 ± 0.052 | 1.02 × 10−6 | 0.119 | 1.25 × 10−7 (Hyper) |
| cg21250978 | 7 | 106,684,541 | PRKAR2B | 0.088 ± 0.060 | −0.026 ± 0.044 | −0.031 ± 0.056 | 4.25 × 10−13 | 0.118 | 6.13 × 10−9 (Hyper) |
| cg22418909 | 8 | 41,166,738 | SFRP1 | 0.091 ± 0.082 | −0.023 ± 0.055 | −0.029 ± 0.052 | 2.38 × 10−9 | 0.118 | 1.22 × 10−10 (Hyper) |
| cg26200585 | 19 | 40,919,245 | PRX | 0.099 ± 0.059 | −0.019 ± 0.040 | −0.019 ± 0.054 | 2.44 × 10−13 | 0.118 | 0 (Hyper) |
| cg24396745 | 15 | 73,660,614 | HCN4 | 0.096 ± 0.098 | −0.022 ± 0.073 | −0.015 ± 0.089 | 3.31 × 10−7 | 0.114 | 1.96 × 10−8 (Hyper) |
| cg04330449 | 5 | 134,871,166 | NEUROG1 | 0.098 ± 0.080 | −0.001 ± 0.061 | −0.019 ± 0.051 | 5.89 × 10−9 | 0.111 | 1.08 × 10−13 (Hyper) |
| cg19589427 | 1 | 173,019,720 | TNFSF18 | 0.076 ± 0.073 | −0.036 ± 0.039 | −0.032 ± 0.051 | 9.08 × 10−10 | 0.11 | 7.78 × 10−10 (Hyper) |
| cg16731240 | 19 | 52,391,250 | ZNF577 | 0.090 ± 0.105 | −0.015 ± 0.072 | −0.022 ± 0.061 | 1.87 × 10−6 | 0.11 | 0 (Hyper) |
| cg03544320 | 4 | 5,894,691 | CRMP1 | 0.088 ± 0.108 | −0.016 ± 0.105 | −0.022 ± 0.101 | 7.22 × 10−6 | 0.108 | 1.61 × 10−10 (Hyper) |
| cg12864235 | 5 | 27,038,782 | CDH9 | 0.092 ± 0.059 | −0.011 ± 0.037 | −0.018 ± 0.040 | 3.56 × 10−12 | 0.108 | 2.37 × 10−13 (Hyper) |
| cg15898840 | 7 | 45,960,834 | IGFBP3 | 0.102 ± 0.095 | −0.001 ± 0.052 | −0.008 ± 0.058 | 4.67 × 10−7 | 0.107 | 2.02 × 10−8 (Hyper) |
| cg08044694 | 19 | 15,391,927 | BRD4 | 0.068 ± 0.072 | −0.029 ± 0.034 | −0.044 ± 0.042 | 1.55 × 10−9 | 0.107 | 1.76 × 10−8 (Hyper) |
| cg03734874 | 14 | 105,071,382 | TMEM179 | 0.099 ± 0.068 | 0.001 ± 0.056 | −0.012 ± 0.055 | 3.06 × 10−10 | 0.106 | 4.39 × 10−13 (Hyper) |
| cg10599444 | 14 | 23,305,941 | MMP14 | 0.064 ± 0.065 | −0.039 ± 0.040 | −0.044 ± 0.056 | 8.35 × 10−11 | 0.106 | 7.42 × 10−7 (Hyper) |
| cg24133115 | 6 | 166,075,520 | PDE10A | 0.096 ± 0.071 | −0.007 ± 0.054 | −0.010 ± 0.046 | 1.50 × 10−9 | 0.105 | 9.66 × 10−10 (Hyper) |
| cg12594641 | 2 | 150,187,223 | LYPD6 | 0.111 ± 0.064 | 0.011 ± 0.071 | 0.004 ± 0.061 | 1.05 × 10−10 | 0.105 | 6.56 × 10−7 (Hyper) |
| cg05724065 | 7 | 56,160,528 | PHKG1 | 0.082 ± 0.053 | −0.017 ± 0.029 | −0.026 ± 0.044 | 3.01 × 10−13 | 0.105 | 4.43 × 10−11 (Hyper) |
| cg19466563 | 4 | 88,450,506 | SPARCL1 | 0.081 ± 0.053 | −0.018 ± 0.027 | −0.027 ± 0.042 | 4.93 × 10−13 | 0.104 | 0 (Hyper) |
| cg24433189 | 16 | 1,128,689 | SSTR5 | 0.092 ± 0.056 | −0.005 ± 0.052 | −0.015 ± 0.064 | 2.58 × 10−12 | 0.104 | 9.78 × 10−9 (Hyper) |
| cg24453664 | 11 | 33,758,413 | CD59 | 0.069 ± 0.066 | −0.031 ± 0.033 | −0.036 ± 0.046 | 3.23 × 10−10 | 0.103 | 9.78 × 10−9 (Hyper) |
| cg26609631 | 13 | 28,366,814 | GSX1 | 0.077 ± 0.081 | −0.025 ± 0.063 | −0.026 ± 0.057 | 4.72 × 10−8 | 0.103 | 1.73 × 10−11 (Hyper) |
| cg10604646 | 1 | 163,172,649 | RGS5 | 0.086 ± 0.041 | −0.029 ± 0.059 | −0.009 ± 0.060 | 4.09 × 10−17 | 0.102 | 2.68 × 10−14 (Hyper) |
| cg03355526 | 5 | 178,368,415 | ZNF454 | 0.073 ± 0.070 | −0.024 ± 0.043 | −0.030 ± 0.061 | 2.48 × 10−9 | 0.101 | 9.13 × 10−13 (Hyper) |
| cg27096144 | 5 | 174,151,779 | MSX2 | 0.074 ± 0.078 | −0.020 ± 0.054 | −0.030 ± 0.056 | 3.60 × 10−8 | 0.101 | 2.11 × 10−7 (Hyper) |
| cg15520279 | 2 | 176,995,088 | HOXD8 | 0.095 ± 0.083 | 0.008 ± 0.048 | −0.013 ± 0.046 | 1.06 × 10−7 | 0.1 | 1.30 × 10−13 (Hyper) |
| cg11733245 | 10 | 6,104,312 | IL2RA | −0.112 ± 0.066 | −0.001 ± 0.028 | −0.016 ± 0.050 | 5.58 × 10−10 | −0.101 | 8.03 × 10−13 (Hypo) |
| cg22325572 | 1 | 111,416,181 | CD53 | −0.102 ± 0.062 | 0.013 ± 0.035 | −0.007 ± 0.048 | 9.63 × 10−11 | −0.102 | 3.52 × 10−12 (Hypo) |
| cg15691199 | 14 | 23,589,419 | CEBPE | −0.102 ± 0.061 | 0.006 ± 0.033 | −0.003 ± 0.052 | 4.72 × 10−11 | −0.102 | 1.33 × 10−9 (Hypo) |
| cg16927606 | 19 | 36,233,324 | U2AF1L4 | −0.086 ± 0.048 | 0.013 ± 0.028 | 0.018 ± 0.044 | 3.10 × 10−14 | −0.103 | 1.79 × 10−8 (Hypo) |
| cg16240480 | 1 | 236,557,473 | EDARADD | −0.128 ± 0.064 | −0.005 ± 0.039 | −0.030 ± 0.049 | 7.69 × 10−11 | −0.106 | 1.59 × 10−9 (Hypo) |
| cg05596756 | 12 | 47,610,220 | FAM113B | −0.102 ± 0.060 | 0.009 ± 0.029 | 0.016 ± 0.047 | 8.28 × 10−13 | −0.116 | 1.53 × 10−10 (Hypo) |
| cg08040471 | 17 | 80,407,779 | C17orf62 | −0.116 ± 0.067 | 0.008 ± 0.036 | 0.004 ± 0.047 | 6.99 × 10−12 | −0.121 | 5.63 × 10−11 (Hypo) |
| cg20622019 | 20 | 43,279,793 | ADA | −0.108 ± 0.072 | 0.020 ± 0.043 | 0.012 ± 0.042 | 3.92 × 10−11 | −0.123 | 1.56 × 10−13 (Hypo) |
| cg05109049 | 17 | 29,641,333 | EVI2B | −0.141 ± 0.081 | 0.007 ± 0.050 | −0.020 ± 0.063 | 1.98 × 10−10 | −0.13 | 2.31 × 10−14 (Hypo) |
| cg07973967 | 17 | 62,009,607 | CD79B | −0.125 ± 0.061 | 0.016 ± 0.047 | 0.002 ± 0.056 | 2.00 × 10−14 | −0.132 | 2.81 × 10−11 (Hypo) |
| (B) Hallmark genes for Cluster II | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Target ID8 | Chrom9 | Position10 | Gene symbol | DNA methylation level in non-cancerous lung tissue (N) samples11 (mean ± SD) | p-value of Welch's t-test (II vs. I and III)12 | Δβ (II-I and III) 13 | p-value of Jonckheere–Terpstra trend test in II14 | ||
| Cluster I | Cluster II | Cluster III | |||||||
| cg14074641 | 16 | 48,181,753 | ABCC12 | −0.002 ± 0.091 | 0.025 ± 0.054 | −0.109 ± 0.105 | 1.01 × 10−10 | 0.101 | 7.05 × 10−2 (Hyper) |
| (C) Hallmark genes for Cluster III | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Target ID 15 | Chrom16 | Position17 | Gene symbol | DNA methylation level in non-cancerous lung tissue (N) samples18 (mean ± SD) | p-Value of Welch's t-test (III vs. I and II)19 | Δβ (III-I and II) 20 | p-Value of Jonckheere–Terpstra trend test in III21 | ||
| Cluster I | Cluster II | Cluster III | |||||||
| cg26606064 | 11 | 125,439,070 | EI24 | 0.020 ± 0.083 | 0.008 ± 0.064 | 0.115 ± 0.105 | 8.57 × 10−10 | 0.101 | 2.36 × 10−2 (Hyper) |
| cg17872476 | 10 | 114,205,654 | VTI1A | −0.034 ± 0.091 | −0.035 ± 0.060 | −0.137 ± 0.120 | 1.61 × 10−8 | −0.102 | 1.51 × 10−2 (Hypo) |
| cg21063899 | 13 | 78,109,801 | SCEL | 0.033 ± 0.088 | 0.013 ± 0.054 | −0.081 ± 0.086 | 3.06 × 10−12 | −0.103 | 1.47 × 10−9 (Hypo) |
| cg14074641 | 16 | 48,181,753 | ABCC12 | −0.002 ± 0.091 | 0.025 ± 0.054 | −0.109 ± 0.105 | 1.40 × 10−12 | −0.121 | 2.44 × 10−1 (Hypo) |
Probe ID for the Infinium HumanMethylation27 Bead Array.
Chromosome.
National Center for Biotechnology Information (NCBI) Database (Genome Build 37).
ΔβN-averageC.
Average βN–C in Cluster I versus average βN–C in Clusters II and III. Such p values were calculated to reveal the hallmark genes of Cluster I that showed DNA methylation statuses significantly different in their N samples in comparison with N samples from other clusters (Clusters II and III).
Average βN–C in Cluster I minus average βN–C in Clusters II and III. If Δβ (I-II and III) was more than 0.1, N samples in Cluster I were considered to show DNA hypermethylation relative to N samples in other clusters, and if Δβ (I-II and III) was less than −0.1, N samples in Cluster I were considered to show DNA hypomethylation relative to N samples in other clusters.
Stepwise DNA hypermethylation (Hyper) and hypomethylation (Hypo) from normal lung tissue samples to N samples, and then to tumorous tissue samples in Cluster I.
Probe ID for the Infinium HumanMethylation27 Bead Array.
Chromosome.
National Center for Biotechnology Information (NCBI) Database (Genome Build 37).
Δβ N-averageC.
Average βN–C in Cluster II versus average βN–C in Clusters I and III. Such p value was calculated to reveal the hallmark gene of Cluster II that showed DNA methylation status significantly different in their N samples in comparison with N samples from other clusters (Clusters I and III).
Average βN–C in Cluster II minus average βN–C in Clusters I and III. If Δβ (II−I and III) was more than 0.1, N samples in Cluster II were considered to show DNA hypermethylation relative to N samples in other clusters.
Stepwise DNA hypermethylation (Hyper) and hypomethylation (Hypo) from normal lung tissue samples to N samples, and then to tumorous tissue samples in Cluster II.
Probe ID for the Infinium HumanMethylation27 Bead Array.
Chromosome.
National Center for Biotechnology Information (NCBI) Database (Genome Build 37).
Δβ N-averageC.
Average βN–C in Cluster III versus average βN–C in Clusters I and II. Such p values were calculated to reveal the hallmark gene of Cluster III that showed DNA methylation statuses significantly different in their N samples in comparison with N samples from other clusters (Clusters I and II).
Average βN–C in Cluster III minus average βN–C in Clusters I and II. If Δβ (III-I and II) was more than 0.1, N samples in Cluster III were considered to show DNA hypermethylation relative to N samples in other clusters and if Δβ (III-I and II) was less than −0.1, N samples in Cluster III were considered to show DNA hypomethylation relative to N samples in other clusters.
Stepwise DNA hypermethylation (Hyper) and hypomethylation (Hypo) from normal lung tissue samples to N samples, and then to tumorous tissue samples in Cluster III.
DNA methylation profiles in the validation cohort
The correlations between the DNA methylation status of hallmark CpG sites for Clusters I, II and III in N samples and clinicopathological parameters of patients in the validation cohort were examined. DNA methylation levels on 17 and 2 hallmark CpG sites for Cluster I were significantly correlated with pleural anthracosis and pulmonary emphysema in the adjacent lung tissue in the validation cohort, respectively (Table 4A), whereas hallmark CpG sites for Clusters II and III never showed such a correlation. In addition, in the validation cohort, DNA methylation levels on 18 hallmark CpG sites for Cluster I were significantly correlated with the presence of AAH, a precancerous lesion for LADCs, in the adjacent lung tissue (Table 4A), even though the correlation between the presence of AAH and epigenetic clustering did not reach statistically significant levels (Table 2). DNA methylation levels on 13 hallmark CpG sites for Cluster I were significantly correlated with tumor anthracosis in LADCs in the validation cohort (Table 4A), whereas hallmark genes for Clusters II and III never showed such a correlation. Hallmark genes for Cluster I showing such correlations with pleural anthracosis, emphysema, presence of AAH or tumor anthracosis are described in Table 3A, and hallmark genes not showing such correlations are described in Supporting Information Table S3.
Table 4.
Correlation between DNA methylation levels of hallmark genes for Clusters I, II and III and the clinicopathological parameters in the validation cohort
| (A) Hallmark genes for Cluster I | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Target ID 1 | Gene symbol | DNA methylation level in non-cancerous lung tissue (N) samples2 (mean ± SD) | |||||||||||
| Pleural anthracosis | Emphysematic change | Atypical adenomatous hyperplasia | Tumor anthracosis | ||||||||||
| G1 | G2-3 | p-Value3 | Negative | Positive | p-Value3 | Absence | Presence | p-Value3 | Negative | Positive | p-Value3 | ||
| cg20249919 | PCSK6 | −0.126 ± 0.049 | −0.049 ± 0.102 | 1.83×10−2 | −0.069 ± 0.067 | −0.049 ± 0.131 | 4.99×10−1 | −0.056 ± 0.101 | −0.096 ± 0.082 | 3.57×10−1 | −0.077 ± 0.085 | −0.047 ± 0.105 | 3.18×10−1 |
| cg23349790 | IGSF21 | −0.044 ± 0.101 | −0.002 ± 0.100 | 4.13×10−1 | −0.035 ± 0.072 | 0.028 ± 0.118 | 3.50×10−2 | −0.005 ± 0.101 | −0.029 ± 0.086 | 5.79×10−1 | −0.054 ± 0.079 | 0.017 ± 0.098 | 1.68×10−2 |
| cg22285621 | SSH3 | −0.073 ± 0.043 | 0.002 ± 0.101 | 1.25×10−2 | −0.001 ± 0.077 | −0.014 ± 0.121 | 6.52×10−1 | −0.004 ± 0.101 | −0.035 ± 0.059 | 3.41×10−1 | −0.015 ± 0.077 | 0.001 ± 0.106 | 5.69×10−1 |
| cg15433631 | IRX2 | −0.041 ± 0.061 | 0.034 ± 0.074 | 4.73×10−2 | 0.025 ± 0.068 | 0.029 ± 0.083 | 8.52×10−1 | 0.026 ± 0.077 | 0.028 ± 0.056 | 9.46×10−1 | 0.010 ± 0.074 | 0.037 ± 0.072 | 2.77×10−1 |
| cg21949305 | ADORA2A,CYTSA | 0.025 ± 0.091 | 0.026 ± 0.060 | 9.73×10−1 | 0.015 ± 0.054 | 0.039 ± 0.069 | 1.91×10−1 | 0.029 ± 0.063 | −0.003 ± 0.036 | 1.28×10−1 | −0.004 ± 0.058 | 0.039 ± 0.061 | 3.48×10−2 |
| cg10942056 | DISP1 | 0.014 ± 0.088 | 0.015 ± 0.068 | 9.71×10−1 | 0.009 ± 0.062 | 0.023 ± 0.077 | 5.00×10−1 | 0.019 ± 0.071 | −0.022 ± 0.027 | 2.48×10−2 | −0.007 ± 0.056 | 0.026 ± 0.072 | 1.08×10−1 |
| cg15149645 | NUPR1 | −0.007 ± 0.124 | 0.013 ± 0.073 | 7.37×10−1 | 0.006 ± 0.070 | 0.015 ± 0.085 | 7.06×10−1 | 0.015 ± 0.079 | −0.036 ± 0.022 | 2.81×10−3 | −0.015 ± 0.081 | 0.021 ± 0.077 | 1.77×10−1 |
| cg06954481 | GBX2 | −0.044 ± 0.031 | 0.013 ± 0.075 | 9.57×10−3 | 0.008 ± 0.062 | 0.003 ± 0.085 | 7.95×10−1 | 0.012 ± 0.073 | −0.047 ± 0.040 | 2.27×10−2 | −0.012 ± 0.058 | 0.016 ± 0.078 | 1.90×10−1 |
| cg21250978 | PRKAR2B | −0.013 ± 0.092 | 0.002 ± 0.058 | 7.44×10−1 | −0.010 ± 0.050 | 0.014 ± 0.070 | 1.95×10−1 | 0.005 ± 0.061 | −0.037 ± 0.033 | 4.81×10−2 | −0.032 ± 0.058 | 0.013 ± 0.059 | 2.63×10−2 |
| cg22418909 | SFRP1 | −0.043 ± 0.076 | 0.002 ± 0.058 | 2.55×10−1 | 0.000 ± 0.065 | −0.003 ± 0.053 | 8.32×10−1 | 0.003 ± 0.061 | −0.041 ± 0.022 | 4.86×10−3 | −0.020 ± 0.057 | 0.007 ± 0.060 | 1.64×10−1 |
| cg26200585 | PRX | 0.020 ± 0.079 | 0.015 ± 0.066 | 8.81×10−1 | 0.013 ± 0.069 | 0.016 ± 0.063 | 8.89×10−1 | 0.019 ± 0.067 | −0.029 ± 0.035 | 3.48×10−2 | −0.006 ± 0.054 | 0.025 ± 0.070 | 1.19×10−1 |
| cg24396745 | HCN4 | −0.056 ± 0.035 | 0.020 ± 0.072 | 3.53×10−3 | 0.017 ± 0.066 | 0.003 ± 0.079 | 4.87×10−1 | 0.015 ± 0.073 | −0.025 ± 0.054 | 1.86×10−1 | −0.005 ± 0.074 | 0.022 ± 0.071 | 2.60×10−1 |
| cg04330449 | NEUROG1 | −0.040 ± 0.033 | 0.010 ± 0.073 | 2.33×10−2 | 0.010 ± 0.065 | −0.005 ± 0.078 | 4.52×10−1 | 0.006 ± 0.072 | −0.020 ± 0.056 | 3.82×10−1 | −0.006 ± 0.044 | 0.012 ± 0.078 | 3.40×10−1 |
| cg19589427 | TNFSF18 | −0.008 ± 0.078 | −0.010 ± 0.070 | 9.65×10−1 | −0.010 ± 0.078 | −0.012 ± 0.057 | 9.26×10−1 | −0.007 ± 0.071 | −0.042 ± 0.036 | 1.10×10−1 | −0.040 ± 0.057 | 0.005 ± 0.069 | 3.13×10−2 |
| cg16731240 | ZNF577 | −0.042 ± 0.037 | 0.014 ± 0.087 | 2.50×10−2 | 0.012 ± 0.1000 | 0.003 ± 0.060 | 7.02×10−1 | 0.007 ± 0.086 | 0.014 ± 0.078 | 8.48×10−1 | −0.015 ± 0.074 | 0.020 ± 0.087 | 1.79×10−1 |
| cg03544320 | CRMP1 | −0.097 ± 0.083 | 0.018 ± 0.094 | 3.14×10−2 | 0.010 ± 0.109 | −0.005 ± 0.084 | 5.67×10−1 | 0.008 ± 0.102 | −0.040 ± 0.036 | 4.56×10−2 | −0.014 ± 0.095 | 0.019 ± 0.097 | 3.04×10−1 |
| cg12864235 | CDH9 | 0.032 ± 0.054 | 0.027 ± 0.057 | 8.51×10−1 | 0.025 ± 0.045 | 0.030 ± 0.066 | 7.86×10−1 | 0.031 ± 0.057 | −0.004 ± 0.019 | 1.29×10−2 | 0.012 ± 0.043 | 0.034 ± 0.060 | 1.77×10−1 |
| cg15898840 | IGFBP3 | −0.043 ± 0.030 | 0.001 ± 0.057 | 2.53×10−2 | −0.004 ± 0.052 | −0.007 ± 0.061 | 8.16×10−1 | −0.002 ± 0.056 | −0.036 ± 0.037 | 1.14×10−1 | −0.003 ± 0.044 | −0.002 ± 0.060 | 9.36×10−1 |
| cg08044694 | BRD4 | −0.067 ± 0.036 | −0.020 ± 0.049 | 3.84×10−2 | −0.021 ± 0.046 | −0.028 ± 0.053 | 6.30×10−1 | −0.022 ± 0.050 | −0.039 ± 0.035 | 3.55×10−1 | −0.041 ± 0.041 | −0.017 ± 0.052 | 1.12×10−1 |
| cg03734874 | TMEM179 | −0.032 ± 0.034 | 0.023 ± 0.074 | 1.62×10−2 | 0.021 ± 0.076 | 0.015 ± 0.068 | 7.56×10−1 | 0.024 ± 0.072 | −0.037 ± 0.039 | 1.89×10−2 | −0.008 ± 0.052 | 0.030 ± 0.075 | 5.63×10−2 |
| cg10599444 | MMP14 | −0.063 ± 0.026 | −0.010 ± 0.060 | 4.85×10−3 | −0.008 ± 0.057 | −0.023 ± 0.060 | 3.87×10−1 | −0.013 ± 0.060 | −0.027 ± 0.041 | 5.25×10−1 | −0.026 ± 0.047 | −0.007 ± 0.059 | 2.66×10−1 |
| cg24133115 | PDE10A | −0.020 ± 0.025 | 0.022 ± 0.060 | 1.58×10−2 | 0.014 ± 0.059 | 0.020 ± 0.057 | 7.02×10−1 | 0.018 ± 0.060 | 0.004 ± 0.027 | 3.88×10−1 | 0.009 ± 0.037 | 0.024 ± 0.063 | 3.25×10−1 |
| cg12594641 | LYPD6 | −0.024 ± 0.041 | 0.036 ± 0.068 | 2.45×10−2 | 0.021 ± 0.062 | 0.036 ± 0.075 | 4.61×10−1 | 0.029 ± 0.071 | 0.013 ± 0.038 | 4.43×10−1 | 0.025 ± 0.045 | 0.035 ± 0.075 | 5.94×10−1 |
| cg05724065 | PHKG1 | 0.016 ± 0.100 | 0.011 ± 0.057 | 9.10×10−1 | 0.014 ± 0.055 | 0.008 ± 0.067 | 7.39×10−1 | 0.016 ± 0.061 | −0.032 ± 0.027 | 1.05×10−2 | −0.002 ± 0.066 | 0.019 ± 0.059 | 3.33×10−1 |
| cg19466563 | SPARCL1 | 0.018 ± 0.081 | 0.012 ± 0.055 | 8.86×10−1 | 0.007 ± 0.052 | 0.021 ± 0.060 | 3.98×10−1 | 0.019 ± 0.056 | −0.035 ± 0.015 | 4.46×10−5 | −0.015 ± 0.047 | 0.025 ± 0.057 | 2.24×10−2 |
| cg24433189 | SSTR5 | 0.024 ± 0.075 | 0.034 ± 0.060 | 7.96×10−1 | 0.031 ± 0.052 | 0.032 ± 0.071 | 9.63×10−1 | 0.035 ± 0.062 | −0.003 ± 0.027 | 2.95×10−2 | 0.026 ± 0.051 | 0.036 ± 0.065 | 5.79×10−1 |
| cg24453664 | CD59 | −0.057 ± 0.039 | 0.000 ± 0.053 | 2.44×10−2 | −0.002 ± 0.054 | −0.009 ± 0.055 | 6.40×10−1 | −0.004 ± 0.054 | −0.021 ± 0.050 | 5.00×10−1 | −0.018 ± 0.050 | 0.001 ± 0.055 | 2.53×10−1 |
| cg26609631 | GSX1 | −0.051 ± 0.058 | 0.006 ± 0.067 | 9.21×10−2 | 0.002 ± 0.066 | −0.005 ± 0.069 | 7.29×10−1 | 0.004 ± 0.068 | −0.050 ± 0.025 | 3.65×10−3 | −0.020 ± 0.054 | 0.010 ± 0.071 | 1.35×10−1 |
| cg10604646 | RGS5 | 0.038 ± 0.039 | 0.033 ± 0.058 | 8.08×10−1 | 0.013 ± 0.062 | 0.056 ± 0.041 | 5.70×10−3 | 0.037 ± 0.056 | −0.014 ± 0.057 | 1.16×10−1 | −0.006 ± 0.063 | 0.049 ± 0.047 | 1.07×10−2 |
| cg03355526 | ZNF454 | −0.061 ± 0.044 | 0.001 ± 0.075 | 2.98×10−2 | 0.003 ± 0.077 | −0.012 ± 0.070 | 4.81×10−1 | 0.002 ± 0.074 | −0.052 ± 0.053 | 8.77×10−2 | −0.007 ± 0.058 | 0.000 ± 0.078 | 7.53×10−1 |
| cg27096144 | MSX2 | −0.066 ± 0.049 | 0.001 ± 0.063 | 3.29×10−2 | −0.006 ± 0.057 | −0.006 ± 0.072 | 9.95×10−1 | −0.003 ± 0.065 | −0.026 ± 0.040 | 2.94×10−1 | −0.023 ± 0.055 | 0.002 ± 0.068 | 2.05×10−1 |
| cg15520279 | HOXD8 | −0.021 ± 0.040 | 0.016 ± 0.070 | 1.15×10−1 | 0.015 ± 0.075 | 0.009 ± 0.056 | 7.16×10−1 | 0.014 ± 0.070 | −0.006 ± 0.025 | 2.05×10−1 | −0.010 ± 0.038 | 0.024 ± 0.075 | 4.76×10−2 |
| cg11733245 | IL2RA | −0.037 ± 0.042 | −0.035 ± 0.051 | 9.29×10−1 | −0.023 ± 0.038 | −0.047 ± 0.060 | 1.10×10−1 | −0.035 ± 0.051 | −0.013 ± 0.023 | 1.09×10−1 | −0.009 ± 0.035 | −0.047 ± 0.050 | 7.07×10−3 |
| cg22325572 | CD53 | −0.024 ± 0.083 | −0.028 ± 0.058 | 9.23×10−1 | −0.017 ± 0.051 | −0.040 ± 0.067 | 1.90×10−1 | −0.029 ± 0.060 | −0.008 ± 0.057 | 4.68×10−1 | 0.003 ± 0.057 | −0.041 ± 0.057 | 2.96×10−2 |
| cg15691199 | CEBPE | −0.029 ± 0.080 | −0.018 ± 0.064 | 7.94×10−1 | −0.016 ± 0.050 | −0.022 ± 0.079 | 7.79×10−1 | −0.022 ± 0.066 | 0.015 ± 0.021 | 1.52×10−2 | −0.002 ± 0.061 | −0.028 ± 0.067 | 2.28×10−1 |
| cg16927606 | U2AF1L4 | −0.012 ± 0.079 | −0.004 ± 0.054 | 8.25×10−1 | −0.005 ± 0.048 | −0.006 ± 0.065 | 9.54×10−1 | −0.010 ± 0.057 | 0.032 ± 0.025 | 1.48×10−2 | 0.009 ± 0.051 | −0.012 ± 0.057 | 2.31×10−1 |
| cg16240480 | EDARADD | −0.040 ± 0.103 | −0.047 ± 0.072 | 8.91×10−1 | −0.032 ± 0.071 | −0.062 ± 0.075 | 1.61×10−1 | −0.048 ± 0.074 | −0.017 ± 0.070 | 3.86×10−1 | 0.001 ± 0.049 | −0.066 ± 0.076 | 1.24×10−3 |
| cg05596756 | FAM113B | −0.010 ± 0.085 | −0.010 ± 0.062 | 9.92×10−1 | 0.000 ± 0.052 | −0.022 ± 0.073 | 2.22×10−1 | −0.014 ± 0.064 | 0.026 ± 0.025 | 2.14×10−2 | 0.011 ± 0.057 | −0.020 ± 0.065 | 1.30×10−1 |
| cg08040471 | C17orf62 | 0.000 ± 0.073 | −0.014 ± 0.066 | 7.11×10−1 | −0.002 ± 0.051 | −0.024 ± 0.080 | 2.80×10−1 | −0.015 ± 0.068 | 0.013 ± 0.029 | 1.21×10−1 | 0.019 ± 0.053 | −0.027 ± 0.066 | 2.03×10−2 |
| cg20622019 | ADA | −0.021 ± 0.073 | −0.033 ± 0.068 | 7.39×10−1 | −0.028 ± 0.069 | −0.035 ± 0.066 | 7.31×10−1 | −0.036 ± 0.069 | 0.008 ± 0.021 | 6.34×10−3 | 0.002 ± 0.051 | −0.047 ± 0.068 | 1.14×10−2 |
| cg05109049 | EVI2B | −0.057 ± 0.106 | −0.041 ± 0.090 | 7.56×10−1 | −0.023 ± 0.093 | −0.062 ± 0.084 | 1.32×10−1 | −0.044 ± 0.090 | −0.009 ± 0.102 | 5.07×10−1 | −0.002 ± 0.054 | −0.058 ± 0.099 | 1.72×10−2 |
| cg07973967 | CD79B | −0.034 ± 0.105 | −0.028 ± 0.075 | 9.16×10−1 | −0.017 ± 0.060 | −0.042 ± 0.094 | 2.83×10−1 | −0.031 ± 0.080 | 0.007 ± 0.021 | 2.06×10−2 | −0.007 ± 0.067 | −0.039 ± 0.081 | 1.69×10−1 |
| (B) Hallmark genes for Cluster II and III | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Target ID 4 | Gene symbol | DNA methylation level in non-cancerous lung tissue (N) samples5 (mean ± SD) | |||||||||||
| Lymphatic invasion | Nodal status | Pathological Tumor-Node-Metastasis stage | |||||||||||
| Negative | Positive | p-Value6 | N0 | N1 | N2-3 | p-Value6 | IA-IB | IIA-IIB | IIIA-IV | p-Value6 | |||
| cg26606064 | EI24 | 0.018 ± 0.098 | 0.098 ± 0.113 | 3.01 × 10−2 | 0.004 ± 0.098 | 0.010 ± 0.094 | 0.118 ± 0.091 | 1.27 × 10−3 | 0.009 ± 0.102 | −0.008 ± 0.082 | 0.118 ± 0.091 | 1.14 × 10−3 | |
| cg17872476 | VTI1A | −0.035 ± 0.134 | −0.126 ± 0.137 | 4.32 × 10−2 | −0.023 ± 0.132 | 0.022 ± 0.127 | −0.149 ± 0.121 | 5.69 × 10−3 | −0.020 ± 0.116 | −0.016 ± 0.166 | −0.149 ± 0.121 | 6.70 × 10−3 | |
| cg21063899 | SCEL | −0.043 ± 0.093 | −0.109 ± 0.110 | 6.08 × 10−2 | −0.024 ± 0.082 | −0.024 ± 0.090 | −0.141 ± 0.096 | 2.81 × 10−4 | −0.021 ± 0.089 | −0.033 ± 0.062 | −0.141 ± 0.096 | 2.60 × 10−4 | |
| cg14074641 | ABCC12 | −0.017 ± 0.118 | −0.083 ± 0.117 | 8.60 × 10−2 | 0.005 ± 0.104 | −0.006 ± 0.112 | −0.118 ± 0.114 | 2.09 × 10−3 | 0.000 ± 0.111 | 0.014 ± 0.084 | −0.118 ± 0.114 | 2.00 × 10−3 | |
Probe ID for the Infinium HumanMethylation27 Bead Array.
ΔβN-averageC.
p values (Welch's t-test) of <0.05 are underlined.
Probe ID for the Infinium HumanMethylation27 Bead Array.
ΔβN-averageC.
p values (Welch's t-test) of <0.05 are underlined.
Hallmark gene ABCC12 was shared between Clusters II and III. The DNA methylation level of ABCC12 was significantly correlated with N stage and TNM stage in the validation cohort (Table 4B). In the learning cohort, the DNA methylation level of the ABCC12 gene was high in Cluster II showing low N and TNM stages, and that of the ABCC12 gene was low in Cluster III showing high N and TNM stages. Therefore, it is feasible that the DNA methylation level of the ABCC12 gene was significantly higher in patients showing lower N and TNM stages in the validation cohort (Table 4B). DNA methylation levels of two of the three remaining hallmark genes (three hallmark genes other than ABCC12) for Cluster III were significantly correlated with lymph vessel invasion in LADCs in the validation cohort, and the DNA methylation levels of all three remaining hallmark genes for Cluster III were significantly correlated with high N and TNM stages (Table 4B). Taken together, correlations between DNA methylation profiles in N samples and clinicopathological characteristics in the adjacent lung tissue or LADCs in the learning cohort were reproduced in the validation cohort.
Discussion
In this study, we focused on DNA methylation profiles in the adjacent non-cancerous lung tissue obtained from patients with LADCs and analyzed the results of methylome analysis of lung tissue samples including 189 N samples at single-CpG resolution. DNA methylation alterations occurred even in N samples relative to C samples, and were inherited by, or strengthened in, T samples (Table 1). These findings are compatible with the “field cancerization” concept in the lung.29 In our previous study using the Infinium assay, we proved that DNA methylation alterations in N samples resulted in silencing of tumor-related genes in tumorous tissue.15 However, the correlation between the results of the Infinium assay in N samples and carcinogenetic factors was not examined in detail.
In this epigenetic clustering of patients with LADCs based on DNA methylation profiles in N samples, many of the patients belonging to Cluster I were heavy smokers. In fact, pleural anthracosis, which mainly reflects the long-term cumulative effects of cigarette smoking, was marked in the lungs of patients belonging to Cluster I. Smoking is known to be a cause of COPD. In fact, many patients in Cluster I actually suffered from obstructive ventilation impairment, and histological findings compatible with emphysema and lung fibrosis were observed in their N samples. Moreover, recurrent inflammation is generally associated with COPD,30 and histological findings compatible with respiratory bronchiolitis20,21 were actually observed in the lungs of patients belonging to Cluster I. Inflammation is known to be one of the major causes of DNA methylation alterations in precancerous conditions in various organs, such as chronic hepatitis16,17 and chronic pancreatitis.31,32 Taken together, the data suggest that the DNA methylation profiles characterizing Cluster I may be established in lung tissue through the long-term cumulative effects of cigarette smoking via chronic inflammation under the conditions of COPD. Unlike the previous study, which revealed aberrant DNA methylation of several tumor-related genes in lung cancers themselves of patients with COPD,33 this study demonstrated for the first time the presence of distinct DNA methylation profiles related to COPD in N samples, based on genome-wide analysis.
The majority of patients belonging to Cluster II were non-smokers, especially young females. DNA methylation profiles characterizing Cluster II may reflect the carcinogenetic pathway that is unrelated to cigarette smoking. Mutation of the EGFR gene is well known to be a driver of LADCs in young female non-smokers, especially in Asia.34 However, Cluster II included LADCs without EGFR gene mutations in non-smokers (data not shown), indicating that DNA methylation profiles in Cluster II were not entirely induced by EGFR mutation.
Although many of the patients belonging to Cluster III were smokers, the average number of cigarettes smoked per day × year index was lower in Cluster III than in Cluster I. In fact severe pleural anthracosis was not so frequently evident in the lungs of patients belonging to Cluster III. In addition, the incidence of emphysematous change and fibrosis was lower in the adjacent lung tissue of patients in Cluster III than in that of patients in Cluster I. DNA methylation profiles in Cluster III did not develop from a background of chronic inflammation in COPD, but may have developed rapidly before the long-term effects of cigarette smoking had accumulated in the adjacent lung tissue (possibly through more direct effects of carcinogens related or unrelated to cigarette smoking). However, to evaluate more precisely the effects of smoking on DNA methylation profiles, detailed DNA methylation analysis should be performed using purified epithelial cells, such as those from the airway epithelium.
Distinct DNA methylation profiles seem to be established in the non-cancerous lung during the carcinogenetic pathway via inflammation in COPD in heavy smokers (Fig. 2a), the carcinogenetic pathway unrelated to cigarette smoking (Fig. 2b), and the carcinogenetic pathway that occurs not via COPD but possibly via more direct effects of carcinogens (Fig. 2c). Each pathway may have distinct target genes as hallmarks for Clusters I, II and III (Table 3 and Supporting Information Table S3). Among 120 hallmark genes for Clusters I, II and III, 119 (one exception, ABCC12, being shared between Clusters II and III) showed ordered differences of DNA methylation from C to N, and then to T samples of the relevant cluster (p < 0.05, Jonckheere–Terpstra trend test, Table 3 and Supporting Information Table S3), indicating that a distinct DNA methylation profile in N samples of each cluster is inherited during progression to Ts.
A proportion of genes described in Table 3 and Supporting Information Table S3 may be simple hallmarks of each cluster (simple target genes of each carcinogenetic pathway). However, at least a proportion of DNA methylation alterations occurring during each carcinogenetic pathway actually result in altered expression of target genes, and may participate in establishment of the clinicopathological characteristics of LADCs in each cluster. The DNA methylation profiles in Cluster I may participate in the generation of locally invasive LADCs, which have a large diameter, a progressed T stage, a high histological grade, frequent pleural invasion and tumor anthracosis. DNA methylation profiles in Cluster II may participate in the generation of clinicopathologically less aggressive LADCs with a favorable outcome. DNA methylation profiles in Cluster III may participate in the generation of the most aggressive LADCs showing frequent lymphatic vessel invasion, blood vessel invasion, a high N stage, a high TNM stage and a poor outcome.
Table 3 includes homeobox genes, such as IRX2 and HOXD8, a gene that has been implicated in cell migration, SPARCL1, and genes that have been implicated in apoptosis, such as RGS5 and EI24. IRX2 is known to participate in early lung development in mouse embryos.35 HOXD8 is known to be methylated and/or down-regulated in human malignancies, especially in metastatic, rather than in primary lesions.36,37 SPARCL1 is an extracellular matrix glycoprotein known to be correlated with cancer invasion.38,39 RGS5 is a member of the family of molecules regulating G protein signaling, and stimulates hypoxia-inducible apoptosis.40 Positive correlations between RGS5 expression and both tumor differentiation and a favorable outcome have been reported.41,42 EI24 is induced by p53, suppresses cell growth and induces apoptosis.43 Reduced expression associated with DNA methylation of IRX2, HOXD8, SPARCL1, RGS5 and EI24 in our cohort of LADCs has been confirmed using expression microarray (data not shown). It is feasible that these target genes of each carcinogenetic pathway participate in determining the clinicopathological characteristics of LADCs in each cluster.
In the validation cohort, the DNA methylation status of hallmark genes identified in N samples of Cluster I was significantly correlated with pleural anthracosis, which reflects the long-term cumulative effects of smoking, and COPD (pulmonary emphysema) in the adjacent lung and tumor anthracosis, which reflect active cancer–stromal interaction in LADCs. The DNA methylation status of the hallmark gene identified in N samples of Cluster II was significantly correlated with lower aggressiveness (low N stage and low TNM stage) of LADCs in the validation cohort. The DNA methylation status of hallmark genes identified in N samples of Cluster III was significantly correlated with aggressiveness of LADCs, such as lymph vessel invasion, a high N stage and a high TNM stage, in the validation cohort. Thus, correlations between distinct DNA methylation profiles in N samples and both carcinogenetic background factors in the adjacent lung tissue and clinicopathological characteristics of LADCs were confirmed in the validation cohort (Table 4).
Receiver operating characteristic curve (ROC) analysis was performed for N samples in the learning cohort, and the thresholds of the representative hallmark genes described in Table 4 were set so that they were nearest to the top left corner of the ROC. Using these thresholds, the sensitivity, specificity and accuracy for prediction of lymphatic vessel involvement, lymph node metastasis, TNM stage and patient outcome (recurrence and death) were calculated in both the learning and validation cohorts (Supporting Information Table S4). Even though Supporting Information Table S4 suggests that the aggressiveness of tumors developing in the same individual patients and patient outcome may be predictable on the basis of DNA methylation status in N samples, further examinations will be needed to set strict criteria for maximal sensitivity, specificity and accuracy.
Although bulk tissue comprising several cell lineages, for a large number of C, N and T samples, was examined in this study, it would be preferable to examine the DNA methylation status of purified cells. Therefore, the DNA methylation status of the representative gene CASP8 (Infinium probe ID: cg26799474), included in Table 1B, was compared between cancer cells and normal peripheral airway epithelial cells obtained by microdissection from formalin-fixed, paraffin-embedded tissues of representative patients with LADCs and patients without primary lung cancers, respectively, using pyrosequencing. The DNA methylation levels in T samples (0.279 ± 0.184) were significantly lower than those in C samples (0.689 ± 0.042) by Infinium assay (p = 3.64 × 10−4). Such a significant difference was reproduced upon comparison with microdissected normal airway epithelium: pyrosequencing showed that the DNA methylation levels in microdissected cancer cells (0.273 ± 0.313) were significantly lower than those in microdissected normal airway epithelial cells (0.765 ± 0.104) (p = 2.74 × 10−3).
Differences in DNA methylation levels among different cell lineages, such as epithelial and stromal components, are also an important issue. Cancer cells and their stromal cells, such as cancer-associated fibroblasts, were again collected separately by microdissection from formalin-fixed, paraffin-embedded tissues from representative patients with LADCs. The DNA methylation levels of representative genes described in Table 1B were evaluated quantitatively by pyrosequencing. In one of the examined genes (CASP8 [Infinium probe ID: cg26799474]), the DNA methylation statuses of cancer cells (0.273 ± 0.313) and stromal cells (0.219 ± 0.094) were almost equal, indicating that both may be affected by carcinogenetic factors. For the other examined gene (LHX1 [Infinium probe ID: cg22660578]), the DNA methylation statuses of cancer cells (0.096 ± 0.141) and stromal cells (0.538 ± 0.486) differed from each other, probably reflecting differences in susceptibility to the effects of carcinogens, or differences in cell lineage.
In summary, DNA methylation profiles reflecting carcinogenetic background factors, such as smoking, inflammation and COPD, appear to be established in adjacent lung tissue in patients with LADCs. Such DNA methylation profiles in adjacent lung tissue may play a role in determining the aggressiveness of tumors developing in the same individual patients, and thus patient outcome.
Acknowledgments
T. Sato is an awardee of a research resident fellowship from the Foundation for Promotion of Cancer Research in Japan.
Glossary
- AAH
atypical adenomatous hyperplasia
- C
normal lung tissue
- COPD
chronic obstructive pulmonary disease
- FDR
false discovery rate
- LADC
lung adenocarcinomas
- N
non-cancerous lung tissue
- ROC
receiver operating characteristic curve
- T
tumorous tissue
- TNM
tumor-node-metastasis
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers—a different disease. Nat Rev Cancer. 2007;7:778–90. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- 3.Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heller G, Zielinski CC, Zochbauer-Muller S. Lung cancer: from single-gene methylation to methylome profiling. Cancer Metastasis Rev. 2010;29:95–107. doi: 10.1007/s10555-010-9203-x. [DOI] [PubMed] [Google Scholar]
- 5.Arai E, Kanai Y. DNA methylation profiles in precancerous tissue and cancers: carcinogenetic risk estimation and prognostication based on DNA methylation status. Epigenomics. 2010;2:467–81. doi: 10.2217/epi.10.16. [DOI] [PubMed] [Google Scholar]
- 6.Kanai Y. Genome-wide DNA methylation profiles in precancerous conditions and cancers. Cancer Sci. 2010;101:36–45. doi: 10.1111/j.1349-7006.2009.01383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arai E, Chiku S, Mori T, et al. Single-CpG-resolution methylome analysis identifies clinicopathologically aggressive CpG island methylator phenotype clear cell renal cell carcinomas. Carcinogenesis. 2012;33:1487–93. doi: 10.1093/carcin/bgs177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zöchbauer-Müller S, Lam S, Toyooka S, et al. Aberrant methylation of multiple genes in the upper aerodigestive tract epithelium of heavy smokers. Int J Cancer. 2003;107:612–6. doi: 10.1002/ijc.11458. [DOI] [PubMed] [Google Scholar]
- 9.Eguchi K, Kanai Y, Kobayashi K, et al. DNA hypermethylation at the D17S5 locus in non-small cell lung cancers: its association with smoking history. Cancer Res. 1997;57:4913–5. [PubMed] [Google Scholar]
- 10.Lamy A, Sesboüé R, Bourguignon J, et al. Aberrant methylation of the CDKN2a/p16INK4a gene promoter region in preinvasive bronchial lesions: a prospective study in high-risk patients without invasive cancer. Int J Cancer. 2002;100:189–93. doi: 10.1002/ijc.10474. [DOI] [PubMed] [Google Scholar]
- 11.Belinsky SA, Palmisano WA, Gilliland FD, et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 2002;62:2370–7. [PubMed] [Google Scholar]
- 12.Bibikova M, Le J, Barnes B, et al. Genome-wide DNA methylation profiling using Infinium(R) assay. Epigenomics. 2009;1:177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- 13.Selamat SA, Chung BS, Girard L, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012;22:1197–211. doi: 10.1101/gr.132662.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lockwood WW, Wilson IM, Coe BP, et al. Divergent genomic and epigenomic landscapes of lung cancer subtypes underscore the selection of different oncogenic pathways during tumor development. PLoS One. 2012;7:e37775. doi: 10.1371/journal.pone.0037775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sato T, Arai E, Kohno T, et al. DNA methylation profiles at precancerous stages associated with recurrence of lung adenocarcinoma. PLoS One. 2013;8:e59444. doi: 10.1371/journal.pone.0059444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagashio R, Arai E, Ojima H, et al. Carcinogenetic risk estimation based on quantification of DNA methylation levels in liver tissue at the precancerous stage. Int J Cancer. 2011;129:1170–9. doi: 10.1002/ijc.26061. [DOI] [PubMed] [Google Scholar]
- 17.Arai E, Ushijima S, Gotoh M, et al. Genome-wide DNA methylation profiles in liver tissue at the precancerous stage and in hepatocellular carcinoma. Int J Cancer. 2009;125:2854–62. doi: 10.1002/ijc.24708. [DOI] [PubMed] [Google Scholar]
- 18.Ushijima T, Hattori N. Molecular pathways: involvement of Helicobacter pylori-triggered inflammation in the formation of an epigenetic field defect, and its usefulness as cancer risk and exposure markers. Clin Cancer Res. 2012;18:923–9. doi: 10.1158/1078-0432.CCR-11-2011. [DOI] [PubMed] [Google Scholar]
- 19.Kanai Y, Hirohashi S. Alterations of DNA methylation associated with abnormalities of DNA methyltransferases in human cancers during transition from a precancerous to a malignant state. Carcinogenesis. 2007;28:2434–42. doi: 10.1093/carcin/bgm206. [DOI] [PubMed] [Google Scholar]
- 20.Garibaldi BT, Illei P, Danoff SK. Bronchiolitis. Immunol Allergy Clin North Am. 2012;32:601–19. doi: 10.1016/j.iac.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 21.Travis WD, Colby TV, Koss MN, et al. Obstructive pulmonary disease. In: King DW, editor. Atlas of nontumor pathology. Non-neoplastic disorders of the lower respiratory tract. Washington, DC: American Registry of Pathology; 2002. pp. 435–71. [Google Scholar]
- 22.Kerr KM. Clinical relevance of the new IASLC/ERS/ATS adenocarcinoma classification. J Clin Pathol. 2013;66:832–8. doi: 10.1136/jclinpath-2013-201519. [DOI] [PubMed] [Google Scholar]
- 23.Noguchi M, Shimosato Y. The development and progression of adenocarcinoma of the lung. Cancer Treat Res. 1995;72:131–42. doi: 10.1007/978-1-4615-2630-8_6. [DOI] [PubMed] [Google Scholar]
- 24.Colby TV, Noguchi M, Henschke C. Adenocarcinoma. In: Travis WD, Brambilla E, Muller-Hermelink HK, Harris CC, et al., editors. World health classification of tumours pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon: IARC Press; 2004. pp. 35–44. [Google Scholar]
- 25.Wang D, Minami Y, Shu Y, et al. The implication of background anthracosis in the development and progression of pulmonary adenocarcinoma. Cancer Sci. 2003;94:707–11. doi: 10.1111/j.1349-7006.2003.tb01506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noguchi M, Morikawa A, Kawasaki M, et al. Small adenocarcinoma of the lung. Histologic characteristics and prognosis. Cancer. 1995;75:2844–52. doi: 10.1002/1097-0142(19950615)75:12<2844::aid-cncr2820751209>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 27.UICC International Union Against Cancer. Lung and pleural tumours. In: Sobin LH, Gospodarowicz MK, Wittekind C, editors. TNM classification of malignant tumours. 7th. Oxford: Wiley-Blackwell; 2009. pp. 138–46. [Google Scholar]
- 28.Hu L, Sekine M, Gaina A, et al. Association of smoking behavior and socio-demographic factors, work, lifestyle and mental health of Japanese civil servants. J Occup Health. 2007;49:443–52. doi: 10.1539/joh.49.443. [DOI] [PubMed] [Google Scholar]
- 29.Kadara H, Kabbout M, Wistuba II. Pulmonary adenocarcinoma: a renewed entity in 2011. Respirology. 2012;17:50–65. doi: 10.1111/j.1440-1843.2011.02095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379:1341–51. doi: 10.1016/S0140-6736(11)60968-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng DF, Kanai Y, Sawada M, et al. DNA methylation of multiple tumor-related genes in association with overexpression of DNA methyltransferase 1 (DNMT1) during multistage carcinogenesis of the pancreas. Carcinogenesis. 2006;27:1160–8. doi: 10.1093/carcin/bgi361. [DOI] [PubMed] [Google Scholar]
- 32.Peng DF, Kanai Y, Sawada M, et al. Increased DNA methyltransferase 1 (DNMT1) protein expression in precancerous conditions and ductal carcinomas of the pancreas. Cancer Sci. 2005;96:403–8. doi: 10.1111/j.1349-7006.2005.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki M, Wada H, Yoshino M, et al. Molecular characterization of chronic obstructive pulmonary disease-related non-small cell lung cancer through aberrant methylation and alterations of EGFR signaling. Ann Surg Oncol. 2010;17:878–88. doi: 10.1245/s10434-009-0739-3. [DOI] [PubMed] [Google Scholar]
- 34.Sharma SV, Bell DW, Settleman J, et al. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–81. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 35.Mummenhoff J, Houweling AC, Peters T, et al. Expression of Irx6 during mouse morphogenesis. Mech Dev. 2001;103:193–5. doi: 10.1016/s0925-4773(01)00353-7. [DOI] [PubMed] [Google Scholar]
- 36.Leshchenko VV, Kuo PY, Shaknovich R, et al. Genomewide DNA methylation analysis reveals novel targets for drug development in mantle cell lymphoma. Blood. 2010;116:1025–34. doi: 10.1182/blood-2009-12-257485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanai M, Hamada J, Takada M, et al. Aberrant expressions of HOX genes in colorectal and hepatocellular carcinomas. Oncol Rep. 2010;23:843–51. [PubMed] [Google Scholar]
- 38.Hurley PJ, Marchionni L, Simons BW, et al. Secreted protein, acidic and rich in cysteine-like 1 (SPARCL1) is down regulated in aggressive prostate cancers and is prognostic for poor clinical outcome. Proc Natl Acad Sci USA. 2012;109:14977–82. doi: 10.1073/pnas.1203525109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turtoi A, Musmeci D, Naccarato AG, et al. Sparc-like protein 1 is a new marker of human glioma progression. J Proteome Res. 2012;11:5011–21. doi: 10.1021/pr3005698. [DOI] [PubMed] [Google Scholar]
- 40.Jin Y, An X, Ye Z, et al. RGS5, a hypoxia-inducible apoptotic stimulator in endothelial cells. J Biol Chem. 2009;284:23436–43. doi: 10.1074/jbc.M109.032664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang G, Song H, Wang R, et al. The relationship between RGS5 expression and cancer differentiation and metastasis in non-small cell lung cancer. J Surg Oncol. 2012;105:420–4. doi: 10.1002/jso.22033. [DOI] [PubMed] [Google Scholar]
- 42.Wang JH, Huang WS, Hu CR, et al. Relationship between RGS5 expression and differentiation and angiogenesis of gastric carcinoma. World J Gastroenterol. 2010;16:5642–6. doi: 10.3748/wjg.v16.i44.5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao X, Ayer RE, Davis SL, et al. Apoptosis factor EI24/PIG8 is a novel endoplasmic reticulum-localized Bcl-2-binding protein which is associated with suppression of breast cancer invasiveness. Cancer Res. 2005;65:2125–9. doi: 10.1158/0008-5472.CAN-04-3377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information