

Abstract
The distal convoluted tubule is the nephron segment that lies immediately downstream of the macula densa. Although short in length, the distal convoluted tubule plays a critical role in sodium, potassium, and divalent cation homeostasis. Recent genetic and physiologic studies have greatly expanded our understanding of how the distal convoluted tubule regulates these processes at the molecular level. This article provides an update on the distal convoluted tubule, highlighting concepts and pathophysiology relevant to clinical practice.
Keywords: distal tubule, Na transport, potassium channels, mineral metabolism, renal physiology
Introduction
The distal convoluted tubule (DCT) is the portion of the nephron that is immediately downstream of the macula densa. Although the DCT is the shortest segment of the nephron, spanning only about 5 mm in length in humans (1), it plays a critical role in a variety of homeostatic processes, including sodium chloride reabsorption, potassium secretion, and calcium and magnesium handling. The DCT has a unique capacity to adapt to changes in hormonal stimuli and the contents of the tubular lumen, and this process contributes to the pathophysiology of a number of clinically relevant scenarios, including loop diuretic resistance and hyperaldosteronism. In recent years, insights gained from Mendelian disorders of BP and electrolyte imbalance, genetically modified animal models, and molecular cloning and study of key components of the machinery that controls the diverse ion transport processes housed in this segment have greatly expanded our understanding of distal tubule function. In this article, we provide a focused overview and update of the molecular physiology of the DCT.
Distal Nephron Nomenclature and Anatomic Considerations
The term distal tubule has been used by anatomists to denote the region of the nephron that extends downstream from the macula densa to the confluence of another tubule (i.e., the collecting system) (Figure 1) (2). It includes two nephron segments, the DCT and the connecting tubule (CNT). The DCT can be further divided into two functionally distinct subsegments, referred to as the DCT1 and the DCT2 (also called the early and late DCTs).
Figure 1.

A human nephron, showing the anatomic location of the distal tubule. The distal convoluted tubule (DCT) is divided into early and late segments, termed DCT1 and DCT2, respectively. The connecting tubule (CNT) is located immediately downstream of the DCT2. Insets compare DCT and CNT cell morphology. The cells of the DCT contain an extensive basolateral membrane system and are mitochondria-rich, indicating high transport activity of the Na+-K+-ATPase. CNT cells, in contrast, contain fewer mitochondria and basolateral membrane invaginations, suggesting that they are less metabolically active.
The DCT1 and DCT2 can be distinguished by their differential responsiveness to the mineralocorticoid aldosterone. Aldosterone is a steroid hormone that is released from the adrenal gland in response to volume depletion or hyperkalemia. It has a similar chemical structure to glucocorticoids, such as cortisol, and both aldosterone and cortisol bind to the mineralocorticoid receptor with nearly equal affinity (3). Although mineralocorticoid receptors are expressed throughout the entire DCT, the DCT2 is sensitive to the actions of aldosterone, because it expresses an enzyme called 11-β hydroxysteroid dehydrogenase 2 (11-βHSD2). 11-βHSD2 metabolizes cortisol to the inactive metabolite cortisone, thereby preventing circulating glucocorticoids from binding to mineralocorticoid receptors expressed in the DCT2 (4). Because the mineralocorticoid receptors in the DCT2 can only be occupied by aldosterone, the DCT2 is more sensitive than the DCT1 to changes in circulating aldosterone levels. 11-βHSD2 is also expressed in other downstream nephron segments, including the CNT and the cortical collecting duct (CCD). For this reason, the DCT2, CNT, and CCD are collectively termed the aldosterone-sensitive distal nephron (5).
Cells of the DCT have a unique morphology that matches their highly active physiology (Figure 1, insets) (6). Their nuclei tend to be positioned to the apical side of the cell because of an extensive basolateral membrane that has numerous deep infoldings. In addition, the cytosol on the basal aspect of DCT cells is packed with mitochondria—in fact, cells of the DCT are among the most mitochondria-rich in the kidney (7). These findings indicate that the DCT engages in processes that require considerable ATP consumption and active transport of electrolytes driven by the basolateral Na+-K+-ATPase.
Mechanism of Na+ Reabsorption in the DCT
Apical Sodium Transport in the Early and Late DCTs
The DCT reabsorbs roughly 5%–10% of the filtered sodium load (8). The thiazide-sensitive NaCl cotransporter (NCC; TSC; SLC12A3) is chiefly responsible for this process. NCC is one of the major targets of thiazide-type diuretics; the other target is sodium-dependent chloride bicarbonate exchanger (NDCBE; SLC4A8), a recently discovered Na+-driven chloride bicarbonate exchanger expressed in the CCD (9). This class of drugs is commonly used to treat hypertension, edema, and nephrolithiasis and include the thiazides chlorothiazide, hydrochlorothiazide, and bendroflumethiazide (only available combined with nadolol in the United States) as well as the thiazide-like diuretics metolazone, indapamide, and chlorthalidone. As shown in Figure 2, in the DCT1, sodium absorption from the tubular lumen is chloride-dependent and entirely mediated by NCC (10). Because a negatively charged chloride anion enters in 1:1 stoichiometry with a sodium cation, this process is electrically silent or electroneutral. Loss-of-function mutations of NCC cause Gitelman syndrome, an autosomal recessive salt wasting disorder that commonly presents with low-to-normal BP, elevated renin levels, hypokalemia, metabolic alkalosis, hypocalciuria, and hypomagnesemia (11). Thus, the phenotype of Gitelman syndrome is similar to the effects of thiazide diuretics. Most Gitelman mutations introduce changes into the NCC polypeptide that cause the cotransporter to misfold (12). This results in enhanced recognition by a molecular chaperone protein called Hsp70, which binds to misfolded NCC as it is being made in the endoplasmic reticulum and targets it for degradation (13,14).
Figure 2.

A model of NaCl reabsorption by cells of the early and late DCTs. Note that the transepithelial voltage is close to zero in the DCT1 and becomes progressively negative in the DCT2. In the early DCT, apical sodium reabsorption is exclusively mediated by thiazide-sensitive NaCl cotransporter (NCC), whereas in the late DCT, both NCC and amiloride-sensitive epithelial sodium channels (ENaCs) are present. The electrogenic transport of sodium by ENaC contributes to the lumen-negative transepithelial voltage. Basolateral sodium efflux is mediated by the Na+-K+-ATPase and aided by Kir4.1-mediated potassium leak currents. Chloride transport is carried out by the chloride channel ClC-Kb and potassium chloride cotransporter 4 (KCC4; SLC12A7).
NCC is expressed throughout the DCT, but the DCT2 also exhibits an amiloride-sensitive sodium transport pathway mediated by the epithelial sodium channel (ENaC) (Figure 2) (15,16). ENaC-mediated sodium transport is electrogenic; thus, in the DCT2, movement of Na+ ions through ENaC without an accompanying anion leaves negative charges in the lumen. Therefore, the transepithelial voltage or sum of membrane potentials on the luminal and basolateral membranes of the DCT epithelium is lumen-negative. As will be discussed below, this electrical gradient provides a driving force for the movement of other ions. Because ENaC is also expressed in the CNT and collecting duct, the transepithelial voltage becomes progressively more lumen-negative downstream of the DCT (17,18).
Basolateral Sodium Transport
In both subsegments of the DCT, after the transapical movement of Na+ through NCC and/or ENaC, Na+ is extruded from the cytosol and into the peritubular fluid by the Na+-K+-ATPase (Figure 2). Because this electrogenic pump exchanges sodium for potassium at a stoichiometry of 3:2, its activity removes one positive charge from the intracellular space, resulting in the generation of a constant negative voltage of −60 to −90 mV across the basolateral membrane (19,20). Along the basolateral membrane, the intracellular voltage becomes more negative with respect to extracellular voltage if the intracellular sodium concentration increases. This finding suggests that, in the DCT, a rise in luminal NCC- or ENaC-mediated sodium transport stimulates basolateral Na+ extrusion through the Na+-K+-ATPase (19). This provides an effective mechanism by which sodium ions can be transported transcellularly (i.e., through cells) and returned back to the plasma.
Basolateral K+ Channels
In order for sodium to be reabsorbed through transcellular mechanisms, basolateral sodium transport through the Na+-K+-ATPase must match the rate of apical sodium flux. Basolateral K+ efflux pathways play an important role in this process (21). By transporting K+ extracellularly, basolateral potassium channels create a “leak” that recycles K+ and helps to maintain the activity of the sodium pump (Figure 2). This so-called “pump leak coupling” maximizes the sodium reabsorptive capacity of an epithelium (22). Although renal physiologists have long appreciated this concept (23), its medical relevance only recently came to light through studies of a rare genetic disorder called EAST/SeSAME (24,25). In 2008, two groups reported that patients with mutations in the DCT-expressed basolateral inward-rectifying K+ channel Kir4.1 (KCNJ10) develop a hereditary salt wasting hypokalemic and hypomagnesemic disorder resembling Gitelman syndrome (24,25). Patients with these mutations typically present in infancy with a constellation of neurologic abnormalities in addition to the renal salt loss; for this reason, the disease has been named EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) by some investigators and SeSAME syndrome (seizures, sensorineural deafness, ataxia, mantal retardation, and electrolyte imbalance) by others. The Gitelman-like phenotype seen in individuals with the syndrome is likely caused by impaired Kir4.1 function and potassium recycling at the basolateral membrane of the DCT (26). In the absence of Kir4.1 activity, the activity of the Na+-K+-ATPase is reduced, resulting in diminished transcellular Na+ reabsorption in the DCT (Figure 3). Recent studies indicate that patients with EAST/SeSAME syndrome develop markedly reduced infoldings of the basolateral membrane of the DCT (26). This reduction in basolateral membrane surface area leads to a decrease in the total number of surface-localized Na+-K+-ATPase molecules, providing a second reason why basolateral sodium transport is reduced in these patients (Figure 3). To date, EAST/SeSAME syndrome is the only clinical distal tubular disorder known to affect a basolateral potassium channel. However, Kir4.1 is not the only potassium channel expressed at the basolateral membrane of the DCT. In fact, a number of K+ channels are expressed basolaterally, including Kir4.2 (KCNJ15) and Kir5.1 (KCNJ16) (21). Kir4.1 may associate in complexes with these alternative K+ transport pathways to form heteromultimers that cooperate in basolateral K+ recycling (27).
Figure 3.
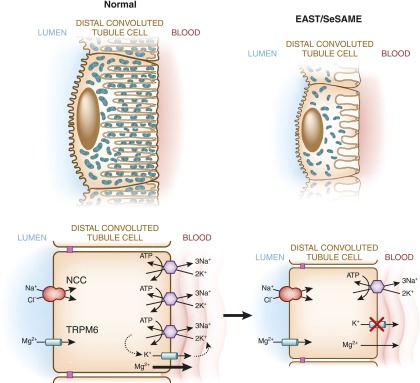
Mechanism of diminished NaCl and Mg2+ reabsorption in EAST/SeSAME syndrome. EAST/SeSAME syndrome is caused by mutations of the basolateral K+ channel (KCNJ10; Kir4.1). This channel mediates leakage of potassium to the peritubular fluid, which provides a supply of potassium ions that drives the activity of the Na+-K+-ATPase, resulting in constant reabsorption of sodium across the basolateral membrane. In EAST/SeSAME syndrome, inactivating mutations of Kir4.1 impair a leak current, which probably reduces activity of the sodium/potassium pump. Patients with the syndrome also display a reduced basolateral membrane surface area, consistent with diminished Na+-K+-ATPase activity. The reduction in basolateral sodium transport reduces the gradient for NCC-mediated Na+ reabsorption and causes salt wasting. Luminal magnesium reabsorption is similarly reduced in these patients, possibly because of diminished basolateral magnesium transport. EAST/SeSAME, epilepsy, ataxia, sensorineural deafness, and tubulopathy/seizures, sensorineural deafness, ataxia, mantal retardation, and electrolyte imbalance.
Cl− Absorption in the DCT
Apical Chloride Transport Pathways
In the early DCT, chloride is transported transcellularly from the tubular lumen with sodium through NCC. Sodium and chloride transport through this cotransporter is interdependent; thus, inhibition of NCC by thiazide diuretics will effectively block chloride reabsorption in the earliest loops of the distal nephron. As mentioned above, on reaching the late DCT, sodium is also transported through amiloride-sensitive ENaCs, which generate a lumen-negative transepithelial potential. This, in turn, serves as a driving force for chloride transport into the peritubular space through paracellular mechanisms (Figure 2). Paracellular transport (i.e., the transport of ions between cells) is a passive process, but cells generally confer specificity to the process by expressing multiprotein complexes at or near the tight junctions that connect adjacent cells. Included among these complexes is a large family of molecules called claudins (28). Current data indicate that different regions of the nephron express specific combinations of claudin proteins, which help to define the paracellular ion selectivities of those nephron segments.
Basolateral Chloride Transport
Because sodium transport in the DCT largely occurs through the coupled transcellular transport of sodium and chloride, basolateral Cl− transport is essential to the development of a gradient for Na+ entry. Cl− exit also helps to lower the intracellular Cl− concentration, which is a potent activator of NCC, and maintains net Na+ reabsorption (29). Cl− exit across the human DCT basolateral membrane is mediated by the chloride channel ClC-Kb. The Na+-K+-ATPase is necessary for this process, because it generates an electrochemical gradient that favors chloride efflux. ClC-Kb channels require an accessory subunit called Barttin to be fully functional (30). Both the ClC-Kb channel and Barttin are also expressed in the thick ascending limb. Thus, it is perhaps not surprising that loss-of-function mutations of either gene cause specific subtypes of Bartter syndrome (30,31). Patients with ClC-Kb channel mutations tend to present with a mixed Bartter/Gitelman phenotype, whereas patients lacking functional Barttin present with a severe neonatal salt wasting disorder that includes sensorineural deafness.
The potassium chloride cotransporter 4 (KCC4; SLC12A7) is expressed in the basolateral membrane of the DCT and mediates coupled electroneutral K-Cl efflux (32). KCC4 activity is stimulated by hypotonicity (33). Because luminal fluid in the DCT is hypotonic relative to plasma (34), the extracellular environment of the DCT theoretically favors KCC activation. This finding is particularly relevant in the DCT2, where tubular fluid is diluted down to 100 mOsM. Thus, a significant fraction of basolateral chloride reabsorption in the late DCT probably occurs through this transport pathway. Although no clinical syndrome involving KCC4 mutations has been described, KCC4 knockout mice develop renal tubular acidosis and deafness (35). This suggests that KCC4 plays an important role in determining renal net acid excretion and K+ secretion into the endolymph of the inner ear, although the mechanisms mediating such processes remain poorly defined.
Regulation of NaCl Transport in the DCT
Although sodium transport in the DCT is mediated by both NCC and ENaC, in this section, we will predominantly focus on the regulation of NCC-mediated Na+ transport. A detailed review of ENaC regulation is covered elsewhere in this series.
Distal Na+ Delivery
NaCl reabsorption in the DCT is dependent on sodium delivery. As the delivered sodium load is increased, the DCT responds by increasing its capacity for sodium transport. These changes are associated with marked alterations in DCT cell morphology, including an increase in basolateral membrane surface area and increased mitochondrial size (36). In concordance with these hypertrophic changes, Na+-K+-ATPase activity and the number of NCC cotransporters expressed per cell both increase dramatically. Thus, luminal sodium seems to be a potent stimulus that induces hypertrophy and activity of the distal tubule. The cellular mechanisms whereby NaCl entry affects NCC transport function and DCT morphology are not known.
The most common clinical scenario in which this phenomenon is encountered is loop diuretic resistance (Figure 4) (37). Chronic treatment with diuretics that inhibit the Na-K-2Cl cotransporter in the loop of Henle (bumetanide-sensitive sodium-potassium-chloride cotransporter [NKCC2, SLC12A1]), such as furosemide, results in a sustained increase in luminal Na+ delivery to the DCT. This induces hypertrophic changes in the DCT that increase NCC-mediated NaCl reabsorption, diminishing the effects of loop diuretics over time (36). Because of these hypertrophic changes, edematous patients who are loop diuretic–resistant and do not have severe CKD will frequently be highly sensitive to the effects of thiazide diuretics. Thus, the addition of even low doses of a thiazide or thiazide-like diuretic often results in a dramatic increase in urinary salt and water excretion.
Figure 4.

Effect of chronic loop diuretic administration on DCT activity and morphology. (Upper panel) Sodium reabsorption normally mediated by the thick ascending limb Na-K-2Cl cotransporter (NKCC2) is blocked by loop diuretics, such as furosemide and bumetanide. This results in enhanced Na+ delivery to the DCT, which likely acts as a stimulus for DCT hypertrophy. (Lower panel) DCT hypertrophy manifests as an increase in mitochondrial size and basolateral membrane infoldings. The increase in NCC-mediated apical sodium transport coupled with enhanced activity of the sodium/potassium pump at the basolateral membrane results in a vectorial increase in sodium reabsorption and diuretic resistance.
The WNK-SPAK/OSR1 Signaling Pathway and NCC
For thiazide-sensitive sodium chloride reabsorption to occur in the DCT, two processes must take place. First, a sufficient number of NCC cotransporters must be present at the DCT apical membrane, where they can come into contact with salt in the tubular lumen and facilitate NaCl cotransport. To get to the apical membrane, NCC must traffic there from a storage pool of vesicles present inside DCT cells (Figure 5). Second, cotransporters present at the DCT lumen must be switched on by phosphorylation. Two related serine threonine kinases, the Ste20-like proline-alanine rich kinase (SPAK) and oxidative stress responsive kinase 1 (OSR1), phosphorylate NCC directly (38). Both of these proteins transfer phosphate groups from ATP to serine and threonine residues present in the intracellular NCC amino terminus (Figure 5). By doing so, SPAK and OSR1 enhance the ability of the cotransporter to transport sodium and chloride, presumably by altering its protein conformation.
Figure 5.

SPAK and OSR1 phosphorylate and activate NCC. For NCC to be active, it must traffic to the plasma membrane from an intracellular storage pool. After it reaches the surface, it is in an inactive state until it is phosphorylated by two kinases, Ste20-like proline-alanine rich kinase (SPAK) and oxidative stress responsive kinase 1 (OSR1).
A family of proteins called With-No-Lysine [amino acid=K] kinases (WNKs) phosphorylates and activates SPAK and OSR1. They can also regulate NCC trafficking by separate mechanisms. These serine threonine kinases were identified as NCC regulators slightly over a decade ago through studies of a rare inherited disorder called Familial Hyperkalemic Hypertension (FHHt; also known as pseudohypoaldosteronism type 2 or Gordon syndrome) (39). FHHt is characterized by the unusual triad of hypertension, hyperkalemia, and normal GFR. Other features include metabolic acidosis and hypercalciuria. Thus, patients with FHHt have a phenotype that is essentially the mirror image of Gitelman syndrome. Moreover, FHHt is exquisitely sensitive to thiazide diuretics, indicating that the disorder is primarily a syndrome of NCC overactivity (40). In 2001, mutations in two WNKs, WNK1 and WNK4, were identified as the cause of FHHt (41). Although mutations in WNKs were the first to be implicated in the pathogenesis of FHHt, they only account for a minority of families affected by the disorder. Most FHHt-affected individuals harbor mutations in two other genes: the E3 ubiquitin ligase Cullin 3 (CUL3) and its adaptor, Kelch-like-3 (KLHL3) (42,43). These two proteins participate in a protein complex that degrades WNK1 and WNK4. A very recent series of papers from several laboratories show that CUL3 covalently attaches ubiquitin molecules to WNK1 and WNK4, marking them for disposal (44–46). To do so, KLHL3 must be present, because it connects CUL3 to the WNKs (Figure 6A).
Figure 6.
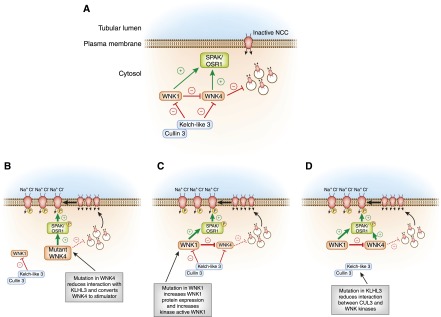
A model of WNK-SPAK/OSR1 regulation of NCC and its role in the pathogenesis of Familial Hyperkalemic Hypertension (FHHt). (A) In the baseline inactive state, WNK4 suppresses NCC trafficking to the plasma membrane, holding the cotransporter in an intracellular storage pool. The kinase active form of WNK1 can reverse this process. The Kelch-like 3/Cullin-3 (KLHL3/CUL3) E3 ubiquitin ligase complex constitutively degrades the WNKs. (B) FHHt-associated mutations in WNK4 reduce binding to KLHL3, increasing WNK4 abundance and triggering NCC activation through the WNK effector kinases SPAK and OSR1. Additionally, FHHt-causing mutations in WNK4 reduce its inhibitory effect on NCC traffic (represented by the hatched bar-headed line), which releases NCC from its intracellular compartment, increasing its trafficking to the cell surface. Thus, FHHt mutations in WNK4 convert it into an NCC stimulator. (C) WNK1 gene mutations increase kinase-active WNK1 expression, which overcomes constitutive degradation by KLHL3/CUL3. Because kinase active WNK1 can inhibit wild-type WNK4 and activate SPAK/OSR1, increased WNK1 expression stimulates NCC surface delivery and phosphorylation. (D) Mutations in KLHL3 either reduce binding of KLHL3/CUL3 to WNK1 and WNK4 or disconnect CUL3 from KLHL3; in either case, the CUL3 E3 ligase is unable to mark WNK signaling complexes for degradation. Increased WNK1 and WNK4 abundance stimulates NCC trafficking to the surface and triggers NCC phosphorylation. FHHt-causing mutations in CUL3 also likely reduce its activity to WNKs, although the mechanism by which this occurs remains unknown. WNK, With-No-Lysine [amino acid=K] kinase.
An elaborate interrelationship between WNK1, WNK4, and the KLHL3/CUL3 complex underlies the pathogenesis of FHHt. WNK1 and WNK4 regulate NCC through distinct mechanisms. According to current models, in the baseline state, where NCC is inactive and WNK4 kinase activity is switched off, WNK4 acts as an inhibitor, suppressing NCC trafficking to the cell surface (Figure 6A) (47–49). FHHt-causing WNK4 mutations are missense mutations that reduce WNK4 binding to KLHL3 (44,46). Total WNK4 protein expression, therefore, is increased in patients who have these mutations (Figure 6B). Disease-causing WNK4 mutants can phosphorylate SPAK and OSR1 (50), but many of them cannot block NCC plasma membrane trafficking (51,52). Thus, increased mutant WNK4 protein expression stimulates NCC phosphorylation and trafficking to the cell surface, resulting in NCC overactivation (Figure 6B) (44,46).
Like WNK4, the kinase active form of WNK1 phosphorylates and activates SPAK and OSR1 (53). In contrast to WNK4, however, WNK1 does not inhibit NCC trafficking. Rather, kinase-active WNK1 reverses the inhibitory effect of WNK4 on NCC traffic (Figure 6, A and C) (48,51). It likely mediates this reversal by binding to and phosphorylating WNK4 (54). Although these complexes cannot block NCC trafficking, they can still stimulate NCC phosphorylation. Thus, high levels of kinase-active WNK1 protein expression facilitate NCC trafficking to the cell surface and enhance NCC phosphorylation status through SPAK/OSR1. FHHt-causing mutations in WNK1 are intron deletions that do not alter the protein structure. Instead, these mutations increase total WNK1 mRNA and protein expression (41). The increased protein expression overrides ubiquitination and degradation by the KLHL3/CUL3 complex, and the net effect of the mutation is to increase total WNK1 kinase activity (55). Thus, in FHHt caused by mutations of WNK1, increased WNK1 protein expression should suppress the inhibitory effect of WNK4 on NCC traffic while stimulating SPAK and OSR1, causing NCC to be overactive (Figure 6C).
FHHt-associated mutations in KLHL3 reduce binding of the CUL3/KLHL3 complex to WNK1 and WNK4 (45) (Figure 6D). Thus, mutations in KLHL3 diminish the amount of CUL3-mediated WNK1 and WNK4 degradation (44–46). This increases WNK1 and WNK4 abundance, resulting in enhanced WNK-dependent signaling and NCC activation (Figure 6D). Mutations in CUL3 always cause skipping of an exon, resulting in an in-frame deletion (42). It seems likely that this specific change in the CUL3 protein structure reduces WNK1 and WNK4 ubiquitination, but this remains to be tested.
Several clinically relevant hormones stimulate salt reabsorption in the distal nephron by increasing NCC activity, and in each of these cases, SPAK and/or OSR1 seem to be involved. These hormones include aldosterone (56,57), angiotensin II (58,59), insulin (60,61), and vasopressin (62,63). All of these hormones have been shown to increase the abundance of phosphorylated active NCC and, in some cases, have also been shown to increase NCC abundance or stimulate the movement of NCC from intracellular vesicles to the luminal membrane (64–66). Because the WNKs are the only known activators of SPAK and OSR1 in the kidney, these findings strongly suggest that hormones that regulate BP and extracellular fluid volume status recruit WNK-dependent signaling processes to activate NCC through SPAK and OSR1 (Figure 7). Indeed, recent work has shown, for example, that angiotensin II–mediated activation of NCC is a WNK4-dependent process (58). Defining the molecular details of how these physiologically relevant stimuli interface with the WNK-SPAK/OSR1 signaling pathway remains an active area of biomedical research.
Figure 7.

A working model for NCC regulation through the WNK-SPAK/OSR1 signaling cascade. To date, a number of hormones have been shown to stimulate NCC phosphorylation at residues that are directly phosphorylated by SPAK and OSR1. These include aldosterone, angiotensin II, insulin, and vasopressin. Tacrolimus also enhances NCC phosphorylation at these sites, resulting in thiazide-sensitive NaCl reabsorption. In all cases, the mechanism likely involves hormonal activation of WNKs, which in turn, activates SPAK/OSR1. In some cases, such as aldosterone (66) and angiotensin II (58,64,65), hormone-induced changes in WNK-SPAK/OSR1-dependent signaling are also associated with increased trafficking of NCC to the plasma membrane.
Patients who are being treated with the immunosuppressant tacrolimus commonly present with hypertension, hyperkalemia, metabolic acidosis, and hypercalciuria—a clinical picture that in most respects resembles FHHt. Recently, it was shown that tacrolimus stimulates the activity of NCC, likely by enhancing its phosphorylation through the WNK-SPAK/OSR1 pathway (Figure 7) (67). Consistent with this finding, renal transplant patients on tacrolimus exhibited a greater urinary fractional excretion of chloride on bendroflumethiazide, indicating that tacrolimus-associated hypertension and hyperkalemia may be highly sensitive to thiazide diuretics (67). Larger clinical studies are currently underway to determine whether thiazides are superior to other agents as first-line therapy for the treatment of tacrolimus-associated hypertension.
K+ Secretion by the DCT
After reabsorption of K+ along the proximal tubule and loop of Henle, approximately 10% of filtered K+ reaches the DCT. As fluid travels down the DCT, the luminal potassium concentration increases, indicating that net K+ secretion occurs along the distal tubule. In the early DCT, the rate of K+ secretion is low, but it increases significantly in the late DCT (68–70). The enhanced K+ secretion parallels the progressive increase in lumen-negative transepithelial voltage observed in the late DCT. Indeed, a substantial component of the increased K+ secretion observed in this segment seems to be voltage-dependent. Voltage-dependent K+ secretion is mediated by the renal outer medullary potassium channel (ROMK; KCNJ1, also known as Kir1.1). ROMK-mediated K+ transport is dependent on the driving force for K+ secretion, which is primarily determined by electrogenic ENaC-mediated sodium reabsorption (71). Thus, as the activity of ENaC increases in the late DCT, more sodium is reabsorbed, which generates a lumen-negative stimulus for K+ efflux through ROMK potassium channels. As one can imagine, enhanced delivery of sodium ions to the late DCT and more downstream ENaC-expressing nephron segments—for example, through the use of loop diuretics—will enhance voltage-dependent K+ secretion.
The transepithelial voltage is not the only factor that determines the rate of K+ secretion in the DCT. Distal potassium secretion is also flow-dependent. Flow-dependent K+ secretion is mediated by the “big” or “maxi”-K+ channel (BK), a large conductance potassium channel that is expressed in all segments of the distal nephron, including the DCT, CNT, and collecting duct. During states of increased distal tubule flow, shear stress along the DCT activates the channel, possibly by increasing intracellular calcium levels and enhancing intracellular nitric oxide production (72,73). Both of the intracellular signals could increase the BK’s open probability and thereby enhance the rate of K+ efflux into the tubular lumen.
These observations have led to an updated model for K+ secretion in the distal nephron (Figure 8) (74,75). At baseline conditions, it has been proposed that the primary channel that mediates K+ secretion across the DCT lumen is ROMK. Under states of high urinary flow, such as during diuretic use, sodium delivery is enhanced, resulting in increased ENaC-mediated Na+ transport, depolarization of the apical membrane, and enhanced voltage-dependent K+ secretion through ROMK. In addition, the enhanced rate of urinary flow will trigger intracellular signaling mechanisms that result in enhanced BK activity. Thus, under low-flow conditions, ROMK is active, whereas under high-flow states, both ROMK and BK are active.
Figure 8.
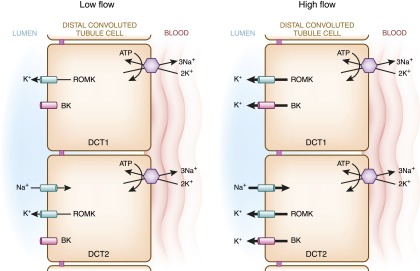
Model of potassium secretion in the DCT. K+ secretion in the distal nephron is voltage- and flow-dependent. In the low-flow state, “big”/“maxi”-K+ channels (BK) are closed, and potassium secretion exclusively occurs through the renal outer medullary potassium channel (ROMK). ROMK is expressed in the early DCT, but its activity there is relatively low because of the transepithelial voltage, which is near zero. In the late DCT, ENaC-mediated Na+ transport generates a driving force for ROMK-mediated K+ secretion. During a high-flow state, both ROMKs and BKs mediate K+ secretion. Increased sodium delivery and tubular fluid flow (for example, by infusing intravenous saline or Na bicarbonate or administering loop diuretics) increases ENaC activity, resulting in enhanced ROMK-mediated K+ secretion. Additionally, the increased flow triggers an intracellular signaling mechanism in the DCT that opens BK channels, facilitating K+ efflux into the tubular lumen.
Additional K+ secretion occurs more distally in the connecting tubule and CCD (71). Classically, the collecting duct has been thought of as the primary nephron segment that mediates K+ secretion. This is perhaps owing to the ease with which collecting ducts can be accessed and studied in the laboratory. In contrast to the collecting duct, distal tubules are difficult to isolate and perfuse, and for years this technical problem may have caused physiologists to overlook the importance of the DCT in renal physiology. However, several studies indicate that the late DCT and CNT mediate a substantial fraction of total distal K+ secretion (71). Additional studies are needed to determine the relative contribution of ROMK and BK in these nephron segments to whole-body potassium homeostasis.
Regulation of Distal Tubule Potassium Transport
Regulation of ROMK Gating
ROMK is an “inward rectifier” potassium channel. This essentially means that the natural tendency of these channels is to transport potassium into cells rather than out of them. However, in the distal tubule, ROMK primarily functions to secrete potassium into the tubular lumen. The reason why ROMK can carry out such a function is because of differences in the chemical and electrical gradients in the DCT, CNT, and CCD. Potassium is the most abundant intracellular cation. Therefore, there is a chemical tendency for potassium to leak outward. Although the voltage on the interior aspect of the luminal membrane is negative, it is not negative enough to counterbalance the high intracellular concentration of positively charged potassium cations. Therefore, K+ preferentially flows outward through ROMK. As described above, other channels that move positive charges inward (such as ENaC) will enhance this net outward flow of K+.
The capacity of a potassium channel to mediate inward rectification is modulated by a number of intrinsic factors, including the plasma membrane phosphoinositide 2 (76) and polyamines (77), but the most clinically relevant of these regulators of ROMK function is magnesium. Mg2+ binds to ROMK at a specific location on its cytoplasmic surface (78–80). By binding to ROMK at this site, Mg2+ blocks the channel’s pore from the inside and prevents K+ from being secreted. In the absence of adequate intracellular magnesium concentrations, potassium is more freely secreted into the tubular lumen (Figure 9). This phenomenon is currently believed to be an important reason why hypomagnesemia is often associated with enhanced distal K+ secretion and refractory hypokalemia (81).
Figure 9.

Role of magnesium in ROMK potassium channel function. Potassium is the most abundant intracellular cation, creating a large chemical gradient that favors the outward flow of K+ through ROMK. (Left panel) Normally, magnesium binds to a cytosol-exposed site in ROMK to limit this outward flow. (Right panel) During hypomagnesemia, fewer Mg2+ ions can bind to this site, and K+ is secreted more freely. Thus, magnesium deficiency causes K+ wasting. This likely explains why magnesium repletion is required to efficiently restore potassium concentrations to normal during concomitant hypomagnesemia and hypokalemia.
Aldosterone, Hyperkalemia, and Potassium Secretion
Hyperkalemia triggers aldosterone release from the adrenal gland. This results in an elevation of circulating aldosterone levels that enhances mineralocorticoid receptor–dependent signaling processes in the late DCT and downstream nephron segments. The mineralocorticoid receptor then triggers a signaling cascade that ultimately affects ROMK biogenesis. Specifically, the receptor translocates to the nucleus, where it stimulates the transcription of serum- and glucocorticoid-regulated kinase 1 (SGK1). SGK1 then phosphorylates a specific residue located in the cytoplasmic tail of ROMK; this event releases the channel from retention in the endoplasmic reticulum, allowing it to traffic more freely to the apical plasma membrane (82–84). Thus, through direct effects of SGK1 on ROMK traffic, aldosterone seems to ensure that an adequate number of potassium channels are available to facilitate K+ secretion in the distal nephron. In addition to its effects on ROMK traffic, aldosterone also stimulates K+ secretion by activating ENaC (85,86). As discussed above, the effect of this enhanced sodium transport through ENaC generates a lumen-negative transepithelial potential difference, which drives ROMK-mediated K+ secretion (Figure 8).
The renal response to hyperkalemia seems to be much more than simply an aldosterone-dependent phenomenon. Potassium loading induced, for example, by administrating a high K+ diet rapidly increases the number of ROMK channels expressed at the plasma membrane of distal nephron cells within hours of treatment; this effect can be seen before the observed increase in circulating aldosterone levels, suggesting that potassium has a direct effect on cells of the DCT. Similar effects have been noted with other components of the ion transport machinery expressed in the DCT, including the BK channel, the Na+-K+-ATPase, and ENaC (87–89). Recent evidence suggests that the aforementioned WNKs participate in the intracellular response of the DCT to hyperkalemia (90,91), and both WNK1 and WNK4 have been shown by several groups to influence the plasma membrane trafficking of both ROMK and BK channels in vitro (92–94). In fact, the bulk of evidence strongly suggests that the WNK signaling pathway seems to regulate transcellular sodium and potassium transport through completely different mechanisms, and it is this differential regulation of the two pathways that leads to the coexistence of hypertension and hyperkalemia seen in patients with FHHt (95). Although exciting progress has been made in this regard, additional studies are needed to define the in vivo relevance of these pathways to whole-body BP and potassium homeostasis in the general population.
Divalent Cation Reabsorption in the DCT
Although less is known about calcium and magnesium handling in the distal tubule, recent discoveries in the molecular mechanisms regulating these processes have expanded our understanding of how the DCT contributes to divalent cation homeostasis.
Calcium
Approximately 7%–10% of filtered calcium is reabsorbed in the DCT. In contrast to other segments of the nephron, which passively reabsorb calcium through paracellular routes, 100% of the calcium that is reabsorbed in the DCT occurs by active transcellular mechanisms. Apical calcium transport is mediated by the transient receptor potential channel subfamily V member 5 (TRPV5) (96) (Figure 10). On entry, Ca2+ associates with the calcium binding protein calbindin-D28K, which helps to buffer intracellular calcium levels and keep free calcium concentrations low (97) (Figure 10). After calcium is shuttled by calbindin-D28K to the basolateral surface, Ca2+ is extruded into the peritubular fluid by a calcium ATPase and the Type 1 sodium calcium exchanger (NCX1; Figure 10).
Figure 10.

Model of calcium reabsorption in the DCT. Apical calcium transport is mediated by transient receptor potential channel subfamily V member 5 (TRPV5) channels, which can be activated by the β-glucuronidase Klotho. Cytosolic calcium is immediately bound by calbindin-D28K, which shuttles calcium to the basolateral aspect of the DCT cell, where it can be transported out by the type 1 sodium calcium exchanger (NCX1) or calcium ATPases. These processes are tightly regulated by hormones, such as parathyroid hormone and 1,25-dihydroxyvitamin D (not shown).
Of these transport processes, apical calcium entry through TRPV5 seems to be the rate-limiting step for calcium reabsorption, and the activity of TRPV5 is regulated by several factors. Parathyroid hormone (PTH) affects TRPV5 channel activity through multiple mechanisms. PTH stimulates transcription of TRPV5 and prevents its degradation by inhibiting its removal from the apical surface of the DCT (98). In addition, PTH stimulates direct phosphorylation of TRPV5 through a protein kinase A–dependent signaling pathway, which alters the gating characteristics of the channel, increasing the likelihood that it will be open (99). 1,25-Dihydroxyvitamin D3 also stimulates TRPV5 and calbindin-D28K expression. More recently, the protein Klotho was shown to regulate calcium homeostasis by increasing the cell surface expression of TRPV5. Klotho is a distal nephron–expressed transmembrane protein with β-glucuronidase enzyme activity. Klotho remodels sugars located on the extracellular loops of the TRPV5 molecule, which slows the rate of the channel’s removal from the plasma membrane by enhancing binding to a secreted sugar binding protein, galectin-1 (100,101). Thus, by increasing the residence time of TRPV5 at the luminal membrane, Klotho increases TRPV5-mediated calcium reabsorption in the DCT (Figure 10). Interestingly, some evidence suggests that Klotho levels drop substantially during CKD (102), and Klotho-deficient mice develop several abnormalities of calcium homeostasis, including nephrocalcinosis, osteopenia, and hypercalciuria (103).
Hypercalcemia is a common side effect of thiazide diuretics (104). Because these drugs act on NCC-mediated salt transport in the DCT, one might assume that they somehow alter distal TRPV5-mediated calcium transport. Interestingly, this assumption turns out not to be the case. Knockout mice lacking TRPV5 are still capable of developing thiazide-induced hypocalciuria, which is probably because of the passive hyper-reabsorption of calcium with sodium and water in the proximal tubule (105) (Figure 11). Thiazide-associated volume depletion may trigger this increase in proximal calcium reabsorption.
Figure 11.

Thiazide-induced hypocalciuria. Administration of thiazide diuretics reduces urinary calcium excretion and can sometimes cause hypercalcemia. The mechanism likely involves an increase in bulk calcium reabsorption with sodium and water in the proximal tubule. Thiazide-induced volume depletion might be the stimulus that triggers this process.
Magnesium
The DCT reabsorbs about 10% of filtered magnesium and is the primary site of active transcellular Mg2+ reabsorption. Apical Mg2+ transport is mediated by transient receptor potential cation channel subfamily M member 6 (TRPM6), a voltage-driven divalent cation channel that is expressed in the early and late DCTs (106) (Figure 12). Like TRPV5, TRPM6 is a member of the transient receptor potential channel superfamily. TRPM6 has a 5-fold preference for magnesium ions over calcium ions, and these transport characteristics allow it to effectively function as a magnesium channel. Mutations in TRPM6 cause hypomagnesemia with secondary hypocalcemia, a rare Mendelian disorder of renal magnesium wasting.
Figure 12.

Model of magnesium reabsorption in the DCT. The magnesium channel transient receptor potential cation channel subfamily M member 6 (TRPM6) mediates luminal magnesium entry. At the luminal membrane, the K+ channel Kv1.1 extrudes K+ ions into the tubular lumen; this process probably generates an electrical driving force for Mg2+ entry through TRPM6. TRPM6 activity is stimulated by the magnesiotropic hormone EGF, which triggers an intracellular signaling cascade in the DCT after cleavage from pro-EGF and binding to basolateral EGF receptors (EGFRs). After transluminal entry, cytosolic Mg2+ is then transported out of the basolateral side of the DCT through unclear mechanisms, although cyclin M2 and SLC41A1 are candidate magnesium transport pathways that might mediate the process. Basolateral membrane voltage generated by the Na+-K+-ATPase is critical for Mg2+ exit, which is illustrated by Kir4.1 mutations in EAST/SeSAME syndrome that reduce pump activity by impaired recycling (Figure 3) or small γ-subunit of the Na+-K+-ATPase (FXYD2) mutations, which alter the pump’s affinity for Na+ and K+.
Other than this fairly recently described magnesium transport pathway, relatively little is known about other transport processes that facilitate transcellular Mg2+ reabsorption. Unlike its fellow divalent cation calcium, evidence does not exist for an intracellular magnesium binding protein that buffers cytosolic magnesium concentrations. With regards to basolateral Mg2+ transport, a mechanism exists for the reclamation of magnesium back into the peritubular fluid and bloodstream; however, the precise molecular identities of these transport processes remain obscure. One putative candidate is cyclin M2 (ancient conserved domain-containing protein 2), a distal nephron-expressed basolateral membrane protein that has been described by some as an Mg2+/metal ion transporter (107) and others as a magnesium sensor (108,109). Another candidate is the basolateral magnesium transporter SLC41A1, which is localized to the DCT. A loss-of-function mutation in this gene was recently shown to cause nephronopthisis, suggesting that it plays an important role in tubular function and morphology (110).
Emerging evidence gained from studies of families with rare inherited disorders of magnesium wasting suggests that the membrane voltage of the DCT plays a critical role in the control of magnesium reabsorption. In isolated dominant hypomagnesemia, mutations in the small γ-subunit of the Na+-K+-ATPase, FXYD2, alter pump function (111–113). Specifically, the absence of this subunit, which is highly expressed in the thick ascending limb of Henle’s loop and DCT, has been shown to alter the affinity of the Na+-K+-ATPase for sodium and potassium (114,115). FXYD2 mutations reduce subunit binding to the pump, although it is still unclear how this reduced interaction causes hypomagnesemia. The aforementioned EAST/SeSAME syndrome (Figure 3) is another hypomagnesemic disorder, in which the basolateral membrane voltage is likely altered. In this case, inactivating mutations of Kir4.1 reduce K+ recycling across the basolateral membrane, which could result in reduced sodium pump function, alteration of the basolateral membrane potential, and diminished transcellular magnesium transport (26).
In 2009, a fascinating new Mendelian disorder was discovered, in which patients develop hypomagnesemia because of a presumed alteration in the apical membrane voltage of the DCT. This autosomal dominant form of hypomagnesemia was attributed to a mutation in the Shaker-related voltage-gated K+ channel Kv1.1 (116). This channel is exclusively expressed at the apical membrane of the early and late DCTs and secretes K+ into the tubular lumen. It is believed that by secreting K+, Kv1.1 channels extrude positive charges into the lumen that provide a driving force for TRPM6-dependent Mg2+ transport (Figure 12). Mutation of Kv1.1 at a specific residue renders the channel nonfunctional, and one mutant allele is sufficient to cause the disease, presumably because of dominant negative inhibition of the remaining wild-type allele (117). A current theory is that the lack of functional Kv1.1 channels decreases the efflux of potassium cations into the lumen, which, in turn, would be expected to make the intracellular voltage on the luminal membrane more positive, decreasing the voltage gradient for luminal Mg2+ entry. Although this hypothesis is provocative, the pathophysiology may not be so simple: as discussed above in the section on ROMK gating, such a change in the luminal membrane potential would be expected to markedly enhance ROMK-mediated K+ secretion and cause hypokalemia, which is not observed in the disorder. Perhaps compensatory changes in ROMK, BK, or even ENaC expression in more downstream nephron segments, where Kv1.1 is not expressed (such as the CNT and CCD) (116), mitigate potassium losses, although to date, this hypothesis has not been tested.
EGF has emerged as an important regulator of Mg2+ handling in the DCT. In a Dutch family with a recessive form of selective hypomagnesemia, affected members had a mutation in the gene encoding the precursor form of EGF (118). This precursor molecule, pro-EGF, is a membrane protein that is expressed at the basolateral membrane of DCT cells. On arrival, the pro-EGF can be proteolytically cleaved to release soluble EGF, which then can interact with the EGF receptor and trigger a signaling cascade that stimulates TRPM6 (Figure 12). The hypomagnesemia-causing mutation results in missorting of pro-EGF, such that its delivery to the basolateral membrane of the DCT is inefficient. Ultimately, this impairs EGF-dependent signaling processes, including EGF-mediated stimulation of TRPM6 (119). The clinical importance of EGF-dependent regulation of TRPM6 is illustrated by the side effect profile of cetuximab, an EGF receptor antagonist used to treat colonic adenocarcinomas. Patients receiving systemic cetuximab can develop profound renal magnesium wasting because of the inhibition of EGF-dependent stimulation of TRPM6 in the DCT.
Hypomagnesemia is a common side effect of thiazide diuretics. Thiazides sharply downregulate TRPM6 expression in the DCT (105), causing decreased magnesium reabsorption and hypomagnesemia. The mechanism by which TRPM6 expression is downregulated by thiazides remains unknown, although several mechanisms have been proposed (105). One attractive hypothesis is that thiazide-induced hypoplasia of DCT cells decreases TRPM6 protein abundance, reducing the total number of channels expressed at the luminal membrane.
Summary
The DCT is a short but critically important nephron segment. The DCT consists of two distinct subsegments; both subsegments are highly metabolically active and play key roles in sodium, potassium, and divalent cation homeostasis. Insights from genetic diseases of BP, potassium, and calcium and magnesium balance have expanded our knowledge of the molecular machinery that mediates these processes. Although it is already known that the DCT plays an important part in certain common pathophysiological states, such as diuretic resistance, the role of the DCT in many other clinically relevant disease states awaits additional investigation. Now that our understanding of the regulatory machinery of the DCT is more complete, such investigations can be pursued with the intent of understanding disease pathogenesis and developing new strategies for the treatment of DCT-related disorders, such as hypertensive and/or edematous states, hyper- or hypokalemic tubulopathies, disorders of divalent ion balance, and nephrolithiasis.
Disclosures
None.
Acknowledgments
This work was supported, in part, by the National Institutes of Health Grants R01-DK51496 (to D.H.E.), R01-DK095841 (to D.H.E.), and P30-DK079307 (to the Pittsburgh Center for Kidney Research), R01-DK098145 (to A.R.S.), and a Mid-Level Veterans Affairs Career Development Grant (to A.R.S.).
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Chabardès D, Gagnan-Brunette M, Imbert-Teboul M, Gontcharevskaia O, Montégut M, Clique A, Morel F: Adenylate cyclase responsiveness to hormones in various portions of the human nephron. J Clin Invest 65: 439–448, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reilly RF, Ellison DH: Mammalian distal tubule: Physiology, pathophysiology, and molecular anatomy. Physiol Rev 80: 277–313, 2000 [DOI] [PubMed] [Google Scholar]
- 3.Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM: Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science 237: 268–275, 1987 [DOI] [PubMed] [Google Scholar]
- 4.Bostanjoglo M, Reeves WB, Reilly RF, Velázquez H, Robertson N, Litwack G, Morsing P, Dørup J, Bachmann S, Ellison DH: 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol 9: 1347–1358, 1998 [DOI] [PubMed] [Google Scholar]
- 5.Meneton P, Loffing J, Warnock DG: Sodium and potassium handling by the aldosterone-sensitive distal nephron: The pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol 287: F593–F601, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Kaissling B, Kriz W: Structural analysis of the rabbit kidney. Adv Anat Embryol Cell Biol 56: 1–123, 1979 [DOI] [PubMed] [Google Scholar]
- 7.Dørup J: Ultrastructure of distal nephron cells in rat renal cortex. J Ultrastruct Res 92: 101–118, 1985 [DOI] [PubMed] [Google Scholar]
- 8.Hierholzer K, Wiederholt M: Some aspects of distal tubular solute and water transport. Kidney Int 9: 198–213, 1976 [DOI] [PubMed] [Google Scholar]
- 9.Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hassan H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, Eladari D: The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120: 1627–1635, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellison DH, Velázquez H, Wright FS: Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am J Physiol 253: F546–F554, 1987 [DOI] [PubMed] [Google Scholar]
- 11.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP: Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24–30, 1996 [DOI] [PubMed] [Google Scholar]
- 12.Kunchaparty S, Palcso M, Berkman J, Velázquez H, Desir GV, Bernstein P, Reilly RF, Ellison DH: Defective processing and expression of thiazide-sensitive Na-Cl cotransporter as a cause of Gitelman’s syndrome. Am J Physiol 277: F643–F649, 1999 [DOI] [PubMed] [Google Scholar]
- 13.Donnelly BF, Needham PG, Snyder AC, Roy A, Khadem S, Brodsky JL, Subramanya AR: Hsp70 and Hsp90 multichaperone complexes sequentially regulate thiazide-sensitive cotransporter endoplasmic reticulum-associated degradation and biogenesis. J Biol Chem 288: 13124–13135, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Needham PG, Mikoluk K, Dhakarwal P, Khadem S, Snyder AC, Subramanya AR, Brodsky JL: The thiazide-sensitive NaCl cotransporter is targeted for chaperone-dependent endoplasmic reticulum-associated degradation. J Biol Chem 286: 43611–43621, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loffing J, Zecevic M, Féraille E, Kaissling B, Asher C, Rossier BC, Firestone GL, Pearce D, Verrey F: Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: Possible role of SGK. Am J Physiol Renal Physiol 280: F675–F682, 2001 [DOI] [PubMed] [Google Scholar]
- 16.Schmitt R, Ellison DH, Farman N, Rossier BC, Reilly RF, Reeves WB, Oberbäumer I, Tapp R, Bachmann S: Developmental expression of sodium entry pathways in rat nephron. Am J Physiol 276: F367–F381, 1999 [DOI] [PubMed] [Google Scholar]
- 17.Wright FS: Increasing magnitude of electrical potential along the renal distal tubule. Am J Physiol 220: 624–638, 1971 [DOI] [PubMed] [Google Scholar]
- 18.Hayslett JP, Boulpaep EL, Kashgarian M, Giebisch GH: Electrical characteristics of the mammalian distal tubule: Comparison of Ling-Gerard and macroelectrodes. Kidney Int 12: 324–331, 1977 [DOI] [PubMed] [Google Scholar]
- 19.Velázquez H, Wright FS: Effects of diuretic drugs on Na, Cl, and K transport by rat renal distal tubule. Am J Physiol 250: F1013–F1023, 1986 [DOI] [PubMed] [Google Scholar]
- 20.Yoshitomi K, Shimizu T, Taniguchi J, Imai M: Electrophysiological characterization of rabbit distal convoluted tubule cell. Pflugers Arch 414: 457–463, 1989 [DOI] [PubMed] [Google Scholar]
- 21.Hamilton KL, Devor DC: Basolateral membrane K+ channels in renal epithelial cells. Am J Physiol Renal Physiol 302: F1069–F1081, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, Hulton SA, Witzgall R, Ben-Zeev B, Howie AJ, Kleta R, Bockenhauer D, Warth R: KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci U S A 107: 14490–14495, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schultz SG: Homocellular regulatory mechanisms in sodium-transporting epithelia: Avoidance of extinction by “flush-through”. Am J Physiol 241: F579–F590, 1981 [DOI] [PubMed] [Google Scholar]
- 24.Scholl UI, Choi M, Liu T, Ramaekers VT, Häusler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP: Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A 106: 5842–5847, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van’t Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R: Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360: 1960–1970, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bandulik S, Schmidt K, Bockenhauer D, Zdebik AA, Humberg E, Kleta R, Warth R, Reichold M: The salt-wasting phenotype of EAST syndrome, a disease with multifaceted symptoms linked to the KCNJ10 K+ channel. Pflugers Arch 461: 423–435, 2011 [DOI] [PubMed] [Google Scholar]
- 27.Lourdel S, Paulais M, Cluzeaud F, Bens M, Tanemoto M, Kurachi Y, Vandewalle A, Teulon J: An inward rectifier K(+) channel at the basolateral membrane of the mouse distal convoluted tubule: Similarities with Kir4-Kir5.1 heteromeric channels. J Physiol 538: 391–404, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balkovetz DF: Claudins at the gate: Determinants of renal epithelial tight junction paracellular permeability. Am J Physiol Renal Physiol 290: F572–F579, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Pacheco-Alvarez D, Cristóbal PS, Meade P, Moreno E, Vazquez N, Muñoz E, Díaz A, Juárez ME, Giménez I, Gamba G: The Na+:Cl- cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem 281: 28755–28763, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Estévez R, Boettger T, Stein V, Birkenhäger R, Otto E, Hildebrandt F, Jentsch TJ: Barttin is a Cl- channel beta-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature 414: 558–561, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Thakker RV: Chloride channels in renal disease. Adv Nephrol Necker Hosp 29: 289–298, 1999 [PubMed] [Google Scholar]
- 32.Velázquez H, Silva T: Cloning and localization of KCC4 in rabbit kidney: Expression in distal convoluted tubule. Am J Physiol Renal Physiol 285: F49–F58, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Mercado A, Song L, Vazquez N, Mount DB, Gamba G: Functional comparison of the K+-Cl- cotransporters KCC1 and KCC4. J Biol Chem 275: 30326–30334, 2000 [DOI] [PubMed] [Google Scholar]
- 34.Gottschalk CW, Mylle M: Evidence that the mammalian nephron functions as a countercurrent multiplier system. Science 128: 594, 1958 [DOI] [PubMed] [Google Scholar]
- 35.Boettger T, Hübner CA, Maier H, Rust MB, Beck FX, Jentsch TJ: Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 416: 874–878, 2002 [DOI] [PubMed] [Google Scholar]
- 36.Ellison DH, Velázquez H, Wright FS: Adaptation of the distal convoluted tubule of the rat. Structural and functional effects of dietary salt intake and chronic diuretic infusion. J Clin Invest 83: 113–126, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellison DH: Diuretic resistance: Physiology and therapeutics. Semin Nephrol 19: 581–597, 1999 [PubMed] [Google Scholar]
- 38.Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR: Activation of the thiazide-sensitive Na+-Cl- cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci 121: 675–684, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Hadchouel J, Delaloy C, Fauré S, Achard JM, Jeunemaitre X: Familial hyperkalemic hypertension. J Am Soc Nephrol 17: 208–217, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Mayan H, Vered I, Mouallem M, Tzadok-Witkon M, Pauzner R, Farfel Z: Pseudohypoaldosteronism type II: Marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J Clin Endocrinol Metab 87: 3248–3254, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Wilson FH, Disse-Nicodème S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP: Human hypertension caused by mutations in WNK kinases. Science 293: 1107–1112, 2001 [DOI] [PubMed] [Google Scholar]
- 42.Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, Tikhonova IR, Bjornson R, Mane SM, Colussi G, Lebel M, Gordon RD, Semmekrot BA, Poujol A, Välimäki MJ, De Ferrari ME, Sanjad SA, Gutkin M, Karet FE, Tucci JR, Stockigt JR, Keppler-Noreuil KM, Porter CC, Anand SK, Whiteford ML, Davis ID, Dewar SB, Bettinelli A, Fadrowski JJ, Belsha CW, Hunley TE, Nelson RD, Trachtman H, Cole TR, Pinsk M, Bockenhauer D, Shenoy M, Vaidyanathan P, Foreman JW, Rasoulpour M, Thameem F, Al-Shahrouri HZ, Radhakrishnan J, Gharavi AG, Goilav B, Lifton RP: Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 482: 98–102, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Louis-Dit-Picard H, Barc J, Trujillano D, Miserey-Lenkei S, Bouatia-Naji N, Pylypenko O, Beaurain G, Bonnefond A, Sand O, Simian C, Vidal-Petiot E, Soukaseum C, Mandet C, Broux F, Chabre O, Delahousse M, Esnault V, Fiquet B, Houillier P, Bagnis CI, Koenig J, Konrad M, Landais P, Mourani C, Niaudet P, Probst V, Thauvin C, Unwin RJ, Soroka SD, Ehret G, Ossowski S, Caulfield M, Bruneval P, Estivill X, Froguel P, Hadchouel J, Schott JJ, Jeunemaitre X; International Consortium for Blood Pressure (ICBP): KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet 44: 456–460, S1–S3, 2012 [DOI] [PubMed] [Google Scholar]
- 44.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP: Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc Natl Acad Sci U S A 110: 7838–7843, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohta A, Schumacher FR, Mehellou Y, Johnson C, Knebel A, Macartney TJ, Wood NT, Alessi DR, Kurz T: The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: Disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem J 451: 111–122, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wakabayashi M, Mori T, Isobe K, Sohara E, Susa K, Araki Y, Chiga M, Kikuchi E, Nomura N, Mori Y, Matsuo H, Murata T, Nomura S, Asano T, Kawaguchi H, Nonoyama S, Rai T, Sasaki S, Uchida S: Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep 3: 858–868, 2013 [DOI] [PubMed] [Google Scholar]
- 47.Subramanya AR, Liu J, Ellison DH, Wade JB, Welling PA: WNK4 diverts the thiazide-sensitive NaCl cotransporter to the lysosome and stimulates AP-3 interaction. J Biol Chem 284: 18471–18480, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Golbang AP, Cope G, Hamad A, Murthy M, Liu CH, Cuthbert AW, O’shaughnessy KM: Regulation of the expression of the Na/Cl cotransporter by WNK4 and WNK1: Evidence that accelerated dynamin-dependent endocytosis is not involved. Am J Physiol Renal Physiol 291: F1369–F1376, 2006 [DOI] [PubMed] [Google Scholar]
- 49.Cai H, Cebotaru V, Wang YH, Zhang XM, Cebotaru L, Guggino SE, Guggino WB: WNK4 kinase regulates surface expression of the human sodium chloride cotransporter in mammalian cells. Kidney Int 69: 2162–2170, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Yang SS, Morimoto T, Rai T, Chiga M, Sohara E, Ohno M, Uchida K, Lin SH, Moriguchi T, Shibuya H, Kondo Y, Sasaki S, Uchida S: Molecular pathogenesis of pseudohypoaldosteronism type II: Generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab 5: 331–344, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Yang CL, Angell J, Mitchell R, Ellison DH: WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest 111: 1039–1045, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson FH, Kahle KT, Sabath E, Lalioti MD, Rapson AK, Hoover RS, Hebert SC, Gamba G, Lifton RP: Molecular pathogenesis of inherited hypertension with hyperkalemia: The Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc Natl Acad Sci U S A 100: 680–684, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vitari AC, Deak M, Morrice NA, Alessi DR: The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391: 17–24, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang CL, Zhu X, Wang Z, Subramanya AR, Ellison DH: Mechanisms of WNK1 and WNK4 interaction in the regulation of thiazide-sensitive NaCl cotransport. J Clin Invest 115: 1379–1387, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vidal-Petiot E, Elvira-Matelot E, Mutig K, Soukaseum C, Baudrie V, Wu S, Cheval L, Huc E, Cambillau M, Bachmann S, Doucet A, Jeunemaitre X, Hadchouel J: WNK1-related Familial Hyperkalemic Hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc Natl Acad Sci U S A 110: 14366–14371, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim GH, Masilamani S, Turner R, Mitchell C, Wade JB, Knepper MA: The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci U S A 95: 14552–14557, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chiga M, Rai T, Yang SS, Ohta A, Takizawa T, Sasaki S, Uchida S: Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int 74: 1403–1409, 2008 [DOI] [PubMed] [Google Scholar]
- 58.Castañeda-Bueno M, Cervantes-Pérez LG, Vázquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G: Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci U S A 109: 7929–7934, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE, McDonough AA: The absence of intrarenal ACE protects against hypertension. J Clin Invest 123: 2011–2023, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Komers R, Rogers S, Oyama TT, Xu B, Yang CL, McCormick J, Ellison DH: Enhanced phosphorylation of Na(+)-Cl- co-transporter in experimental metabolic syndrome: Role of insulin. Clin Sci (Lond) 123: 635–647, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sohara E, Rai T, Yang SS, Ohta A, Naito S, Chiga M, Nomura N, Lin SH, Vandewalle A, Ohta E, Sasaki S, Uchida S: Acute insulin stimulation induces phosphorylation of the Na-Cl cotransporter in cultured distal mpkDCT cells and mouse kidney. PLoS ONE 6: e24277, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saritas T, Borschewski A, McCormick JA, Paliege A, Dathe C, Uchida S, Terker A, Himmerkus N, Bleich M, Demaretz S, Laghmani K, Delpire E, Ellison DH, Bachmann S, Mutig K: SPAK differentially mediates vasopressin effects on sodium cotransporters. J Am Soc Nephrol 24: 407–418, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pedersen NB, Hofmeister MV, Rosenbaek LL, Nielsen J, Fenton RA: Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney Int 78: 160–169, 2010 [DOI] [PubMed] [Google Scholar]
- 64.Sandberg MB, Riquier AD, Pihakaski-Maunsbach K, McDonough AA, Maunsbach AB: ANG II provokes acute trafficking of distal tubule Na+-Cl(-) cotransporter to apical membrane. Am J Physiol Renal Physiol 293: F662–F669, 2007 [DOI] [PubMed] [Google Scholar]
- 65.San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, Chari D, Kahle KT, Leng Q, Bobadilla NA, Hebert SC, Alessi DR, Lifton RP, Gamba G: Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci U S A 106: 4384–4389, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rozansky DJ, Cornwall T, Subramanya AR, Rogers S, Yang YF, David LL, Zhu X, Yang CL, Ellison DH: Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J Clin Invest 119: 2601–2612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoorn EJ, Walsh SB, McCormick JA, Fürstenberg A, Yang CL, Roeschel T, Paliege A, Howie AJ, Conley J, Bachmann S, Unwin RJ, Ellison DH: The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 17: 1304–1309, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schnermann J, Steipe B, Briggs JP: In situ studies of distal convoluted tubule in rat. II. K secretion. Am J Physiol 252: F970–F976, 1987 [DOI] [PubMed] [Google Scholar]
- 69.Stanton BA, Giebisch GH: Potassium transport by the renal distal tubule: Effects of potassium loading. Am J Physiol 243: F487–F493, 1982 [DOI] [PubMed] [Google Scholar]
- 70.Wade JB, Fang L, Coleman RA, Liu J, Grimm PR, Wang T, Welling PA: Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietary potassium. Am J Physiol Renal Physiol 300: F1385–F1393, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Giebisch G: Renal potassium transport: Mechanisms and regulation. Am J Physiol 274: F817–F833, 1998 [DOI] [PubMed] [Google Scholar]
- 72.Liu W, Xu S, Woda C, Kim P, Weinbaum S, Satlin LM: Effect of flow and stretch on the [Ca2+]i response of principal and intercalated cells in cortical collecting duct. Am J Physiol Renal Physiol 285: F998–F1012, 2003 [DOI] [PubMed] [Google Scholar]
- 73.Kudlacek PE, Pluznick JL, Ma R, Padanilam B, Sansom SC: Role of hbeta1 in activation of human mesangial BK channels by cGMP kinase. Am J Physiol Renal Physiol 285: F289–F294, 2003 [DOI] [PubMed] [Google Scholar]
- 74.Holtzclaw JD, Grimm PR, Sansom SC: Role of BK channels in hypertension and potassium secretion. Curr Opin Nephrol Hypertens 20: 512–517, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sansom SC, Welling PA: Two channels for one job. Kidney Int 72: 529–530, 2007 [DOI] [PubMed] [Google Scholar]
- 76.Huang CL, Feng S, Hilgemann DW: Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 391: 803–806, 1998 [DOI] [PubMed] [Google Scholar]
- 77.Ficker E, Taglialatela M, Wible BA, Henley CM, Brown AM: Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science 266: 1068–1072, 1994 [DOI] [PubMed] [Google Scholar]
- 78.Nichols CG, Ho K, Hebert S: Mg(2+)-dependent inward rectification of ROMK1 potassium channels expressed in Xenopus oocytes. J Physiol 476: 399–409, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu Z, MacKinnon R: Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature 371: 243–246, 1994 [DOI] [PubMed] [Google Scholar]
- 80.Welling PA, Ho K: A comprehensive guide to the ROMK potassium channel: Form and function in health and disease. Am J Physiol Renal Physiol 297: F849–F863, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang CL, Kuo E: Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 18: 2649–2652, 2007 [DOI] [PubMed] [Google Scholar]
- 82.Yoo D, Fang L, Mason A, Kim BY, Welling PA: A phosphorylation-dependent export structure in ROMK (Kir 1.1) channel overrides an endoplasmic reticulum localization signal. J Biol Chem 280: 35281–35289, 2005 [DOI] [PubMed] [Google Scholar]
- 83.Yoo D, Kim BY, Campo C, Nance L, King A, Maouyo D, Welling PA: Cell surface expression of the ROMK (Kir 1.1) channel is regulated by the aldosterone-induced kinase, SGK-1, and protein kinase A. J Biol Chem 278: 23066–23075, 2003 [DOI] [PubMed] [Google Scholar]
- 84.O’Connell AD, Leng Q, Dong K, MacGregor GG, Giebisch G, Hebert SC: Phosphorylation-regulated endoplasmic reticulum retention signal in the renal outer-medullary K+ channel (ROMK). Proc Natl Acad Sci U S A 102: 9954–9959, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D: WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J 15: 2371–2380, 1996 [PMC free article] [PubMed] [Google Scholar]
- 86.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Münster C, Chraïbi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O: Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J 20: 7052–7059, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Palmer LG, Antonian L, Frindt G: Regulation of apical K and Na channels and Na/K pumps in rat cortical collecting tubule by dietary K. J Gen Physiol 104: 693–710, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Estilo G, Liu W, Pastor-Soler N, Mitchell P, Carattino MD, Kleyman TR, Satlin LM: Effect of aldosterone on BK channel expression in mammalian cortical collecting duct. Am J Physiol Renal Physiol 295: F780–F788, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Silva P, Hayslett JP, Epstein FH: The role of Na-K-activated adenosine triphosphatase in potassium adaptation. Stimulation of enzymatic activity by potassium loading. J Clin Invest 52: 2665–2671, 1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cheng CJ, Baum M, Huang CL: Kidney-specific WNK1 regulates sodium reabsorption and potassium secretion in mouse cortical collecting duct. Am J Physiol Renal Physiol 304: F397–F402, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu Z, Wang HR, Huang CL: Regulation of ROMK channel and K+ homeostasis by kidney-specific WNK1 kinase. J Biol Chem 284: 12198–12206, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lazrak A, Liu Z, Huang CL: Antagonistic regulation of ROMK by long and kidney-specific WNK1 isoforms. Proc Natl Acad Sci U S A 103: 1615–1620, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kahle KT, Wilson FH, Leng Q, Lalioti MD, O’Connell AD, Dong K, Rapson AK, MacGregor GG, Giebisch G, Hebert SC, Lifton RP: WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet 35: 372–376, 2003 [DOI] [PubMed] [Google Scholar]
- 94.Wade JB, Fang L, Liu J, Li D, Yang CL, Subramanya AR, Maouyo D, Mason A, Ellison DH, Welling PA: WNK1 kinase isoform switch regulates renal potassium excretion. Proc Natl Acad Sci U S A 103: 8558–8563, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Welling PA, Chang YP, Delpire E, Wade JB: Multigene kinase network, kidney transport, and salt in essential hypertension. Kidney Int 77: 1063–1069, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hoenderop JG, van der Kemp AW, Hartog A, van de Graaf SF, van Os CH, Willems PH, Bindels RJ: Molecular identification of the apical Ca2+ channel in 1, 25-dihydroxyvitamin D3-responsive epithelia. J Biol Chem 274: 8375–8378, 1999 [DOI] [PubMed] [Google Scholar]
- 97.Hemmingsen C: Regulation of renal calbindin-D28K. Pharmacol Toxicol 87[Suppl 3]: 5–30, 2000 [PubMed] [Google Scholar]
- 98.Cha SK, Wu T, Huang CL: Protein kinase C inhibits caveolae-mediated endocytosis of TRPV5. Am J Physiol Renal Physiol 294: F1212–F1221, 2008 [DOI] [PubMed] [Google Scholar]
- 99.de Groot T, Lee K, Langeslag M, Xi Q, Jalink K, Bindels RJ, Hoenderop JG: Parathyroid hormone activates TRPV5 via PKA-dependent phosphorylation. J Am Soc Nephrol 20: 1693–1704, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG: The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310: 490–493, 2005 [DOI] [PubMed] [Google Scholar]
- 101.Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro-O M, Huang CL: Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci U S A 105: 9805–9810, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hu MC, Kuro-o M, Moe OW: Klotho and chronic kidney disease. Contrib Nephrol 180: 47–63, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alexander RT, Woudenberg-Vrenken TE, Buurman J, Dijkman H, van der Eerden BC, van Leeuwen JP, Bindels RJ, Hoenderop JG: Klotho prevents renal calcium loss. J Am Soc Nephrol 20: 2371–2379, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duarte CG, Winnacker JL, Becker KL, Pace A: Thiazide-induced hypercalcemia. N Engl J Med 284: 828–830, 1971 [DOI] [PubMed] [Google Scholar]
- 105.Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ: Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 115: 1651–1658, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schlingmann KP, Weber S, Peters M, Niemann Nejsum L, Vitzthum H, Klingel K, Kratz M, Haddad E, Ristoff E, Dinour D, Syrrou M, Nielsen S, Sassen M, Waldegger S, Seyberth HW, Konrad M: Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet 31: 166–170, 2002 [DOI] [PubMed] [Google Scholar]
- 107.Goytain A, Quamme GA: Functional characterization of ACDP2 (ancient conserved domain protein), a divalent metal transporter. Physiol Genomics 22: 382–389, 2005 [DOI] [PubMed] [Google Scholar]
- 108.Stuiver M, Lainez S, Will C, Terryn S, Günzel D, Debaix H, Sommer K, Kopplin K, Thumfart J, Kampik NB, Querfeld U, Willnow TE, Němec V, Wagner CA, Hoenderop JG, Devuyst O, Knoers NV, Bindels RJ, Meij IC, Müller D: CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. Am J Hum Genet 88: 333–343, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.de Baaij JH, Stuiver M, Meij IC, Lainez S, Kopplin K, Venselaar H, Müller D, Bindels RJ, Hoenderop JG: Membrane topology and intracellular processing of cyclin M2 (CNNM2). J Biol Chem 287: 13644–13655, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hurd TW, Otto EA, Mishima E, Gee HY, Inoue H, Inazu M, Yamada H, Halbritter J, Seki G, Konishi M, Zhou W, Yamane T, Murakami S, Caridi G, Ghiggeri G, Abe T, Hildebrandt F: Mutation of the Mg2+ transporter SLC41A1 results in a nephronophthisis-like phenotype. J Am Soc Nephrol 24: 967–977, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kantorovich V, Adams JS, Gaines JE, Guo X, Pandian MR, Cohn DH, Rude RK: Genetic heterogeneity in familial renal magnesium wasting. J Clin Endocrinol Metab 87: 612–617, 2002 [DOI] [PubMed] [Google Scholar]
- 112.Meij IC, Koenderink JB, van Bokhoven H, Assink KF, Groenestege WT, de Pont JJ, Bindels RJ, Monnens LA, van den Heuvel LP, Knoers NV: Dominant isolated renal magnesium loss is caused by misrouting of the Na(+),K(+)-ATPase gamma-subunit. Nat Genet 26: 265–266, 2000 [DOI] [PubMed] [Google Scholar]
- 113.Cairo ER, Friedrich T, Swarts HG, Knoers NV, Bindels RJ, Monnens LA, Willems PH, De Pont JJ, Koenderink JB: Impaired routing of wild type FXYD2 after oligomerisation with FXYD2-G41R might explain the dominant nature of renal hypomagnesemia. Biochim Biophys Acta 1778: 398–404, 2008 [DOI] [PubMed] [Google Scholar]
- 114.Béguin P, Wang X, Firsov D, Puoti A, Claeys D, Horisberger JD, Geering K: The gamma subunit is a specific component of the Na,K-ATPase and modulates its transport function. EMBO J 16: 4250–4260, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Arystarkhova E, Wetzel RK, Asinovski NK, Sweadner KJ: The gamma subunit modulates Na(+) and K(+) affinity of the renal Na,K-ATPase. J Biol Chem 274: 33183–33185, 1999 [DOI] [PubMed] [Google Scholar]
- 116.Glaudemans B, van der Wijst J, Scola RH, Lorenzoni PJ, Heister A, van der Kemp AW, Knoers NV, Hoenderop JG, Bindels RJ: A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest 119: 936–942, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van der Wijst J, Glaudemans B, Venselaar H, Nair AV, Forst AL, Hoenderop JG, Bindels RJ: Functional analysis of the Kv1.1 N255D mutation associated with autosomal dominant hypomagnesemia. J Biol Chem 285: 171–178, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Groenestege WM, Thébault S, van der Wijst J, van den Berg D, Janssen R, Tejpar S, van den Heuvel LP, van Cutsem E, Hoenderop JG, Knoers NV, Bindels RJ: Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J Clin Invest 117: 2260–2267, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Thebault S, Alexander RT, Tiel Groenestege WM, Hoenderop JG, Bindels RJ: EGF increases TRPM6 activity and surface expression. J Am Soc Nephrol 20: 78–85, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]