

1. Introduction
The indole alkaloids, ranging from lysergic acid to vincristine, have long inspired organic synthesis chemists. Interest in developing new methods for indole synthesis has burgeoned over the past few years. These new methods have been fragmented across the literature of organic chemistry. In this review, we present a framework for the classification of all indole syntheses.
As we approach the classification of routes for the preparation of indoles, we are mindful that the subject has occupied the minds of organic chemists for more than a century. There have been many reviews of indole synthesis.1 We were also aware that much more could be said than we have written. We have only briefly covered the conversion of indolines into indoles, and the reduction of oxindoles to indoles. We have not covered the extensive literature on the modification of existing indoles. Throughout, our interest has been to be illustrative, not exhaustively inclusive. It is apparent, however, that every indole synthesis must fit one or the other of the nine strategic approaches adumbrated here. The web of scientific citations unites and organizes the world-wide research effort. It is our intention that the system put forward here for classifying indole syntheses will be universally understood. As authors conceive of new approaches to the indole nucleus, they will be able to classify their approach, and so readily discover both the history and the current state of the art with that strategy for indole construction. In addition to avoiding duplication, it is also our hope that efforts will then be directed toward the very real challenges that remain to be overcome. It is noteworthy that, in the most recent year we have covered, 2009, significant new contributions were reported for each of these nine strategies. We have highlighted these at the end of each section.
There are four bonds in the five-membered indole ring. In classifying methods for synthesis (Fig. 1), we have focused on the last bond formed. We have also differentiated, in distinguishing Type 1 versus Type 2 and Type 3 versus Type 4, between forming a bond to a functionalized aromatic carbon, and forming a bond to an aromatic carbon occupied only by an H. Type 5 has as the last step C–N bond formation, while with Type 6 the last step is C–C bond formation. In Type 7, the benzene ring has been derived from an existing cyclohexane, and in Type 8, the benzene ring has been built onto an existing pyrrole. Finally, in Type 9, both rings have been constructed.
Fig 1.
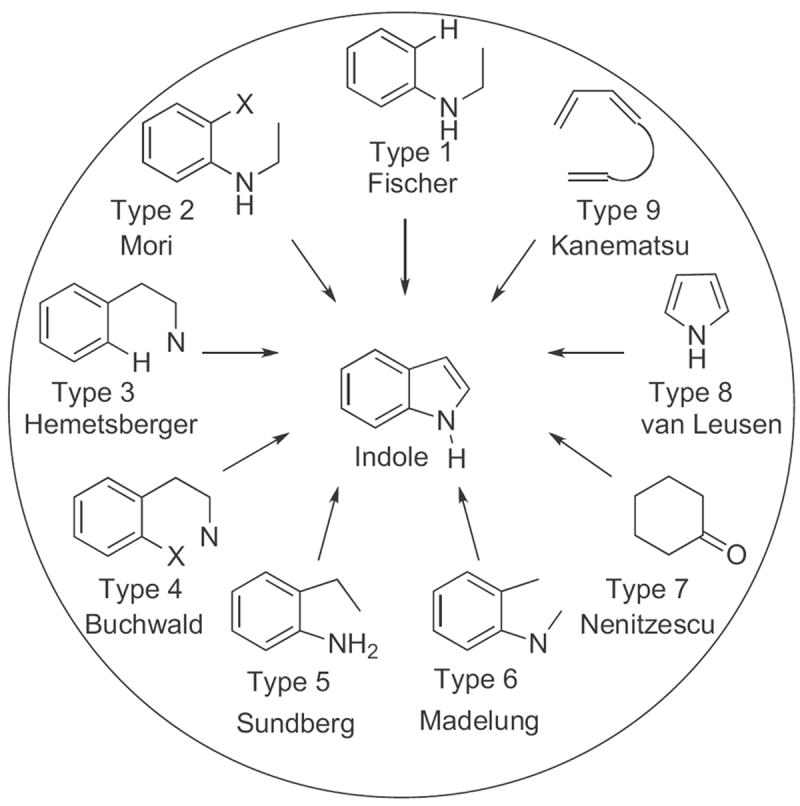
The nine types of indole synthesis.
There are several name reactions associated with indole synthesis. We have tried to note these in context, and to group examples of a particular name reaction together. For convenience, the ‘name reaction’ indole syntheses mentioned in this review are:
Bartoli indole synthesis—Type 1
Bischler indole synthesis—Type 5
Fischer indole synthesis—Type 1
Hemetsberger indole synthesis—Type 3
Julia indole synthesis—Type 5
Larock indole synthesis—Type 5
Leimgruber–Batcho indole synthesis—Type 5
Madelung indole synthesis—Type 6
Nenitzescu indole synthesis—Type 7
Reissert indole synthesis—Type 5
Sundberg indole synthesis—Type 5
While it might be sufficient to merely label the nine strategies 1–9, for ease of recollection we have also associated each strategy with the name of an early or well-known practitioner. The division of strategies is strictly operational. Thus, the Fischer indole synthesis is classified as Type 1, Ar–H to C2, since that is the way it is carried out, even though the last bond formed, as the reaction proceeds, is in fact N to C1.
2. Type 1
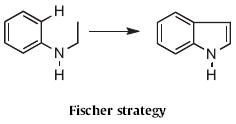
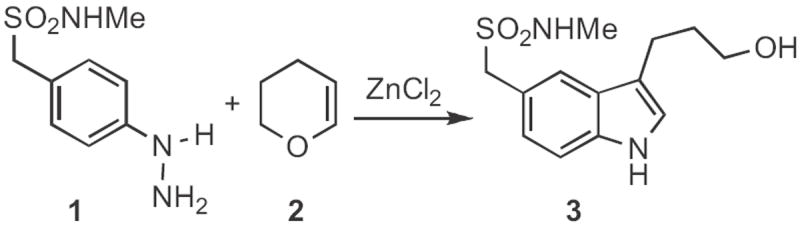
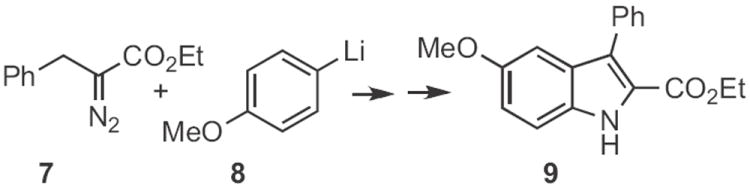
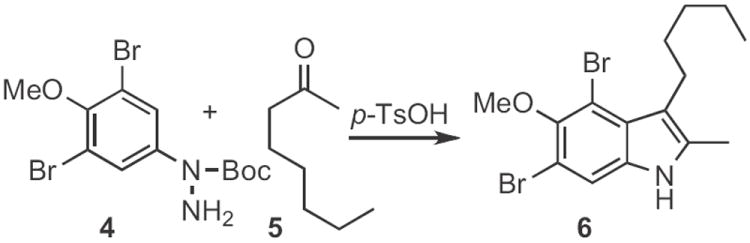
Type 1 synthesis (Scheme 1-17) involves aromatic C–H functionalization. Although C–H activation is thought of as a modern topic, the venerable Fischer indole synthesis (still under active development, Schemes 1-3) falls under this heading. Paul R. Brodfuehrer and Shaopeng Wang of Bristol-Myers Squibb described2 the convenient (Scheme 1) reaction of an aryl hydrazine 1 with dihydropyran 2 to give the 3-hydroxypropylindole 3. Stephen L. Buchwald of MIT developed3 an elegant (Scheme 2) amination of aryl iodides to give Boc-protected aryl hydrazines, such as 4. Acid-mediated condensation of 4 with the ketone 5 delivered the indole 6. The condensation of 4 and 5 proceeded with high regioselectivity. Norio Takamura of Musashino University, Tokyo presented4 a complementary approach (Scheme 3), the addition of an aryllithium 8 to an α-diazo ester 7, followed by acid-mediated cyclization. The ester of 9 is easily manipulated, and can also be removed altogether. Several other useful variations on the Fischer indole synthesis have been reported.5-7
Scheme 1.
Scheme 17.
Scheme 3.
Scheme 2.
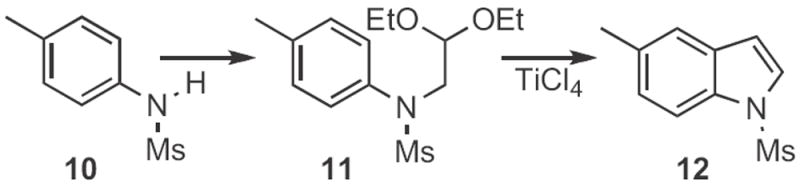
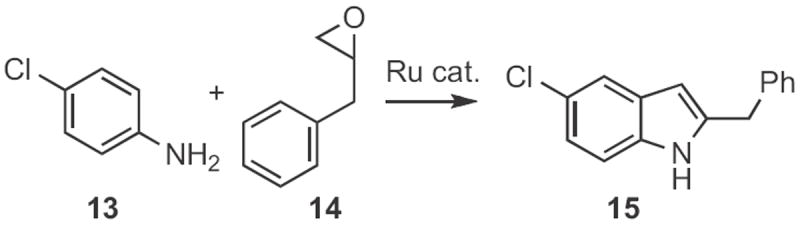
Indoles can also be formed by acid-mediated cyclization of aldehydes. Richard J. Sundberg of the University of Virginia described8 the preparation from 10 (Scheme 4) and cyclization of acetals, such as 11 to give the indole 12. The Bischler indole synthesis9a,b is a variation on this approach. Chan Sik Cho and Sang Chul Shim of Kyungpook National University, Taegu devised9c a route to indoles (Scheme 5) based on Ru-mediated addition of an aniline 13 to an epoxide 14. An interesting oxidatione–reduction cascade led to the 2-alkyl indole 15, probably via a Bischler-like tautomerization.
Scheme 4.
Scheme 5.
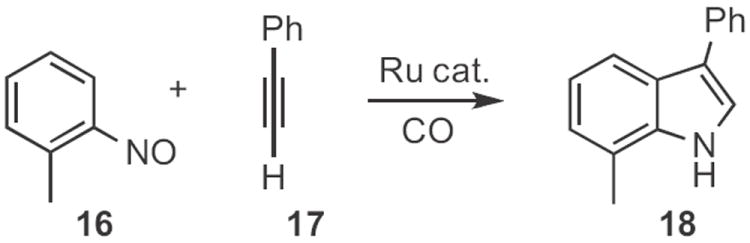
Other transition-metal-mediated protocols for indole synthesis have been developed. In a variant on the Bartoli indole synthesis, Kenneth M. Nicholas of the University of Oklahoma reported10 the Ru-catalyzed reductive coupling of a nitrosoaromatic, such as 16 (Scheme 6) with an alkyne 17 to give the indole 18. Akio Saito and Yuji Hanzawa of Showa Pharmaceutical University described11 the Rh-catalyzed cyclization of 19 (Scheme 7) to 20. The reaction was thought to proceed via the allene 21.
Scheme 6.
Scheme 7.
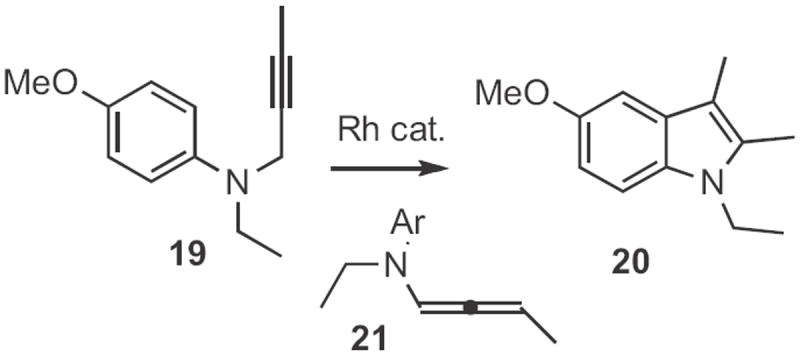
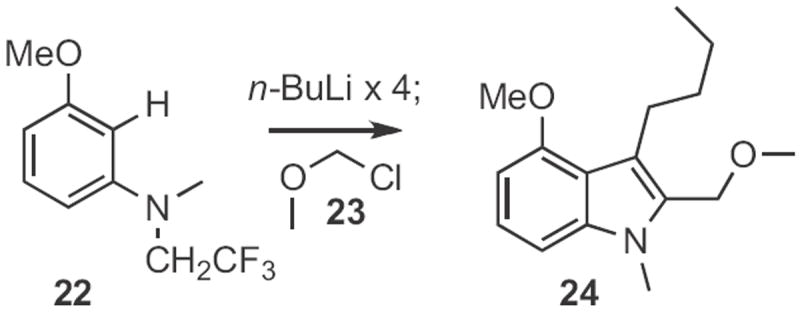
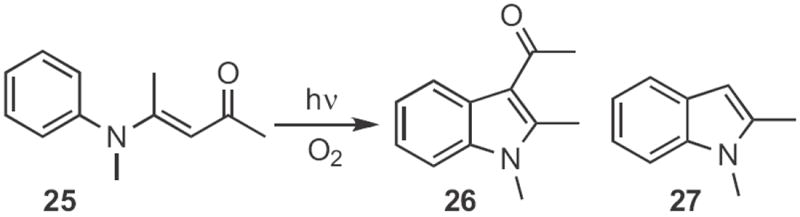
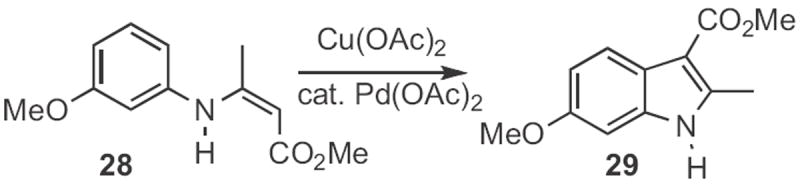
Several other Type 1 indole syntheses have been described. In the examples cited so far, only one regioisomeric aryl H could be substituted. In an ortho-metalation approach, Francis Johnson of SUNY Stony Brook showed12 that (Scheme 8) the anion from cyclization of 22 could be alkyated with an electrophile, such as 23 to give the indole 24. Darrell Watson and D.R. Dillin at the University of Mary Hardin-Baylor reported13 a photochemical route (Scheme 9) to indoles. Irradiation of 25 in an oxygen atmosphere led to 26. When the photolysis was carried out under nitrogen, the product was 27. Frank Glorius of the Universität Münster devised14 a related catalytic oxidation of enamines, such as 28 (Scheme 10) to the indole 29. Just recently, Yan-Guang Wang of Zhejian University, Hangzhou described15 the coupling (Scheme 11) of a wide range of anilides, such as 30 with ethyl diazoacetate 31 to give the indole 32.
Scheme 8.
Scheme 9.
Scheme 10.
Scheme 11.
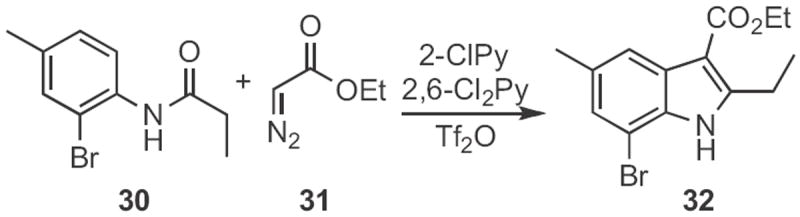
Indoles, such as 35 (Scheme 12) can also be prepared from oxindoles, such as 34, prepared from 33. Wendell Wierenga, then at Upjohn, optimized16 both the Gassman synthesis of oxindoles from anilines, and the subsequent reduction. This is a net Type 1 synthesis.
Scheme 12.
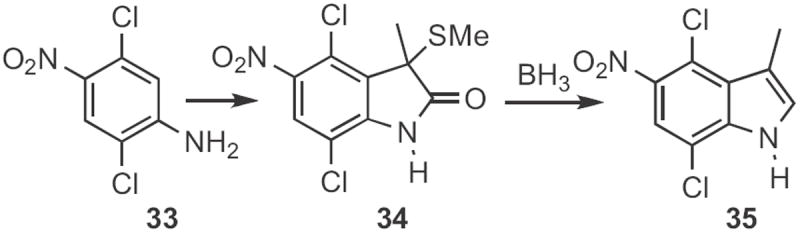
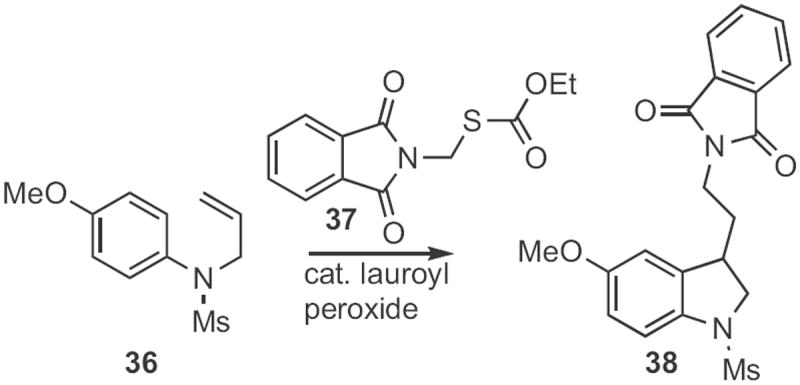
Samir Z. Zard of Ecole Polytechnique described17 the cyclization (Scheme 13) of allyl anilines, such as 36 to the indoline 38 using 37. As indolines can be converted into indoles by oxidation18 or by base-mediated elimination of an N-sulfonyl group19 this is also a net Type 1 indole synthesis.
Scheme 13.
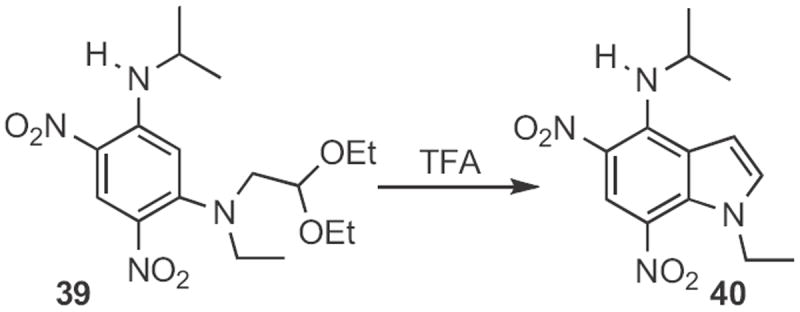
In 2009, four interesting new examples of Type 1 indole synthesis were described. It had been thought that the cyclization of an acetal (Scheme 4) to the indole would only work with electron-rich aromatic rings. Dali Yin of the Institute of Materia Medica, Beijing20 observed that 39 (Scheme 14), readily prepared by sequential displacement on the corresponding difluorodintrobenzene, smoothly cyclized to 40.
Scheme 14.
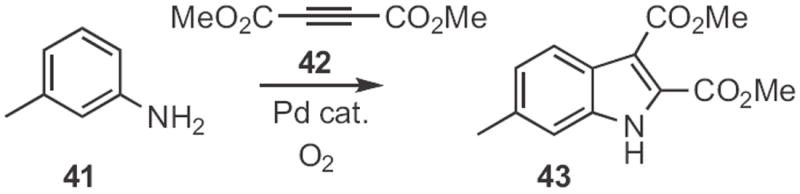
Following up on the work of Glorius (Scheme 10), Ning Jiao of Peking University found21 that, under oxidizing conditions, an aniline derivative, such as 41 (Scheme 15) could be condensed with the diester 42 to give the indole 43. Note that the cyclization proceeded with high regioselectivity. The product was easily hydrolyzed and decarboxylated to give the 2,3-unsubstituted indole.
Scheme 15.
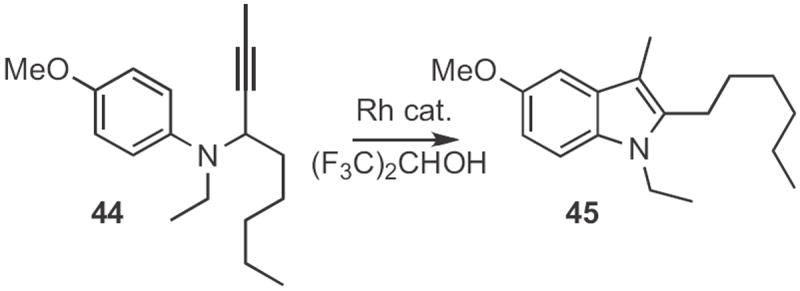
Akio Saito and Yuji Hanazawa of Showa Pharmaceutical University published22 a full account of the Rh-mediated cyclization of propargylaniline derivatives, such as 44 (Scheme 16) that they developed. This reaction is apparently proceeding via rearrangement to an intermediate o-allenylaniline, that then cyclizes to the product, 45.
Scheme 16.
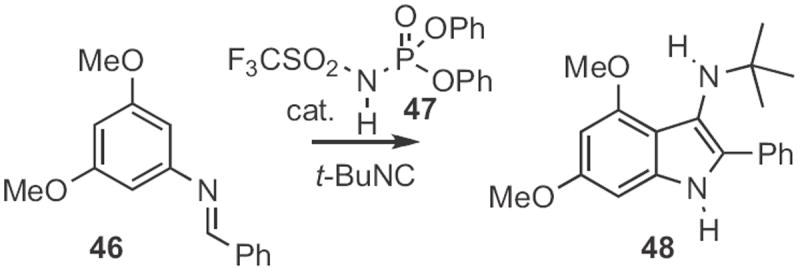
Erik J. Sorensen of Princeton University uncovered23 a route to indoles (Scheme 17) based on an interrupted Ugi reaction, the combination of 46 and tert-butyl isocyanide to give the aminoindole 48. The acid 47 was particularly effective at mediating this reaction.
3. Type 2
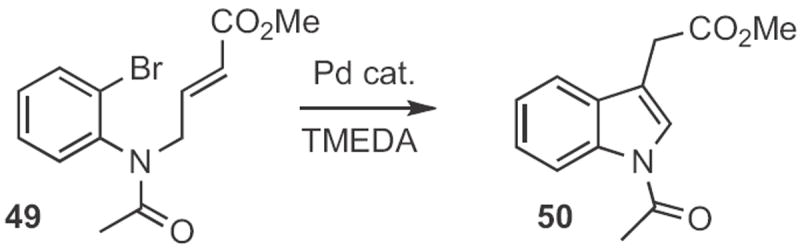
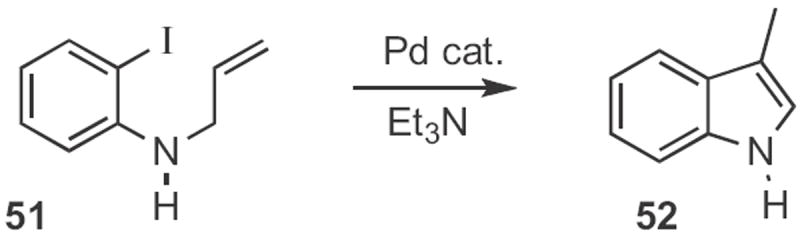
In a landmark paper in 1977, Miwako Mori, working with Yoshio Ban at Hokkaido University, reported24 the first intramolecular Heck cyclization, converting the 2-bromoaniline derivative 49 (Scheme 18) into the N-acetyl indole 50 with a Pd catalyst. In 1980, Louis S. Hegedus at Colorado State University showed25 that iodides were superior to bromides for the cyclization, and that free amines, such as 51 (Scheme 19) were compatible with the reaction conditions, forming 52.
Scheme 18.
Scheme 19.
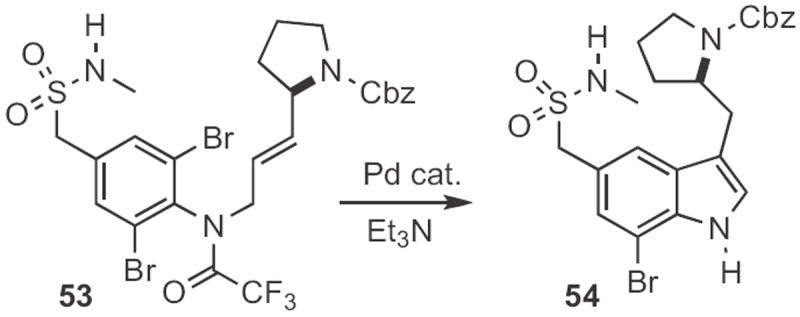
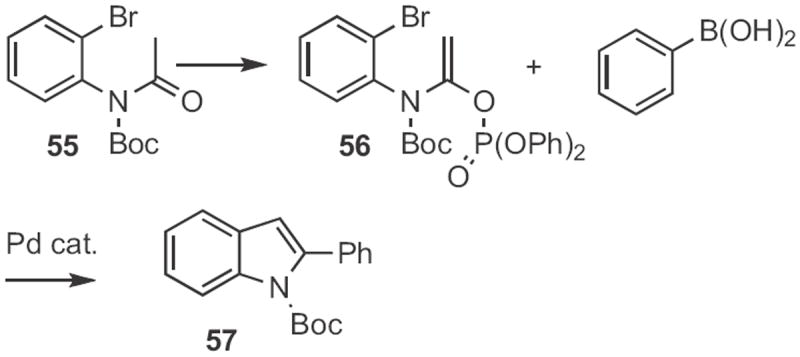
This approach has been extended in several directions. John E. Macor at Pfizer found26 that cyclization of the dibromide 53 to 54 (Scheme 20) was more efficient than cyclization of the corresponding monobromide. Note that the potentially labile allylic carbamate survived the Pd reaction conditions. Haruhiko Fuwa and Makoto Sasaki of Tohoku University devised27 the conversion of the N-acetyl aniline 55 (Scheme 21) into the enol phosphonate 56. Consecutive Suzuki coupling followed by Heck cyclization delivered the indole 57.
Scheme 20.
Scheme 21.
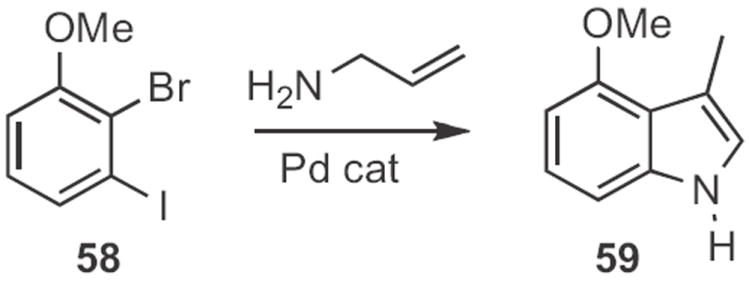
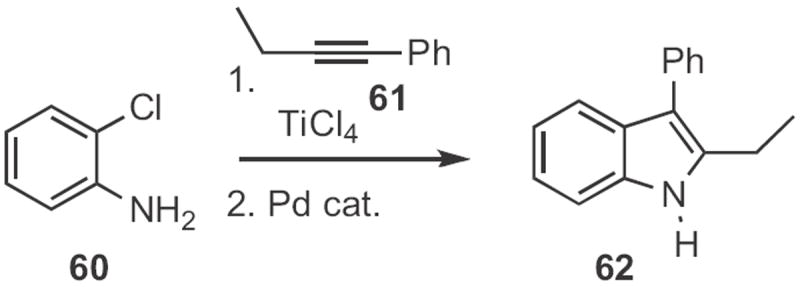
Morten Jørgensen of H. Lundbeck A/S, Denmark took advantage28 of the more facile oxidative addition of aryl iodides compared to aryl bromides to accomplish sequential N-arylation and Heck cyclization, converting 58 (Scheme 22) into the indole 59. Lutz Ackermann of the Ludwigs-Maxmilian-Universität München effected29 regioselective Ti-mediated hydroamination of the alkyne 61 (Scheme 23) with the aniline 60. Pd-mediated cyclization of the nucleophilic enamine so formed gave the indole 62.
Scheme 22.
Scheme 23.
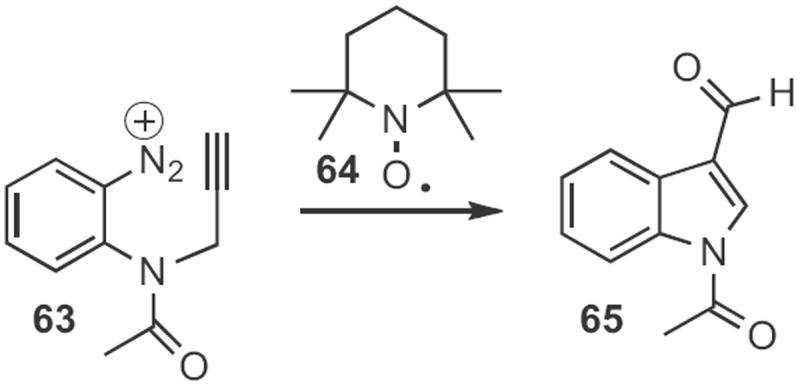
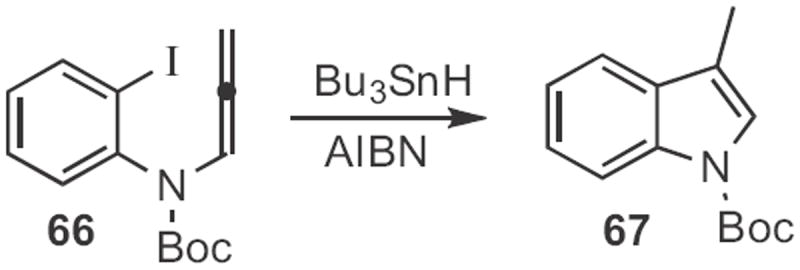
Indoles can also be prepared by free radical cyclization. Athelstan L. J. Beckwith of the University of Adelaide cleverly employed30 the nitroxide 64 (Scheme 24) to effect first reduction, to facilitate loss of N2 from the diazonium salt 63, then radical cyclization, then radical-radical coupling with the nitroxide, followed by loss of the amine to give the indole aldehyde 65. Richard P. Hsung, now at the University of Wisconsin, demonstrated31 that a more conventional reductive cyclization of the allenylaniline 66 to form 67 (Scheme 25) was also effective.
Scheme 24.
Scheme 25.
Lanny S. Liebeskind of Emory University showed32 that ortho-bromo allyl anilines, such as 68 (Scheme 26) could, on transmetalation, be induced to cyclize to the indoline anion. The anion could be trapped with a variety of electrophiles. The product indoline was readily oxidized to the indole 69. Professor Buchwald generated33 from 70 (Scheme 27) a zirconocene benzyne complex that inserted into the pendent alkene. Iodination delivered the indoline 71, that via elimination and bromination was carried on to the indole 72.
Scheme 26.
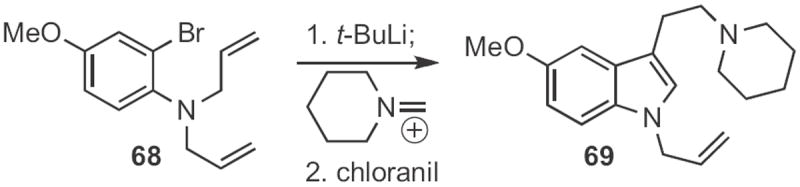
Scheme 27.
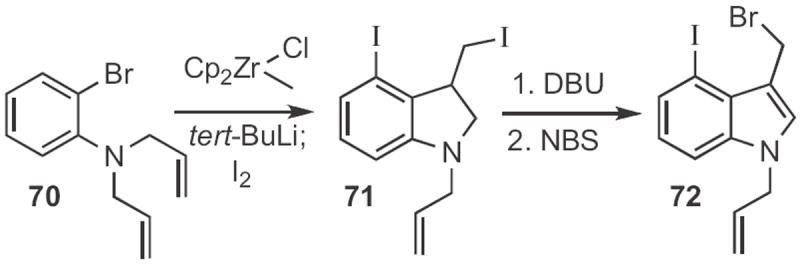
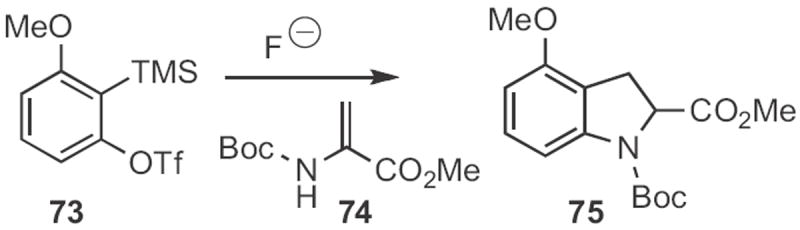
Brian M. Stoltz of Caltech added34 the anion derived from 74 (Scheme 28) to the benzyne derived from 73 to give the indoline 75. The authors did not oxidize 75 to the corresponding indole, but this should be straightforward.
Scheme 28.
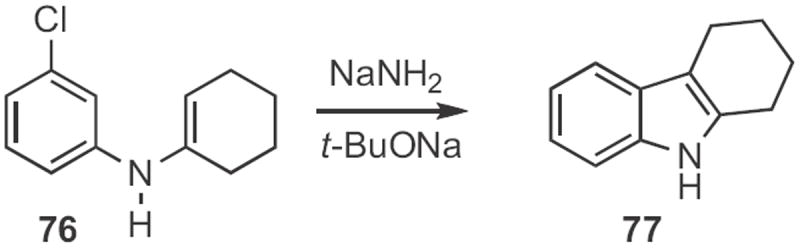
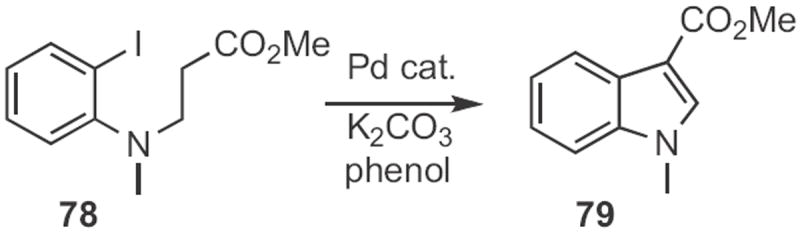
As described35 by Brigitte Jamart-Grégoire of the Université de Nancy, a benzynewas also the intermediate in the cyclization of the anion derived from 76 (Scheme 29) to the indole 77. Daniel Solé of the Universitat de Barcelona effected36 the conceptual alternative, the Pd-mediated arylation of the anion derived from 78 (Scheme 30). Depending on the reaction conditions, the dominant product could be either the indoline, or the indole 79.
Scheme 29.
Scheme 30.
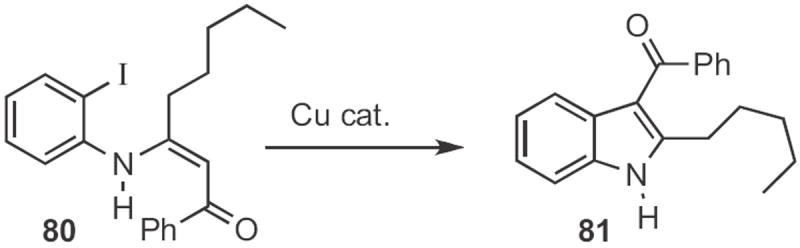
Among the several Type 2 indole syntheses reported in 2009, two were particularly interesting. Sandro Cacchi of the Università degli Studi ‘La Sapienza’, Roma, prepared37 the enaminone 80 (Scheme 31) by condensation of the iodoaniline with the acetylenic ketone. On exposure to a Cu catalyst, 80 cyclized to the indole 81.
Scheme 31.
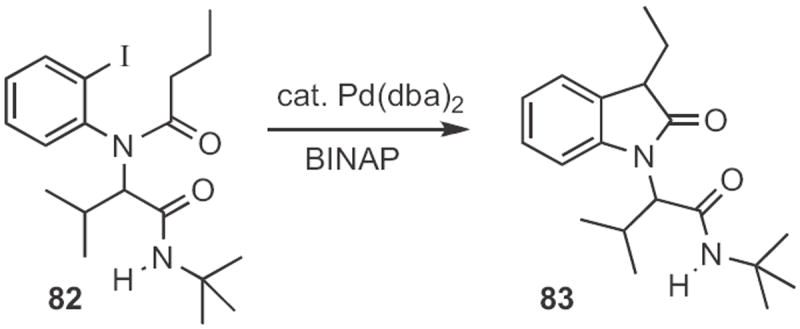
Luc Neuville and JZhu of CNRS Gif-sur-Yvette assembled38 (Scheme 32) the amide 82 by a four-component coupling. With the proper choice of ligand, 82 could be cyclized to 83. The conversion of an oxindole into the indole is described in the preceding section.
Scheme 32.
4. Type 3
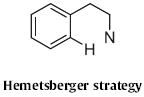
The lead Type 3 approach is the Hemetsberger39 indole synthesis, as, for instance, employed40 by John K. MacLeod of Australia National University in his synthesis (Scheme 33) of cis-trikentrin A. The aldehyde 84 was homologated to the azido ester 85, that was then heated to convert it into the indole 86.
Scheme 33.
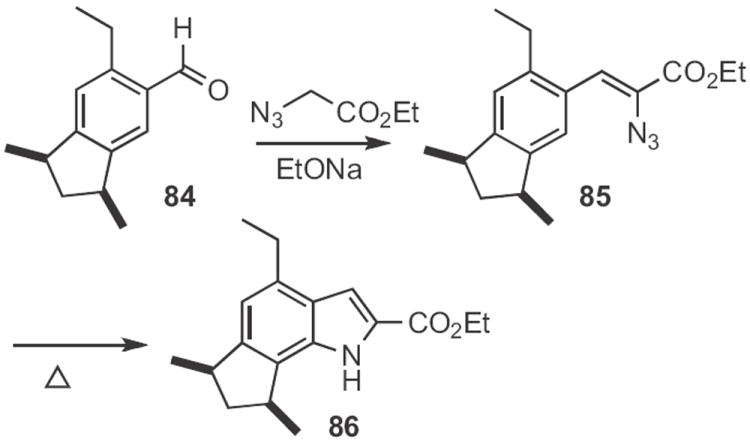
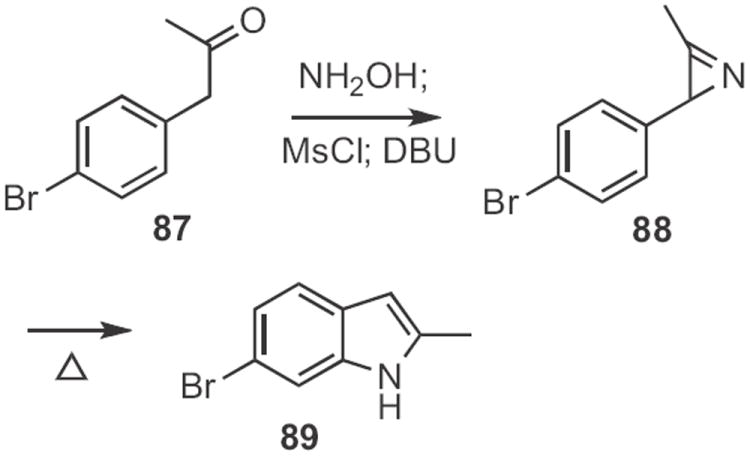
The thermal conversion of azido styrenes, such as 85 into the indole had been shown39 to proceed by way of the azirine. We therefore developed41 a general method for the conversion of an α-aryl ketone, such as 87 (Scheme 34) into the azirine 88. Thermolysis of the azirine gave the indole 89. Subsequently, Koichi Narasaka of the University of Tokyo demonstrated42 that Rh trifluoroacetate catalyzed the conversion of azirines, such as 88 into indoles at room temperature. Tom G. Driver of the University of Illinois, Chicago later found43 that the same catalyst converted azido styrenes, such as 85 (Scheme 33) into the indole, also at room temperature.
Scheme 34.
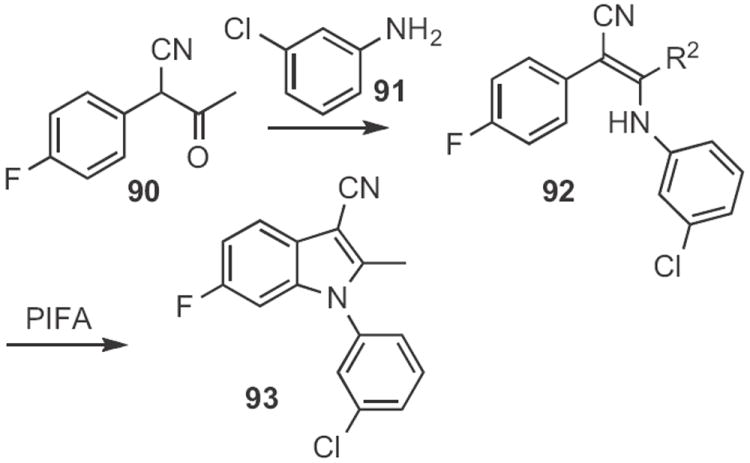
Kang Zhao of Tianjin University established44 that PIFA oxidation of an enamine, such as 92 (Scheme 35), prepared from 90 and 91, offered a convenient route to the N-aryl indole 93. This cyclization may likely also be proceeding by way of the intermediate azirine.
Scheme 35.
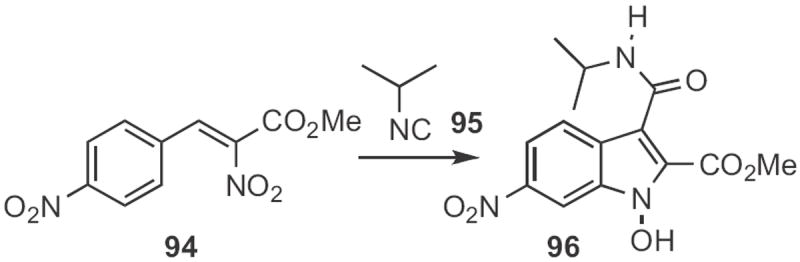
H. Person of the Université de Rennes found45 that exposure of a β-nitro styrene 94 (Scheme 36) to an isonitrile 95 led to the N-hydroxy indole 96. Glen A. Russell of Iowa State University reported46 a related reductive cyclization of a β-nitro styrene with triethyl phosphite.
Scheme 36.
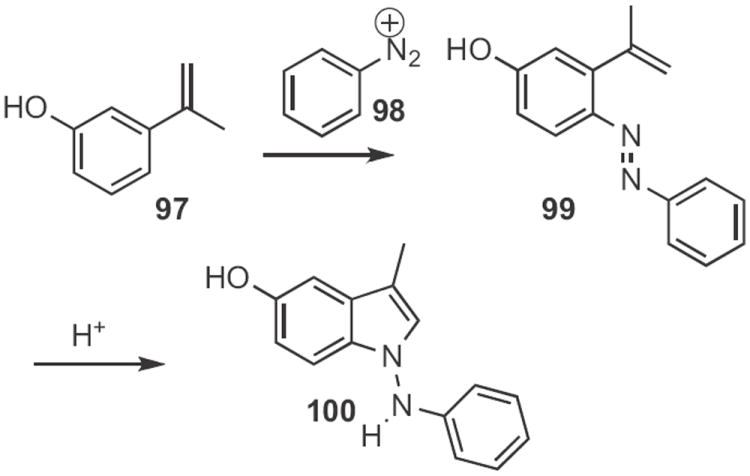
The coupling of a phenol 97 (Scheme 37) with a diazonium salt 98 is a well-known process. Masato Satomura of Fuji Photo Film Co. discovered47 that exposure of the adduct 99 to mild acid led to cyclization to the indole 100. The N–N bond was readily cleaved by Raney nickel to give the free amine.
Scheme 37.
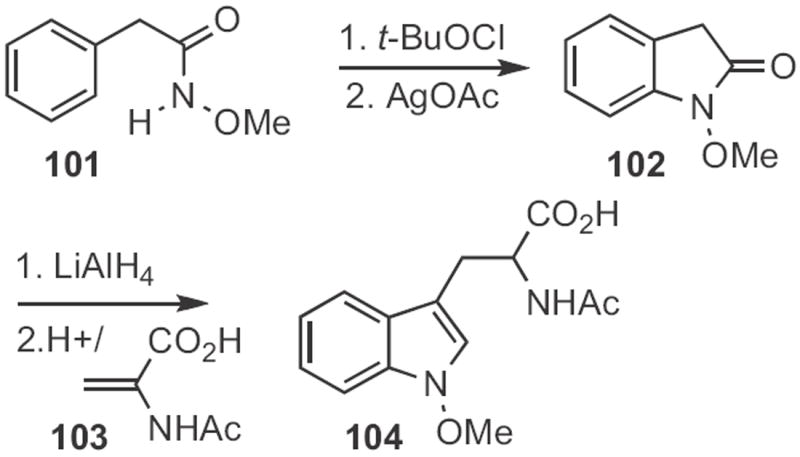
The cyclic heptadepsipeptide HUN-7293 contains the N-methoxy tryptophan 104 (Scheme 38). To prepare 104, Dale L. Boger of Scripps/La Jolla took advantage48 of the Kikugawa oxindole synthesis to convert 101 into 102. Reduction followed by acid-catalyzed condensation with the enamide 103 then delivered 104.
Scheme 38.
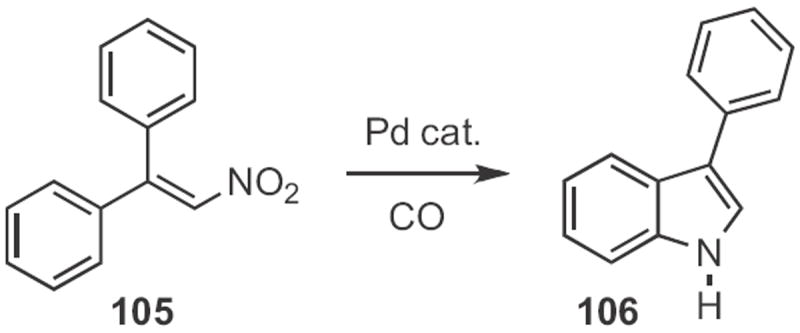
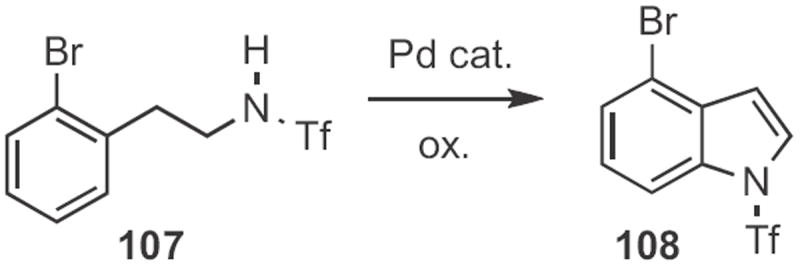
In 2009, Vy M. Dong of the University of Toronto found49 that CO could serve (Scheme 39) as the reductant for the cyclization of a β-nitro styrene 105 to the indole 106. Jin-Quan Yu, also of Scripps/La Jolla, developed50 an oxidant that enabled the Pd-mediated cyclization of 107 (Scheme 40) to the indole 108.
Scheme 39.
Scheme 40.
5. Type 4
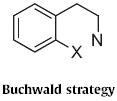
The development of transition-metal-mediated aryl halide amination opened the way to Type 4 indole synthesis. In 1998, Stephen L. Buchwald of MIT reported51 that on exposure to benzylamine in the presence of a Pd catalyst, the dibromide 109 (Scheme 41) smoothly cyclized to the indoline 110. Ammonium formate in the presence of Pd/C converted 110 into the indole 111.
Scheme 41.
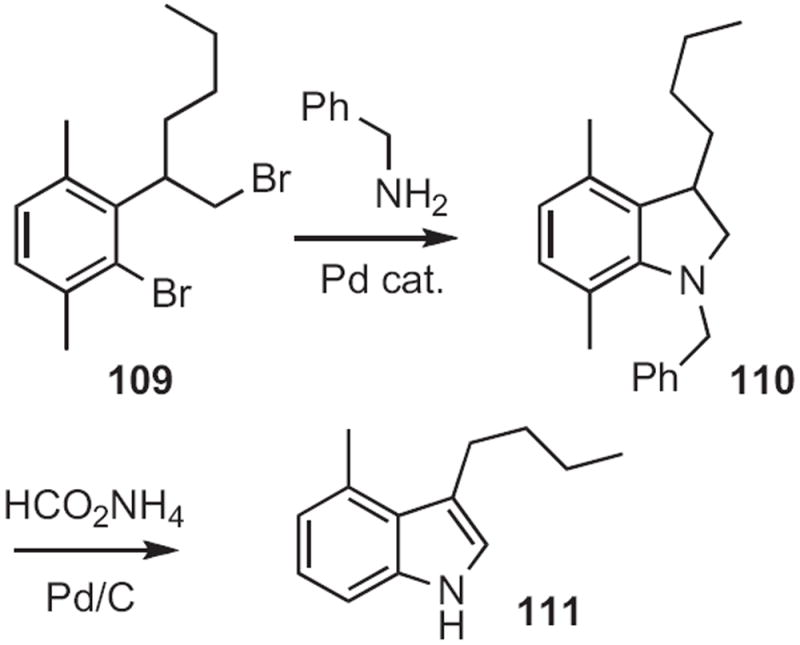
In the course of a synthesis of the duocarmycins, Tohru Fukuyama of the University of Tokyo employed52 a similar approach, cyclizing 112 (Scheme 42) to 113. By that time, the Cu catalysts for aryl halide amination had been developed.
Scheme 42.
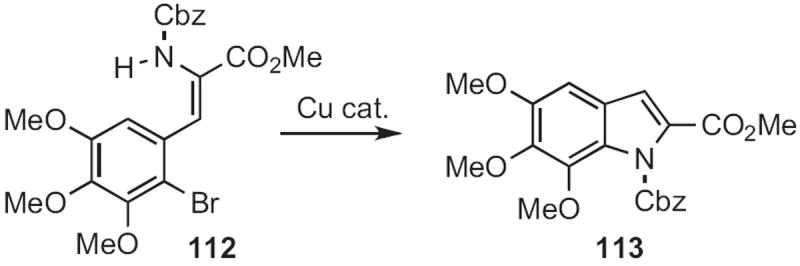
José Barluenga of the University of Oviedo took advantage53 of the greater reactivity of an aryl bromide compared to the chloride as he developed the convergent coupling of 114 (Scheme 43) with 115 to give the indole 116. For this coupling, a Pd catalyst was required.
Scheme 43.
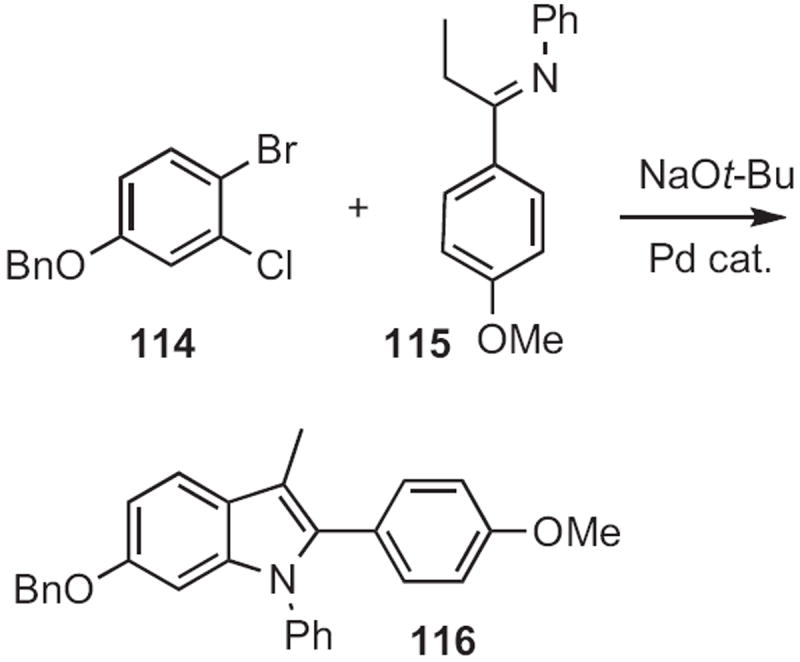
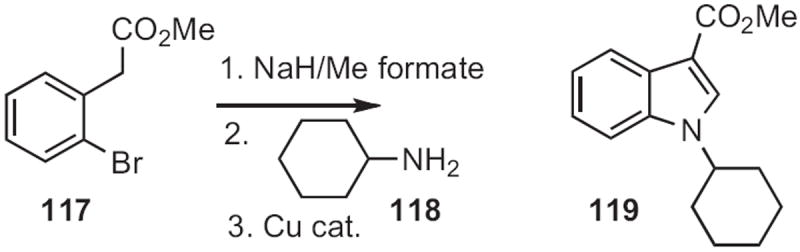
Alexander V. Karchava of Moscow State University devised54 a route to indoles from ortho-bromophenylacetic acid esters, such as 117 (Scheme 44). Formylation followed by condensation with an amine 118 set the stage for the Cu-mediated intramolecular amination to give the indole 119.
Scheme 44.
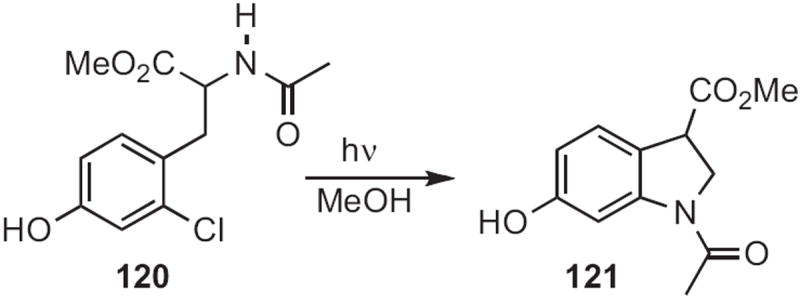
Phenols, under photolysis, can activate meta-substituted halides for nucleophilic displacement. Nien-chu C. Yang of the University of Chicago devised55 an indoline synthesis based on this effect, irradiating 120 (Scheme 45) to give 121.
Scheme 45.
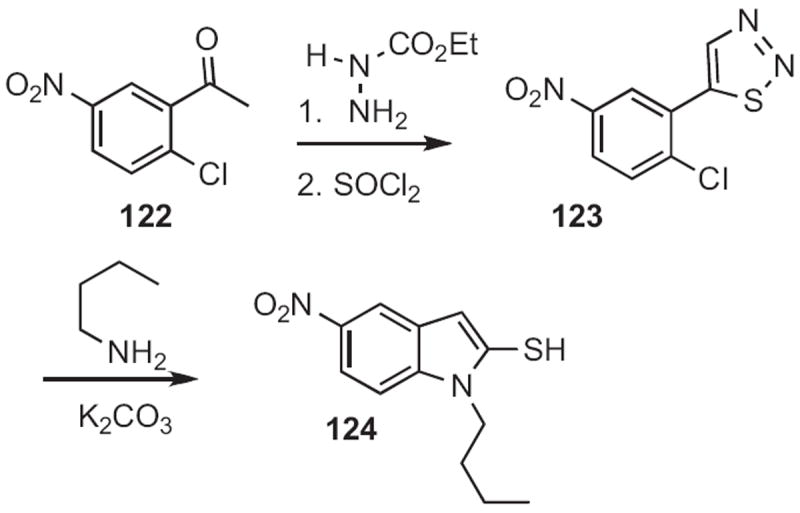
Similarly, nitro groups can activate para-substituted halides for nucleophilic displacement. Douglas C. Neckers of Bowling Green State University observed56 that exposure to a primary amine converted the thiadiazole 123 (Scheme 46), prepared from 122, into the indole-2-thiol 124. The reaction is thought to be proceeding by way of the alkyne thiol.
Scheme 46.
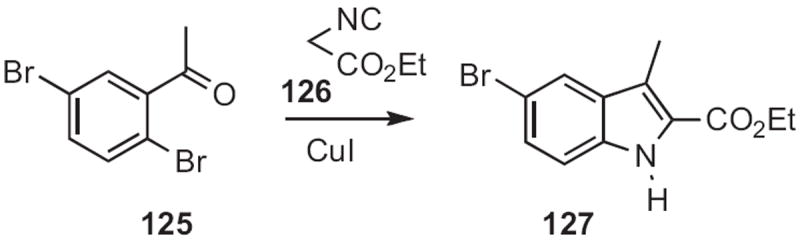
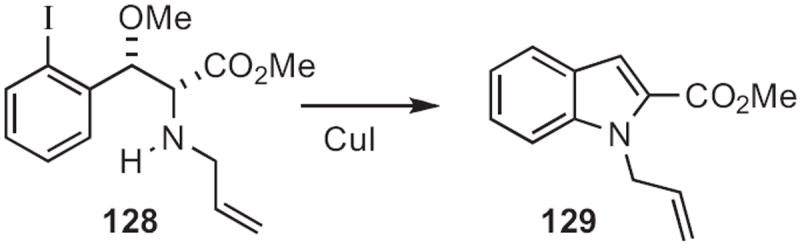
In 2009, Qian Cai and Ke Ding of the Institute of Biological Chemistry, Guangzhou described57 (Scheme 47) the CuI-mediated condensation of the isocyano ester 126 with o-halo aromatic ketones and aldehydes, such as 125 to give directly the corresponding indole 127. Stuart L. Schreiber of Harvard University took58 a related approach, cyclizing 128 (Scheme 48), prepared via the corresponding aziridine, to the indole 129.
Scheme 47.
Scheme 48.
6. Type 5
In 1969, Richard J. Sundberg of the University of Virginia reported59 that ortho-azido styrenes, such as 130 (Scheme 49) were converted on thermolysis into the corresponding indole 131. He later found60 that heating ortho-nitro styrenes, such as 132 (Scheme 50) with P(OEt)3 also delivered the indole. Aryl migration dominated over alkyl migration, leading to 133. Recently, Tom G. Driver of the University of Illinois, Chicago showed61 that the azide version of the Sundberg indole synthesis could be carried out at lower temperature with a Rh catalyst.
Scheme 49.
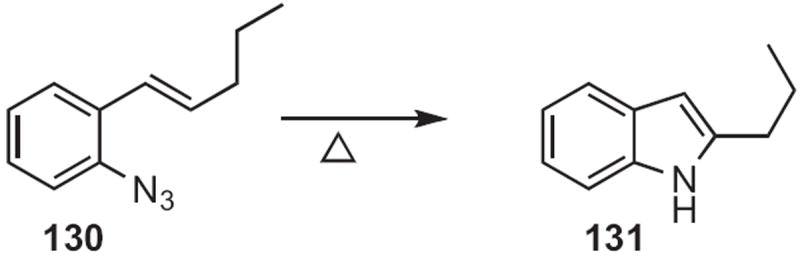
Scheme 50.
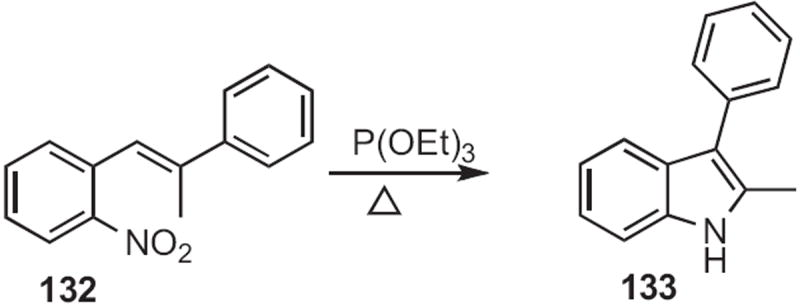
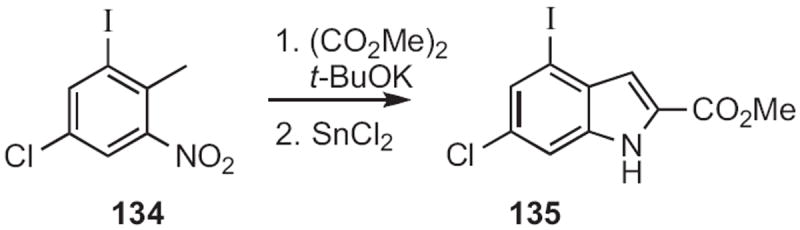
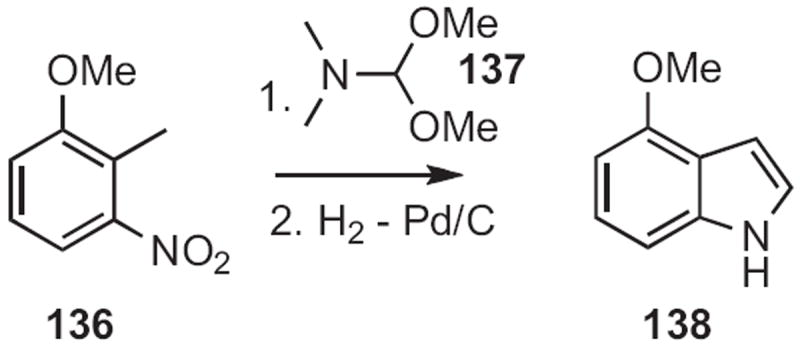
Benzylic methyl groups are acidic enough to be deprotonated, especially when there is an ortho-nitro group. This is the basis for the Reissert indole synthesis62 (134 to 135, Scheme 51) and the Leimgruber–Batcho indole synthesis63 (136 to 138, Scheme 52).
Scheme 51.
Scheme 52.
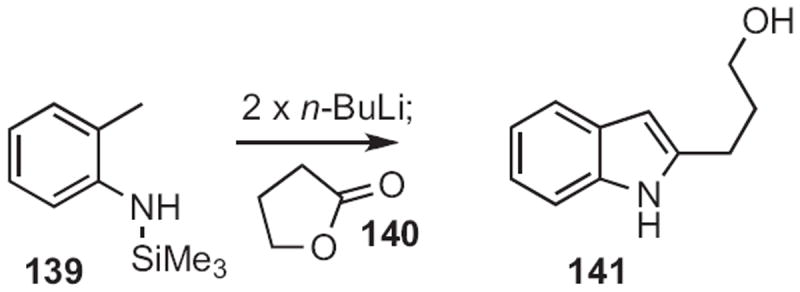
Amos B. Smith III of the University of Pennsylvania took advantage64 of the acidity of 139 (Scheme 53). Double deprotonation followed by condensation with 140 delivered the indole 141.
Scheme 53.
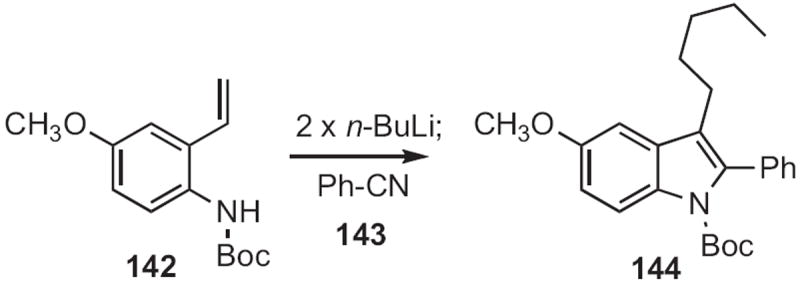
Donal F. O’Shea of University College Dublin demonstrated65 that an alkyllithium first deprotonated 142 (Scheme 54), and then added to the pendent alkene. Benzonitrile 143 was added to the resulting carbanion to give the indole 144.
Scheme 54.
It is clear that any synthetic route to ortho-amino or ortho-nitro α-aryl ketones or aldehydes can be used to prepare indoles. Joseph F. Bunnett of the University of California, Santa Cruz observed66 that, under SRN1 conditions, acetone enolate displaced the bromide of 145 (Scheme 55), leading to the indole 146. Viresh H. Rawal of the University of Chicago arylated67 the silyl enol ether 147 (Scheme 56) with an ortho-nitrophenyl iodinium salt (NPIF) to give, after reduction, the indole 148. M. Mahmoun Hossain of the University of Wisconsin, Milwaukee inserted68 ethyl diazoacetate into the aldehyde 149 (Scheme 57), converting it, via reduction, into the indole 150.
Scheme 55.
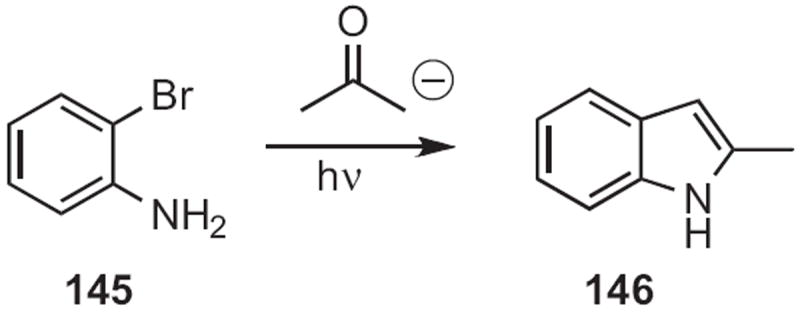
Scheme 56.
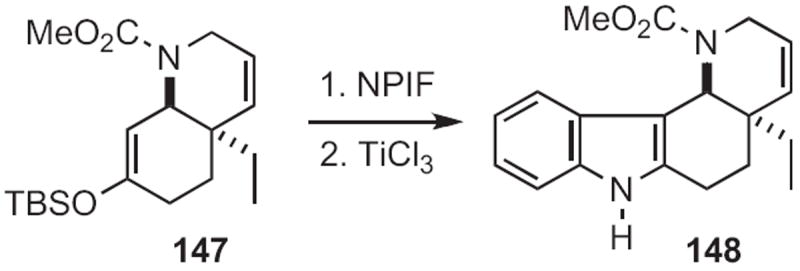
Scheme 57.
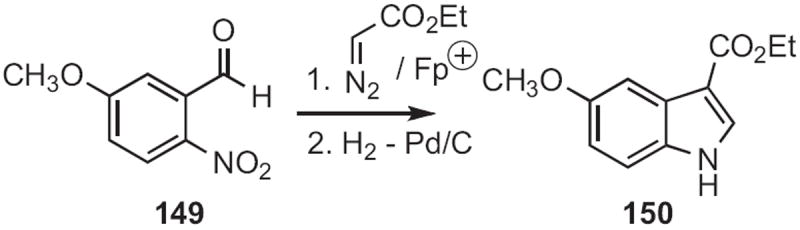
As demonstrated69 by Ken-ichi Fujita and Ryohei Yamaguchi of Kyoto University, in situ oxidation of the alcohol 151 (Scheme 58) led to the indole 152. With added 2-propanol, an alcohol with an ortho-nitro group was also converted into the indole.
Scheme 58.
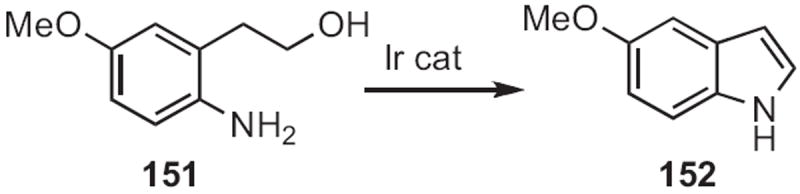
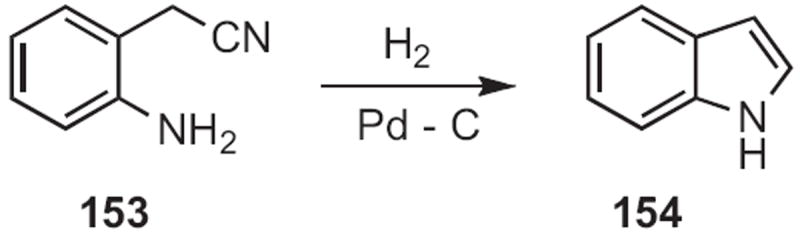
Hironao Sajiki and Kosaku Hirota of Gifu Pharmaceutical University showed70 that reduction of an ortho-amino nitrile, such as 153 (Scheme 59) delivered the indole 154, presumably by trapping of the intermediate imine. It may well be that an ortho-nitro substituent would work as well, but such a transformation was not included in this report.
Scheme 59.
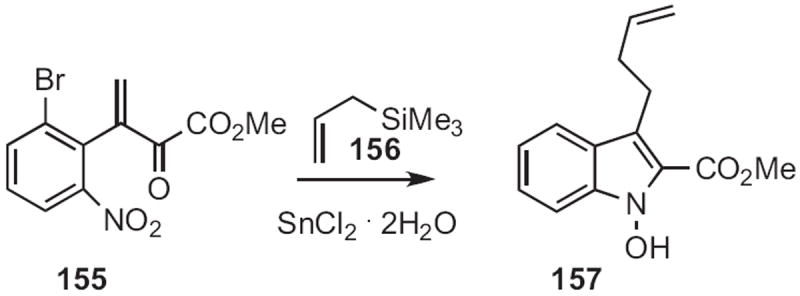
K. C. Nicolaou of Scripps/La Jolla prepared71 the enone 155 (Scheme 60) from the intermediate in the Bischler indole synthesis. Reduction of 155 gave an intermediate that reacted with mild nucleophiles, such as the allylsilane 156 to give the indole 157.
Scheme 60.
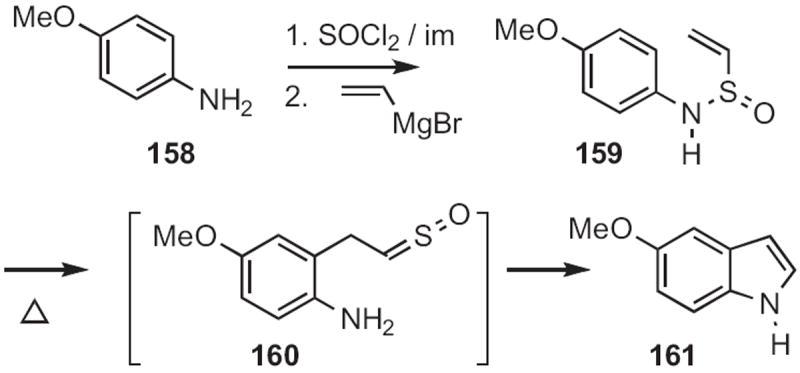
In 1986, Sylvestre A. Julia of the Ecole Normale Supérieure, Paris reported72 that sulfinamides, such as 159 (Scheme 61), readily prepared from the aniline 158, were converted by heating into the indole 161 via 160. It is striking that the Julia indole synthesis has been little used since it was reported.
Scheme 61.
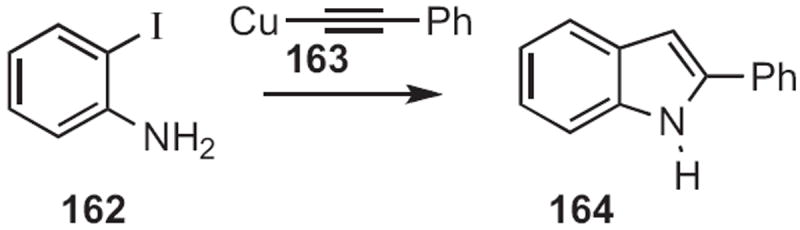
The preparation of indoles from ortho-haloanilines by condensation with an alkyne goes back at least to 1963, when C. E. Castro of the University of California, Riverside, observed73 (Scheme 62) that coupling of 162 with 163 led not to the diaryl alkyne, but to the indole 164.
Scheme 62.
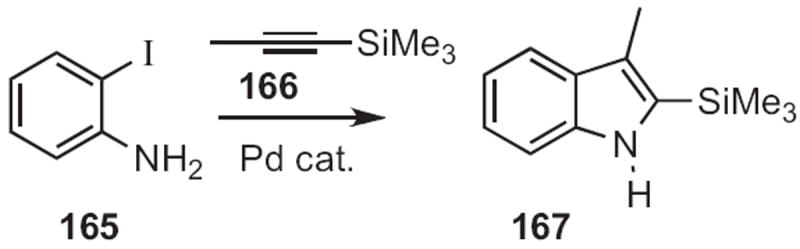
In 1985, Edward C. Taylor of Princeton University and Alexander McKillop of the University of East Anglia showed74 that Pd was effective at cyclizing ortho-alkynylanilines to the corresponding indole. This led to the 1989 report75 by J. K. Stille of Colorado State University that the two-step coupling described by Castro (Scheme 62) could be carried out at much lower temperature using Pd catalysis. With this precedent, in 1991 Richard C. Larock of Iowa State University disclosed76 that, using Pd catalysis (Scheme 63), internal alkynes, such as 166 could be condensed with an ortho-iodoaniline 165 under Pd catalysis to give the 2,3-disubstituted indole 167 with high regiocontrol. One of the advantages of the Larock indole synthesis is the malleability of the 2-silyl substituent on the product indole.
Scheme 63.
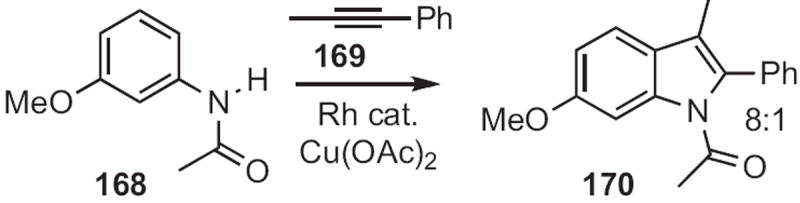
More recently, (the late) Keith Fagnou of the University of Ottawa demonstrated77 that Rh catalysis could effect ortho functionalization of acetanilides, such as 168 (Scheme 64). Subsequent coupling with internal alkynes, such as 169 led to the indole 170 with high regiocontrol.
Scheme 64.
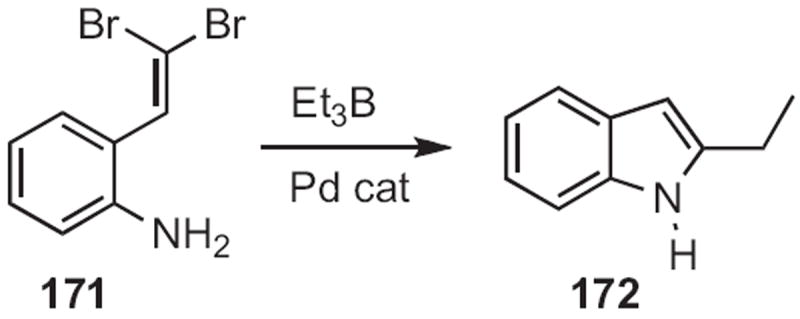
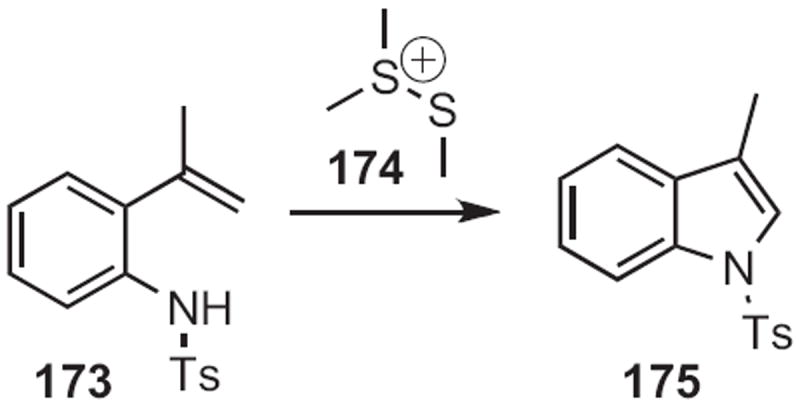
Several other flexible routes to indoles have been developed. Mark Lautens of the University of Toronto established78 that ortho dihaloalkylidene anilines, such as 171 (Scheme 65) could be condensed with alkyl, alkenyl or aryl boranes or boronic acids to give the 2-substituted indole, in this case 172. Kentaro Okuma of Fukuoka University found79 that the sulfonium salt 174 (Scheme 66) effected cyclization of an ortho alkenyl aniline, such as 173 to the indole 175. Jeffrey N. Johnston, now at Vanderbilt University, effected80 free radical reductive cyclization of halides, such as 176 (Scheme 67), to give the indole 177.
Scheme 65.
Scheme 66.
Scheme 67.
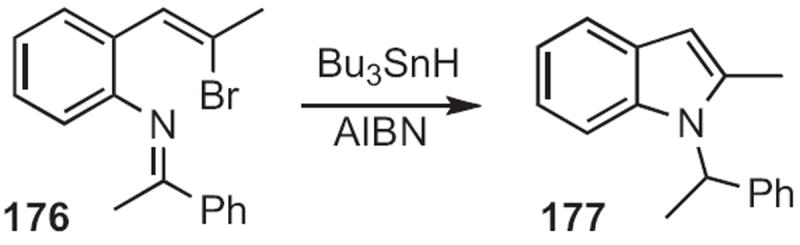
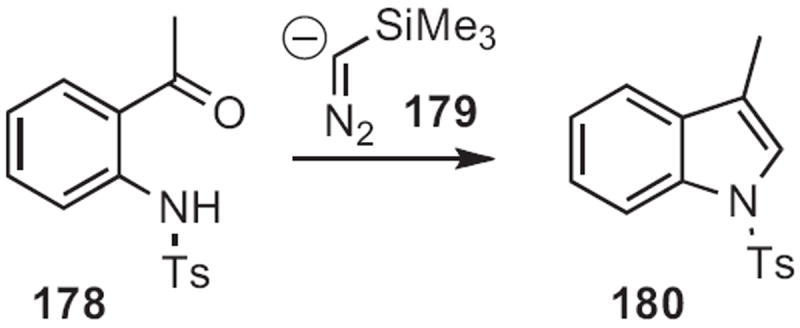
Toyohiko Aoyama of Nagoya City University reacted81 ortho-acylanilines, such as 178 (Scheme 68) with lithio TMS diazomethane 179 to give an alkylidene carbene, that inserted into the adjacent NH to give the indole 180. Bartolo Gabriele of the Università della Calabria added82 acetylides, such as 182 (Scheme 69) to ortho-acylanilines, such as 181 to give alkynyl alcohols, that underwent carbonylative cyclization with Pd catalysis to give the indole 183.
Scheme 68.
Scheme 69.
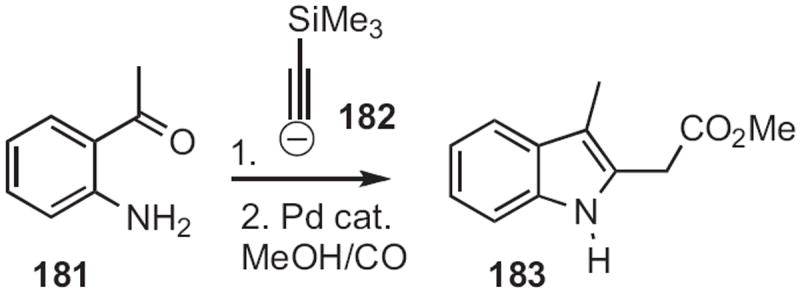
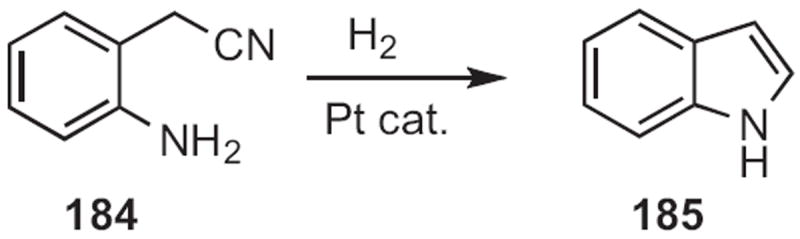
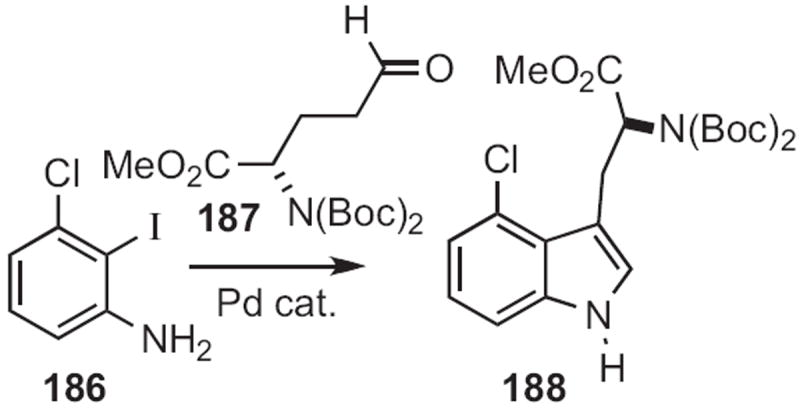
In 2009, Hideo Nagashima of Kyushu University reported83 that an o-nitrophenyl acetonitrile 184 could indeed (Scheme 70) be reductively cyclized to the indole 185. Yanxing Jia of Peking University prepared84 188, a key intermediate in the synthesis of (−)-cis-clavicipitic acid, by selective condensation of the aldehyde 187 (Scheme 71) with the iodoaniline 186.
Scheme 70.
Scheme 71.
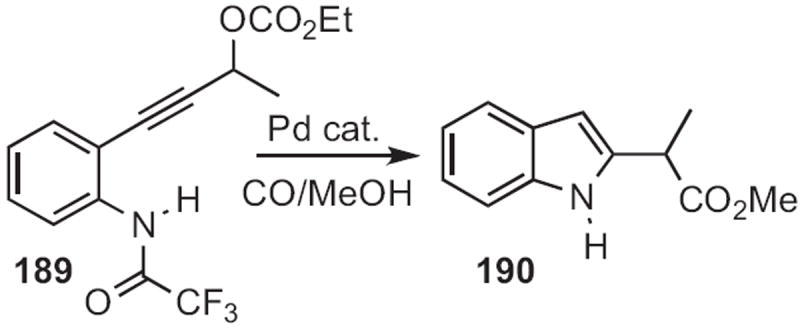
Sandro Cacchi of the Università ‘La Sapienza’, Rome, extended85 the Gabriele approach, cyclizing (Scheme 72) the propargylic carbonate 189 to 190. This transformation may be proceeding by way of the intermediate allene. Two related approaches to indole synthesis86,87 also appeared.
Scheme 72.
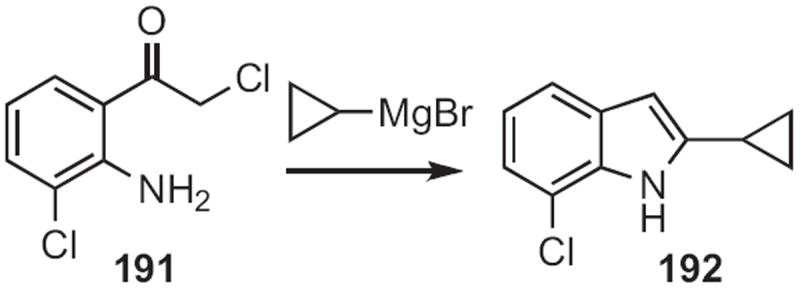
Tao Pei of Merck Rahway developed88 a powerful new approach to substituted indoles, based on the addition (Scheme 73) of an organometallic to a chloro ketone 191. The conversion into 192 proceeded by 1,2-migration of the arene with nucleophilic displacement of chloride.
Scheme 73.
7. Type 6
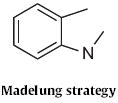
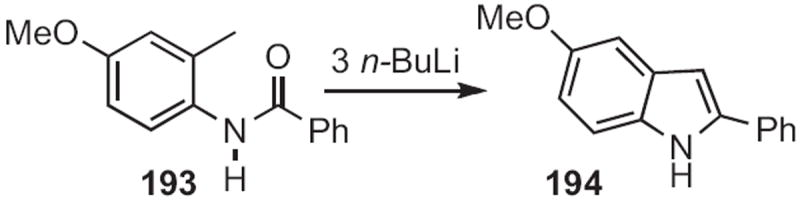
The Madelung indole synthesis, as exemplified by the cyclization (Scheme 74) of 193 to 194, was originally carried out at elevated temperature with bases, such as NaNH2. Willam J. Houlihan of Sandoz, Inc. (now Novartis) showed89 that, with BuLi, the cyclization of 193 to 194 was facile below room temperature. D. N. Reinhoudt of the University of Twente found90 that phenylacetonitriles, such as 195 (Scheme 75) could be cyclized under even milder conditions, to form 196.
Scheme 74.
Scheme 75.
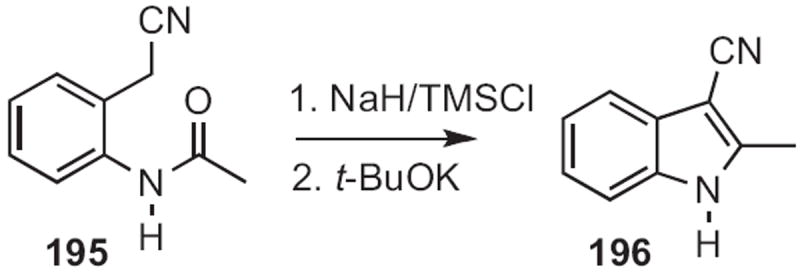
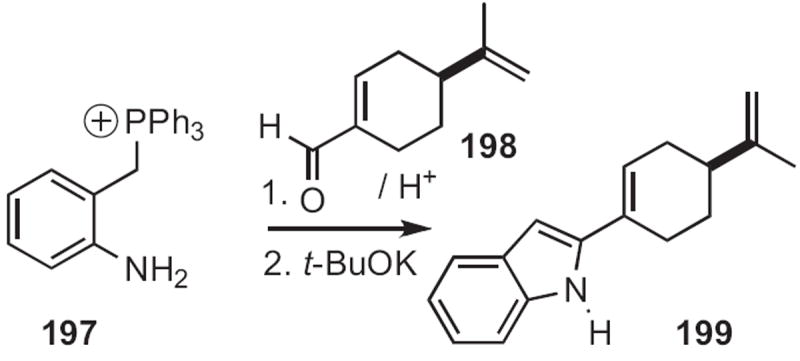
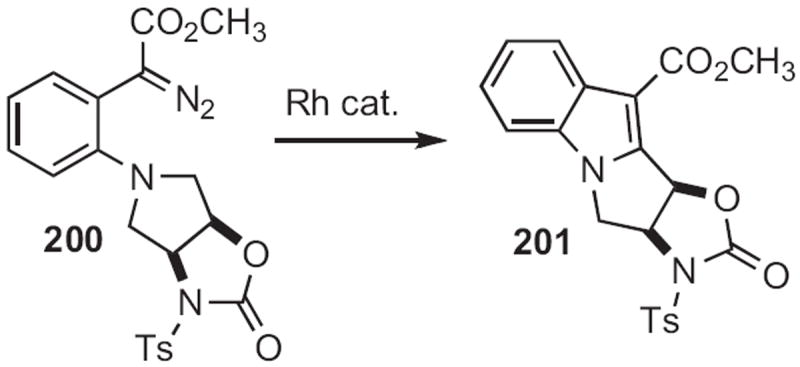
George A. Kraus of Iowa State University described91 a conceptually related cyclization (Scheme 76). Condensation of an aldehyde 198 with the aniline 197 gave the imine, that on exposure to strong base gave the indole 199. Gary A. Sulikowski, now at Vanderbilt University, showed92 that cyclization of the carbene derived from 200 (Scheme 77) proceeded to give 201 with high regiocontrol.
Scheme 76.
Scheme 77.
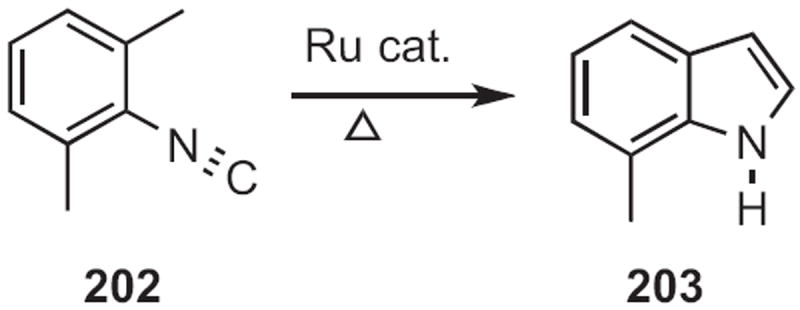
Bond formation in the opposite direction has also been developed. William D. Jones reported93 that a Ru complex catalyzed the conversion of the isonitrile 202 (Scheme 78) into the indole 203. This reaction may be proceeding by way of the Ru vinylidene complex.
Scheme 78.
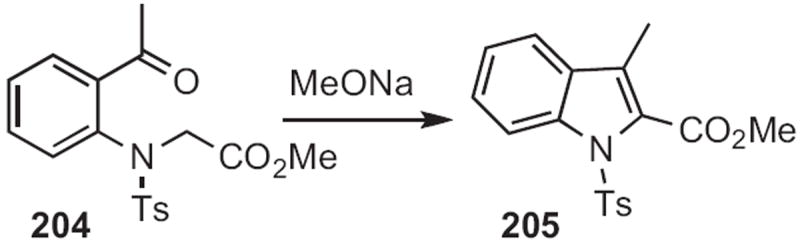
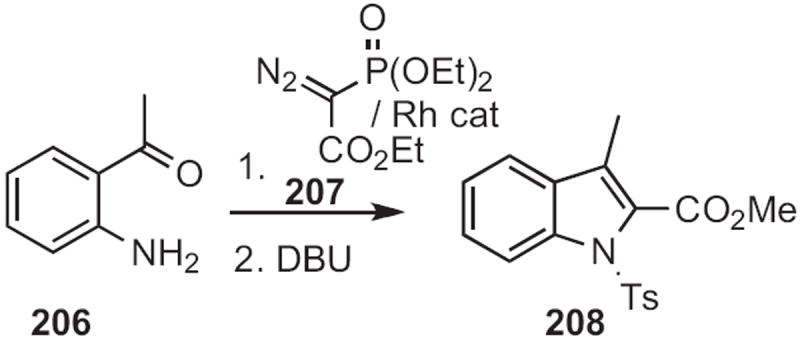
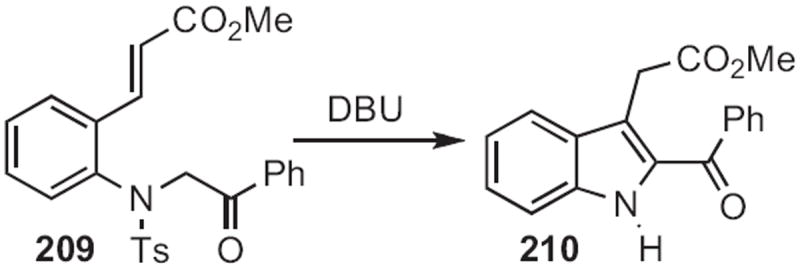
Charles D. Jones of Lilly described94 an anionic cyclization in this direction, converting 204 (Scheme 79) into 205. Yoshinori Nakamura of the Tanabe Seiyaku Co. contributed95 the Rh-mediated coupling of the diazophosphonate 207 (Scheme 80) to an ortho-acylaniline, such as 206, to give, after cyclization, the indole 208. Note that, in the cyclization of 209 (Scheme 81) developed96 by RodneyW. Stevens of Pfizer Nagoya, re-aromatization to the indole 210 was achieved by elimination of arenesulfinate.
Scheme 79.
Scheme 80.
Scheme 81.
In 1994, Tohru Fukuyama, now at the University of Tokyo, disclosed97a the cascade radical cyclization of the isonitrile 211 (Scheme 82) to the indole 212. Later, he applied97b a variant of this cyclization in the total synthesis of a complex indole alkaloid. Jon D. Rainier, now at the University of Utah, has explored97c related radical cyclizations.
Scheme 82.
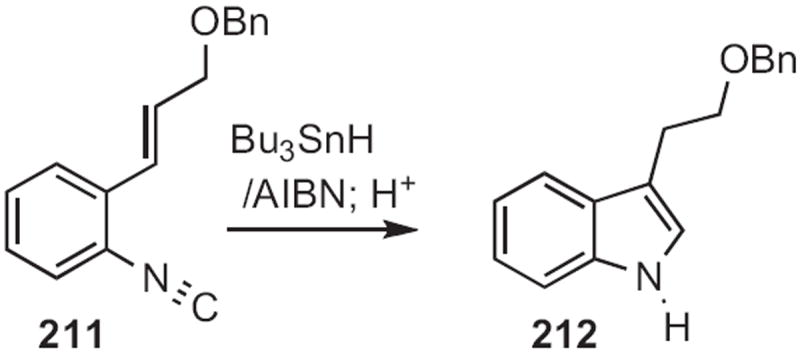
Alois Fürstner of the Max-Planck-Institute Mülheim developed98a, b a reductive coupling of acyl anilides, such as 213 to give 214 (Scheme 83). In the presence of a silyl chloride, the reaction was catalytic in Ti. Bruce C. Lu of Boehringer Ingelheim employed98c this reductive coupling in a combinatorial route to indoles.
Scheme 83.
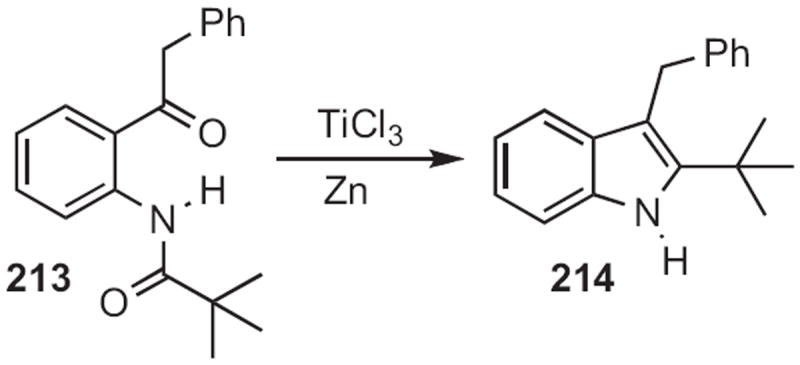
In 2009, Professor Doyle reported99 an alternative (Scheme 84) diazo-based approach to indoles, Lewis acid-mediated cyclization of 215 to 216.
Scheme 84.
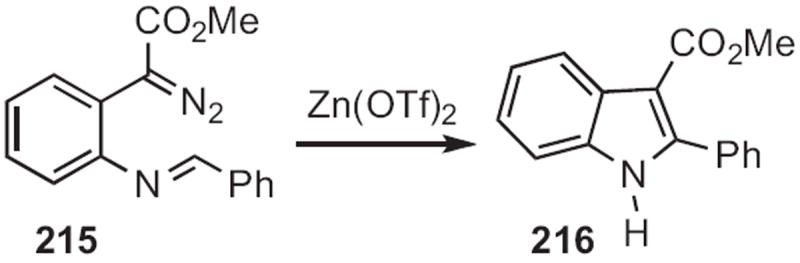
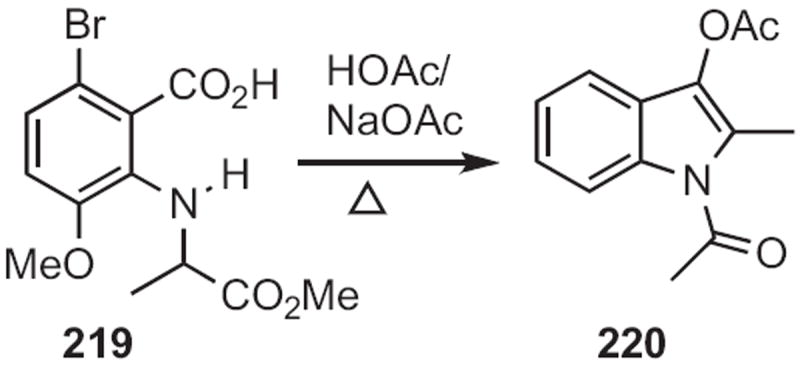
Churl Min Seong of the Korea Research Institute of Chemical Technology described100 the facile cyclization (Scheme 85) of an o-cyano N-benzyl aniline 217 to the indole 218. Andrew D. Hamilton employed101 a related protocol (Scheme 86), the cyclization of 219 to 220.
Scheme 85.
Scheme 86.
8. Type 7
Type 7 includes all routes to indoles from cycloalkane derivatives. The earliest such approach is the Nenitzescu indole synthesis, exemplified (Scheme 87) in a modern manifestation102 by Daniel M. Ketcha of Wright State University and Lawrence J. Wilson of Procter & Gamble. The combination of the benzoquinone 221 with the resin-bound enamine 222 gave, after release from the resin, the indole 223.
Scheme 87.
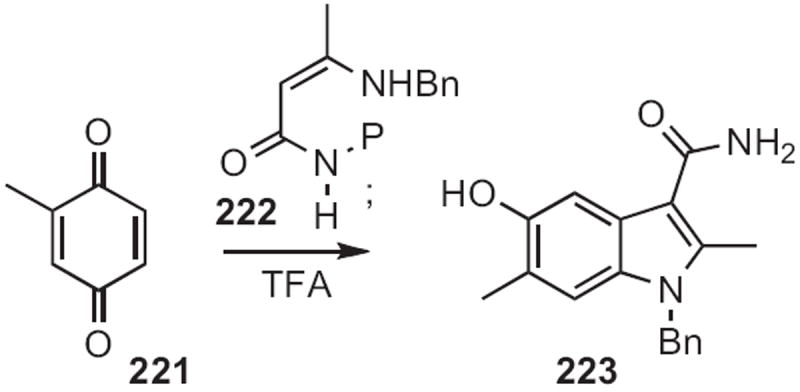
Michael A. Kerr of the University of Western Ontario developed103 (Scheme 88) a complementary protocol for the conversion of a benzoquinone into the indole. Diels–Alder cycloaddition of the imine 224 to the diene 225 gave the adduct 226. Protection followed by oxidative cleavage and condensation delivered the indole 227.
Scheme 88.
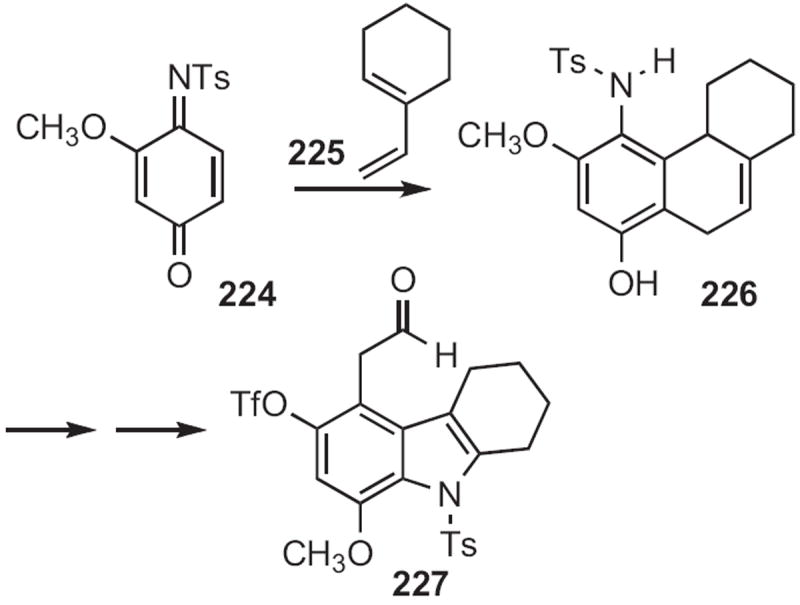
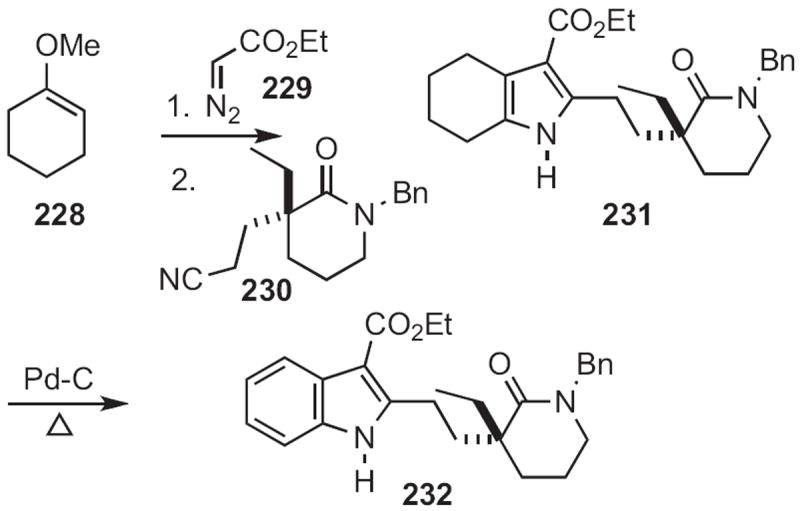
Fused pyrroles, such as 231 (Scheme 89) and 235 (Scheme 90) are readily aromatized. Brian L. Pagenkopf of the University of Western Ontario established104 a pyrrole synthesis from cyclohexanone, by cyclopropanation of the enol ether 228 followed by condensation with the nitrile 230. The aromatization of 231 to 232 was accomplished by heating with Pd/C in mesitylene.
Scheme 89.
Scheme 90.
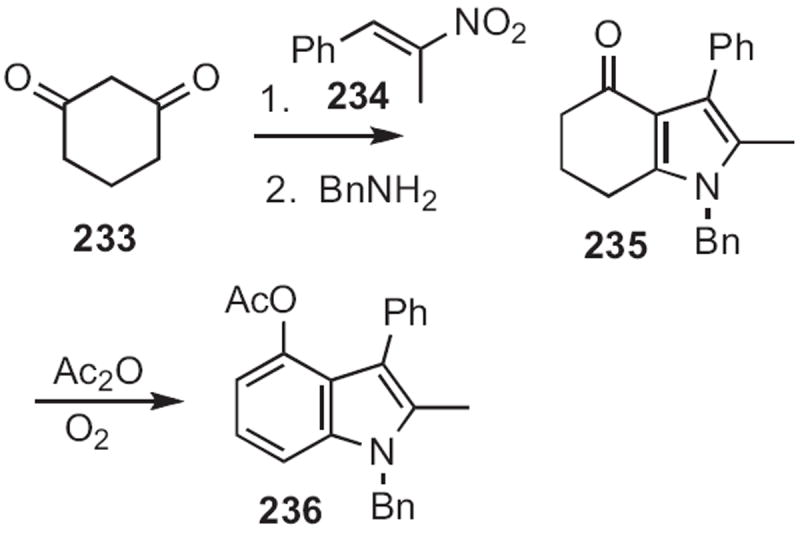
Teruhiko Ishikawa and Seiki Saito of Okayama University condensed105 (Scheme 90) cyclohexane-1,3-dione 233 with the nitroalkene 234, leading after exchange with benzylamine to the pyrrole 235. Aromatization gave the 4-oxygenated indole 236. Chihiro Kibiyashi of the Tokyo College of Pharmacy reported106 a related approach to 4-oxygenated indoles.
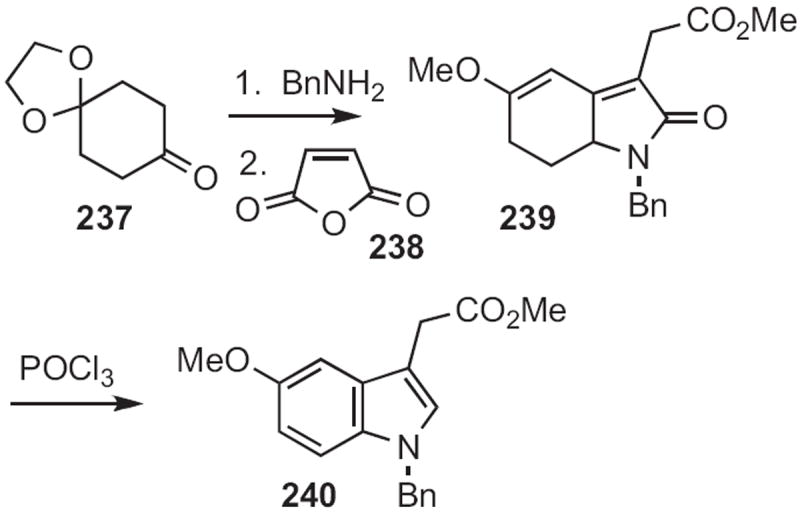
Michel Pfau of ESPCI Paris devised107 an intriguing protocol for indole construction, starting with the benzyl imine of the monoprotected cyclohexane-1,4-dione 237 (Scheme 91). Metalation of the imine followed by condensation with maleic anhydride 238, with methanol workup, delivered the lactam 239. Exposure of 239 to POCl3 effected aromatization to the 5-methoxyindole 240.
Scheme 91.
In 2009, Yong-Qiang Tu of Lanzhou University described108 the ring expansion (Scheme 92) of 241 to 242. The aromatization of 242 to the indole should be facile. Tsutomu Inokuchi of Okayama University showed109 that reduction (Scheme 93) of the Michael adduct 243 followed by aromatization delivered the indole 244.
Scheme 92.
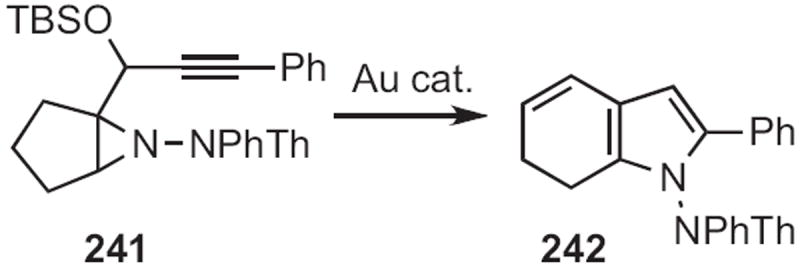
Scheme 93.
9. Type 8
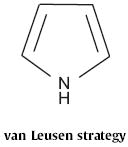
Type 8 indole syntheses include all those that proceed by way of the preformed N-containing five-membered ring. In 1986, Albert M. van Leusen of Groningen University established110 a route to highly substituted indoles, based on the condensation of isonitriles, such as 245 (Scheme 94) with unsaturated ketones, such as 246 to give the 2,3-bisalkenylpyrrole 247. Heating followed by aromatization with DDQ completed the synthesis of the indole 248.
Scheme 94.
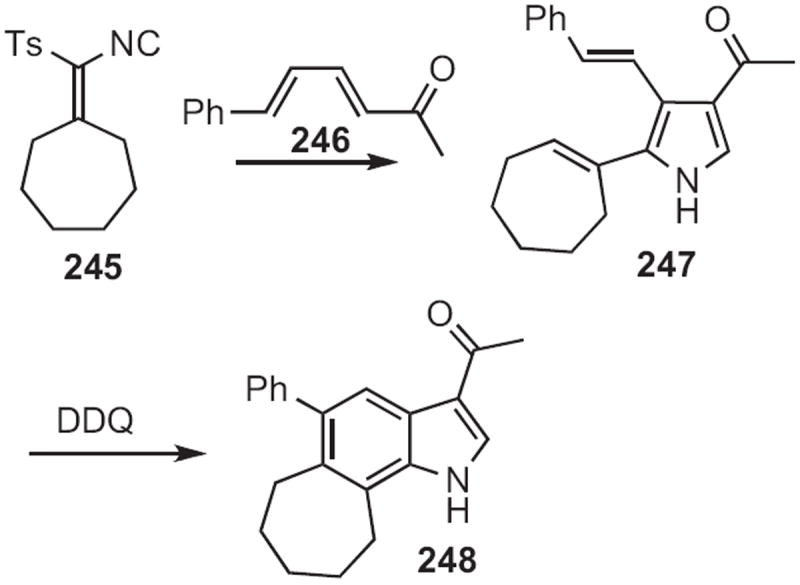
Hiroyuki Ishibashi of Kyoto Pharmaceutical University demonstrated111 (Scheme 95) a route to 4-substituted indoles from pyrrole itself. Condensation of 249 with the chlorosulfide followed by saponification and intramolecular Friedel–Crafts acylation delivered the versatile intermediate 250. Oxidation gave the indole 251. The addition of nucleophiles to 250 followed by dehydration gave the 4-alkylindole (not illustrated).
Scheme 95.
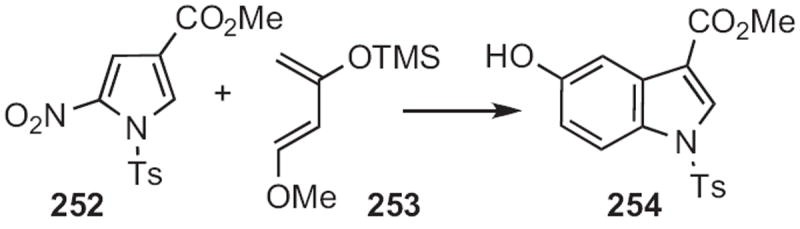
Pedro Mancini of the Universidad Nacional de Litoral showed112 that nitropyrroles, such as 252 (Scheme 96) were effective Diels–Alder dienophiles. Regiocontrol was poor with isoprene, whereas addition to the more activated diene 253 proceeded to give the 5-hydroxyindole 254 with complete regiocontrol.
Scheme 96.
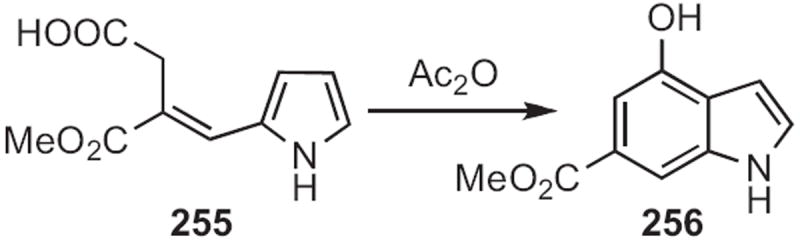
Edwin Vedejs of the University of Michigan optimized113 the acetic anhydride-mediated cyclization of the Stobbe condensation product 255 (Scheme 97) to the indole 256. Although this cyclization had been reported earlier, Vedejs found that the conditions originally described also delivered substantial quantities of an indolizidine by-product.
Scheme 97.
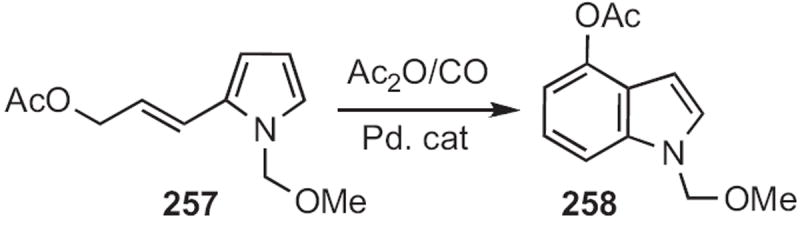
Masanobu Hidai of the University of Tokyo developed114 the Pd-catalyzed cyclocarbonylation of the allylic acetate 257 (Scheme 98) to the 4-acetoxyindole 258. It seems likely that a more highly substituted version of 257 would cyclize with equal facility.
Scheme 98.
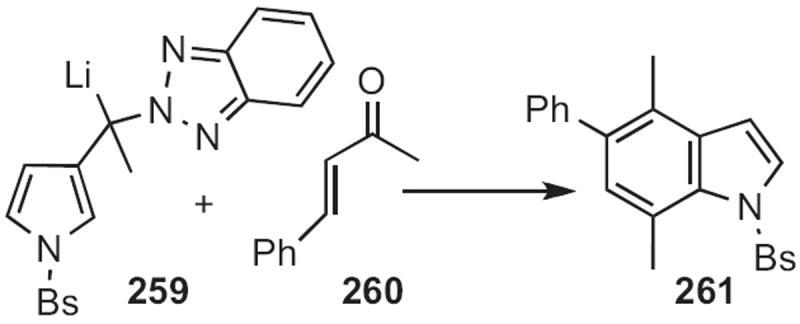
Alan R. Katrizky of the University of Florida devised115 an approach to indoles with more highly substituted benzene rings. Addition of the benzotriazolyl anion 259 (Scheme 99) to an enone, such as 260 followed by acid-catalyzed dehydrative cyclization delivered the indole 261.
Scheme 99.
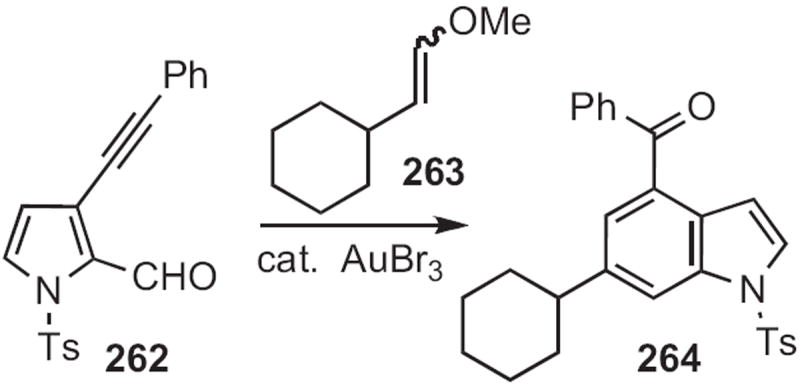
Naoki Asao of Tohoku University found116 that AuBr3 was an effective catalyst for the cyclocondensation (Scheme 100) of 262 with 263 to give the indole 264. F. Dean Toste of the University of California, Berkeley uncovered117 a related Au-catalyzed cyclization leading to indoles.
Scheme 100.
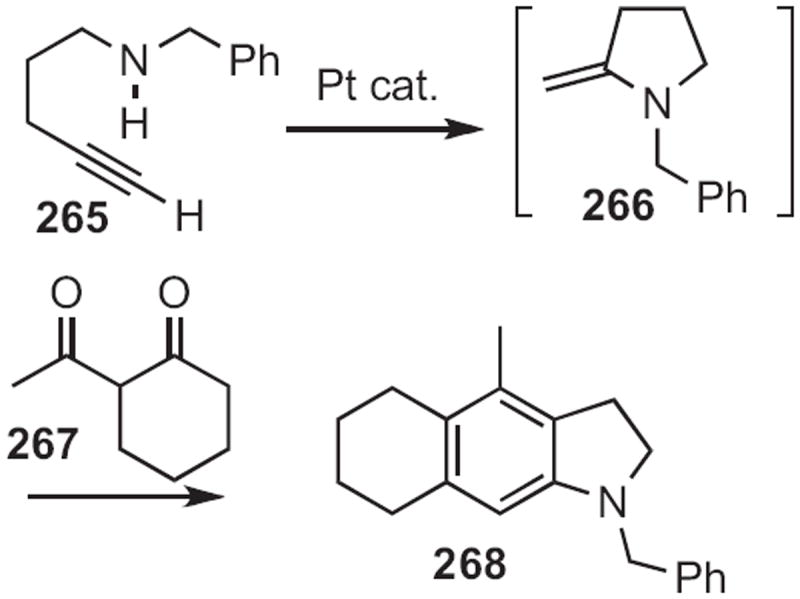
In 2009, Chi-Meng Che of the University of Hong Kong118 described (Scheme 101) the Pt-mediated intramolecular hydroamination of the alkyne 265. Condensation of the cyclic enamine 266 so prepared with a β-diketone 267 proceeded with high regioselectivity to give the indoline 268. For the aromatization of a similar N-benzyl indoline, see Scheme 41.
Scheme 101.
10. Type 9
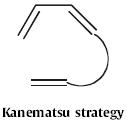
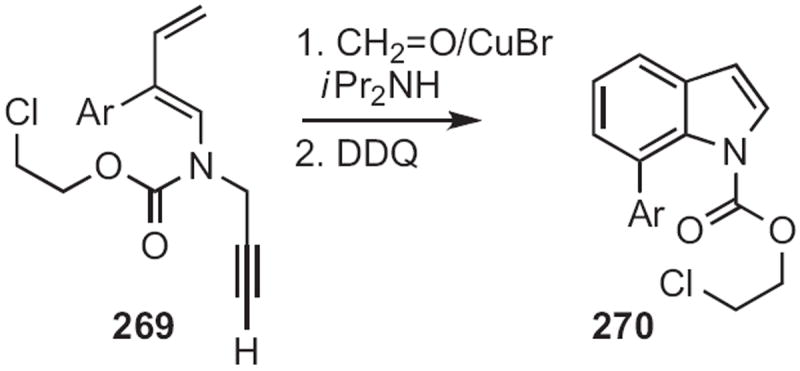
The least developed approach to indoles is Type 9, the simultaneous construction of both rings of the indole. This route was pioneered in 1986119 by Ken Kanematsu of Kyushu University. Homologation of 269 (Scheme 102) to the allene led to the intramolecular Diels–Alder cyclization product, that was readily aromatized to the indole 270.
Scheme 102.
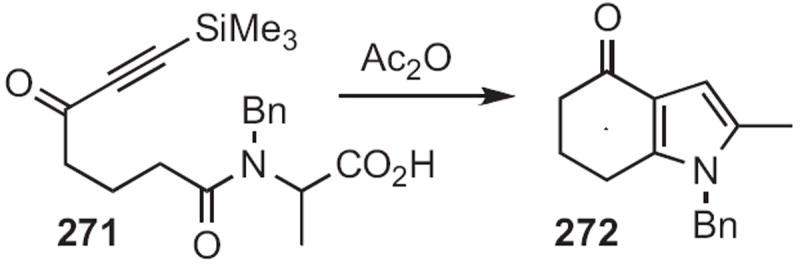
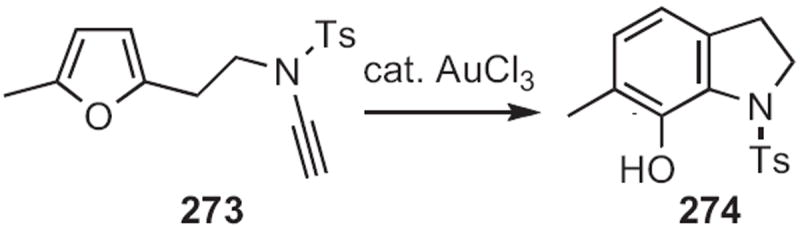
Three related approaches have been put forward since that time. Michael J. Martinelli, then at Lilly, established120 that acetic anhydride-mediated decarboxylation of 271 (Scheme 103) led to a 1,3-dipole, that added in an intramolecular fashion to the alkyne, delivering the dihydro indole 272. In a complementary approach, A. Stephen K. Hashmi of Ruprecht-Karls-Universität Heidelberg found121 that with catalytic AuBr3, 273 (Scheme 104) cyclized efficiently to 274. As outlined earlier in this review, both 272 and 274 would be readily aromatized to the corresponding indoles.
Scheme 103.
Scheme 104.
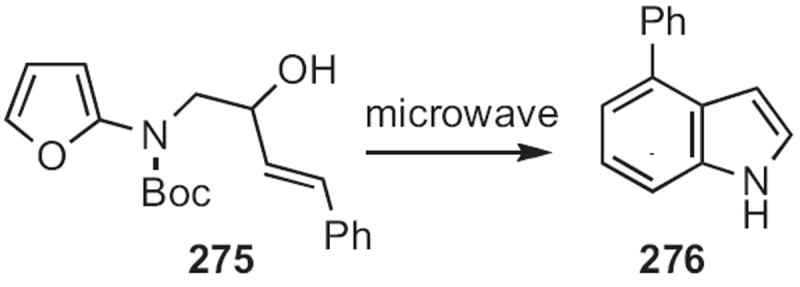
In 2009, Peter Wipf of the University of Pittsburgh described122 the intramolecular Diels—Alder cyclization (Scheme 105) of the allylic alcohol 275. Microwave heating led directly to the doubly aromatized product 276.
Scheme 105.
11. Conclusions
In this review, we have tried to be inclusive, but certainly not comprehensive. We hope that the scheme outlined here for the classification of synthetic routes to indoles will be useful to future practitioners of the art, and will stimulate new thinking in the field.
Acknowledgments
The authors thank Professor Gordon W. Gribble for his advice and encouragement. PKT thanks Randy W. Jackson for his understanding and support.
Biographies

Douglass F. Taber was born in 1948 in Berkeley, California. He earned a B.S. in Chemistry with Honors from Stanford University in 1970, and a Ph.D. in Organic Chemistry from Columbia University in 1974 (G. Stork). After a postdoctoral year at the University of Wisconsin (B.M. Trost), Taber accepted a faculty position at Vanderbilt University. He moved to the University of Delaware of Delaware in 1982, where he is currently Professor of Chemistry. Taber is the author of more than 200 research papers on organic synthesis and organometallic chemistry. He is also the author of the weekly Organic Highlights published at http://www.organic-chemistry.org/

Pavan K. Tirunahari was born inWarangal, A.P, India in 1968. He received his Bachelor of Science and Master of Science degrees from Osmania University, Hyderabad. He then joined the group of Dr. B. G. Hazra at National Chemical Laboratory, Pune, Maharastra. He received his Ph.D degree in Organic Chemistry from the University of Pune. He did his postdoctoral studies in the group of Professor James. P. Morken at the University of North Carolina. Currently he is working at Accel Synthesis, Inc., Garnet Valley, PA. His research interests include process research, synthetic methodologies, medicinal chemistry, and pharmacology.
References and notes
- 1.For recent reviews of indole synthesis, see Patil SA, Patil R, Miller DD. Curr Med Chem. 2011;18:615–637. doi: 10.2174/092986711794480195.; Cacchi S, Fabrizi G, Goggiamani A. Org Biomol Chem. 2011;9:641–652. doi: 10.1039/c0ob00501k.; Song JJ, Reeves JT, Fandrick DR, Tan Z, Yee NK, Senanayake CH. ARKIVOC. 2010:390–449.; Palmisano G, Penoni A, Sisti M, Tibiletti F, Tollari S, Nicholas KM. Curr Org Chem. 2010;14:2409–2441.; Patil SA, Patil R, Miller DD. Curr Med Chem. 2009;16:2531–2565. doi: 10.2174/092986709788682010.; Barluenga J, Rodriguez F, Fananas FJ. Chem—Asian J. 2009;4:1036–1048. doi: 10.1002/asia.200900018.; Russel JS, Pelkey ET. Prog Heterocycl Chem. 2009;20:122–151.; Krüger K, Tillack A, Beller M. Adv Synth Catal. 2008;350:2153–2167.; Humphrey GR, Kuethe JT. Chem Rev. 2006;106:2875–2911. doi: 10.1021/cr0505270.; Gribble GW. Pure Appl Chem. 2003;75:1417–1432.; Gribble GW. J Chem Soc Perkin Trans. 2000;1:1045–1075.
- 2.Brodfuehrer PR, Chen B-C, Sattelberg TR, Sr, Smith PR, Reddy JP, Stark DR, Quinlan SL, Reid JG, Thottathil JK, Wang S. J Org Chem. 1997;62:9192–9202. [Google Scholar]
- 3.Chae J, Buchwald SL. J Org Chem. 2004;69:3336–3339. doi: 10.1021/jo035819k. [DOI] [PubMed] [Google Scholar]
- 4.Yasui E, Wada M, Takamura N. Tetrahedron Lett. 2006;47:743–746. [Google Scholar]
- 5.Maruoka K, Oishi M, Yamamoto H. J Org Chem. 1993;58:7638–7639. [Google Scholar]
- 6.Cao C, Shi Y, Odom AL. Org Lett. 2002;4:2853–2856. doi: 10.1021/ol0201052. [DOI] [PubMed] [Google Scholar]
- 7.Wagaw S, Yang BH, Buchwald SL. J Am Chem Soc. 1999;121:10251–10263. [Google Scholar]
- 8.Sundberg RJ, Laurino JP. J Org Chem. 1984;49:249–254. [Google Scholar]
- 9.For a modern procedure for the Bischler synthesis, see Sridharan V, Perumal S, Avendaño C, Menéndez JC. Synlett. 2006:91–95.; For a variant leading to 3-substituted indoles, see Pchalek K, Jones AW, Wekking MMT, Black DSC. Tetrahedron. 2005;61:77–82.Cho CS, Kim JH, Choi H-J, Kim T-J, Shim SC. Tetrahedron Lett. 2003;44:2975–2977.
- 10.Penoni A, Volkmann J, Nicholas KM. Org Lett. 2002;4:699–701. doi: 10.1021/ol017139e. [DOI] [PubMed] [Google Scholar]
- 11.Saito A, Kanno A, Hanzawa Y. Angew Chem Int Ed. 2007;46:3931–3933. doi: 10.1002/anie.200605162. [DOI] [PubMed] [Google Scholar]
- 12.Johnson F, Subramanian R. J Org Chem. 1986;51:5040–5041. [Google Scholar]
- 13.Watson D, Dillin DR. Tetrahedron Lett. 1980;21:3969–3970. [Google Scholar]
- 14.Würtz S, Rakshit S, Neumann JJ, Dröge T, Glorius F. Angew Chem Int Ed. 2008;47:7230–7233. doi: 10.1002/anie.200802482. [DOI] [PubMed] [Google Scholar]
- 15.Cui S-L, Wang J, Wang Y-G. J Am Chem Soc. 2008;130:13526–13527. doi: 10.1021/ja805706r. [DOI] [PubMed] [Google Scholar]
- 16.Wierenga W, Griffin J, Warpehoski MA. Tetrahedron Lett. 1983;24:2437–2440. [Google Scholar]
- 17.Quiclet-Sire B, Zard SZ. Org Lett. 2008;10:3279–3282. doi: 10.1021/ol801162m. [DOI] [PubMed] [Google Scholar]
- 18.Chandra T, Zou S, Brown KL. Tetrahedron Lett. 2004;45:7783–7786. [Google Scholar]
- 19.Samet AV, Zakharov EP, Semenov VV, Buchanan AC, III, Gakh AA. Synth Commun. 2001;31:1441–1445. [Google Scholar]
- 20.Liu K, Yin D. Org Lett. 2009;11:637–639. doi: 10.1021/ol8027187. [DOI] [PubMed] [Google Scholar]
- 21.Shi Z, Zhang C, Li S, Pan D, Ding S, Cui Y, Jiao N. Angew Chem Int Ed. 2009;48:4572–4576. doi: 10.1002/anie.200901484. [DOI] [PubMed] [Google Scholar]
- 22.Saito A, Oa S, Fukaya H, Hanzawa Y. J Org Chem. 2009;74:1517–1524. doi: 10.1021/jo8022523. [DOI] [PubMed] [Google Scholar]
- 23.Schneekloth JS, Jr, Kim J, Sorensen EJ. Tetrahedron. 2009;65:3096–3101. doi: 10.1016/j.tet.2008.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mori M, Chiba K, Ban Y. Tetrahedron Lett. 1977:1037–1040. [Google Scholar]
- 25.Odle R, Blevins B, Ratcliff M, Hegedus LS. J Org Chem. 1980;45:2709–2710. [Google Scholar]
- 26.Macor JE, Ogilvie RJ, Wythes MJ. Tetrahedron Lett. 1996;37:4289–4292. [Google Scholar]
- 27.Fuwa H, Sasaki M. Org Lett. 2007;9:3347–3350. doi: 10.1021/ol071312a. [DOI] [PubMed] [Google Scholar]
- 28.Jensen T, Pedersen H, Bang-Andersen B, Madsen R, Jørgensen M. Angew Chem Int Ed. 2008;47:888–890. doi: 10.1002/anie.200703763. [DOI] [PubMed] [Google Scholar]
- 29.Ackermann L, Kaspar LT, Gschrei CJ. Chem Commun. 2004:2824–2825. doi: 10.1039/b411571f. [DOI] [PubMed] [Google Scholar]
- 30.Beckwith ALJ, Meijs GFJCS. Chem Commun. 1981:595–597. [Google Scholar]
- 31.Shen L, Hsung RP. Org Lett. 2005;7:775–778. doi: 10.1021/ol047572z. [DOI] [PubMed] [Google Scholar]
- 32.Zhang D, Liebeskind LS. J Org Chem. 1996;61:2594–2595. doi: 10.1021/jo960304x. [DOI] [PubMed] [Google Scholar]
- 33.Tidwell JH, Peat AJ, Buchwald SL. J Org Chem. 1994;59:7164–7168. [Google Scholar]
- 34.Gilmore CD, Allan KM, Stoltz BM. J Am Chem Soc. 2008;130:1558–1559. doi: 10.1021/ja0780582. [DOI] [PubMed] [Google Scholar]
- 35.Caubère C, Caubère P, Renard P, Bizot-Espiart J-G, Jamart-Grégoire B. Tetrahedron Lett. 1993;34:6889–6892. [Google Scholar]
- 36.Solé D, Serrano O. J Org Chem. 2008;73:2476–2479. doi: 10.1021/jo800034m. [DOI] [PubMed] [Google Scholar]
- 37.Bernini R, Cacchi S, Fabrizi G, Filisti E, Sferrazza A. Synlett. 2009:1480–1484. [Google Scholar]
- 38.Erb W, Neuville L, Zhu J. J Org Chem. 2009;74:3109–3115. doi: 10.1021/jo900210x. [DOI] [PubMed] [Google Scholar]
- 39.Hemetsberger H, Knittel D, Weidmann H. Monatsh Chem. 1970;101:161–165. [Google Scholar]
- 40.MacLeod JK, Monahan LC. Tetrahedron Lett. 1988;29:391–392. [Google Scholar]
- 41.Taber DF, Tian W. J Am Chem Soc. 2006;128:1058–1059. doi: 10.1021/ja058026j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiba S, Hattori G, Narasaka K. Chem Lett. 2007;36:52–53. [Google Scholar]
- 43.Stokes BJ, Dong H, Leslie BE, Pumphrey AL, Driver TG. J Am Chem Soc. 2007;129:7500–7501. doi: 10.1021/ja072219k. [DOI] [PubMed] [Google Scholar]
- 44.Du Y, Liu R, Linn G, Zhao K. Org Lett. 2006;8:5919–5922. doi: 10.1021/ol062288o. [DOI] [PubMed] [Google Scholar]
- 45.Person H, Del Aguila Pardo M, Foucaud A. Tetrahedron Lett. 1980;21:281–284. [Google Scholar]
- 46.Russell GA, Yao C-F, Tashtoush HI, Russell JE, Dedolph DE. J Org Chem. 1991;56:663–669. [Google Scholar]
- 47.Satomura M. J Org Chem. 1993;58:3757–3760. [Google Scholar]
- 48.Boger DL, Keim H, Oberhauser B, Schreiner EP, Foster CA. J Am Chem Soc. 1999;121:6197–6205. [Google Scholar]
- 49.Hsieh THH, Dong VM. Tetrahedron. 2009;65:3062–3068. [Google Scholar]
- 50.Mei T-S, Wang X, Yu J-Q. J Am Chem Soc. 2009;131:10806–10807. doi: 10.1021/ja904709b. [DOI] [PubMed] [Google Scholar]
- 51.Aoki K, Peat AJ, Buchwald SL. J Am Chem Soc. 1998;120:3068–3073. [Google Scholar]
- 52.Yamada K, Kurokawa T, Tokuyama H, Fukuyama T. J Am Chem Soc. 2003;125:6630–6631. doi: 10.1021/ja035303i. [DOI] [PubMed] [Google Scholar]
- 53.Barluenga J, Jiménez-Aquino A, Valdés C, Aznar F. Angew Chem Int Ed. 2007;46:1529–1532. doi: 10.1002/anie.200604407. [DOI] [PubMed] [Google Scholar]
- 54.Melkonyan FS, Karchava AV, Yurovskaya MA. J Org Chem. 2008;73:4275–4278. doi: 10.1021/jo800630v. [DOI] [PubMed] [Google Scholar]
- 55.Zhang B, Zhang J, Yang D-DH, Yang N-cC. J Org Chem. 1996;61:3236–3237. [Google Scholar]
- 56.Androsov DA, Neckers DC. J Org Chem. 2007;72:5368–5373. doi: 10.1021/jo0707784. [DOI] [PubMed] [Google Scholar]
- 57.Cai Q, Li Z, Wei J, Ha C, Pei D, Ding K. Chem Commun. 2009:7581–7583. doi: 10.1039/b918345k. [DOI] [PubMed] [Google Scholar]
- 58.Taylor AM, Schreiber SL. Tetrahedron Lett. 2009;50:3230–3233. doi: 10.1016/j.tetlet.2009.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sundberg RJ, Lin L-S, Blackburn DE. J Heterocycl Chem. 1969;6:441–441. [Google Scholar]
- 60.Sundberg RJ, Yamazaki T. J Org Chem. 1967;32:290–294. [Google Scholar]
- 61.Shen M, Leslie BE, Driver TG. Angew Chem Int Ed. 2008;47:5056–5059. doi: 10.1002/anie.200800689. [DOI] [PubMed] [Google Scholar]
- 62.Katayama S, Ae N, Nagata R. J Org Chem. 2001;66:3474–3483. doi: 10.1021/jo010002h. [DOI] [PubMed] [Google Scholar]
- 63.Siu J, Baxendale IR, Ley SV. Org Biomol Chem. 2004;2:160–167. doi: 10.1039/b313012f. [DOI] [PubMed] [Google Scholar]
- 64.Smith AB, III, Visnick M. Tetrahedron Lett. 1985;26:3757–3760. [Google Scholar]
- 65.Coleman CM, O’Shea DF. J Am Chem Soc. 2003;125:4054–4055. doi: 10.1021/ja034283h. [DOI] [PubMed] [Google Scholar]
- 66.Bard RN, Bunnett JF. J Org Chem. 1980;45:1547–1548. [Google Scholar]
- 67.Kozmin SA, Rawal VH. J Am Chem Soc. 1998;120:13523–13524. [Google Scholar]
- 68.Islam MS, Brennan C, Wang Q, Hossain MM. J Org Chem. 2006;71:4675–4677. doi: 10.1021/jo0601821. [DOI] [PubMed] [Google Scholar]
- 69.Fujita K-i, Yamamoto K, Yamaguchi R. Org Lett. 2002;4:2691–2694. doi: 10.1021/ol026200s. [DOI] [PubMed] [Google Scholar]
- 70.Sajiki H, Ikawa T, Hirota K. Org Lett. 2004;6:4977–4980. doi: 10.1021/ol047871o. [DOI] [PubMed] [Google Scholar]
- 71.Nicolaou KC, Estrada AA, Lee SH, Freestone GC. Angew Chem Int Ed. 2006;45:5364–5368. doi: 10.1002/anie.200601808. [DOI] [PubMed] [Google Scholar]
- 72.Baudin J-B, Julia SA. Tetrahedron Lett. 1986;27:837–840. [Google Scholar]
- 73.(a) Castro CE, Stevens RD. J Org Chem. 1963;28:2163–2163. [Google Scholar]; (b) Castro CE, Gaughan EJ, Owsley DC. J Org Chem. 1966;31:4071–4078. [Google Scholar]
- 74.Taylor EC, Katz AH, Salgado-Zamora H. Tetrahedron Lett. 1985;26:5963–5966. [Google Scholar]
- 75.Rudisill DE, Stille JK. J Org Chem. 1989;54:5856–5866. [Google Scholar]
- 76.(a) Larock RC, Yum EK. J Am Chem Soc. 1991;113:6689–6690. [Google Scholar]; (b) Larock RC, Yum EK, Refvik MD. J Org Chem. 1998;63:7652–7662. [Google Scholar]
- 77.Stuart DR, Bertrandf-Laperle M, Burgess KMN, Fagnou K. J Am Chem Soc. 2008;130:16474–16475. doi: 10.1021/ja806955s. [DOI] [PubMed] [Google Scholar]
- 78.Fang Y-Q, Lautens M. J Org Chem. 2008;73:538–549. doi: 10.1021/jo701987r. [DOI] [PubMed] [Google Scholar]
- 79.Okuma K, Takeshita I, Yasuda T, Shioji K. Chem Lett. 2006;35:1122–1123. [Google Scholar]
- 80.Prabhakaran EN, Nugent BM, Williams AL, Nailor KE, Johnston JN. Org Lett. 2002;4:4197–4200. doi: 10.1021/ol027064u. [DOI] [PubMed] [Google Scholar]
- 81.Miyagi T, Hari Y, Aoyama T. Tetrahedron Lett. 2004;45:6303–6305. [Google Scholar]
- 82.Gabriele B, Mancuso R, Salerno G, Lupinacci E, Ruffolo G, Costa M. J Org Chem. 2008;73:4971–4977. doi: 10.1021/jo8006495. [DOI] [PubMed] [Google Scholar]
- 83.Motoyama Y, Kamo K, Nagashima H. Org Lett. 2009;11:1345–1348. doi: 10.1021/ol9001366. [DOI] [PubMed] [Google Scholar]
- 84.Xu Z, Li Q, Zhang L, Jia Y. J Org Chem. 2009;74:6859–6862. doi: 10.1021/jo9012755. [DOI] [PubMed] [Google Scholar]
- 85.Cacchi S, Fabrizi G, Filisti E. Synlett. 2009:1817–1821. [Google Scholar]
- 86.Ohta Y, Chiba H, Oishi S, Fujii N, Ohno H. J Org Chem. 2009;74:7052–7058. doi: 10.1021/jo901328q. [DOI] [PubMed] [Google Scholar]
- 87.Mitra T, Das S, Basak A. Tetrahedron Lett. 2009;50:5846–5849. [Google Scholar]
- 88.Pei T, Tellers DM, Streckfuss EC, Chen C-y, Davies IW. Tetrahedron. 2009;65:3285–3291. [Google Scholar]
- 89.Houlihan WJ, Parrino VA, Uike Y. J Org Chem. 1981;46:4511–4515. [Google Scholar]
- 90.Orlemans EOM, Schreuder AH, Conti PGM, Verboom W, Reinhoudt DN. Tetrahedron. 1987;43:3817–3826. [Google Scholar]
- 91.Kraus GA, Guo H. Org Lett. 2008;10:3061–3063. doi: 10.1021/ol801034x. [DOI] [PubMed] [Google Scholar]
- 92.Le S, Lee W-M, Sulikowski GA. J Org Chem. 1999;64:4224–4225. [Google Scholar]
- 93.Jones WD, Kosar WP. J Am Chem Soc. 1986;108:5640–5641. [Google Scholar]
- 94.(a) Jones CD, Suárez T. J Org Chem. 1972;37:3622–3623. [Google Scholar]; (b) Jones CD. J Org Chem. 1972;37:3624–3625. [Google Scholar]
- 95.Nakamura Y, Ukita T. Org Lett. 2002;4:2317–2320. doi: 10.1021/ol025916k. [DOI] [PubMed] [Google Scholar]
- 96.Nakao K, Murata Y, Koike H, Uchida C, Kawamura K, Mihara S, Hayashi S, Stevens RW. Tetrahedron Lett. 2003;44:7269–7271. [Google Scholar]
- 97.(a) Fukuyama T, Chen X, Peng G. J Am Chem Soc. 1994;116:3127–3128. [Google Scholar]; (b) Reding MT, Fukuyama T. Org Lett. 1999;1:973–976. [Google Scholar]; (c) Ranier JD, Kennedy AR. J Org Chem. 2000;65:6213–6216. doi: 10.1021/jo000831n. [DOI] [PubMed] [Google Scholar]
- 98.(a) Fürstner A, Hupperts A, Ptock A, Janssen E. J Org Chem. 1994;59:5215–5229. [Google Scholar]; (b) Fürstner A, Hupperts A. J Am Chem Soc. 1995;117:4468–4475. [Google Scholar]; (c) Ding F, Zhang Y, Qu B, Li G, Farina V, Lu BZ, Senanayake CH. Org Lett. 2008;10:1067–1070. doi: 10.1021/ol7029905. [DOI] [PubMed] [Google Scholar]
- 99.Zhou L, Doyle MP. J Org Chem. 2009;74:9222–9224. doi: 10.1021/jo902089e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Seong CM, Park CM, Choi J, Park NS. Tetrahedron Lett. 2009;50:1029–1031. [Google Scholar]
- 101.Wyrembak PN, Hamilton AD. J Am Chem Soc. 2009;131:4566–4567. doi: 10.1021/ja809245t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ketcha DM, Wilson LJ, Portlock DE. Tetrahedron Lett. 2000;41:6253–6257. [Google Scholar]
- 103.Lebold TP, Kerr MA. Org Lett. 2008;10:997–1000. doi: 10.1021/ol703085f. [DOI] [PubMed] [Google Scholar]
- 104.Morales CL, Pagenkopf BL. Org Lett. 2008;10:157–159. doi: 10.1021/ol702376j. [DOI] [PubMed] [Google Scholar]
- 105.Arai M, Miyauchi Y, Miyahara T, Ishikawa T, Saito S. Synlett. 2008:122–126. [Google Scholar]
- 106.Iida H, Yuasa Y, Kibayashi C. Tetrahedron Lett. 1982;23:3591–3594. [Google Scholar]
- 107.Revial G, Jabin I, Lim S, Pfau M. J Org Chem. 2002;67:2252–2256. doi: 10.1021/jo0110597. [DOI] [PubMed] [Google Scholar]
- 108.Zhao X, Zhang E, Tu Y-Q, Zhang Y-Q, Yuan D-Y, Cao K, Fan C-A, Zhang F-M. Org Lett. 2009;11:4002–4004. doi: 10.1021/ol901649p. [DOI] [PubMed] [Google Scholar]
- 109.Ma J-L, Li X-X, Kusuyama T, El-Tantawy El-Sayed I, Inokuchi T. J Org Chem. 2009;74:9218–9221. doi: 10.1021/jo902068m. [DOI] [PubMed] [Google Scholar]
- 110.Moskal J, van Leusen AM. J Org Chem. 1986;51:4131–4139. [Google Scholar]
- 111.Ishibashi H, Tabata T, Hanaoka K, Iriyama H, Akamatsu S, Ikeda M. Tetrahedron Lett. 1993;34:489–492. [Google Scholar]
- 112.Della Rossa C, Kneeteman M, Mancini P. Tetrahedron Lett. 2007;48:1435–1438. [Google Scholar]
- 113.Kim M, Vedejs E. J Org Chem. 2004;69:6945–6948. doi: 10.1021/jo040191e. [DOI] [PubMed] [Google Scholar]
- 114.Iwasaki M, Kobayashi Y, Li J-P, Matsuzaka H, Ishii Y, Hidai M. J Org Chem. 1991;56:1922–1927. [Google Scholar]
- 115.Katritzky AR, Ledoux S, Nair SK. J Org Chem. 2003;68:5728–5730. doi: 10.1021/jo026853m. [DOI] [PubMed] [Google Scholar]
- 116.Asao N, Aikawa H. J Org Chem. 2006;71:5249–5253. doi: 10.1021/jo060597m. [DOI] [PubMed] [Google Scholar]
- 117.Zhao J, Hughes CO, Toste FD. J Am Chem Soc. 2006;128:7436–7437. doi: 10.1021/ja061942s. [DOI] [PubMed] [Google Scholar]
- 118.Liu X-Y, Che C-M. Angew Chem Int Ed. 2009;48:2367–2371. doi: 10.1002/anie.200805383. [DOI] [PubMed] [Google Scholar]
- 119.(a) Hayakawa K, Yasukouchi T, Kanematsu K. Tetrahedron Lett. 1986;27:1837–1840. [Google Scholar]; (b) Hayakawa K, Yasukouchi T, Kanematsu K. Tetrahedron Lett. 1987;28:5895–5898. [Google Scholar]
- 120.Hutchison DR, Nayyar NK, Martinelli MJ. Tetrahedron Lett. 1996;37:2887–2890. [Google Scholar]
- 121.Hashmi ASK, Rudolph M, Bats JW, Frey W, Rominger F, Oeser T. Chem —Eur J. 2008;14:6672–6678. doi: 10.1002/chem.200800210. [DOI] [PubMed] [Google Scholar]
- 122.Petronijevic F, Timmons C, Cuzzupe A, Wipf P. Chem Commun. 2009:104–106. doi: 10.1039/b816989f. [DOI] [PMC free article] [PubMed] [Google Scholar]