

Abstract
Background
Hyperparathyroidism-jaw tumor syndrome (HPT-JT) is a rare autosomal dominant disease secondary to germline inactivating mutations of the tumor suppressor gene HRPT2/CDC73. The aim of the present study is to determine the optimal surgical approach to parathyroid disease in patients with HPT-JT.
Method
A retrospective analysis of clinical and genetic features, parathyroid operative outcomes, and disease outcomes in seven unrelated HPT-JT families.
Results
Seven families had five distinct germline HRPT2/CDC73 mutations. Sixteen affected family members (median age of 30.7 years) were diagnosed with primary hyperparathyroidism. Fifteen of the 16 patients underwent preoperative tumor localization studies and uncomplicated bilateral neck exploration at initial operation - all were in biochemical remission at most recent follow up. 31% of patients had multiglandular involvement. 37.5% of patients developed parathyroid carcinoma (median overall survival 8.9 years; median follow-up 7.4 years). Long-term follow-up showed 20% of patients had recurrent primary hyperparathyroidism.
Conclusions
Given the high risk of malignancy and multiglandular involvement in our cohort, we recommend bilateral neck exploration and en-bloc resection of parathyroid tumors suspicious for cancer and life-long postoperative follow-up.
Keywords: hyperparathyroidism-jaw tumor syndrome, parathyroidectomy, parathyroid cancer, parafibromin
Introduction
Primary hyperparathyroidism (PHPT), a common disorder, results from parathyroid adenomas (single or involving multiple glands), or carcinoma. Approximately 5% of all cases of PHPT are associated with hereditary syndromes that include multiple endocrine neoplasia types 1 and 2A (MEN1, MEN2A), familial isolated hyperparathyroidism (FIHP), and the hyperparathyroidism–jaw tumor (HPT–JT) syndrome (1, 2).
HPT-JT is a rare autosomal dominant disorder with incomplete penetrance and variable expression. The syndrome is characterized by the development of PHPT secondary to parathyroid tumors in approximately 90% of carriers. Approximately 35% of patients with HPT-JT may also develop ossifying fibromas of the mandible and/or maxilla (3). Less common manifestations of the disease include renal lesions (Wilm's tumors, polycystic disease, hamartomas and adenocarcinomas), and uterine tumors (1, 3).
The susceptibility gene for HPT-JT is HRPT2/CDC73, a putative tumor suppressor gene, located on chromosome 1q31.2. HRPT2/CDC73 encodes the ubiquitously expressed nuclear protein parafibromin (4). A number of reports have linked HPT-JT to germline inactivating mutations in HRPT2/CDC73, with an associated loss of parafibromin expression and/or function in associated parathyroid tumors (1, 2, 5). While loss of parafibromin protein expression has been observed in sporadic parathyroid carcinomas, non-HPT-JT-related familial or sporadic benign parathyroid adenomas do not have loss of parafibromin expression (6). Parafibromin is believed to function as an inhibitor of cellular proliferation via cell cycle arrest, and as a transcriptional regulator through interactions with the RNA polymerase II-associated factor 1 (PAF1) complex. Parafibromin may also function in the Wnt signaling pathway (7).
PHPT in the context of HPT-JT has been previously characterized as a more aggressive disease relative to sporadic PHPT, with frequent multiglandular involvement, increased risk of persistent/recurrent disease, and a higher frequency of parathyroid carcinoma and metastasis (5). However, many of these studies were conducted prior to the identification of the HRPT2/CDC73 gene, and were limited by small study cohorts and incomplete clinical data. Following the identification of HRPT2/CDC73, more recent studies examining larger kindreds have identified lower rates of synchronous multiglandular involvement (13.2%), biochemical recurrence (17.6–22.3%), longer disease-free intervals (mean 13.7 years), and fewer cases of parathyroid carcinoma (4.4-24.3%) (4, 8).
There is a dearth of literature examining and characterizing HPT-JT disease specific features. Furthermore, the optimal surgical approach and follow-up for patients with HPT-JT with PHPT remains controversial. A number of small studies have recommended extensive parathyroidectomy (subtotal) given the syndrome's more aggressive features and higher risk of cancer (8-10). However, Iacobone and colleagues evaluated 3 large HPT-JT families and advocated for selective parathyroidectomy in all cases and unilateral exploration in cases with concordant preoperative imaging localizing studies showing single-gland disease. In their series of 17 patients, 82.4% of patients with pathologically confirmed parathyroid adenoma had single-gland involvement and no patients had synchronous multiglandular involvement (5). In comparison, Sarquis and colleagues in their series of 11 patients noted 45.4% of patients had single gland-involvement and 54.5% of patients had synchronous multiglandular involvement (8). Given the wide variation reported in the frequency of synchronous multiglandular involvement, the issue of whether a unilateral or bilateral approach should be used for parathyroidectomy in patients with known HPT-JT remains controversial.
The aim of the present study is to describe the clinical, pathological, demographic, and genetic features and surgical outcomes of 16 affected individuals from 7 families with HPT-JT, in order to provide recommendations for optimal management with regards to preoperative localization and surgical exploration.
Methods
Patients
Demographics, genetic tests, pathology, radiology, and operative history were reviewed in patients who were evaluated at the National Institutes of Health (NIH) Warren Magnuson Clinical Center on an Institutional Review Board approved clinical protocol (91-DK-0085, Studies of Hyperparathyroidism and Related Disorders). The study population consisted of seven families with 16 affected members (ten males, six females), followed at the NIH between July 1986 and January 2014. Previously, four of the seven families have been reported and have been included in this study (1). All patients were initially identified and referred to the NIH following biochemical evidence of hypercalcemia. While all patients in the cohort received genetic screening for the HRPT2 mutation, asymptomatic members of affected families were not screened for the HRPT2 mutation.
Clinical Investigation
HPT-JT was diagnosed when patients had biochemical evidence of PHPT as defined by hypercalcemia, inappropriately normal or increased intact parathyroid hormone (iPTH) levels, and normal or increased 24-hour urinary calcium with normal renal function, histologic evidence of an abnormal parathyroid gland, and the presence of a germ-line HRPT2/CDC73 mutation. Definitive diagnosis also required negative screening for MEN1 or MEN2 via absence of clinical manifestations, personal or family history, and negative genetic testing (5). Genetic testing for germline HRPT2/CDC73 mutations was performed on leukocyte DNA via direct sequencing of exons 1-17 and all intron-exon junctions of the HRPT2/CDC73 gene and, in families with suspected HPT-JT in which no mutation was found, by deletion/duplication analysis of the HRPT2/CDC73 gene via targeted array comparative genomic hybridization.
Imaging workup included an ultrasound of the neck, sestamibi scan, orthopantomographic X-rays and/or computed tomography (CT) of the mandible and maxilla for identification of jaw tumors, and abdominal ultrasound and/or CT for evaluation of kidney and uterine abnormalities. Biochemical workup included testing of serum total calcium, ionized calcium, phosphate, 25OH-vitamin D levels, and iPTH levels.
Inpatient and outpatient medical records were reviewed for demographics, clinical presentation, biochemical, genetic and imaging workup, operative management (all surgical data was limited to initial operation), postoperative course, pathology, and follow-up care. All patients were contacted via telephone to assess their most recent clinical and laboratory data.
Remission was defined as postoperative normalization of serum calcium and iPTH levels for at least 6 months following parathyroidectomy; persistent disease was defined as hypercalcemia occurring within 6 months after operation; recurrent disease was defined as hypercalcemia developing after surgery with remission for at least 6 months (5). For patients with parathyroid carcinoma, overall survival (OS) was defined as time to disease-related mortality or last follow-up.
The histological diagnosis was confirmed according to the World Health Organization guidelines (11). The diagnosis of adenoma was based on the finding of a typical encapsulated lesion consisting of small uniform cells arranged with a delicate capillary network, with a rim of normal or atrophic parathyroid tissue evident outside the capsule. Parathyroid carcinoma was defined histologically by a trabecular arrangement of tumor cells, fibrous bands, mitoses, and capsular or blood vessels and/or surrounding soft tissue invasion (12). Atypical adenoma was defined as having necrosis, trabecular arrangement of tumor cells, fibrous bands and or mitoses but with no evidence of vascular or capsular invasion. Patients with parathyroid adenoma or atypical parathyroid adenoma were grouped as a benign parathyroid disease.
Immunohistochemistry
Representative formalin fixed paraffin embedded tissue sections from normocellular parathyroids and parathyroid adenomas from 9 patients with germline HRPT2/CDC73 mutation were stained. Immunohistochemical studies were performed on a Leica BondMax automated stainer using the BondMax detection kit (Richmond, VA, USA). Antigen retrieval was performed using high pH Bond buffer H2 for 25 min. Primary antibody to Parafibromin (Santa Cruz, CA, SC-33638, mouse monoclonal 2H1) was used at a dilution of 1:400. Diaminobenzidine was used for detection, followed by a light hematoxylin counterstain.
Statistical Analyses
Statistical analysis was performed using Mann-Whitney U test and Fischer's exact test, as appropriate. A P value of <0.05 was considered statistically significant. All calculations were performed using GraphPad Software (La Jolla, CA, USA).
Results
Sixteen affected patients from 7 unrelated families with HPT-JT presented with PHPT. Genetic analysis of germline DNA identified five distinct germline mutations of the HRPT2/CDC73 gene (Table 1). Whole gene deletion of HRPT2/CDC73 was identified in 7 of 16 patients (from one family), duplication of two nucleotides (c.687_688dupAG) in exon 7 resulting in a frameshift mutation was identified in 5 of 16 patients (from three families), substitution of a serine for tyrosine (p.Tyr55Ser) in exon 2 was identified in 2 of 16 patients (from one family), a substitution of one nucleotide (c.664C>T) in exon 7 resulting in protein truncation (p.Arg222X) was identified in a single patient, and a substitution of one nucleotide (c.226C>T) in exon 2 resulting in protein truncation (p.Arg76X) was identified in a single patient.
Table 1. Clinical, biochemical and genetic data for cohort.
| Benign Parathyroid Disease |
Parathyroid Carcinoma |
Entire Cohort | |
|---|---|---|---|
| Gender | |||
| Male/Female | 7/3 | 3/3 | 10/6 |
| Age at Diagnosis (years) | |||
| Median | 26.4 | 29.9 | 30.7 |
| Range | 18-38 | 31-49 | 18-49 |
| Signs/Symptoms at Presentation | |||
| Psychiatric/Neurologic * | 4/10 (40.0%) | 3/6 (50%) | 7/16 (43.8%) |
| Fatigue | 5/10 (50.0%) | 1/6 (16.7%) | 6/16 (37.5%) |
| GastrointestinaI symptoms ** | 2/10 (20.0%) | 3/6 (50%) | 5/16 (31.3%) |
| Renal *** | 2/10 (20.0%) | 1/6 (16.7%) | 3/16 (18.8%) |
| Bony pain | 3/10 (30.0%) | 4/6 (66.7%) | 3/16 (18.8%) |
| Fractures | 1/10 (10.0%) | 1/6 (16.7%) | 2/16 (12.5%) |
| Neck mass | 0/10 (0%) | 1/6 (16.7%) | 1/16 (6.3%) |
| Laboratory Findings | |||
| Average total calcium § | 11.7 mg/dl | 13.1 mg/dl | 12.2 mg/dl |
| Intact PTH (benign parathyroid disease)? | 212.6 pg/ml | 356.5 pg/ml | 236.6 pg/ml |
| Associated Pathology | |||
| Uterine tumor | 0/3 (0%) | 2/3 (66.7%) | 2/6 (33.3%) |
| Renal lesions | 0/10 (0%) | 3/6 (50.0%) | 3/16 (18.8%) |
| Jaw tumor | 0/10 (0%) | 2/6 (12.5%) | 2/16 (12.5%) |
| Genetics/Mutation Type | |||
| CDC73 (whole gene deletion) | 3/10 (30.0%) | 4/6 (66.7%) | 7/16 (43.8%) |
| c.687_688dupAG (exon 7) | 3/10 (30.0%) | 2/6 (33.3%) | 5/16 (31.3%) |
| Y552 (exon 2) | 2/10 (20.0%) | 0/6 (0%) | 2/16 (12.5%) |
| R222X (exon 7) | 1/10 (10.0%) | 0/6 (0%) | 1/16 (6.3%) |
| R76X (exon 2) | 1/10 (10.0%) | 0/6 (0%) | 1/16 (6.3%) |
| Post-operative Complications | |||
| Hypoparathyroidism (permanent) | 1/10 (10.0%) | 1/6 (16.7%) | 2/16 (12.5%) |
| Permanent recurrent laryngeal nerve injury ¶# | 0/10 (0%) | 0/10 (0%) | 0/16 (0%) |
| Hematoma | 0/10 (0%) | 0/10 (0%) | 0/16 (0%) |
| Follow-Up (years) | |||
| Median | 2.3 | 7.4 | 3.7 |
| Range | 0.05-13.4 | 0.04-11.4 | 0.05-13.4 |
Headaches, memory loss/changes, difficulty concentrating
Abdominal pain, constipation, Gastroesophageal Reflux Disease (GERD)
Polyuria, nephrolithiasis
Total calcium reference range (8.5 – 10.2 mg/dl)
Intact PTH reference range (10 – 55 pg/ml)
At initial operation.
1 patient had RLN injury during the 2nd operation.
Clinical and demographic information is summarized in Table 1 and Figure 1. All 16 patients had symptomatic PHPT on presentation. The average total calcium was 12.2 mg/dl (median 12.0 mg/dl, ref. range 8.5 – 10.2 mg/dl) and the average iPTH was 236.6 pg/ml (median 190.4 pg/ml, ref. range 10 – 55 pg/ml). Neck ultrasound and/or sestamibi scan were performed in 14 of the 16 patients. Imaging correctly identified 11 patients with single-gland disease and two patients with multigland disease, as confirmed by pathology. One patient had preoperative imaging which suggested single-gland disease, and was found to have multigland disease intraoperatively. The patient was in biochemical remission postoperatively and the intraoperative findings were confirmed on pathology. Sestamibi scan was completely correct for preoperative lateralization in 46.2% of patients.
Figure 1.
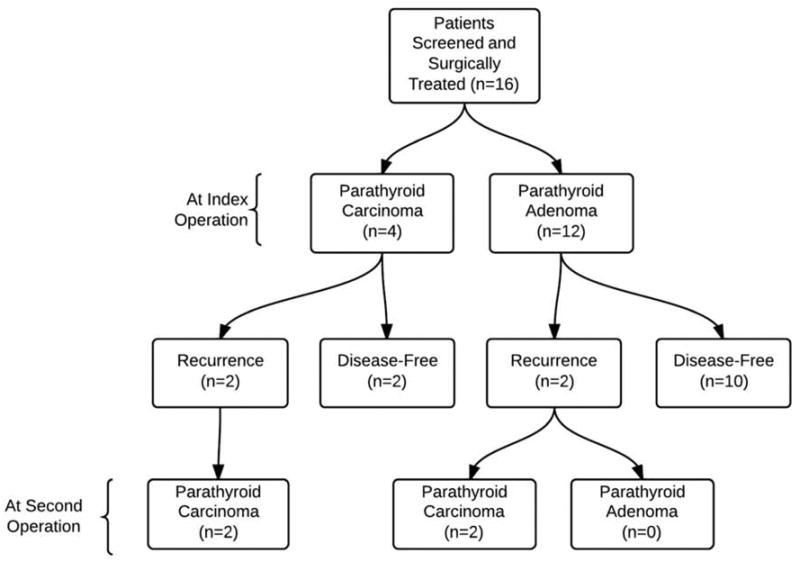
Pathologic and operative findings and patient outcome.
Fifteen of the 16 patients underwent uncomplicated bilateral neck exploration at initial operation and all were in biochemical remission postoperatively. Ten patients had intraoperative PTH monitoring. Nine of the ten patients had a decrease of ≥75% from their baseline PTH on IOPTH monitoring and all had evidence of biochemical remission (Figure 2). Three of the 16 patients had recurrent PHPT and were all ultimately diagnosed with parathyroid carcinoma either at initial operation or subsequent operation. One patient who did not have intraoperative PTH monitoring on initial operation was found to have persistent PHPT, with final pathology revealing parathyroid carcinoma. Five patients had multiglandular disease, and six patients underwent subtotal parathyroidectomy (3.5 glands). There were no cases of recurrent laryngeal nerve injury, and/or neck hematoma following the initial operation. However, one patient had a case of permanent recurrent laryngeal nerve injury during a reoperation for recurrent PHPT. Ten of the 16 patients had postoperative transient hypoparathyroidism following initial operation, requiring calcium supplementation. Two patients had permanent hypoparathyroidism with both patients having undergone a subtotal parathyroidectomy.
Figure 2.
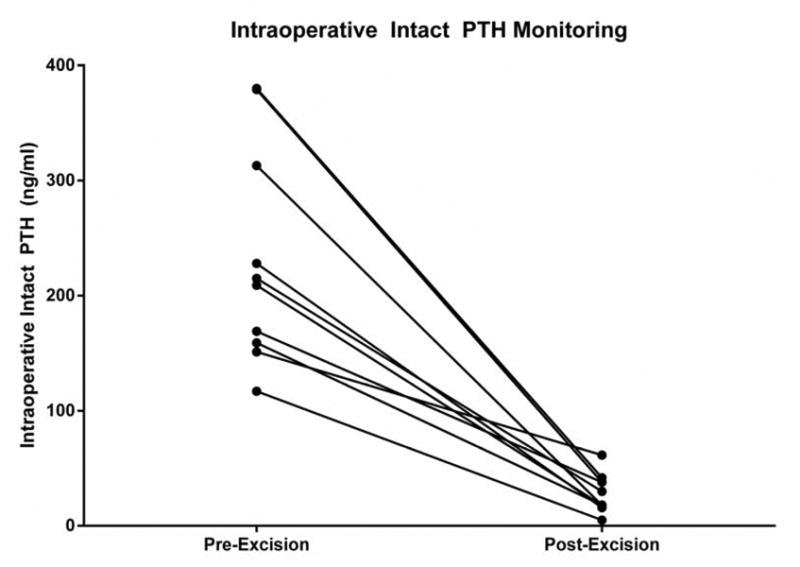
Pair wise comparison of pre-excision and post-excision intact PTH levels in patients undergoing parathyroidectomy.
On final pathology, 37.5% percent of patients were found to have parathyroid carcinoma of which two-thirds subsequently developed bone, lung, and /or liver metastases. At initial surgery, only one of the six patients with parathyroid carcinoma had a palpable neck mass. Intraoperatively, patients with parathyroid carcinoma demonstrated features of malignancy, including enlarged gland size, firm texture, grey/white-color and/or gross adherence/invasion of adjacent tissue. Median OS following initial operation for these patients was 8.9 years (median follow up of 7.4 years), with 3 of the 6 patients dying secondary to complications from parathyroid carcinoma. Total calcium levels, iPTH levels and parathyroid gland size were compared between patients with benign parathyroid disease and parathyroid carcinoma. Average total calcium levels for patients with benign parathyroid disease was 11.7 mg/dl compared to 13.1 mg/dl (Figure 3A, p=0.056). Intact PTH levels for patients with benign parathyroid disease was 212.6 pg/dl compared to 356.5 pg/dl for patients with parathyroid carcinoma (p=0.12). Average parathyroid gland size was 19.3 mm for patients with benign parathyroid disease compared to 26.3 mm for patients with parathyroid carcinoma (Figure 3B, p=0.23). The average age of diagnosis for parathyroid carcinomas was 35.4 years, compared to 30.5 years for benign parathyroid disease (p=0.38).
Figure 3.
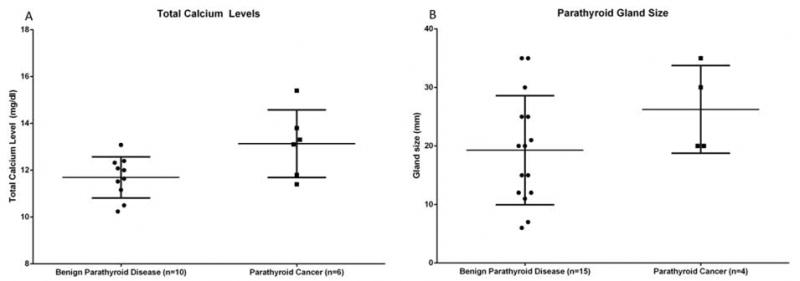
Comparison of patients with parathyroid adenoma and parathyroid carcinoma with respect to (a) total calcium level (p=0.056), and (b) parathyroid gland size (p=0.230).
Parafibromin staining was performed on 9 parathyroid adenomas from HRPT2 patients and was matched to normocellular parathyroids from the same patients, which were stained as control tissue. All normocellular parathyroids showed nuclear positivity throughout the gland (Figure 4A). In 7 cases there was diffuse loss of nuclear parafibromin staining in the adenomatous component with preservation of staining in adjacent normal parathyroid (where present) and within fibrovascular septa (Figure 4B). Interestingly, in two adenoma cases (both with a substitution of a serine for tyrosine (p.Tyr55Ser)) preservation of nuclear parafibromin was present (Figure 4C).
Figure 4.
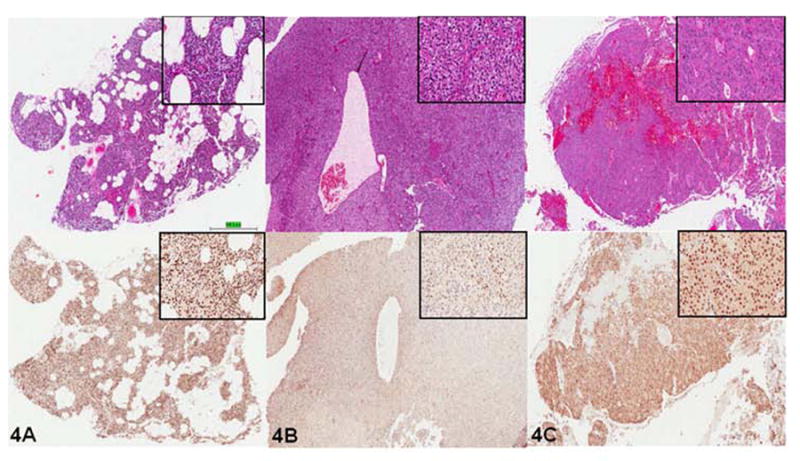
Immunohistochemical analysis of parafibromin expression in patients with HPT-JT syndrome. The upper panels were stained with Hematoxylin and Eosin, and the lower panels were stained for parafibromin. (a) Normocellular parathyroid tissue showing strong diffuse nuclear immunostaining. (b) Benign parathyroid adenoma showing loss of positivity in the same HPT-JT patient. (c) Benign parathyroid adenoma from patient with substitution of a serine for tyrosine (p.Tyr55Ser) showing retained positive nuclear immunostaining. Note magnified (40x) inserts of tissue samples on top right.
Discussion
HPT-JT syndrome is a rare autosomal dominant disease characterized by PHPT, fibroosseous jaw tumors, uterine tumors, and renal lesions. Genetic mutation analysis, preoperative localization studies, operative data, and postoperative findings showed that there is a high frequency of multiglandular disease and parathyroid carcinoma.
Given the familial history and genetic predisposition to parathyroid adenomas, nearly all of the patients underwent bilateral neck exploration at their initial operation regardless of preoperative localization studies. HPT-JT, classically described as a disease with frequent multiglandular involvement, has a 21.3% prevalence of multiglandular disease from previous published cases in the literature (Table 2). Sarquis and colleagues reported that 54.5% of patients had synchronous multiglandular involvement (8) compared to no synchronous multiglandular involvement in a cohort reported by Iacobone and colleagues. Iacobone and colleagues recommended unilateral exploration in patients with concordant preoperative localizing studies suggesting single-gland disease (5). In this series of patients, the data show 31.3% of patients with synchronous multiglandular involvement. In a familial syndrome such as HPT-JT with a high rate of multiglandular disease, and a predisposition to parathyroid carcinoma, a high failure rate may result with limited neck exploration. Many of these patients undergo multiple operations due to recurrences and parathyroid carcinoma, which exposes them to an increased risk for complications like recurrent laryngeal nerve injury and hypoparathyroidism. Thus, the initial operation should be performed to not only provide the best chance for definitive treatment, but to avoid the need for reoperation and associated morbidity. However, normal parathyroid glands should not be removed as 2 of the 16 patients had permanent hypoparathyroidism when an initial subtotal parathyroidectomy was performed.
Table 2. Review of the literature focusing on HRPT2-related HPT (period 2002-2013).
| Reference | Family (n) | Affected patients (n) | Patients with PHPT (n) | Single-gland involvement (n) | Synchronous multiglandular involvement (n) | Recurrences (n) | Jaw-Tumor (n) | Parathyro id Carcinom a (n) | Renal lesions (n) | Uterine lesions (n) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Carpten (1) | 14 | 66 | 66/66 | NA | NA | NA | 30/66 | 11/66 | 18/66 | NA | |||
| Shattuck (19) | 3 | 3 | 3/3 | NA | NA | NA | NA | 3/3 | NA | NA | |||
| Howell (20) | 3 | 7 | 7/7 | NA | NA | 0/7 | 0/7 | 3/7 | 0/7 | NA | |||
| Simonds (21) | 1 | 4 | 4/4 | 4/4 | 0/4 | 0/4 | 0/4 | 1/4 | 0/4 | NA | |||
| Cetani (22) | 2 | 4 | 4/4 | 3/4 | 1/4 | NA | 0/4 | 0/4 | 0/4 | NA | |||
| Villablanca (23) | 2 | 9 | 9/9 | 7/9 | 2/9 | 3/9 | 0/9 | 0/9 | 0/9 | NA | |||
| Cavaco (24) | 6 | 11 | 9/11 | 5/9* | 1/9* | 0/9 | 2/11 | 0/11 | 2/11 | NA | |||
| Howell (25) | 1 | 2 | 2/2 | 2/2 | 0/2 | 0/2 | 1/2 | 0/2 | NA | NA | |||
| Gimm (26) | 1 | 3 | 3 | NA | NA | 1/3 | NA | 1/3 | NA | NA | |||
| Bradley (3) | 2 | 11 | 9 | NA | NA | NA | 0/11 | 2/11 | 0/11 | 6/7 | |||
| Moon (27) | 1 | 2 | 2 | 2/2 | 0/2 | NA | 1/2 | 2/2 | NA | NA | |||
| Mizusawa (28) | 3 | 7 | 7/7 | 6/7** | NA** | 1/7 | 1/7 | 1/7 | 0/7 | 0/3 | |||
| Aldred (29) | 1 | 3 | 3/3 | 3/3 | 0/3 | 0/3 | 2/3 | 0/3 | NA | NA | |||
| Bradley (30) | 5 | 5 | 5/5 | 4/5 | 1/5 | NA | 2/5 | 0/5 | 0/5 | 1/4 | |||
| Juhlin (31) | 1 | 1 | 1/1 | 1/1 | 0/1 | NA | NA | 0/1 | NA | NA | |||
| Guarnieri (32) | 1 | 5 | 4/5 | 4/4 | 0/4 | 1/4 | NA | 1/5 | 0/4 | 2/3 | |||
| Kelly (33) | 1 | 2 | 2/2 | 1/2 | 1/2 | 2/2 | NA | 2/2 | NA | NA | |||
| Yamashita (34) | 1 | 1 | 1/1 | 1/1 | 0/1 | 0/1 | 1/1 | 0/1 | NA | NA | |||
| Cetani (35) | 1 | 1 | 1/1 | 1/1 | 0/1 | 1/1 | 0/1 | 0/1 | 0/1 | NA | |||
| Cetani (36) | 2 | 3 | 3/3 | NA | NA | NA | NA | 3/3 | NA | NA | |||
| Raue (37) | 1 | 2 | 2/2 | 1/2 | 1/2 | NA | 1/2 | 1/2 | NA | NA | |||
| Cetani (38) | 1 | 1 | 1/1 | 1/1 | 0/1 | NA | 0/1 | 1/1 | NA | NA | |||
| Sarquis (8) | 3 | 11 | 11/11 | 5/11 | 6/11 | 8/11 | 1/11 | 1/11 | 4/11 | 5/6 | |||
| Guarnieri (39) | 4 | 9 | 6/9 | 6/6 | 0/6 | 3/6 | 0/9 | 3/9 | 3/9 | NA | |||
| Howell (40) | 1 | 1 | 1/1 | 1/1 | 0/1 | 0/1 | NA | NA | NA | NA | |||
| Silveira (18) | 1 | 9 | 9/9 | 3/9 | 6/9 | 6/9 | 0/9 | 1/9 | 4/9 | 5/9 | |||
| Iacobone (5) | 3 | 17 | 16/17 | 15/16*** | 0/16*** | 3/16 | 1/17 | 1/17 | 1/17 | 8/13 | |||
| Rekik (41) | 1 | 1 | 1/1 | 1/1 | 0/1 | 0/1 | 1/1 | 0/1 | 0/1 | 1/1 | |||
| Panicker (42) | 1 | 6 | 5/6 | NA | NA | 0/5 | 1/6 | 0/6 | 0/6 | 1/2 | |||
| Cavaco (43) | 2 | 2 | 2/2 | 2/2 | 0/2 | 1/2 | 0/2 | 2/2 | 0/2 | 0/2 | |||
| Pichardo-Lowden (16) | 1 | 1 | 1/1 | 1/1 | 0/1 | 1/1 | 0/1 | 0/1 | 1/1 | NA | |||
| Domingues (44) | 1 | 1 | 1/1 | 1/1 | 0/1 | 0/1 | 0/1 | 0/1 | 0/1 | NA | |||
| Bricaire (45) | 15 | 13 | 12/13 | NA | NA | NA | 3/15 | 2/15 | 2/15 | 2/6 | |||
| Total | 87 | 224 | 213/224 | 78.7% | 21.3 | 29.5% | 23.5% | 18.7% | 17.4% | 50% | |||
| Present series | 7 | 16 | 16/16 | 11/16 (68.8%) | 5/16 (31.3%) | 4/16 (25.0%) | 2/16 (12.5%) | 6/16 (37.5%) | 3/16 (18.8%) | 2/6 (33.3%) | |||
Three patients with known PHPT were not operated on; pathology (gland no., histology) unknown.
Information concerning gland involvement not available for the patient with PHPT and parathyroid carcinoma.
One patient with known PHPT was not operated on; pathology (gland no., histology) unknown
Iacobone and colleagues recommended unilateral exploration with concordant preoperative imaging studies showing single-gland disease (5). This approach assumes that preoperative localization studies are accurate and have a low false-positive rate. However, in this cohort, the preoperative studies in patients with multiglandular disease were not as reliable. Preoperative localization studies were available for three out of the five patients with synchronous multiglandular involvement. The studies were accurate in two out of the three patients. The patient with inaccurate preoperative localization had an ultrasound and a Sestamibi scan showing only single gland disease, and upon exploration was found to have synchronous multiglandular disease. Given the high rate of multiglandular disease in these patients, their genetic predisposition, and the risk of reoperation, initial surgical management should include a bilateral neck exploration.
Although parathyroid carcinoma is a rare tumor causing less than one percent of cases of PHPT in the Western world, parathyroid carcinoma has been reported to be relatively common in patients with HPT-JT syndrome (13). Iacobone and colleagues reported that 11.8% of their cohort was diagnosed with parathyroid carcinoma and an extensive review of the literature by that study revealed that the prevalence of parathyroid carcinoma was 18.7% (5). In comparison, 37.5% of the patients in this cohort developed parathyroid carcinoma either at initial operation and/or during follow-up. Evidence from literature shows total calcium levels and the size of gland as potential markers of parathyroid carcinoma (14). No significant difference in gland size between benign parathyroid tumors and parathyroid carcinoma was found in this cohort. Total calcium levels were higher in patients with parathyroid carcinoma, but the findings were not statistically significant. Previous studies have reported high levels of total calcium in patients with parathyroid carcinoma associated with increased renal (56% nephrolithiasis and 84% renal insufficiency) and skeletal (greater than 40%) involvement compared to patients with benign primary hyperparathyroidism (less than 20% renal and less than 5% bone involvement)(15). In this series, patients with parathyroid carcinoma had less renal involvement (16.7%) and increased bone involvement (66.7%). Furthermore, one of the patients on initial operation had a 30 mm right inferior parathyroid adenoma and an unremarkable right superior parathyroid gland. Subsequently, the patient developed recurrent PHPT within two years and on reoperation was found to have a right superior parathyroid carcinoma. The high incidence of parathyroid carcinoma in this cohort indicates the need for surgical intervention in cases of even mild PHPT. For parathyroid glands suspicious for malignancy based on intraoperative findings such as large gland size, firm texture, grey/whitecolor and/or gross adherence to adjacent tissue, we recommend en-bloc resection of the tumor with a hemithyroidectomy.
Given their high susceptibility to parathyroid cancer, there is also clearly a need for longterm follow-up in patients with HPT-JT syndrome and frequent monitoring to detect the potential development of parathyroid carcinoma. Family members of affected patients should be offered genetic counseling and testing if a CDC73 germline mutation is identified. The youngest reported case of PHPT in a family diagnosed with HPT-JT syndrome is in a 7 year old child. Therefore, we recommend genetic screening within affected families at 5 years of age (16). For those found to be asymptomatic carriers, current recommendations include lifelong serum testing for biochemical evidence of PHPT every 6 to 12 months, with panoramic dental imaging and renal ultrasound at least every 5 years after identification of germline mutation. Asymptomatic female carriers should also have close gynecologic follow-up to address the risk of benign and malignant uterine tumors (17). Iacobone and colleagues advocate the use of parafibromin immunostaining as a first –line screening tool in cases of suspected familial non-MEN HPT, suggesting that it serves as a distinguishing feature of HPT-JT (5). However, in this series, immunostaining in two of nine adenoma cases (both with a substitution of a serine for tyrosine (p.Tyr55Ser)) showed preservation of nuclear parafibromin. Consequently, while parafibromin immunostaining may prove a cost-effective strategy for initial screening, physicians and researchers should be mindful of the significant risk for false-negative results. Given the high rate of recurrence and parathyroid carcinoma in HPT-JT patients, we advocate physicians proceed to genetic testing for HRPT2/CDC73 mutations despite parafibromin staining if clinical suspicion is high.
Previous studies have reported a high rate of PHPT recurrence in patients with the HRPT2/CDC73 germline mutation. A recent study by Silveira and colleagues showed a high rate of recurrence (4/9) and/or persistent (2/9) PHPT. The authors speculated that aggressive treatment may be warranted and genotype-phenotype data may be able to direct the surgeon (18). The recurrence rate of PHPT in this cohort was 25.0%. Of the four patients who had recurrent disease, all four patients were ultimately diagnosed with parathyroid carcinoma and had either whole gene deletion of CDC73 or duplication of two nucleotides (c.687_688dupAG) in exon 7 resulting in a frameshift mutation. Unfortunately, the small sample size did not allow sufficient power to correlate between the type of genetic mutation and phenotypic expression of parathyroid carcinoma. Furthermore, a literature review showed that the rate of recurrence of PHPT was 14.2%. Although the low rate of recurrence may lead the clinician to offer a less aggressive initial operation, given the risk of recurrence and parathyroid carcinoma, patients would likely benefit from a more complete exploration and parathyroidectomy in the initial setting of any abnormal glands.
The type of operation did not influence whether the patient had a longer disease-free interval. Immunohistochemistry from a single patient undergoing a subtotal parathyroidectomy comparing a normal parathyroid gland and an abnormal parathyroid adenoma showed discordant results. The abnormal parathyroid gland showed absence of nuclear staining, while positive parafibromin nuclear staining was identified in the unaffected gland. Postoperatively, this patient subsequently developed permanent hypoparathyroidism. Given these findings, routine subtotal or total parathyroidectomy confers no benefit and likely leads to increased risk of permanent hypoparathyroidism. Furthermore, total parathyroidectomy with autotransplantation may also allow the seeding and dissemination of parathyroid cancer cells via the autotransplantation.
HPT-JT syndrome is a rare genetic disorder resulting in PHPT, a high likelihood of multiglandular disease, and a high risk for parathyroid carcinoma. Given the genetic predisposition and high cancer risk, bilateral neck exploration to assess all four parathyroid glands with en-bloc resection of enlarged glands suspicious for cancer would serve these patients best.
Acknowledgments
We thank Drs. Wang and Mietinenn for their assistance with the immunohistochemistry staining. We also thank the physicians and nurses who participated in the clinical care of these patients, and the patients and their families.
Financial Support: This research was supported by the intramural research programs of the Center for Cancer Research, National Cancer Institute, National Institutes of Health and the National Institute of Digestive and Diabetes and Kidney Diseases; as well as the National Institutes of Health (NIH) Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH from Pfizer Inc, The Doris Duke Charitable Foundation, The Alexandria Real Estate Equities, Inc. and Mr. and Mrs. Joel S. Marcus, and the Howard Hughes Medical Institute, as well as other private donors. For a complete list, please visit the Foundation website at: http://fnih.org/work/education-training-0/medical-research-scholars-program
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002 Dec;32(4):676–80. doi: 10.1038/ng1048. Research Support, Non-U.S. Gov't, Research Support, U.S. Gov't, P.H.S. [DOI] [PubMed] [Google Scholar]
- 2.Arnold A, Marx SJ. Familial hyperparathyroidism (including MEN, FHH, and HPT-JT) In: Cea Rosen., editor. Primer on the Metabolic Bone Diseases and Mineral Metabolism. 8. John Wiley & Sons, Inc; 2013. [Google Scholar]
- 3.Bradley KJ, Hobbs MR, Buley ID, Carpten JD, Cavaco BM, Fares JE, et al. Uterine tumours are a phenotypic manifestation of the hyperparathyroidism-jaw tumour syndrome. J Intern Med. 2005 Jan;257(1):18–26. doi: 10.1111/j.1365-2796.2004.01421.x. Research Support, Non-U.S. Gov't, Research Support, U.S. Gov't, P.H.S. [DOI] [PubMed] [Google Scholar]
- 4.Yart A, Gstaiger M, Wirbelauer C, Pecnik M, Anastasiou D, Hess D, et al. The HRPT2 tumor suppressor gene product parafibromin associates with human PAF1 and RNA polymerase II. Mol Cell Biol. 2005 Jun;25(12):5052–60. doi: 10.1128/MCB.25.12.5052-5060.2005. Research Support, Non-U.S. Gov't. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iacobone M, Masi G, Barzon L, Porzionato A, Macchi V, Ciarleglio FA, et al. Hyperparathyroidism-jaw tumor syndrome: a report of three large kindred. Langenbecks Arch Surg. 2009 Sep;394(5):817–25. doi: 10.1007/s00423-009-0511-y. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 6.Gill AJ, Clarkson A, Gimm O, Keil J, Dralle H, Howell VM, et al. Loss of nuclear expression of parafibromin distinguishes parathyroid carcinomas and hyperparathyroidismjaw tumor (HPT-JT) syndrome-related adenomas from sporadic parathyroid adenomas and hyperplasias. Am J Surg Pathol. 2006 Sep;30(9):1140–9. doi: 10.1097/01.pas.0000209827.39477.4f. [DOI] [PubMed] [Google Scholar]
- 7.Masi G, Barzon L, Iacobone M, Viel G, Porzionato A, Macchi V, et al. Clinical, genetic, and histopathologic investigation of CDC73-related familial hyperparathyroidism. Endocr Relat Cancer. 2008 Dec;15(4):1115–26. doi: 10.1677/ERC-08-0066. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 8.Sarquis MS, Silveira LG, Pimenta FJ, Dias EP, Teh BT, Friedman E, et al. Familial hyperparathyroidism: surgical outcome after 30 years of follow-up in three families with germline HRPT2 mutations. Surgery. 2008 May;143(5):630–40. doi: 10.1016/j.surg.2007.12.012. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 9.Barry MK, van Heerden JA, Grant CS, Thompson GB, Khosla S. Is familial hyperparathyroidism a unique disease? Surgery. 1997 Dec;122(6):1028–33. doi: 10.1016/s0039-6060(97)90205-1. [DOI] [PubMed] [Google Scholar]
- 10.Huang SM, Duh QY, Shaver J, Siperstein AE, Kraimps JL, Clark OH. Familial hyperparathyroidism without multiple endocrine neoplasia. World J Surg. 1997 Jan;21(1):22–8. doi: 10.1007/s002689900188. Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, Non-P.H.S. discussion 9. [DOI] [PubMed] [Google Scholar]
- 11.DeLellis RA, L R, Heitz PU, Heng C. World Health Organization Classification of Tumours Pathology and Genetics: tumours of endocrine organs. IARC; Lyon: 2004. [Google Scholar]
- 12.Schantz A, Castleman B. Parathyroid carcinoma. A study of 70 cases. Cancer. 1973 Mar;31(3):600–5. doi: 10.1002/1097-0142(197303)31:3<600::aid-cncr2820310316>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 13.DeLellis RA. Parathyroid carcinoma: an overview. Adv Anat Pathol. 2005 Mar;12(2):53–61. doi: 10.1097/01.pap.0000151319.42376.d4. Review. [DOI] [PubMed] [Google Scholar]
- 14.Obara T, Okamoto T, Kanbe M, Iihara M. Functioning parathyroid carcinoma: clinicopathologic features and rational treatment. Semin Surg Oncol. 1997 Mar-Apr;13(2):134–41. doi: 10.1002/(sici)1098-2388(199703/04)13:2<134::aid-ssu9>3.0.co;2-a. Review. [DOI] [PubMed] [Google Scholar]
- 15.Mittendorf EA, McHenry CR. Parathyroid carcinoma. J Surg Oncol. 2005 Mar 1;89(3):136–42. doi: 10.1002/jso.20182. [DOI] [PubMed] [Google Scholar]
- 16.Pichardo-Lowden AR, Manni A, Saunders BD, Baker MJ. Familial hyperparathyroidism due to a germline mutation of the CDC73 gene: implications for management and age-appropriate testing of relatives at risk. Endocr Pract. Jul-Aug;17(4):602–9. doi: 10.4158/EP10337.RA. [DOI] [PubMed] [Google Scholar]
- 17.Rich TA, Hu MI, Martin JW, Perrier ND, Waguespack SG. CDC73-Related Disorders. 1993 [PubMed] [Google Scholar]
- 18.Silveira LG, Dias EP, Marinho BC, Gomez RS, De Marco L, Sarquis MS. HRPT2-related familial isolated hyperparathyroidism: could molecular studies direct the surgical approach? Arq Bras Endocrinol Metabol. 2008 Nov;52(8):1211–20. doi: 10.1590/s0004-27302008000800003. Case Reports] [Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 19.Shattuck TM, Valimaki S, Obara T, Gaz RD, Clark OH, Shoback D, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003 Oct 30;349(18):1722–9. doi: 10.1056/NEJMoa031237. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 20.Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, Chen J, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003 Sep;40(9):657–63. doi: 10.1136/jmg.40.9.657. Research Support, Non-U.S. Gov't. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simonds WF, Robbins CM, Agarwal SK, Hendy GN, Carpten JD, Marx SJ. Familial isolated hyperparathyroidism is rarely caused by germline mutation in HRPT2, the gene for the hyperparathyroidism-jaw tumor syndrome. J Clin Endocrinol Metab. 2004 Jan;89(1):96–102. doi: 10.1210/jc.2003-030675. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 22.Cetani F, Pardi E, Borsari S, Viacava P, Dipollina G, Cianferotti L, et al. Genetic analyses of the HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. J Clin Endocrinol Metab. 2004 Nov;89(11):5583–91. doi: 10.1210/jc.2004-0294. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 23.Villablanca A, Calender A, Forsberg L, Hoog A, Cheng JD, Petillo D, et al. Germline and de novo mutations in the HRPT2 tumour suppressor gene in familial isolated hyperparathyroidism (FIHP) J Med Genet. 2004 Mar;41(3):e32. doi: 10.1136/jmg.2003.012369. Research Support, Non-U.S. Gov't. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cavaco BM, Guerra L, Bradley KJ, Carvalho D, Harding B, Oliveira A, et al. Hyperparathyroidismjaw tumor syndrome in Roma families from Portugal is due to a founder mutation of the HRPT2 gene. J Clin Endocrinol Metab. 2004 Apr;89(4):1747–52. doi: 10.1210/jc.2003-031016. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 25.Howell VM, Zori RT, Stalker HJ, Williams C, Jesse N, Nelson AE, et al. A molecular diagnosis of hyperparathyroidism-jaw tumor syndrome in an adolescent with recurrent kidney stones. J Pediatr. 2004 Oct;145(4):567. doi: 10.1016/j.jpeds.2004.04.023. Case Reports] [Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 26.Gimm O, L K, Nguyen Thanh P, et al. Das Familiäre Nebenschilddrüsenkarzinom. Indikation zur prophylaktischen Parathyreoidektomie? Chirurg. 2006;77:15–24. doi: 10.1007/s00104-005-1110-2. [DOI] [PubMed] [Google Scholar]
- 27.Moon SD, Park JH, Kim EM, Kim JH, Han JH, Yoo SJ, et al. A Novel IVS2-1G>A mutation causes aberrant splicing of the HRPT2 gene in a family with hyperparathyroidism-jaw tumor syndrome. J Clin Endocrinol Metab. 2005 Feb;90(2):878–83. doi: 10.1210/jc.2004-0991. Case Reports] [Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 28.Mizusawa N, Uchino S, Iwata T, Tsuyuguchi M, Suzuki Y, Mizukoshi T, et al. Genetic analyses in patients with familial isolated hyperparathyroidism and hyperparathyroidism-jaw tumour syndrome. Clin Endocrinol (Oxf) 2006 Jul;65(1):9–16. doi: 10.1111/j.1365-2265.2006.02534.x. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 29.Aldred MJ, Talacko AA, Savarirayan R, Murdolo V, Mills AE, Radden BG, et al. Dental findings in a family with hyperparathyroidism-jaw tumor syndrome and a novel HRPT2 gene mutation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006 Feb;101(2):212–8. doi: 10.1016/j.tripleo.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 30.Bradley KJ, Cavaco BM, Bowl MR, Harding B, Cranston T, Fratter C, et al. Parafibromin mutations in hereditary hyperparathyroidism syndromes and parathyroid tumours. Clin Endocrinol (Oxf) 2006 Mar;64(3):299–306. doi: 10.1111/j.1365-2265.2006.02460.x. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 31.Juhlin C, Larsson C, Yakoleva T, Leibiger I, Leibiger B, Alimov A, et al. Loss of parafibromin expression in a subset of parathyroid adenomas. Endocr Relat Cancer. 2006 Jun;13(2):509–23. doi: 10.1677/erc.1.01058. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 32.Guarnieri V, Scillitani A, Muscarella LA, Battista C, Bonfitto N, Bisceglia M, et al. Diagnosis of parathyroid tumors in familial isolated hyperparathyroidism with HRPT2 mutation: implications for cancer surveillance. J Clin Endocrinol Metab. 2006 Aug;91(8):2827–32. doi: 10.1210/jc.2005-1239. Case Reports] [Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 33.Kelly TG, Shattuck TM, Reyes-Mugica M, Stewart AF, Simonds WF, Udelsman R, et al. Surveillance for early detection of aggressive parathyroid disease: carcinoma and atypical adenoma in familial isolated hyperparathyroidism associated with a germline HRPT2 mutation. J Bone Miner Res. 2006 Oct;21(10):1666–71. doi: 10.1359/jbmr.060702. Case Reports] [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 34.Yamashita Y, Akiyama T, Mizusawa N, Yoshimoto K, Goto M. A case of hyperparathyroidism-jaw tumour syndrome found in the treatment of an ossifying fibroma in the maxillary bone. Int J Oral Maxillofac Surg. 2007 Apr;36(4):365–9. doi: 10.1016/j.ijom.2006.08.007. Case Reports. [DOI] [PubMed] [Google Scholar]
- 35.Cetani F, Pardi E, Ambrogini E, Viacava P, Borsari S, Lemmi M, et al. Different somatic alterations of the HRPT2 gene in a patient with recurrent sporadic primary hyperparathyroidism carrying an HRPT2 germline mutation. Endocr Relat Cancer. 2007 Jun;14(2):493–9. doi: 10.1677/ERC-06-0092. Case Reports] [Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 36.Cetani F, Ambrogini E, Viacava P, Pardi E, Fanelli G, Naccarato AG, et al. Should parafibromin staining replace HRTP2 gene analysis as an additional tool for histologic diagnosis of parathyroid carcinoma? Eur J Endocrinol. 2007 May;156(5):547–54. doi: 10.1530/EJE-06-0720. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 37.Raue F, Haag C, Frank-Raue K. Hyperparathyroidism-jaw tumor syndrome. A hereditary form of primary hyperparathyroidism with parathyroid carcinoma. Dtsch Med Wochenschr. 2007 Jul 29;132(27):1459–62. doi: 10.1055/s-2007-982052. Case Reports. [DOI] [PubMed] [Google Scholar]
- 38.Cetani F, Pardi E, Ambrogini E, Banti C, Viacava P, Borsari S, et al. Hyperparathyroidism 2 gene (HRPT2, CDC73) and parafibromin studies in two patients with primary hyperparathyroidism and uncertain pathological assessment. J Endocrinol Invest. 2008 Oct;31(10):900–4. doi: 10.1007/BF03346439. Case Reports] [Research Support, Non-U.S. Gov't. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guarnieri V, Bisceglia M, Bonfitto N, Cetani F, Marcocci C, Minisola S, et al. Re: Familial hyperparathyroidism: surgical outcome after 30 years of follow-up in three families with germline HRPT2 mutations. Surgery. 2008 Nov;144(5):839–40. doi: 10.1016/j.surg.2008.08.008. Case Reports, Comment Letter] [Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 40.Howell VM, Gill A, Clarkson A, Nelson AE, Dunne R, Delbridge LW, et al. Accuracy of combined protein gene product 9.5 and parafibromin markers for immunohistochemical diagnosis of parathyroid carcinoma. J Clin Endocrinol Metab. 2009 Feb;94(2):434–41. doi: 10.1210/jc.2008-1740. Evaluation Studies Multicenter Study] [Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 41.Rekik N, Ben Naceur B, Mnif M, Mnif F, Mnif H, Boudawara T, et al. Hyperparathyroidism-jaw tumor syndrome: a case report. Ann Endocrinol (Paris) 2010 Mar;71(2):121–6. doi: 10.1016/j.ando.2009.09.004. Case Reports. [DOI] [PubMed] [Google Scholar]
- 42.Panicker LM, Zhang JH, Dagur PK, Gastinger MJ, Simonds WF. Defective nucleolar localization and dominant interfering properties of a parafibromin L95P missense mutant causing the hyperparathyroidism-jaw tumor syndrome. Endocr Relat Cancer. 2010 Jun;17(2):513–24. doi: 10.1677/ERC-09-0272. Research Support, N.I.H., Intramural. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cavaco BM, Santos R, Felix A, Carvalho D, Lopes JM, Domingues R, et al. Identification of de novo germline mutations in the HRPT2 gene in two apparently sporadic cases with challenging parathyroid tumor diagnoses. Endocr Pathol. 2011 Mar;22(1):44–52. doi: 10.1007/s12022-011-9151-1. Case Reports. [DOI] [PubMed] [Google Scholar]
- 44.Domingues R, Tomaz RA, Martins C, Nunes C, Bugalho MJ, Cavaco BM. Identification of the first germline HRPT2 whole-gene deletion in a patient with primary hyperparathyroidism. Clin Endocrinol (Oxf) 2012 Jan;76(1):33–8. doi: 10.1111/j.1365-2265.2011.04184.x. Case Reports] [Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 45.Bricaire L, Odou MF, Cardot-Bauters C, Delemer B, North MO, Salenave S, et al. Frequent large germline HRPT2 deletions in a French National cohort of patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2013 Feb;98(2):E403–8. doi: 10.1210/jc.2012-2789. [DOI] [PubMed] [Google Scholar]