

Abstract
Modular protein interaction domains that recognize linear peptide motifs are found in hundreds of proteins within the human genome. Some protein interaction domains such as SH2, 14-3-3, Chromo and Bromo domains serve to recognize post-translational modification of amino acids (such as phosphorylation, acetylation, methylation etc.) and translate these into discrete cellular responses. Other modules such as SH3 and PDZ domains recognize linear peptide epitopes and serve to organize protein complexes based on localization and regions of elevated concentration. In both cases, the ability to nucleate specific signaling complexes is in large part dependent on the selectivity of a given protein module for its cognate peptide ligand. High throughput analysis of peptide-binding domains by peptide or protein arrays, phage display, mass spectrometry or other HTP techniques provides new insight into the potential protein-protein interactions prescribed by individual or even whole families of modules. Systems level analyses have also promoted a deeper understanding of the underlying principles that govern selective protein-protein interactions and how selectivity evolves. Lastly, there is a growing appreciation for the limitations and potential pitfalls of high-throughput analysis of protein-peptide interactomes. This review will examine some of the common approaches utilized for large-scale studies of protein interaction domains and suggest a set of standards for the analysis and validation of datasets from large-scale studies of peptide-binding modules. We will also highlight how data from large-scale studies of modular interaction domain families can provide insight into systems level properties such as the linguistics of selective interactions.
1. Introduction
The ability of every cell to respond to extrinsic and intrinsic cues depends upon the coordinated association and disassociation of multi-protein complexes [1]. Cellular communication thus relies upon a complex network of transient protein-protein interactions. Proteins involved in this intricate dance of cellular signaling tend to be modular in nature, being composed of multiple independently folding domains that convey either a catalytic function or act to recognize other proteins and thereby promote the formation of transient signaling complexes [2]. Among the large and growing class of modular protein interaction domains (PIDs) are a sizable fraction that recognize short motifs within larger polypeptides. In most cases, binding to a linear peptide motif results in a relatively small contact interface of sufficient affinity to promote assembly without sacrificing rapid reversibility. Secondary contacts promote specificity as well as increased stability of the complex which may be necessary for sustained signaling. Nonetheless, association between a modular interaction domain and its cognate peptide ligand, usually in the nanomolar to low micromolar range, is generally a necessary prerequisite event in complex formation. Thus, understanding the selectivity of these interactions is invaluable for assembling potential interactomes and mapping signaling networks. The advent of genome sequencing has provided extensive sets of PIDs and lead in turn to efforts to map the interactions of large families of domains. In particular, the SH2, PTB, 14-3-3, PDZ, and SH3 domains have been the subject of a variety of large-scale studies aimed at identifying potential interacting partners and discovering the motifs and engineering principals that underlie selective interactions. The approaches used include phage display, peptide libraries and arrays, protein microarrays, and mass-spectrometry approaches. This review aims to provide a brief overview of modular peptide-binding domains; outline the strengths and limitations of various high-throughput approaches to determining interactions; suggest a common set of principals when analyzing and validating HTP datasets; and highlight how these studies can yield novel insights regarding the underlying language of selective peptide recognition, the evolution of specificity and the scope of the potential cellular interactome.
2. Why study protein-peptide interactions?
The discovery of the Src homology 2 (SH2) domain by the Pawson and Hanafusa labs in the mid-1980’s revealed that proteins commonly contain independently folding domains of conserved sequence that allowed selective non-catalytic interactions with other proteins [3-5]. The decades since this paradigm-shift finding have seen the identification of thousands of conserved sequence regions cataloged in databases such as PFam [6], SMART [7], and CDD [8]. Dozens of these have been described as specific modular protein interactions domains (PIDs) [9, 10]. The ability to study PIDs in isolation from their host proteins is the foundation of an extremely powerful reductionist approach that underlies much of our current understanding of protein function and the protein interaction networks that control much of cell biology, including signal transduction. Reductionist approaches inherently sacrifice contextual breadth for pointed confidence. By abstracting the biological context of the full-length protein and its multitude of inter-related interactions that occur within the crowded and physicochemically diverse milieu of the cell down to the level of an isolated PID interacting with short peptides in order to confidently assess an interaction, we can presume that much contextual information is lost. This is balanced against the ability to control for the precise nature of the interacting partners and manipulate them at will to tease apart molecular mechanisms. In addition, there are purely practical advantages relating to the ability to biophysically measure interactions directly and quantitatively that favor reductionism. The relevancy of the reductionist approach to study PIDs has been proven time and again in their ability to explain in vivo behavior of proteins in signal transduction networks. Indeed, given the complex issues associated with more “physiological” experiments, it is not surprising that much of our mechanistic understanding of protein-protein interactions is based upon reductionist approaches. Both the design and interpretation of cell-based experiments commonly relies upon in vitro studies of the component pieces.
2.1 History and importance of peptide binding protein modules
As noted above, the SH2 domain was among the first protein-interaction modules to be described in detail [3, 5]. In many ways, the SH2 domain remains archetypal of the large and growing group of protein-protein interactions domains and so it is worth briefly reviewing the general properties of this module. The SH2 domain is an approximately 100 amino acid residue domain that displays a recognizable pattern of conserved amino acids and a relatively conserved structural fold [11]. Early on, it was recognized that the SH2 domain sequence is conserved among a number of oncoproteins such as Src, Abl and Crk [5, 12] involved in tyrosine kinase signaling. This led to the realization that SH2 domains could bind to specific phosphotyrosine (pY)–containing peptide motifs [13, 14]. We now know that the human genome encodes a total of 121 SH2 domains in 111 proteins and that the SH2 domain is the primary mediator for pTyr signaling in animal cells [15, 16]. Together with a subset of PTB domains and few isolated other protein domains, the SH2 domains function to couple tyrosine phosphorylation to coordinate formation of multi-protein complexes involved in signal transduction. Specificity in recognizing pTyr peptide motifs is thus crucial for high fidelity signaling. Mutations in SH2 domain containing proteins are associated with a large number of human diseases including cancer, diabetes and immune disorders [15, 17]. SH2 domains have thus been tapped as molecular tools for profiling disease states, particularly in human cancers [18, 19].
A wide variety of modular PIDs have been identified in the quarter century since the identification of the SH2 domain. More than 70% of the human proteome contains at least one identifiable domain of some sort [20]. At this time, approximately a hundred specialized PIDs have been identified that recognize a plethora of motifs [10, 21]. Modular PIDs can regulate a multitude of cellular processes from assembly of protein complexes to selectively regulating enzymatic activity, allosteric regulation of protein function, proper targeting of protein localization and orientation of protein configuration [1, 22]. Domains can be subclassed according to the various ligands present in the cell as binding to post-translational modifications (PTMs), short linear peptide motifs, phospholipids or DNA (Figure 1). In this review we focus only on those domains that recognize linear peptide motifs including those that bind specific PTMs such as protein phosphorylation, methylation and acetylation.
Figure 1. Modular Protein Interaction Domains recognize short linear peptide motifs.
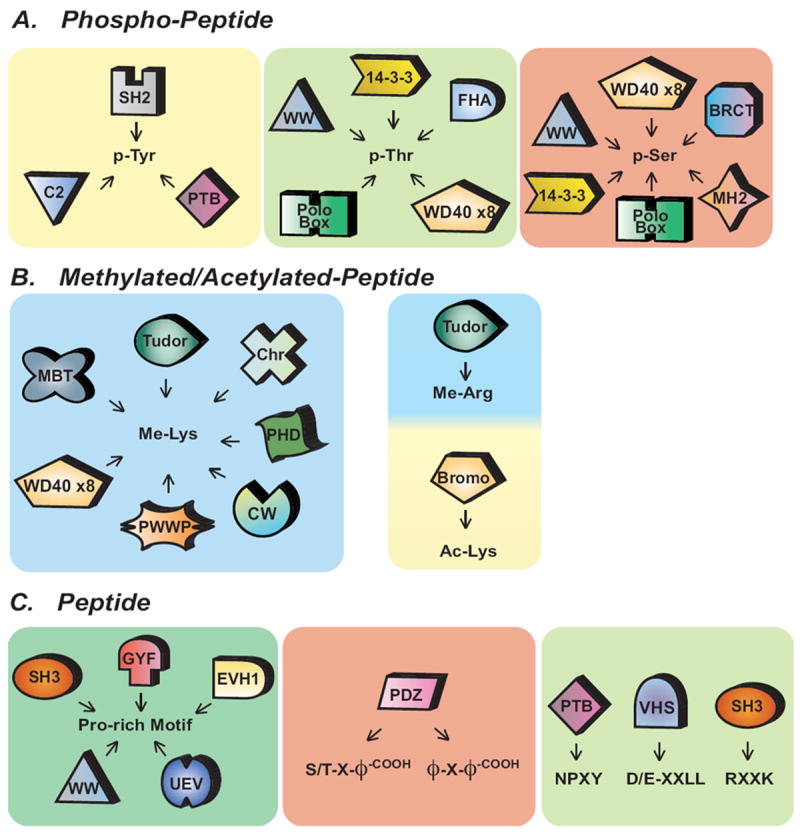
(A) Phosphorylation on serines (pSer), threonines (pThr), and tyrosines (pTyr) can be recognized by a subset of specialized PIDs. (B) Post-translational modifications on lysine by methylation (Me-Lys) mediate interactions with a large set of interaction modules whereas lysine acetylation (Ac-Lys) and arginine methylation (Me-Arg) are restricted to only Bromo and Tudor domains, respectively. (C) Modular PIDs that recognize short linear motifs. (X, represents any natural amino; ϕ, hydrophobic amino acids; -COOH, carboxyl terminus of amino acids)
2.2 PID basics
While protein domains can have enzymatic properties such as kinases, phosphatases, GTPases and methylases, a large number of domains mediate protein-protein interactions and are thus termed protein interaction domains (PIDs). Among the PIDs, a number of modules are defined to recognize linear peptide sequences including those that specifically recognize PTMs [2] (Figure 1). Such PIDs convey interactions that may provide localization cues, allosteric regulation, and coordinate the formation of multi-protein complexes that function as molecular machines [23]. Domains are often duplicated throughout evolution and undergo evolutionary divergence that drives functional adaptation (reviewed by Chothia and Gough)[24]. Clustering by domain organization provides insight into the development of organismal complexity and specialization of tissue types that is a hallmark of evolved complexity in animals [25].
Post-translation modifications such as protein phosphoryation on the hydroxyl side chains of tyrosine, serine and threonine residues serve to distinguish docking sites for PIDs such as the SH2, 14-3-3, and a subset of PTB, Polo Box, BRCT, FHA and WW domains (Figure 1A). While a majority of SH2, 14-3-3, Polo-Box, BRCT and FHA are dedicated phospho-binding domains [26], it is worth remembering that PIDs are quite adaptable. This is clear if we consider PTB and WW domains, which are by no means exclusive phospho-binding domains and can in many instances bind to phospho-independent ligands (Figures 1A,C). As we will discuss later, this is important to keep in mind in designing HTP studies of ligand specificity as experimental constraints may ignore potential ligand space. Other PTMs on linear peptides including acetylation and methylation similarly have domains dedicated to their recognition. Acetylation on lysine residues and methylation on both lysine and arginine residues function as marks for coordinating complex formation on histones and piRNA complexes [27]. These PTM marks can be recognized by the subset of domains including CW, Chromo, MBT, Tudor, and Bromo domains (Figure 1B). Other common PTMs, not directly discussed in this review, such as lysine ubiquitylation and sumoylation, are also important for numerous cellular processes and are recognized by a large subset of selective PIDs though generally in a manner that does not identify the substrate peptide to which they are anchored (reviewed in [28, 29]). Other types of PTMs including lysine succinylation, crotonylation, and malonylation have been reported, though it remains to be seen whether there are specific PIDs that recognize these modifications [30-32].
Linear peptide motifs that do not depend upon PTMs are also commonly utilized to coordinate protein-protein interactions. Such short linear peptides sequences of are commonly found in exposed unstructured regions between domains or in loops within folded domains [33]. Domains that recognize these short linear peptides include the Src-homology 3 (SH3), WW, glycine-tyrosine-phenylalanine (GYF), and PSD-95/Discs-large/ZO-1 (PDZ) domains (Figure 1C). As noted above, PIDs often display remarkable plasticity in terms of their cognate ligands. For example, SH3 domains generally bind to Pro-rich peptides [34], but specific SH3 domains have evolved to interact with quite divergent peptide motifs (such as R-x-x-K, where x represents any natural amino acid) [35], or even with the surface of ubiquitin [36]. Similarly, PDZ domains generally bind to C-terminal peptide motifs [37] but in certain cases also recognize specific phospholipids. For example, the PDZ domains (PDZ1 and PDZ2) of syntenin-1 cooperate to support PtdIns-4,5-P2 binding and membrane localization [38, 39]. Reductionist approaches have allowed the detailed biochemical study of domains, with extensive use of techniques such as affinity purification by GST pull-down or co-immunoprecipitation, phage display and crystallographic studies to define interactions. Thus it was recognized early on that SH3 domains generally bind to Pro-rich peptides that form a left-handed poly-proline type II helix, with the minimal consensus Pro-x-x-Pro. Each Pro is usually preceded by an aliphatic residue. Each pair of aliphatic-Pro pairs residues binds within a hydrophobic pocket on the surface of the SH3 domain. Early studies recognized two major classes of SH3 domain ligands: Class I and Class 2 ligands, which recognize RK-x-x-P-x-x-P and P-x-x-P-x-R motifs, respectively. This view of SH3 domains was expanded with the recognition of a small subclass of SH3 domains that recognize RxxK motifs while other SH3 domains recognize KKPP or PxxDY [40, 41] and at least one SH3 domain binds to the ubiquitin polypeptide [36]. Some studies were biased by the assumption that SH3 domains would prefer PxxP-containing peptide ligands and the need to constrain library variability and thus used peptides centered around a conserved PxxP motif to define SH3 specificity [42], potentially overlooking novel binding modalities. While the results of these studies remain informative, and reducing ligand complexity is often necessary to experimental design, such examples serve as a caution that with reductionist approaches it is easy to oversimplify assumptions regarding ligand preference.
Protein domains generally select their ligands through an exposed surface interface that recognizes one or more primary determinants, with flanking residues creating additional contacts that provide the element of selectivity. For example, a near universal feature of SH2 domains is phosphorylated tyrosine (pTyr or pY) recognition accommodated by a conserved pocket on the SH2 domain in which an arginine residue that coordinates the phosphate. Specificity of one SH2 domain over another is determined through the residues that surround the pTyr residue, particularly those immediately C-terminal at positions +1 to +5. Thus, the SH2 domain of Crk, recognizes a core motif pY-x-x-P/L [43, 44]. Additional contacts expand this to pY-x-V-L/P-R/K. Other domains that recognize phosphorylated motifs, including 14-3-3 [45], WW [46], FHA [47], and PTB [48]domains, similarly recognize adjacent residues to allow discrimination between specific phosphorylated sites. Despite inherent flexibility in ligand preference, the ability to classify domains according to relatively specific motifs that define binding preference suggests that specificity, while flexible, is generally limited by biological function. Thus, large scale or systems-level studies may provide sufficiently detailed information on specificity preferences to determine generalized rules of specificity as well as a necessary level of understanding of the linguistics of contextual ligand selectivity.
While PIDs have a remarkable ability to recognize their cognate peptide ligands, the transient nature of protein domain-peptide interactions implies that these modules must perform something of a balancing act. An individual domain may potentially engage several distinct ligands, either simultaneously or at distinct points in a complex spatiotemporal signaling matrix (Figure 2A, E). Likewise, an individual peptide ligand may potentially engage multiple domains at various points in time and space (Figure 2A, D). Thus, the affinity for a particular binding site must presumably not be so high that binding becomes essentially irreversible over the time spans of protein-mediated and enzymatic signaling events. Lower affinity interactions may actually be crucial for achieving selectivity by allowing the rapid sampling of multiple ligands as well as to allow spatiotemporal organization of signaling networks to be achieved. The equilibrium dissociation constants for domain-peptide interactions with their cognate physiological ligands typically fall in the range of low micromolar to nanomolar affinities. At the tighter end of this are often domain-peptide interactions that are regulated through PTMs. In addition to promoting direct binding of a peptide to a domain, PTMs can also be used to inhibit interactions. So while SH2, FHA, 14-3-3, Chromo, Bromo and other domains do not bind stably until the peptide has acquired an appropriate PTM, binding of certain SH3, PDZ and other domains is negatively regulated by phosphorylation of the peptide ligand. For example, the interaction of the NR2B subunit of the NMDA receptor with PSD-95 is negatively modulated by phosphorylation of the PDZ ligand at the -2 position serine residue by CK2 [49]. Likewise, PTMs on PIDs can also destabilize or disrupt peptide interactions. Thus SAP-97 directly associates with NR2A through its PDZ1 domain, and phosphorylation of Ser-232 in SAP-97 by CaMKII disrupts NR2A interaction both in vitro and in vivo [50]. In a related manner phosphorylation of the tandem SH2 domains of p85-alpha (PI3K1), by the phorbol ester stimulated PKC, at conserved positions S361 and S652 within the pTyr binding pocket of the SH2 domain results in repulsion of the negatively charged pTyr peptide [51]. Such complexity is not limited to simple Michaelian interactions and allows binary protein-protein interactions to display emergent properties such as ultrasensitivity. The WD40 repeat module of Cdc4 interacts with multiple low affinity peptide binding sites on Sic1 in a such a manner that a threshold of at least 6 out of 9 sites must be phosphorylated and available for binding in order to promote an interaction of sufficient stability to allow both a detectable biochemical interactions and the biological outcome of ubiquitination [52, 53]. Multiple sampling of low affinity sites may in fact be a general mechanism for promoting switch-like binding events [54]. Such complexity is not easily captured using HTP methods. Low affinity interactions, multiple PTMs, negative regulatory effects and emergent properties present particular challenges and it is important to keep these caveats in mind both in the design and interpretation of HTP studies as well as in the use of HTP data sets for modeling complex cellular events.
Figure 2. Protein domain interactions in time and space.
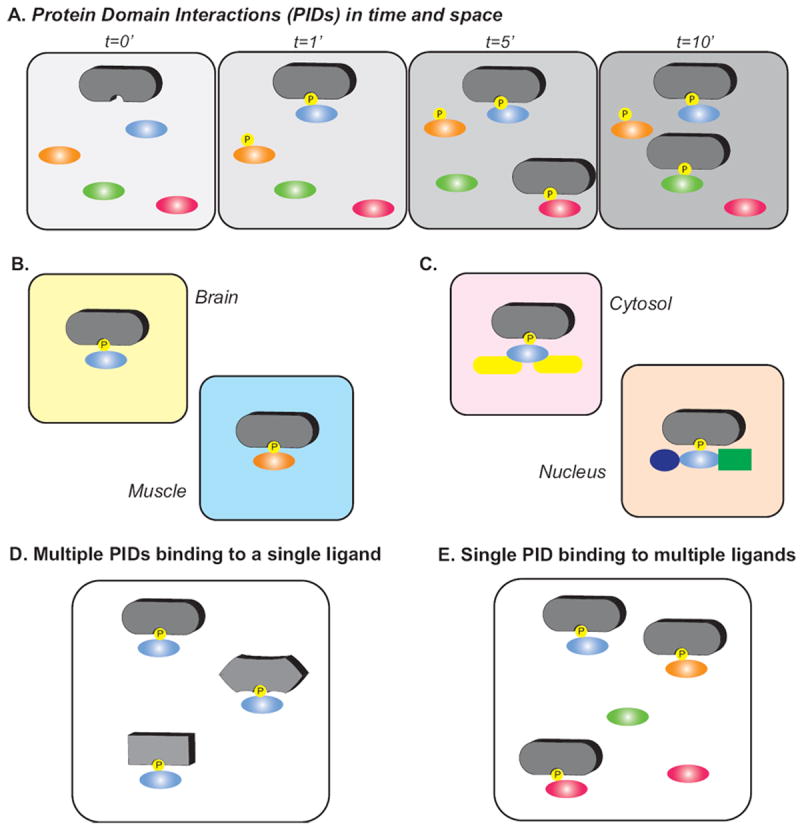
(A) Protein interactions domains (PIDs) (gray shapes) can coordinate multiple different interactions partners (oval shapes) in a temporal manner. For example, temporal phosphorylation of substrates by protein kinases may influence the time dependent protein domain interactions. The cellular context plays an important role in determine PIDs in specific cell types with co-expressed ligands (B) and spatial localization within the cell (C). Multiple PIDs can bind a single ligand (D) and vice versa, a single PID can recognize multiple ligands as PIDs coordinate transient protein-protein interactions (E).
2.3 PID-Peptide Interactions Direct Information Flow
Peptide binding protein modules or PIDs recognize unique peptide motifs that impart the primary level of interaction specificity by constraining potential interaction partners. For example, the SH2 domain recognizes phosphorylated tyrosine residues (pY) in the context of a stretch of adjacent amino acids (usually C terminal) to the pY, depicted as pY-x-x-x-x-x. There are generally multiple members of a given module class and in most cases multiple peptide motifs that can bind to a given PID. Even simple motifs may comprise thousands of potential binding peptides. There are, for instance, 8000 unique sequences within the three variable positions of the motif pY-x-x-x. Individual PID-peptide interactions therefore face the dual challenge of achieving biologically relevant affinities and some measure of uniqueness, or specificity relative to other module-motif interactions within the same family in order to direct information flow within a cell.
Protein modules that recognize short peptide sequences differ from other protein-protein interactions in that they typically involve relatively small interfacial contact areas. A recent analysis of a non-redundant set of protein-peptide interactions confirmed that the change in accessible surface area (ΔASA) upon binding was half that of protein-protein interactions and almost a third of IUP-protein interactions [55]. Given that a) at the level of individual amino acids a large ΔASA seems to be necessary for a large contribution to binding energy [56]; and b) The entropic penalty associated with binding an unstructured peptide would generally be greater than the entropic penalty associated with binding a folded protein [57], PID-peptide interactions may be driven to maximize the binding energy that single amino acids can contribute and minimize the entropic penalties associated with ordering an unconstrained peptide. Indeed, it seems that peptides tend to maximize their enthalpic potential within the binding site, utilize clefts whenever possible to maximize ΔASA, and minimize the conformational change of their protein binding partner and therefore the overall entropic penalty due to binding [55]. Taking an example from SH2 domain-phosphopeptide interactions, it has been shown recently that artificially restricting the rotamers of the pY residue doesn’t appreciably increase the affinity of a model SH2-phosphopeptide interaction [55], implying that the binding energy is not very sensitive to entropic penalties on the part of the peptide, at least at the level of the primary interaction hot spot (the pY-SH2 domain interaction provides roughly 1/2 of the binding energy for SH2-phosphopeptide interactions [58, 59]). This finding supports the idea that peptide binding modules possess the ability to minimize entropic penalties [55], and is especially interesting considering the prevalence of peptide motifs within disordered regions of proteins [60] where a minimization of entropic penalties would likely be important. These biophysical studies support the validity of studying PID-peptide interactions. Moreover, high throughput investigations, by virtue of their ability to generate large sets of sometimes quantitative or semi-quantitative interactions, can in turn inform our understanding of the general biophysical principals that underlie these interactions.
Regardless of the biophysical mechanisms by which a protein-peptide interaction is achieved, a consequence of the small binding interface of PID-peptide interactions relative to protein-protein interactions is the exaggerated power of individual interfacial residues to influence binding [61]. Amino acid residues within defined motifs serve as hotspots for cognate interactions [55]. Thus, single amino acid substitutions can impart large changes in affinity and corresponding module specificity [43, 62, 63]. The specificity of SH2 domains has been employed to pan the proteome, resulting in unique binding patterns among protein module family members [19]. The specificity of module-peptide interaction principles is also leveraged synthetically to evolve specific and highly affine interactions with protein, peptide, and nucleic acid targets [64, 65]. PID-peptide interactions utilize subtle motif variations to achieve selectivity [9], resulting in impressive specificities within domain families [66-68]. This results in a remarkably complex language for PID-peptide interactions that will be discussed in detail below (section 5.3).
Despite the potential for highly affine and specific interactions, most PID-peptide interactions have affinities in the micromolar range. This may be a result of pressure for dynamic interactions in signaling networks as a necessary component of cellular plasticity. Nevertheless, contextual factors, defined as either secondary contacts (interactions outside of the primary motif-PID binding interface) or cellular context (localization, expression levels etc) play a critical role. There have been many examples of the role secondary contacts play in potentiating interactions but the primary contact between PID and peptide remains the dominant and constant feature essential for the interaction. Binding between short peptide motifs and PIDs on average accounts for 80% of the total binding energy[69]. Consistent with this, a recent report by Bae et al highlights the necessary role a secondary binding site plays in mediating the interaction between the N-terminal SH2 domain of PLC-γ and FGFR1 pY-766 [70]. Estimating binding energies from the reported dissociation constants suggests that the secondary contact imparts roughly 15% of the binding energy, while the primary pTyr motif binding accounts for roughly 85% of the total binding energy [70]. As this example highlights, PID-peptide motif interactions reported in the literature are generally necessary for signal propagation, and in many instances are sufficient for functional interactions. This is reinforced not only from dominant negative mutants [71] and specificity switching point mutations [61], but from investigations of pathogenic, oncogenic, and even synthetic proteins which prove capable of rewiring signaling pathways [20, 72, 73]. In these cases, the signaling pathways are ’hijacked’ by ectopic proteins which present high affinity peptide motifs that outcompete cognate interactions. For example, the interaction between the pYDEV motif within the enteropathogenic E. coli protein Tir and the Nck1 SH2 domain leads to drastic cytoskeletal rearrangements [74]. In a related vein, a chimeric Grb2 SH2 domain-FADD death effector domain (DED) fusion is capable of switching cellular responses so that mitogenic signals result in activation of an apoptotic pathway [75] thereby connecting distinct signaling pathways together via novel synthetic domain associations. In such cases, native secondary contacts are not possible because one of the cognate interaction partners is missing. It is possible in the case of virally mediated ectopic manipulation that different secondary contacts are involved, but there is less rationale for their involvement in completely synthetic signaling cascades. Such work reinforces the notion that the PID-peptide interaction is the primary binding determinant, with non-motif contextual factors playing a secondary role. Beyond secondary contacts, signaling networks employ combinatorial module-peptide and module-motif interactions to potentiate binding events [2, 76], employ multi-motif/single module interaction gating [52, 77, 78], and spatial regulation, leveraging the unique physicochemical environments within a cell [79-82]. As an example of the latter mechanism, recent work has demonstrated the ability of a module motif interaction to potentiate the activity of a kinase, whose substrate is immobilized within a membrane [83]. PID-peptide interactions are in most cases necessary, and in many cases sufficient to define protein-protein interactions. They define specificity and contribute significantly to the biophysical properties of interactions. As a result they are excellent surrogates for biological protein-protein interactions that are inherently amenable to high throughput study.
2.4 PID-Peptide interactions underpin the robustness and evolvability of interaction networks
The prevalence of PIDs in the human proteome is a direct result of rapid expansion of many PID families over the span of metazoan evolution [20]. Mechanisms such as gene duplication and domain shuffling have allowed independently folding modules to efficiently become inserted into novel protein contexts, and in doing so promote the evolution of cellular functions that provide a fitness advantage [21]. This has resulted in the coincident rapid evolution of cellular function and the evolution of robust signaling networks - defined both as an ability of a system to respond with fidelity to noisy inputs and as organism fitness in response to gene deletion. A common architectural feature of these networks, largely resulting from the gene duplication events that constructed them, is the integration of multiple signals into central modules for information processing, resulting in a scale free network topology [84, 85]. This scale free or highly interconnected topology confers redundancy in terms of gene function while also providing for signal amplification or dampening at the central information processing hubs. Taking an example from protein tyrosine kinase networks, the hub protein p130Cas has 16 confirmed tyrosine phosphorylation sites, resulting in a daunting combinatorial problem assuming all 16 can be phosphorylated at the same time [86]. As the p130Cas example implies, the peptide interaction partners of modules tend to reside in disordered regions of proteins [60] which evolve more rapidly than ordered regions [87, 88], and are overrepresented in signaling hubs [89, 90]. While central information processing hubs allow robustness, modularity, and plasticity, they are also weak spots in the network susceptible to ectopic manipulation. Indeed, the targeting of nodes with therapeutic agents in a combinatorial and rational fashion has been suggested as a promising biomedical approach [40, 41]. Given the importance of PID-peptide interactions within this systemic context, interaction specificities, affinities and general principles are a requisite step towards understanding the emergent properties of cellular systems, the architectures that endow them, and mechanisms by which to perturb them.
3. HTP delineation of interactions
Issues of emergent properties and complex interactions aside, HTP techniques are capable of probing interaction specificity on a broad scale, and in doing so they provide the breadth of data necessary to ascertain the underlying rules and engineering principals by which PIDs operate at a systems level. Each method has particular strengths and weaknesses. In this section we will outline the major approaches used to map the specificity of PIDs and some of the advantages and weaknesses of each (Table 1).
Table 1. List of high-throughput methods for characterizing binding specificity, their advantages and disadvantages.
| Method | Peptide Library Size | Quantitative | Advantages | Disadvantage |
|---|---|---|---|---|
| Peptide Array | 10-1000’s | Semi-Quantitative |
|
|
| Protein Microarrays | 10-100’s | Quantitative |
|
|
| Phage Display | 1×10ˆ10 | Not quantitative |
|
|
| Mass Spectrometry | 10-100’s | Not quantitative |
|
|
3.1 Peptide arrays
High-density peptide arrays of synthetic peptides are a powerful tool for mapping domain-mediated protein-peptide interactions at the proteome level. The initial concept of parallel synthesis of multiple components on a solid support was pioneered by Ronald Frank and Mario Geysen [91, 92] with the demonstration of parallel synthesis on cellulose discs packed in a column and peptides on plastic pins, respectively. Nearly a decade later, Dr. Frank and colleagues extended this approach to establish the SPOT synthesis method. In SPOT synthesis, sub-microliter droplets of activated FMOC-amino acids are spotted onto the planar surface of a porous activated cellulose membrane effectively generating an open reactor for synthesis of cellulose-bound peptides. Automation of the technique made it accessible enough to be widely used [93]. One of the benefits of SPOT peptide synthesis is that it circumvents any limitation as to the types and modifications of amino acids that may be employed. As with other forms of solid phase peptide synthesis (SPPS), SPOT synthesis allows the use of unnatural amino acids as well as modified amino acids as building blocks in the synthesis. This is highly advantageous for the study of interactions that depend upon post-translational modification of the peptide ligand. The SPOT method can be modified to utilize different chemical approaches. In the standard SPPS, the peptide is coupled with the C-terminus immobilized to the solid support with a free N-terminus. This presented a challenge for studying PDZ domains, which recognizes C-terminal peptides. A method of peptide cyclization in order to free the C-terminus allows studies on PDZ domain interactions [94].
Because it is relatively easy to synthesize hundreds or even thousands of individual peptides on a single cellulose membrane, the SPOT method allows the interrogation of genome-wide sets of peptide sequences, yielding extremely large sets of binary interaction data. PID studies can easily detect peptide binding using large sets of purified recombinant PIDs incubated individually with the SPOT membranes and subsequently detected using antibodies against specific expression tags (eg GST, His) or by using radiolabeled or fluorescently labeled protein (Figure 3A). Another advantage of this technique is the ability to observe weak binding or non-binding peptides and thus generate data not only about the peptides that bind, but also those that do not. In some cases, knowing the certain peptides that do not bind to a PID may be as informative as knowing ones that do.
Figure 3. High-throughput Methods for studying protein domains.
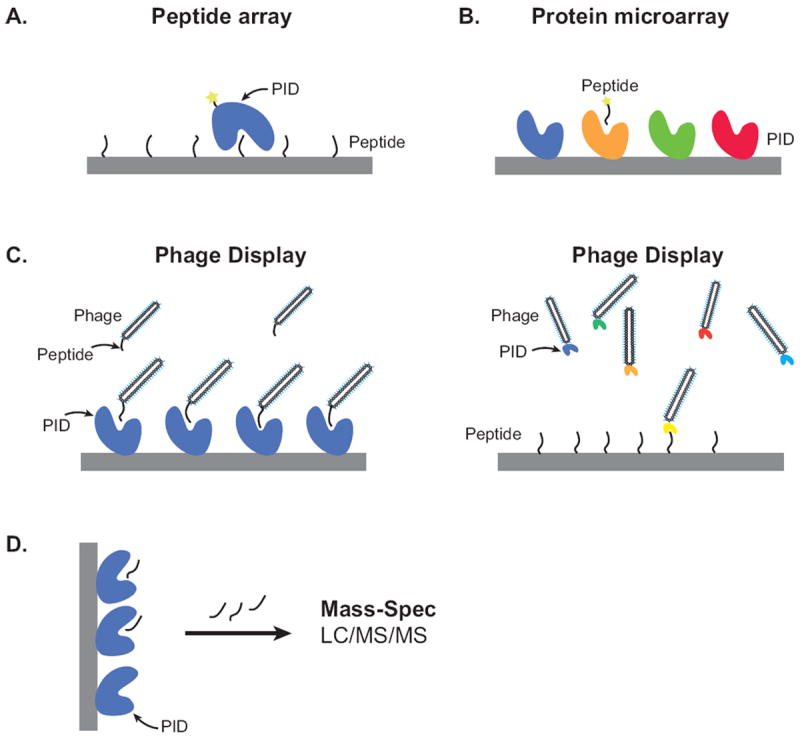
(A) Peptide arrays consist of a diverse set of peptides synthesized onto cellulose membranes using the SPOT method or isolated peptides arrayed in large scale onto glass slides using a microarray printer. Soluble protein interaction domains (PIDs) flowed across the membrane can be detected using antibody detection or fluorescent labeling of proteins (indicated with a star). (B) Protein microarrays require soluble PIDs to be printed onto various solid surfaces. Labeled peptides are flowed across the surface and detected using a fluorescent CCD camera. (C) Phage display allows one to create a highly random set of peptides expressed on the surface of bacteriophage. After selection and binding to soluble PIDs, captured phage can then be sequenced to determine the peptide sequence (left panel). In a reverse approach, a diverse library of PIDs expressed on the phage coat can be screened for binding to select peptides (right panel). (D) Mass spectrometry allows one to capture proteins or peptides using a bait (eg PID). Captured proteins or peptides are digested and subsequently sequenced using MS/MS for peptide and protein identification.
Designing arrays of defined peptides generally requires some degree of a priori knowledge of the binding profile for the domain of interest. Oriented peptide library arrays synthesize using the SPOT approach require less understanding of binding preference but generally work best when at least one critical determinant for binding is fixed. SPOT-synthesis peptide libraries fix individual positions (eg +1, +2 or +3) while using a randomized mixture of amino acids at other positions [95]. Library arrays are commonly used to identify favorable residues at various positions that allow the generation of general specificity profiles. This approach appears has been successful with domains where a major factor is fixed such as pTyr for SH2 domains or PxxP for SH3 domains [41, 66, 95]. The relatively high affinity of SH2 domains for pTyr peptides means that library arrays need only fix the pTyr and other positions may be varied, whereas domains such as SH3 and PDZ domains that bind at one magnitude lower affinity generally require at least 2 fixed residues.
Regardless of the application, SPOT peptide arrays are generally a semi-quantitative method and specific interactions require verification or quantification by other methods [96]. Since peptides on the peptide array do not undergo purification, the yield and efficiency of peptide synthesis on a solid-support is not uniform and may be difficult to assess. Synthesis failure or low yield of a peptide could thus result in false negative results. There are a number of fairly obvious measures that should be considered by anyone using peptide arrays to map specific interactions. As these overlap with the techniques below they are discussed in section 4.
3.2 Protein Microarrays
Protein microarrays are the complement to peptide arrays [97, 98]. Also referred to as ‘protein chips’, these consist of purified proteins arrayed in a high-density format. Protein microarrays are typically prepared by immobilizing proteins onto a solid substrate such as a modified glass microscope slide using a contact or a noncontact microarrayer [99]. Developing the type of surface, attachment method, detection method, and necessary controls are likely key determinants in the success of a given protein microarray system. Soluble proteins immobilized on the slide can be probed for a variety of functions such as binding to other proteins or peptides visualized by fluorescent dye or radioisotopic labeling (Figure 3B). In theory, protein microarrays can be spotted in large scale and probed with increasing concentrations of labeled material, producing semi-quantitative or even quantitative information. In practice, protein microarrays often accrue a high rate of false-positive and false negative data and remain difficult to judge as we discuss below. In terms of studying protein domain interactions with short peptide sequences, protein-domain microarrays provide a means to identify novel protein-protein interactions using limited subset of protein domains and peptide ligands in a manner analogous to peptide arrays [100, 101]. Proteins are inherently complex given their tertiary structure and may not maintain activity on the array surface. The use of protein microarrays remains in its infancy in terms of the methods of fabrication and analysis but it is clear that non-selective interactions are a problem with arrayed proteins on substrates in interfacial assays. A study of PDZ domain binding by glass substrate protein microarrays indicated extensive false-positives with a false-positive rate close (approximately 50%) and poor correspondence of estimated Kd values in comparison to solution binding measured by fluorescence polarization. The same study identified a significant rate of false negatives. [67]. At present protein microarrays may be best suited for low-throughput and carefully controlled studies. One example of lower-throughput protein array technique is the Rosette assay in which increasing concentrations of peptide or lysate are spotted onto a membrane support and then probed with SH2 domains [19]. Rosette produces semi-quantitative data indicating relative binding preferences and is of particular use along side SH2-domain profiling experiments using the same sets of SH2 domains. With the application of stringent protein activity tests, extensive use of positive and negative control peptides, and adequate methods to screen out false-positive interactions, protein microarrays may be used to generate high quality data in larger scale studies (see section 4).
3.3 Phage-display peptide libraries
Phage display has proven to be a powerful and versatile method for studying PIDs. Several variations have been applied to study protein interactions and these are reviewed in detail elsewhere [102, 103]. In the most common implementation used to identify optimal binding peptides, a phage display library undergoes multiple rounds of panning against a purified PID (Figure 3C, left panel). Random peptide libraries can be generated using randomized DNA oligonucleotides incorporated into the genome of the phage for expression on the coat proteins (P8) of bacteriophage. These libraries can achieve diversity of greater than 10 billion random peptides [104]. Phage bound to individual domains can be readily recovered and sequenced to determine the peptide sequence preference. The list of peptide sequence information can then be used to generate a position-weighted matrix (PWM) to define binding motifs. One clear benefit of phage display is that a vast number of chemically diverse peptide sequences can be generated efficiently and inexpensively as part of the phage display library. This is balanced by the limitation that without complex utilization of alternate codons, only natural amino acids are included in the library peptides. This makes using phage display somewhat of a challenge for studying domains that recognize PTMs or alternative amino acids. Phage display is not, of course, limited to studying peptides as it may equally be used to display a library of sequence varied domains on the phage coat (Figure 3C, right panel). This provides the opportunity to explore the variability and evolvability of PIDs to recognize specific ligand sequences. For example, phage display mutagenesis of a single PDZ domain allows the generation of a wide variety of ligand-binding preferences that encompass both naturally evolved ligands but also explore ligand binding space outside of that for which PDZ domains have naturally evolved [105]. Thus phage display is also a powerful tool for PID engineering and molecular evolution [105, 106]. Stable expression of certain domains and sequences on phage may be limited, however, potentially posing a challenge for genome-wide screening. As with other HTP techniques controls are essential (see section 4)
3.4 Mass spectrometry
Mass-spectrometry (MS) has become a powerful tool for studying protein-protein interactions as well as for the identification of PTM sites (reviewed in [107, 108]). When PIDs are used as capture reagents to precipitate interacting proteins or peptides, tandem- MS/MS is an efficient means of identifying the interaction partners (Figure 3D). The data obtained is limited in part by the set-up of the capture step. This may constrain interactions to proteins expressed in specific cells or tissues and those interactions that are of sufficient affinity (or sufficiently slow off-rate) or expressed at sufficiently high level to allow capture and identification [109](Figure 2B). Thus, the number of peptide interactions may be limited for a given PID. Such information may not be sufficient for a detailed analysis of domain specificity but it can provide a very useful context for physiological interactions. A study with 10 GST-tagged WW domains identified a list of 148 protein interactions from Jurkat cell lysates [110]. The proteins identified in their screen were enriched into 2 clusters based on known preferred WW motifs, PY and PPLP or Pro/Arg. Another study using purified Plk1 Polo-Box domain identified over 600 bound proteins from G1/S arrested U2OS cells [111], revealing the power and sensitivity of MS. To extend the ability to profile interactions, other approaches include using a library set of peptides to identify domains that recognize a specific type of post-translational modification. A recent study by Christofk et al., utilized mass spectrometry and peptide libraries to identify proteins that could specifically recognize only phosphorylated peptide libraries [112]. Thus, MS may be powerful complementary approach to the three methods mentioned above to study large-scale PIDs in a physiological setting.
3.5 Other HTP techniques: Plate-based and biophysical assays
In addition to the widely used approaches noted above, there are a wide variety of biophysical assays that are potentially amenable to scaling up to some form of HTP assay. There are many solution-based binding assays that are available in plate-based detection formats that may be scaled up to become useful for HTP analysis of PIDs. Fluorescence-based binding assays in particular allow for automated set-up and detection. Fluorescence anisotropy or fluorescence polarization is widely used to measure the binding of labeled peptide ligands to domains [43, 67]. 384-well plate-based detectors allow hundreds of interactions to be quantitatively measured and equilibrium dissociation values ascertained. Intrinsic tryptophan fluorescence can be used to detect binding in cases such as SH3 domains that contain a tryptophan residue in the peptide binding cleft [113]. These methods are limited both by the need to synthesize peptides and purify active PIDs, as well as the general issues of ensuring data quality described below. They do, however produce potentially rich quantitative datasets and thus have a clear role.
Yeast two-hybrid has been used very successfully to detect interaction partners for PIDs such as PDZ and SH3 domains [114, 115], however it is less able to accommodate PTM-dependent interactions. Yeast almost entirely lack tyrosine phosphorylation making yeast 2-hybrid ineffective for the study of pTyr-binding domains. Mammalian 2-hybrid systems have been developed and used in select instances [116, 117]. Related systems such as the Lumier method (luminescence-based mammalian interactome mapping) assay originally described by Dr. Jeffery Wrana and colleagues detects Renilla luciferase tagged prey proteins captured by flag-tagged bait proteins allow interactions to be studied from mammalian cell lysates, albeit under conditions of ectopic overexpression [118]. PIDs that recognize reversible PTBs can also be used to profile cellular activation states. SH2 domains in particular serve as a means of probing the global state of tyrosine phosphoproteome. Thereby using SH2 domains as a detection reagent, similar to that of an antibody for a western blot, the lysates of various cell lines or cell lysates from various times points post-stimulation, can be used to read out the presence of specific phosphotyrosine motifs to which each SH2 domain can bind [18, 19]. Most of these systems are focused on protein-protein interactions, but in specific instances these systems are amenable to the study of PID-peptide interactions. Fluorescence-based PID-peptide interactions in particular stand out in this regard. As with the other HTP techniques described, there is a clear need for stringent standards, controls and validation in order to obtain high quality and reproducible data.
4 The critical importance of controls and validation
Hypothesis-driven studies that reveal the details of a particular interaction are required to include extensive controls, replicates and orthologous validation in order to pass muster. Yet large-scale or HTP studies do not always provide these basic tenants of the scientific method, instead relying on the novelty of the approach and the sheer overwhelming quantity of data. Because of the amount of data generated, it is difficult to validate any but a small number of interactions and because the methods are often novel it is not always clear what the correct controls should be or whether replication is even feasible. Yet the data generated may be extensively utilized as hypothesis generating, or for developing complex network models, and so it is essential that data quality be as high as possible and limitations revealed up-front. Here we will outline a number of controls and validation approaches that may be applied across multiple experimental platforms with the aim of producing high quality data-sets for PID-peptide interactions using HTP approaches.
4.1 Garbage in, garbage out: Positive controls for every protein and/or peptide
The quality of the protein and peptide are clearly critical to obtaining valid interaction data. Recombinant expressed and purified proteins do not always behave as we would like due to misfolding, degradation, denaturation etc. To the degree possible in a given HTP experimental set efforts should always be made to ensure that the proteins and peptides being used are correctly expressed, folded and active at the time of the actual assay. Much of this can be done through the development of a solid set of internal controls. The wealth of available data in the literature developed from carefully controlled hypothesis-driven experiments (see below) should provide that many of the proteins and peptides in a given HTP experiment to have at least one known positive control for functional binding activity. For instance, if the Grb2 SH2 domain is used, a pY-V-N-V motif-containing peptide might serve as a control to ensure that in each iteration of the experiment that the Grb2 protein used is functionally active. This provides at least a minimal level of confidence that at the time of that particular experiment each protein is capable of binding to a cognate peptide ligand. If the study is quantitative then additional validation information is available in the form of the measured binding affinity. Most discovery-based experiments will utilize at least a few well-described proteins or peptides, so that while not every peptide or protein will have an control partner, a representative fraction will have. A set of binary control pairs also provides a measure of the assumed failure rate for proteins or peptides in a given experimental set-up. This in turn can be used to estimate the number of replicates needed to ensure a complete data set.
4.3 Filtering the noise
Non-selective or false-positive interactions are a potentially major confounding factor in any large-scale data set [67]. Yet because data from large-scale studies is easily accessible, these datasets are widely used by computational modelers as the basis for signaling network models and analysis. Removing false-positives need not be difficult in large HTP approaches as the large quantity of data may itself suggest apparent highly populated interaction hubs that in fact represent “sticky” peptides or proteins that are simply non-selective. Removal of non-selective interaction hubs may lose a small number of real interactions, but serves to vastly improve the overall data quality by removing large numbers of non-selective interactions. For example, we recently completed a study of interactions between 192 peptides and 50 SH2 domains [43]. Our initial interaction set comprised 905 binary interaction pairs but when examining the specificity of SH2 domains, about which a fair amount is known [119] it became clear that certain peptides were interacting in a non-selective manner with large numbers of SH2 domains such that they defied known binding motifs. It would be extremely difficult to pick out all of the non-selective interactions based on assumed parameters governing binding, and in any case this would contaminate the dataset with preconceived assumptions that might not be valid. But as it turns out, the data structure itself may often provide clues as to the identity of non-selective interaction hubs. When we graphed the degree of binding for all 50 SH2 domains for each peptide, selective interacting peptides exhibited the expected bi-modal pattern in which most SH2 domains did not bind at all (very low binding signal intensity) and a small number bound strongly (high binding signal intensity). Non-selective interacting peptides exhibited a skewed profile with many peptides binding with medium to high intensity and almost none with very low intensity. This recognizable binding pattern coincided perfectly with the set of peptides that bound non-selectively to GST with signal intensity above the mean. In practice this meant that apparent “hub” peptides were, in fact, often hubs for non-selective interactions and filtering them out reduced the number of binary interaction pairs identified from 905 to 523. This resulted in a final dataset that overlapped almost perfectly with the literature-validated set of 39 interactions and orthologous validation set of 55 interactions [120].
Mass spectrometry based proteomics experiments similarly identify large numbers of non-selective binding partners. Again, large data sets themselves may be utilized to self-filter by identifying proteins that appear over and over again as interacting partners for multiple types of proteins. This is the basis for the contaminant repository for affinity purifications (CRAP) database developed by the Gingras and Nesvizhskii groups. Certain proteins identified in this manner are likely to be common across many experimental set-ups and represent common laboratory proteins, accidental contaminants from dust or physical contact, and proteins commonly used as molecular weight standards. But in a single large-scale HTP study of many PIDs, the data specific to a single study can be used to identify non-specific interactions specific that the experimental system and improve data quality by eliminating these interactions.
In various ways, these and related methods utilize the large quantities of data generated by HTP studies to enable statistical approaches and pattern analyses to be employed. These may be very effective for identifying potential false-positive interactions and in some cases allow the assigning of scores reflecting confidence in specific interactions. HTP approaches are particularly amenable to the use of a range of statistical approaches. Probabilistic scoring of affinity purification-mass spectrometry data is the basis for ‘significance analysis of interactome’ (SAINT), a computational tool that assigns confidence scores to protein-protein interaction data generated using affinity purification-mass spectrometry (AP-MS) [122]. Other approaches such as normalized spectral abundance factor (NSAF)[123], CompPASS [124] and Annotator [125] are just some examples of tools that provide statistical tests to determine significance of mass spectrometry data.
4.4 Literature validation
It is relatively simple to compare one database of interactions from a large-scale interaction study with another. But if either or both studies suffer from large false-positive or false-negative rates the datasets will only overlap to a small degree. This is exactly what has been reported in several instances [126-128], and is essentially uninformative beyond suggesting that one or both studies have data quality issues. Yet there is an immense and growing body of literature carefully detailing specific interactions. The major hurdle is that protein interaction databases are largely populated by data from HTP studies and the majority of interactions described by traditional hypothesis-driven research remains largely inaccessible on the scale necessary to validate HTP studies. In the case of PID-peptide interactions, even databases that do actively curate primary literature interactions such as the Human Protein Reference Database (HPRD), BioGRID and Molecular Interaction (MINT) only rarely provide details regarding the specific PID and peptides responsible for interactions [129-131]. This is unfortunate as this exactly the sort of carefully controlled orthologous data that is likely to be the best test of HTP datasets. It is therefore the responsibility of the investigator and the community as a whole to develop such datasets. HTP studies provide a unique opportunity to populate such datasets with both their own data and whatever data they are able to glean from the literature to validate their results. Literature validation does not suggest infallibility of published studies, but significant discordance between HTP data and existing literature should certainly be cause for concern while a high degree of concordance serves as convincing validation.
4.5 Motifs matter
Related to the theme of using existing knowledge to validate HTP approaches, there are many instances in which binding motifs have been described for protein-peptide interactions. In the case of SH2 domains, for instance, motif data is available from Scansite and SMALI in the form of PSSMs and regular expression motifs [66, 119]. In many cases, structural data supports the fundamental assertions of these motifs. For instance, we know from both Scansite as well as a series of elegant structural studies that the Grb2 SH2 domain has a very strong preference for an asparagine residue at the +2 position C-terminal to the pTyr residue to accommodate a beta-turn required by a tryptophan residue in the SH2 domain that obstructs the peptide-binding channel. Similarly, the Crk SH2 domain has a very strong preference for a proline or leucine at the +3 position of its cognate peptide ligands that has been repeatedly confirmed in multiple independent studies [43, 44]. As already noted, motifs are by no means absolute and in many instances simplify binding data to a level that ignores contextual information, but they do provide an excellent test of HTP data sets. If, for instance, a HTP study indicates ligands for the Grb2 domain that do not conform to the pY-x-N motif, this suggests that there may be an issue with non-selective interactions (false-positives) and that at the very least additional validation is required for those ligands. Similarly well-established and largely invariant motifs for other PIDs serve as an excellent test of data quality but also to identify potentially novel interactions that should be subject to orthologous validation. Early studies using protein microarrays contain numerous examples of apparent binding peptides that do not conform to established motifs [132]. As these early studies also did not contain positive controls for either proteins or peptides and lacked extensive orthologous validation, so it is difficult to establish the precise issues that led to such apparent false-positives. It is now clear that the protein microarray technology employed in these studies has a high level of false-positives and that the apparent dissociation constants reported often do no correlate with equilibrium dissociation constant values measured in carefully controlled solution phase binding experiments [67]. The controls and validation strategies noted above are in part drawn from such lessons learned in the past decade of HTP studies of PID-peptide interactions but are only a first step towards using HTP studies to generate high quality interaction data.
4.6 Spatiotemporal considerations
In cases where large-scale studies are conducted in cells or using cell lysates, spatiotempral considerations become a major factor in experimental design. For example, upon stimulation receptor and non-receptor kinases become activated and engage substrates resulting in phosphorylation of specific proteins that may then serve as binding partners for their cognate PID binding partners. Of course stimulation and downstream phosphorylation events follow their own very specific time-course, such that a different phosphosite may appear or disappear at various times points and spatial regions of the cell (Figure 2A, C) [1, 133, 134]. To make matters more complex, there are significant differences in expression profiles of both the PID-containing proteins and the phosphorylated targets in different tissues [135]. Thus the same pTyr site might engage a different SH2 domain in brain than it does in muscle to initiate a different signaling cascade downstream of the same stimulation. Or the same SH2 domain protein might find a different set of available pTyr ligands available in different tissues (Figure 2B). Likewise, a given PID may find a very different set of potential ligands in the nucleus than it does in the cytoplasm (Figure 2C). Prolonged stimulation with super-physiological concentrations of a growth factor or other stimulation may result in profoundly different patterns of phosphorylation and downstream signaling than would occur within the bounds of normal physiological signal transduction [136]. Considering the stimulations conditions, tissue or cell lines uses and the time-course of the experiment may significantly alter the spatiotemporal signaling and thus the binding partners that may be recovered.
5. Systems-Level Analysis of Protein Interaction Domains
Data from HTP studies of PID-peptide interactions provides a unique opportunity to develop both models that extend beyond the source data as well as to glean new insight into the global engineering principals that guide the selectivity, competition, cooperation and evolution of PID families and their host proteins (Table 2). It is impossible to outline all of the uses for HTP analysis of PID-peptide interactions in a single manuscript, so in the interest of space we will note just two examples – one predictive and one at the level of general engineering principals governing binding.
Table 2. High-throughput Studies of Protein Interaction Domains.
| Domain | Ligand | Method | Reference |
|---|---|---|---|
| SH2 | pTyr | Peptide Array | [43, 66] |
| Protein Microarray | [132] | ||
| Reverse Phase Protein Arrays | [19] | ||
| SH3 | Proline rich motifs | Peptide Array | [148] |
| Phage Display | [115], 149] | ||
| Yeast Two-Hybrid | [115] | ||
| PDZ | C-terminal motifs | Phage Display | [104] |
| Protein Microarray/FP | [67] | ||
| Yeast Two-Hybrid | [150] | ||
| PTB | pTyr and NPxY motifs | Peptide Array | [151] |
| Protein Microarray | [132] | ||
| WW | Proline rich motifs | Protein Array | [152] |
| Mass Spectrometry | [110] | ||
| Polo Box | pSer/Thr motifs | Mass Spectrometry | [111] |
| 14-3-3 | pSer motifs | Peptide Array | [145] |
| Tudor, MBT, Chromo | Methylated Histone motifs | Protein Array | [100] |
5.1 Binding: motifs to prediction
One of the first uses for the data generated from large-scale analysis of PID-peptide interactions was to develop predictive models for PID ligand selectivity. Binding data from various types of peptide library approaches provides insight into the preferred amino acids at positions surrounding the conserved central motif. Early studies of ligand specificity, particularly that of SH2 domains has been evaluated using the synthetic peptide library approach developed by Songyang and Cantley [44, 137, 138]. In the case of SH2 domains, this identifies favorable residues within the peptide ligand surrounding the conserved pTyr that support binding by a particular SH2 domain [44, 95]. This takes the form of a probability that a given amino acid occupies a position x within a motif x-pY-x-x-x-x. Likewise, studies of SH3 domains might generate data for an x-x-P-x-x-P-x-x motif. Probability information of this type is often described using position weight matrices (PWM) such as the position-specific scoring matrices (PSSM) that underlie widely used ligand prediction tools such as ScanSite (http://scansite.mit.edu/) and Scoring Matrix-Assisted Ligand Identification (SMALI, http://lilab.uwo.ca/SMALI.htm) [66, 119]. PDZ domain binding preferences obtained from randomized phage-peptide libraries have been used in a similar manner to generate PWMs [104, 139]. PWMs do an admirable job of identifying primary factors required for binding and identify key residues that may form direct contacts or otherwise be necessary for a peptide ligand to fit into the binding cleft of a given PID. PWM-based prediction methods are simple to run and generalizable across any short linear peptide sequence. The more flexible Hidden-Markov Models (HMMs) commonly used to identify longer sequence stretches such as PIDs themselves [140-142], lack sufficient power when confronted by such very short peptide motifs [143], but remain powerful tools for longer stretches of sequence. Even very simple regular expression-based queries can be used in cases of well-defined motifs [144]. As a general rule, all of these computational methods lack the ability to discern contextual information embedded in peptide sequences. For instance, the preference for residues at adjacent positions may differ according to the surrounding residues. And with a few exceptions non-permissive residues or so-called anti-motifs are not part of the standard models either because the models do not easily accommodate such information or, more commonly, there is a paucity of such data.
In silico prediction can provide testable hypotheses but remains unable to predict interactions with high accuracy and precision. This is in part because recognition events are complex, even at the level of isolated SH2 domains and synthetic peptides. For instance, SH2 domains recognize peptide targets not only through permissive residues adjacent to the pTyr that constitute binding motifs, but also by making use of contextual sequence information and non-permissive residues [43] that constitute anti-motifs. Some rules such as non-tolerated residues and contextual dependence of the residues at one position on those at another position are not accommodated PSSM. Results using ad hoc rules such as 1) eliminating non-tolerating residues at each position and 2) requiring the presence of a favorable residue at least one position [145] can improve domain-peptide predictions.
5.2 Anti-motifs and Negative selection in Binding Specificity
Given the extensive use of degenerate peptide libraries and phage display to discern optimal binding peptides, much of the information generated to date focuses on residues that are positively selected for in these methods. In contrast, residues that are disfavored or prohibited at certain positions within the canonical binding motif remain poorly defined. Yet exclusion of specific amino acid residues at specific positions represents another layer of information that a PID can interpret to determine binding. Several recent systems-level studies have shown that residues that are non-permissive to binding act as an anti-motif overlapping the canonical binding motif and that the recognition of both motif and anti-motif provides a higher level of selectivity (Figure 4). SH2 domains utilize anti-motifs extensively to provide selectivity particularly in cases where several domains have similar primary binding motifs (Figure 4A)[43]. For example both Crk and Brk recognize the generalized motif of pY-x-x-L/P, yet the presence or absence of anti-motifs can determine the selectivity for one SH2 over the other. This suggests the recognition of even short peptides can be highly nuanced in a manner analogous to the natural language. Similarly, SH3, 14-3-3 and Chromo domains also sense non-permissive residues or “anti-motifs” to fine-tune specificity between related domains in a family [145, 146]. Two recent studies on 14-3-3 and Chromo domains using peptide arrays were able to identity anti-motifs found within shared consensus motifs that play an important role in achieving appropriately high selectivity [145, 146]. The presence of either Asp at -1 and or Pro/Arg at +1 perturbs 14-3-3 binding even though positive motifs, Arg at -3 and -4 or Pro at +2, are present (Figure 4B). Similarly, using peptide-scanning mutagenesis of the various positions surrounding methylated lysines on histones 9 and 27 revealed that the presence of either Thr or Ala at the -3 position was critical for determining the specificity between the HP1 or the Pc class of Chromo domains (Figure 4C). Lastly, the SH3 domains Bem1p and Nbp2p from yeast share the consensus motif, ψ-x-P-x-R-x-A-P-x-x-P (ψ representing hydrophobic amino acids) (Figure 4D, left panel) [62]. Conserved residues within the SH3 domain sense the sequence context of the peptide to prevent nonspecific interactions both in vitro and in vivo [62]. The use of addressable peptide arrays in each of these studies was essential to developing the data necessary to understand this principle as peptide arrays provide not only positive binding information but also inform as to interactions that are disfavored. This highlights an interesting problem that large data sets can make use of, which is that data about interactions that fail to occur despite clearly favorable motifs is itself useful in understanding more complex rules governing selectivity. It is clear the protein-protein interactions have evolved to make use of available information. At a practical level, a deeper understanding of these rules is clearly important for developing better predictive tools.
Figure 4. Anti-motifs drive negative selection for optimizing specificity.
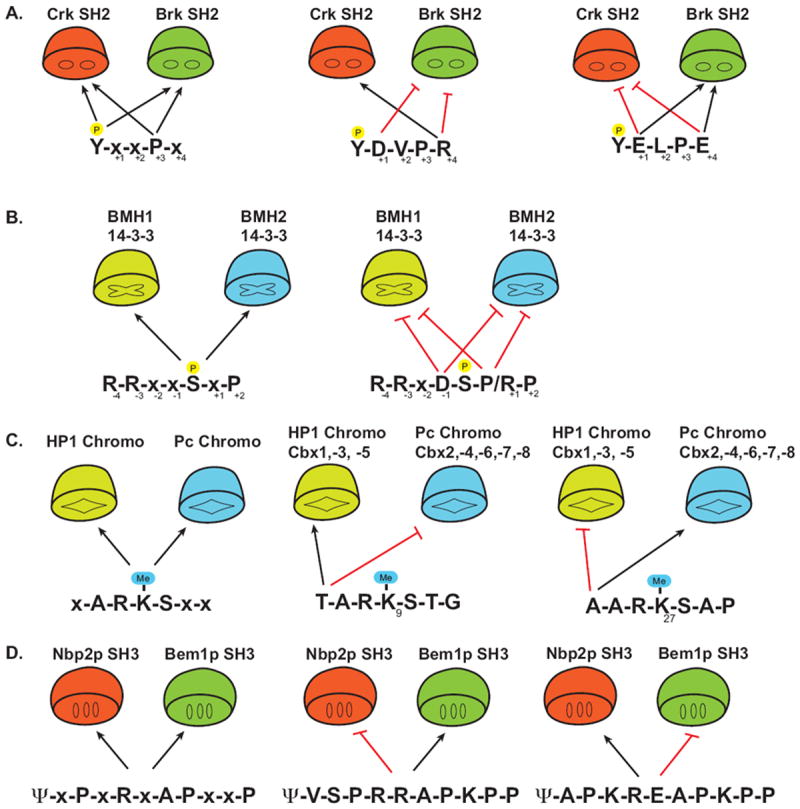
(A) The SH2 domain of Crk and Brk both recognize the motif pY-x-x-L/P (the P in yellow circle above the Y represents a phosphate). Black arrows indicate permissive or positive preferential residues. Anti-motifs (red hash) of select peptides determine the selectivity for either Crk or Brk. (x denotes any natural amino acid) (B) Yeast 14-3-3 proteins BMH1 and BMH2 recognize the consensus motif R-R-x-x-pS-x-P (left panel), however sequences which contain an Asp at -1 or Pro/Lys at +1 disrupt binding despite having permissive factors.
(C) Recognition of the methylated lysines (Me-K) on the histone tail by distinct Chromo domains is determine by anti-motifs. Me-K(9) and Me-K(27) both contain the core consensus x-A-R-K-S-x-x. Recognition by either the HP-1 subclass or the Pc subclass of Chromo domains is determined by the amino acid at the -3 position of the Me-K. (D) The yeast SH3 domains of Bem1p and Nbp2p recognize the consensus motif ψ-x-P-x-R-x-A-P-x-x-P (ψ represents a hydrophobic residue, x is any natural amino acid). The sensing of the sequence context by these SH3 domains either inhibits (red hash line) or allows binding (black arrows), thereby preventing non-specific interactions.
5.3 Problems with current systems
HTP studies of PIDs are capable of generating a large wealth of peptide-domain interaction data. The design of these studies can utilize physiological (found in potential in vivo binding partners) and non-physiological peptides as well as highly diverse (and largely non-physiological) peptide libraries. Approaches using peptide libraries or non-physiological peptides run the risk of determining optimal binding solutions that are not found in nature, while limiting experiments to physiological peptides reduces the potential data set to a point where it may be difficult to discern the underlying rules that govern selectivity. Virtually all HTP studies of PIDs are limited to determining binary interactions between a protein domain and it’s ligand, yet many interactions depend upon larger multi-protein complexes or are regulated in ways that may not be easily recapitulated in vitro. Certain PIDs can be found in multiple conformations that alter ligand binding. The INAD PDZ domain (PDZ5) can be found in a redox-dependent equilibrium between two conformations [147]. The reduced form has a structure similar to that of other PDZ domains while the oxidized form assumes a distorted ligand-binding site as a result of a strong intramolecular disulfide bond formation. Without prior knowledge of domains behaving in various manners, interactions captured in an in vitro HTP study may be limited.
Studies using protein microarrays and peptide arrays have generated a wealth of potential interaction data. However, many of these studies have limited orthogonal validation and fail to compare their results to the “known knowns” of the literature or canonical motifs. This ignores the issues associated with any large-scale and single-method study and results in datasets of unknown quality. When binding events do not match the known motifs they should be scrutinized carefully. The notion that testing a hand-picked small fraction of novel interactions in a cell-based system validates the remainder of the data from a large-scale study is absurd and should be dismissed out of hand. Such validation certainly can lead to exciting and novel mechanistic understanding and is certainly important, but it should very clearly not be taken to validate unrelated interactions. Only random validation of a significant number of the potential novel interactions by orthogonal methods can even provide an estimate of the data quality. LTP direct experimental measurement of select binding partners typically focuses on specific interactions driven by hypotheses relating to the signaling events under investigation. This yields a set of high quality, but inevitably sparse data. Thus while we are aware that pTyr signaling is extensively used in a range of critical signaling networks in metazoans, certain pTyr proteins and SH2 domains are well studied while others are left untouched. Yet as incomplete as the literature is, it does provide a solid foundation for validating high-throughput techniques. Failure to identify well-characterized interactions may indicate false-negatives. High-quality specific interactions are an essential step in determing protein functions and the molecular mechanisms that underpin cellular behavior. There is a clear need to investigate less well-studied proteins and HTP experiments can be a rich source for discovery data that drives future hypotheses so long as the appropriate experimental design ensures high quality data. Finally, in silico methods for prediction (Scansite, SMALI) do a remarkable job of pointing out potential binding pairs and ignoring irrelevant pairs because motifs matter [66, 119]. Their fail at the ultimate prize of accurate prediction is simply a reflection of our incomplete understanding of the complexity of the underlying interactions. HTP studies serve to provide the large and rich data to improve these methods and develop new computational approaches to modeling protein-protein interaction networks.
6. Future directions
We are in the early days of a HTP analysis of PID-peptide interactions. The HTP methods discussed have only scratched the surface yet the data generated has already proven invaluable in mapping novel signaling networks as well as understanding the mechanistic principals used to establish selective interactions. As in any maturing field, technology development and proof-of-principal gives way to carefully controlled experiments that produce high-quality data. Some of the HTP approaches are already mature as new ones continue to be developed. With carefully designed studies, HTP techniques hold the promise of developing interaction and PTM site data that is codified by both temporal and spatial parameters in order to understand the spatiotemporal organization of protein-protein interaction networks. By combining interaction datasets with large-scale genomic and expression pattern data, it will increasingly be possible to establish defined interaction networks that are specific to cell types, developmental lineages and disease states. This in turn holds the promise of deeper mechanistic understanding of signaling dynamics. Carefully controlled and well designed HTP studies of PID interactions can therefore leverage the power of reductionist biochemistry with systems level understanding of interactions and the networks that they construct.
Acknowledgments
We thank members of the Pawson lab (Samuel Lunenfeld Research Institute, Toronto), Brian Kay (U of Illinois, Chicago) for helpful comments and suggestions. The work is been supported by the National Science Foundation (MCB-0819125); The University of Chicago Cancer Research Center; a pilot project grant from the University of Chicago Diabetes Research Training Center, and The Cancer Research Foundation to P.D.N. B.A.L. is supported by the Canadian Health Institute Research (CIHR) postdoctoral fellowship. B.W.E. is funded in part by the NIH TG-GM07183.
References
- 1.Scott JD, Pawson T. Cell signaling in space and time: where proteins come together and when they’re apart. Science. 2009;326:1220–1224. doi: 10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 3.Sadowski I, Stone JC, Pawson T. A noncatalytic domain conserved among cytoplasmic protein-tyrosine kinases modifies the kinase function and transforming activity of Fujinami sarcoma virus P130gag-fps. Mol Cell Biol. 1986;6:4396–4408. doi: 10.1128/mcb.6.12.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadowski I, Pawson T. Catalytic and non-catalytic domains of the Fujinami sarcoma virus P130gag-fps protein-tyrosine kinase distinguished by the expression of v-fps polypeptides in Escherichia coli. Oncogene. 1987;1:181–191. [PubMed] [Google Scholar]
- 5.Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332:272–275. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- 6.Sammut SJ, Finn RD, Bateman A. Pfam 10 years on: 10,000 families and still growing. Brief Bioinform. 2008;9:210–219. doi: 10.1093/bib/bbn010. [DOI] [PubMed] [Google Scholar]
- 7.Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marchler-Bauer A, Panchenko AR, Shoemaker BA, Thiessen PA, et al. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 2002;30:281–283. doi: 10.1093/nar/30.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhattacharyya RP, Remenyi A, Yeh BJ, Lim WA. Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu Rev Biochem. 2006;75:655–680. doi: 10.1146/annurev.biochem.75.103004.142710. [DOI] [PubMed] [Google Scholar]
- 10.Seet BT, Dikic I, Zhou MM, Pawson T. Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol. 2006;7:473–483. doi: 10.1038/nrm1960. [DOI] [PubMed] [Google Scholar]
- 11.Kuriyan J, Cowburn D. Modular peptide recognition domains in eukaryotic signaling. Annu Rev Biophys Biomol Struct. 1997;26:259–288. doi: 10.1146/annurev.biophys.26.1.259. [DOI] [PubMed] [Google Scholar]
- 12.DeClue JE, Sadowski I, Martin GS, Pawson T. A conserved domain regulates interactions of the v-fps protein-tyrosine kinase with the host cell. Proc Natl Acad Sci U S A. 1987;84:9064–9068. doi: 10.1073/pnas.84.24.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson D, Koch CA, Grey L, Ellis C, et al. Binding of SH2 domains of phospholipase C gamma 1, GAP, and Src to activated growth factor receptors. Science. 1990;250:979–982. doi: 10.1126/science.2173144. [DOI] [PubMed] [Google Scholar]
- 14.Moran MF, Koch CA, Anderson D, Ellis C, et al. Src homology region 2 domains direct protein-protein interactions in signal transduction. Proc Natl Acad Sci U S A. 1990;87:8622–8626. doi: 10.1073/pnas.87.21.8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu BA, Jablonowski K, Raina M, Arce M, et al. The human and mouse complement of SH2 domain proteins-establishing the boundaries of phosphotyrosine signaling. Mol Cell. 2006;22:851–868. doi: 10.1016/j.molcel.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Liu BA, Shah E, Jablonowski K, Stergachis A, et al. The SH2 Domain-Containing Proteins in 21 Species Establish the Provenance and Scope of Phosphotyrosine Signaling in Eukaryotes. Sci Signal. 2011;4:ra83. doi: 10.1126/scisignal.2002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lappalainen I, Thusberg J, Shen B, Vihinen M. Genome wide analysis of pathogenic SH2 domain mutations. Proteins. 2008;72:779–792. doi: 10.1002/prot.21970. [DOI] [PubMed] [Google Scholar]
- 18.Machida K, Eschrich S, Li J, Bai Y, et al. Characterizing tyrosine phosphorylation signaling in lung cancer using SH2 profiling. PLoS One. 2010;5:e13470. doi: 10.1371/journal.pone.0013470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Machida K, Thompson CM, Dierck K, Jablonowski K, et al. High-throughput phosphotyrosine profiling using SH2 domains. Mol Cell. 2007;26:899–915. doi: 10.1016/j.molcel.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 20.Pawson T, Warner N. Oncogenic re-wiring of cellular signaling pathways. Oncogene. 2007;26:1268–1275. doi: 10.1038/sj.onc.1210255. [DOI] [PubMed] [Google Scholar]
- 21.Jin J, Xie X, Chen C, Park JG, et al. Eukaryotic protein domains as functional units of cellular evolution. Sci Signal. 2009;2:ra76. doi: 10.1126/scisignal.2000546. [DOI] [PubMed] [Google Scholar]
- 22.Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 23.Pawson T, Nash P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000;14:1027–1047. [PubMed] [Google Scholar]
- 24.Chothia C, Gough J. Genomic and structural aspects of protein evolution. Biochem J. 2009;419:15–28. doi: 10.1042/BJ20090122. [DOI] [PubMed] [Google Scholar]
- 25.Vogel C, Chothia C. Protein family expansions and biological complexity. PLoS Comput Biol. 2006;2:e48. doi: 10.1371/journal.pcbi.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohammad DH, Yaffe MB. 14-3-3 proteins, FHA domains and BRCT domains in the DNA damage response. DNA Repair (Amst) 2009;8:1009–1017. doi: 10.1016/j.dnarep.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen C, Nott TJ, Jin J, Pawson T. Deciphering arginine methylation: Tudor tells the tale. Nat Rev Mol Cell Biol. 2011;12:629–642. doi: 10.1038/nrm3185. [DOI] [PubMed] [Google Scholar]
- 28.Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol. 2009;10:659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010;11:861–871. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng C, Lu Z, Xie Z, Cheng Z, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics. 2011 doi: 10.1074/mcp.M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan M, Luo H, Lee S, Jin F, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Z, Tan M, Xie Z, Dai L, et al. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol. 2011;7:58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linding R, Russell RB, Neduva V, Gibson TJ. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003;31:3701–3708. doi: 10.1093/nar/gkg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zarrinpar A, Bhattacharyya RP, Lim WA. The structure and function of proline recognition domains. Sci STKE. 2003;2003:RE8. doi: 10.1126/stke.2003.179.re8. [DOI] [PubMed] [Google Scholar]
- 35.Berry DM, Nash P, Liu SK, Pawson T, McGlade CJ. A high-affinity Arg-X-X-Lys SH3 binding motif confers specificity for the interaction between Gads and SLP-76 in T cell signaling. Curr Biol. 2002;12:1336–1341. doi: 10.1016/s0960-9822(02)01038-2. [DOI] [PubMed] [Google Scholar]
- 36.Stamenova SD, French ME, He Y, Francis SA, et al. Ubiquitin binds to and regulates a subset of SH3 domains. Mol Cell. 2007;25:273–284. doi: 10.1016/j.molcel.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- 38.Zimmermann P, Meerschaert K, Reekmans G, Leenaerts I, et al. PIP(2)-PDZ domain binding controls the association of syntenin with the plasma membrane. Mol Cell. 2002;9:1215–1225. doi: 10.1016/s1097-2765(02)00549-x. [DOI] [PubMed] [Google Scholar]
- 39.Wu H, Feng W, Chen J, Chan LN, et al. PDZ domains of Par-3 as potential phosphoinositide signaling integrators. Mol Cell. 2007;28:886–898. doi: 10.1016/j.molcel.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 40.Mongiovi AM, Romano PR, Panni S, Mendoza M, et al. A novel peptide-SH3 interaction. EMBO J. 1999;18:5300–5309. doi: 10.1093/emboj/18.19.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jia CY, Nie J, Wu C, Li C, Li SS. Novel Src homology 3 domain-binding motifs identified from proteomic screen of a Pro-rich region. Mol Cell Proteomics. 2005;4:1155–1166. doi: 10.1074/mcp.M500108-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Sparks AB, Rider JE, Hoffman NG, Fowlkes DM, et al. Distinct ligand preferences of Src homology 3 domains from Src, Yes, Abl, Cortactin, p53bp2, PLCgamma, Crk, and Grb2. Proc Natl Acad Sci U S A. 1996;93:1540–1544. doi: 10.1073/pnas.93.4.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu BA, Jablonowski K, Shah EE, Engelmann BW, et al. SH2 domains recognize contextual peptide sequence information to determine selectivity. Mol Cell Proteomics. 2010;9:2391–2404. doi: 10.1074/mcp.M110.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 45.Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 46.Lu PJ, Zhou XZ, Shen M, Lu KP. Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science. 1999;283:1325–1328. doi: 10.1126/science.283.5406.1325. [DOI] [PubMed] [Google Scholar]
- 47.Durocher D, Henckel J, Fersht AR, Jackson SP. The FHA domain is a modular phosphopeptide recognition motif. Mol Cell. 1999;4:387–394. doi: 10.1016/s1097-2765(00)80340-8. [DOI] [PubMed] [Google Scholar]
- 48.Kavanaugh WM, Turck CW, Williams LT. PTB domain binding to signaling proteins through a sequence motif containing phosphotyrosine. Science. 1995;268:1177–1179. doi: 10.1126/science.7539155. [DOI] [PubMed] [Google Scholar]
- 49.Chung HJ, Huang YH, Lau LF, Huganir RL. Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J Neurosci. 2004;24:10248–10259. doi: 10.1523/JNEUROSCI.0546-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gardoni F, Mauceri D, Fiorentini C, Bellone C, et al. CaMKII-dependent phosphorylation regulates SAP97/NR2A interaction. J Biol Chem. 2003;278:44745–44752. doi: 10.1074/jbc.M303576200. [DOI] [PubMed] [Google Scholar]
- 51.Lee JY, Chiu YH, Asara J, Cantley LC. Inhibition of PI3K binding to activators by serine phosphorylation of PI3K regulatory subunit p85{alpha} Src homology-2 domains. Proc Natl Acad Sci U S A. 2011;108:14157–14162. doi: 10.1073/pnas.1107747108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nash P, Tang X, Orlicky S, Chen Q, et al. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- 53.Dueber JE, Mirsky EA, Lim WA. Engineering synthetic signaling proteins with ultrasensitive input/output control. Nat Biotechnol. 2007;25:660–662. doi: 10.1038/nbt1308. [DOI] [PubMed] [Google Scholar]
- 54.Mittag T, Marsh J, Grishaev A, Orlicky S, et al. Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase. Structure. 2010;18:494–506. doi: 10.1016/j.str.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.London N, Movshovitz-Attias D, Schueler-Furman O. The structural basis of peptide-protein binding strategies. Structure. 2010;18:188–199. doi: 10.1016/j.str.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 56.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 57.Finkelstein AV, Janin J. The price of lost freedom: entropy of bimolecular complex formation. Protein Eng. 1989;3:1–3. doi: 10.1093/protein/3.1.1. [DOI] [PubMed] [Google Scholar]
- 58.Bradshaw JM, Mitaxov V, Waksman G. Investigation of phosphotyrosine recognition by the SH2 domain of the Src kinase. J Mol Biol. 1999;293:971–985. doi: 10.1006/jmbi.1999.3190. [DOI] [PubMed] [Google Scholar]
- 59.Gan W, Roux B. Binding specificity of SH2 domains: insight from free energy simulations. Proteins. 2009;74:996–1007. doi: 10.1002/prot.22209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fuxreiter M, Tompa P, Simon I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics. 2007;23:950–956. doi: 10.1093/bioinformatics/btm035. [DOI] [PubMed] [Google Scholar]
- 61.Marengere LE, Songyang Z, Gish GD, Schaller MD, et al. SH2 domain specificity and activity modified by a single residue. Nature. 1994;369:502–505. doi: 10.1038/369502a0. [DOI] [PubMed] [Google Scholar]
- 62.Gorelik M, Stanger K, Davidson AR. A Conserved residue in the yeast Bem1p SH3 domain maintains the high level of binding specificity required for function. J Biol Chem. 2011;286:19470–19477. doi: 10.1074/jbc.M111.229294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alexander J, Lim D, Joughin BA, Hegemann B, et al. Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context-dependent mitotic signaling. Science signaling. 4:ra42. doi: 10.1126/scisignal.2001796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bradbury AR, Sidhu S, Dubel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nature biotechnology. 29:245–254. doi: 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Friesen WJ, Darby MK. Specific RNA binding proteins constructed from zinc fingers. Nature structural biology. 1998;5:543–546. doi: 10.1038/794. [DOI] [PubMed] [Google Scholar]
- 66.Huang H, Li L, Wu C, Schibli D, et al. Defining the specificity space of the human SRC homology 2 domain. Mol Cell Proteomics. 2008;7:768–784. doi: 10.1074/mcp.M700312-MCP200. [DOI] [PubMed] [Google Scholar]
- 67.Stiffler MA, Chen JR, Grantcharova VP, Lei Y, et al. PDZ domain binding selectivity is optimized across the mouse proteome. Science. 2007;317:364–369. doi: 10.1126/science.1144592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaneko T, Huang H, Zhao B, Li L, et al. Loops govern SH2 domain specificity by controlling access to binding pockets. Science signaling. 3:ra34. doi: 10.1126/scisignal.2000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stein A, Aloy P. Contextual specificity in peptide-mediated protein interactions. PLoS One. 2008;3:e2524. doi: 10.1371/journal.pone.0002524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bae JH, Lew ED, Yuzawa S, Tome F, et al. The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell. 2009;138:514–524. doi: 10.1016/j.cell.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tanaka M, Gupta R, Mayer BJ. Differential inhibition of signaling pathways by dominant-negative SH2/SH3 adapter proteins. Mol Cell Biol. 1995;15:6829–6837. doi: 10.1128/mcb.15.12.6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bashor CJ, Helman NC, Yan S, Lim WA. Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science (New York, N Y) 2008;319:1539–1543. doi: 10.1126/science.1151153. [DOI] [PubMed] [Google Scholar]
- 73.Pawson T. Dynamic control of signaling by modular adaptor proteins. Curr Opin Cell Biol. 2007;19:112–116. doi: 10.1016/j.ceb.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 74.Gruenheid S, DeVinney R, Bladt F, Goosney D, et al. Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat Cell Biol. 2001;3:856–859. doi: 10.1038/ncb0901-856. [DOI] [PubMed] [Google Scholar]
- 75.Howard PL, Chia MC, Del Rizzo S, Liu FF, Pawson T. Redirecting tyrosine kinase signaling to an apoptotic caspase pathway through chimeric adaptor proteins. Proc Natl Acad Sci U S A. 2003;100:11267–11272. doi: 10.1073/pnas.1934711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gureasko J, Galush WJ, Boykevisch S, Sondermann H, et al. Membrane-dependent signal integration by the Ras activator Son of sevenless. Nature structural & molecular biology. 2008;15:452–461. doi: 10.1038/nsmb.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klein P, Pawson T, Tyers M. Mathematical modeling suggests cooperative interactions between a disordered polyvalent ligand and a single receptor site. Curr Biol. 2003;13:1669–1678. doi: 10.1016/j.cub.2003.09.027. [DOI] [PubMed] [Google Scholar]
- 78.Borg M, Mittag T, Pawson T, Tyers M, et al. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc Natl Acad Sci U S A. 2007;104:9650–9655. doi: 10.1073/pnas.0702580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kholodenko BN. Four-dimensional organization of protein kinase signaling cascades: the roles of diffusion, endocytosis and molecular motors. The Journal of experimental biology. 2003;206:2073–2082. doi: 10.1242/jeb.00298. [DOI] [PubMed] [Google Scholar]
- 80.Kholodenko BN. Cell-signalling dynamics in time and space. Nature reviews. 2006;7:165–176. doi: 10.1038/nrm1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kholodenko BN, Hoek JB, Westerhoff HV. Why cytoplasmic signalling proteins should be recruited to cell membranes. Trends in cell biology. 2000;10:173–178. doi: 10.1016/s0962-8924(00)01741-4. [DOI] [PubMed] [Google Scholar]
- 82.Groves JT, Kuriyan J. Molecular mechanisms in signal transduction at the membrane. Nature structural & molecular biology. 17:659–665. doi: 10.1038/nsmb.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li J, Nayak S, Mrksich M. Rate enhancement of an interfacial biochemical reaction through localization of substrate and enzyme by an adaptor domain. The journal of physical chemistry. 114:15113–15118. doi: 10.1021/jp102820e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Amit I, Wides R, Yarden Y. Evolvable signaling networks of receptor tyrosine kinases: relevance of robustness to malignancy and to cancer therapy. Molecular systems biology. 2007;3:151. doi: 10.1038/msb4100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barabasi AL, Albert R. Emergence of scaling in random networks. Science (New York, N Y) 1999;286:509–512. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 86.Hlavacek WS, Faeder JR, Blinov ML, Perelson AS, Goldstein B. The complexity of complexes in signal transduction. Biotechnology and bioengineering. 2003;84:783–794. doi: 10.1002/bit.10842. [DOI] [PubMed] [Google Scholar]
- 87.Brown CJ, Takayama S, Campen AM, Vise P, et al. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol. 2002;55:104–110. doi: 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]
- 88.Neduva V, Russell RB. Linear motifs: evolutionary interaction switches. FEBS Lett. 2005;579:3342–3345. doi: 10.1016/j.febslet.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 89.Haynes C, Oldfield CJ, Ji F, Klitgord N, et al. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol. 2006;2:e100. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, et al. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Frank R, Heikens W, Heisterberg-Moutsis G, Blocker H. A new general approach for the simultaneous chemical synthesis of large numbers of oligonucleotides: segmental solid supports. Nucleic Acids Res. 1983;11:4365–4377. doi: 10.1093/nar/11.13.4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Geysen HM, Meloen RH, Barteling SJ. Use of peptide synthesis to probe viral antigens for epitopes to a resolution of a single amino acid. Proc Natl Acad Sci U S A. 1984;81:3998–4002. doi: 10.1073/pnas.81.13.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Frank R. The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports--principles and applications. J Immunol Methods. 2002;267:13–26. doi: 10.1016/s0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- 94.Boisguerin P, Leben R, Ay B, Radziwill G, et al. An improved method for the synthesis of cellulose membrane-bound peptides with free C termini is useful for PDZ domain binding studies. Chem Biol. 2004;11:449–459. doi: 10.1016/j.chembiol.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 95.Rodriguez M, Li SS, Harper JW, Songyang Z. An oriented peptide array library (OPAL) strategy to study protein-protein interactions. J Biol Chem. 2004;279:8802–8807. doi: 10.1074/jbc.M311886200. [DOI] [PubMed] [Google Scholar]
- 96.Weiser AA, Or-Guil M, Tapia V, Leichsenring A, et al. SPOT synthesis: reliability of array-based measurement of peptide binding affinity. Anal Biochem. 2005;342:300–311. doi: 10.1016/j.ab.2005.04.033. [DOI] [PubMed] [Google Scholar]
- 97.Wolf-Yadlin A, Sevecka M, MacBeath G. Dissecting protein function and signaling using protein microarrays. Curr Opin Chem Biol. 2009;13:398–405. doi: 10.1016/j.cbpa.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.MacBeath G. Protein microarrays and proteomics. Nat Genet. 2002;32(Suppl):526–532. doi: 10.1038/ng1037. [DOI] [PubMed] [Google Scholar]
- 99.Austin J, Holway AH. Contact printing of protein microarrays. Methods Mol Biol. 2011;785:379–394. doi: 10.1007/978-1-61779-286-1_25. [DOI] [PubMed] [Google Scholar]
- 100.Kim J, Daniel J, Espejo A, Lake A, et al. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Espejo A, Cote J, Bednarek A, Richard S, Bedford MT. A protein-domain microarray identifies novel protein-protein interactions. Biochem J. 2002;367:697–702. doi: 10.1042/BJ20020860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Smith GP, Petrenko VA. Phage Display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 103.Sidhu SS, Fairbrother WJ, Deshayes K. Exploring protein-protein interactions with phage display. Chembiochem. 2003;4:14–25. doi: 10.1002/cbic.200390008. [DOI] [PubMed] [Google Scholar]
- 104.Tonikian R, Zhang Y, Sazinsky SL, Currell B, et al. A specificity map for the PDZ domain family. PLoS Biol. 2008;6:e239. doi: 10.1371/journal.pbio.0060239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ernst A, Sazinsky SL, Hui S, Currell B, et al. Rapid evolution of functional complexity in a domain family. Sci Signal. 2009;2:ra50. doi: 10.1126/scisignal.2000416. [DOI] [PubMed] [Google Scholar]
- 106.Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, et al. High-resolution mapping of protein sequence-function relationships. Nat Methods. 2010;7:741–746. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003;21:255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- 108.Gingras AC, Gstaiger M, Raught B, Aebersold R. Analysis of protein complexes using mass spectrometry. Nat Rev Mol Cell Biol. 2007;8:645–654. doi: 10.1038/nrm2208. [DOI] [PubMed] [Google Scholar]
- 109.Ahrens CH, Brunner E, Qeli E, Basler K, Aebersold R. Generating and navigating proteome maps using mass spectrometry. Nat Rev Mol Cell Biol. 2010;11:789–801. doi: 10.1038/nrm2973. [DOI] [PubMed] [Google Scholar]
- 110.Ingham RJ, Colwill K, Howard C, Dettwiler S, et al. WW domains provide a platform for the assembly of multiprotein networks. Mol Cell Biol. 2005;25:7092–7106. doi: 10.1128/MCB.25.16.7092-7106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lowery DM, Clauser KR, Hjerrild M, Lim D, et al. Proteomic screen defines the Polo-box domain interactome and identifies Rock2 as a Plk1 substrate. EMBO J. 2007;26:2262–2273. doi: 10.1038/sj.emboj.7601683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Christofk HR, Wu N, Cantley LC, Asara JM. Proteomic screening method for phosphopeptide motif binding proteins using peptide libraries. J Proteome Res. 2011;10:4158–4164. doi: 10.1021/pr200578n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weng Z, Rickles RJ, Feng S, Richard S, et al. Structure-function analysis of SH3 domains: SH3 binding specificity altered by single amino acid substitutions. Mol Cell Biol. 1995;15:5627–5634. doi: 10.1128/mcb.15.10.5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Song E, Gao S, Tian R, Ma S, et al. A high efficiency strategy for binding property characterization of peptide-binding domains. Mol Cell Proteomics. 2006;5:1368–1381. doi: 10.1074/mcp.M600072-MCP200. [DOI] [PubMed] [Google Scholar]
- 115.Tong AH, Drees B, Nardelli G, Bader GD, et al. A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science. 2002;295:321–324. doi: 10.1126/science.1064987. [DOI] [PubMed] [Google Scholar]
- 116.Lievens S, Lemmens I, Tavernier J. Mammalian two-hybrids come of age. Trends Biochem Sci. 2009;34:579–588. doi: 10.1016/j.tibs.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lievens S, Vanderroost N, Van der Heyden J, Gesellchen V, et al. Array MAPPIT: high-throughput interactome analysis in mammalian cells. J Proteome Res. 2009;8:877–886. doi: 10.1021/pr8005167. [DOI] [PubMed] [Google Scholar]
- 118.Barrios-Rodiles M, Brown KR, Ozdamar B, Bose R, et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science. 2005;307:1621–1625. doi: 10.1126/science.1105776. [DOI] [PubMed] [Google Scholar]
- 119.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu BA, Jablonowski K, Engelmann BW, Higginbotham K, Nash PD. Src Homology 2 Domain Binding Sites in Insulin, IGF-1 and FGF Receptor Mediated Signaling Networks Reveal an Extensive Potential Interactome. 2012 doi: 10.1186/1478-811X-10-27. publication in preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fenyo D, Eriksson J, Beavis R. Mass spectrometric protein identification using the global proteome machine. Methods Mol Biol. 2010;673:189–202. doi: 10.1007/978-1-60761-842-3_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Choi H, Larsen B, Lin ZY, Breitkreutz A, et al. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat Methods. 2011;8:70–73. doi: 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sardiu ME, Cai Y, Jin J, Swanson SK, et al. Probabilistic assembly of human protein interaction networks from label-free quantitative proteomics. Proc Natl Acad Sci U S A. 2008;105:1454–1459. doi: 10.1073/pnas.0706983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sylvester JE, Bray TS, Kron SJ. Annotator: Post-processing Software for generating function-based signatures from quantitative mass spectrometry. J Proteome Res. 2012 doi: 10.1021/pr200460s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ito T, Chiba T, Ozawa R, Yoshida M, et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci U S A. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Huang H, Jedynak BM, Bader JS. Where have all the interactions gone? Estimating the coverage of two-hybrid protein interaction maps. PLoS Comput Biol. 2007;3:e214. doi: 10.1371/journal.pcbi.0030214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hart GT, Ramani AK, Marcotte EM. How complete are current yeast and human protein-interaction networks? Genome Biol. 2006;7:120. doi: 10.1186/gb-2006-7-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Prasad TS, Kandasamy K, Pandey A. Human Protein Reference Database and Human Proteinpedia as discovery tools for systems biology. Methods Mol Biol. 2009;577:67–79. doi: 10.1007/978-1-60761-232-2_6. [DOI] [PubMed] [Google Scholar]
- 130.Stark C, Breitkreutz BJ, Reguly T, Boucher L, et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zanzoni A, Montecchi-Palazzi L, Quondam M, Ausiello G, et al. MINT: a Molecular INTeraction database. FEBS Lett. 2002;513:135–140. doi: 10.1016/s0014-5793(01)03293-8. [DOI] [PubMed] [Google Scholar]
- 132.Jones RB, Gordus A, Krall JA, MacBeath G. A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature. 2006;439:168–174. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- 133.Bisson N, James DA, Ivosev G, Tate SA, et al. Selected reaction monitoring mass spectrometry reveals the dynamics of signaling through the GRB2 adaptor. Nat Biotechnol. 2011;29:653–658. doi: 10.1038/nbt.1905. [DOI] [PubMed] [Google Scholar]
- 134.Olsen JV, Vermeulen M, Santamaria A, Kumar C, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 135.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Borisov N, Aksamitiene E, Kiyatkin A, Legewie S, et al. Systems-level interactions between insulin-EGF networks amplify mitogenic signaling. Mol Syst Biol. 2009;5:256. doi: 10.1038/msb.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vetter SW, Zhang ZY. Probing the phosphopeptide specificities of protein tyrosine phosphatases, SH2 and PTB domains with combinatorial library methods. Curr Protein Pept Sci. 2002;3:365–397. doi: 10.2174/1389203023380594. [DOI] [PubMed] [Google Scholar]
- 138.Songyang Z, Shoelson SE, McGlade J, Olivier P, et al. Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Mol Cell Biol. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tonikian R, Zhang Y, Boone C, Sidhu SS. Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries. Nat Protoc. 2007;2:1368–1386. doi: 10.1038/nprot.2007.151. [DOI] [PubMed] [Google Scholar]
- 140.Baldi P, Chauvin Y, Hunkapiller T, McClure MA. Hidden Markov models of biological primary sequence information. Proc Natl Acad Sci U S A. 1994;91:1059–1063. doi: 10.1073/pnas.91.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Krogh A, Brown M, Mian IS, Sjolander K, Haussler D. Hidden Markov models in computational biology. Applications to protein modeling. J Mol Biol. 1994;235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- 142.Sonnhammer EL, Eddy SR, Birney E, Bateman A, Durbin R. Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 1998;26:320–322. doi: 10.1093/nar/26.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Horan K, Shelton CR, Girke T. Predicting conserved protein motifs with Sub-HMMs. BMC Bioinformatics. 2010;11:205. doi: 10.1186/1471-2105-11-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Puntervoll P, Linding R, Gemund C, Chabanis-Davidson S, et al. ELM server: A new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Res. 2003;31:3625–3630. doi: 10.1093/nar/gkg545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Panni S, Montecchi-Palazzi L, Kiemer L, Cabibbo A, et al. Combining peptide recognition specificity and context information for the prediction of the 14-3-3-mediated interactome in S. cerevisiae and H. sapiens. Proteomics. 2011;11:128–143. doi: 10.1002/pmic.201000030. [DOI] [PubMed] [Google Scholar]
- 146.Kaustov L, Ouyang H, Amaya M, Lemak A, et al. Recognition and specificity determinants of the human cbx chromodomains. J Biol Chem. 2011;286:521–529. doi: 10.1074/jbc.M110.191411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mishra P, Socolich M, Wall MA, Graves J, et al. Dynamic scaffolding in a G protein-coupled signaling system. Cell. 2007;131:80–92. doi: 10.1016/j.cell.2007.07.037. [DOI] [PubMed] [Google Scholar]
- 148.Carducci M, Perfetto L, Briganti L, Paoluzi S, et al. The protein interaction network mediated by human SH3 domains. Biotechnol Adv. 2011 doi: 10.1016/j.biotechadv.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 149.Tonikian R, Xin X, Toret CP, Gfeller D, et al. Bayesian modeling of the yeast SH3 domain interactome predicts spatiotemporal dynamics of endocytosis proteins. PLoS Biol. 2009;7:e1000218. doi: 10.1371/journal.pbio.1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lenfant N, Polanowska J, Bamps S, Omi S, et al. A genome-wide study of PDZ-domain interactions in C. elegans reveals a high frequency of non-canonical binding. BMC Genomics. 2010;11:671. doi: 10.1186/1471-2164-11-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Smith MJ, Hardy WR, Murphy JM, Jones N, Pawson T. Screening for PTB domain binding partners and ligand specificity using proteome-derived NPXY peptide arrays. Mol Cell Biol. 2006;26:8461–8474. doi: 10.1128/MCB.01491-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hu H, Columbus J, Zhang Y, Wu D, et al. A map of WW domain family interactions. Proteomics. 2004;4:643–655. doi: 10.1002/pmic.200300632. [DOI] [PubMed] [Google Scholar]