

Abstract
Arrays of subnanoliter wells (nanowells) provide a useful system to isolate single cells and analyze their secreted proteins. Two general approaches have emerged: one that uses open arrays and local capture of secreted proteins, and a second (called microengraving) that relies on closed arrays to capture secreted proteins on a solid substrate, which is subsequently removed from the array. However, the design and operating parameters for efficient capture from these two approaches to analyze single-cell secretion have not been extensively considered. Using numerical simulations, we analyzed the operational envelope for both open and closed formats, as a function of the spatial distribution of capture ligands, their affinities for the protein, and the rates of single-cell secretion. Based on these analyses, we present a modified approach to capture secreted proteins in-well for highly active secreting cells. This simple method for in-well detection should facilitate rapid identification of cell lines with high specific productivities.
The number of techniques for single-cell analysis that rely on microfabricated systems has expanded rapidly. Several approaches have been developed to monitor cell–cell interactions,1,2 cell proliferation and differentiation,3 and protein secretion4−7 with single-cell resolution. In particular, the use of arrays of subnanoliter wells (nanowells) for the characterization of cells secreting antibodies or other proteins have shown great potential for accelerating the discovery of novel antigen-specific antibodies2,8,910 evaluating immunological responses,11 and selecting clonal cell lines with high specific productivities.12,13
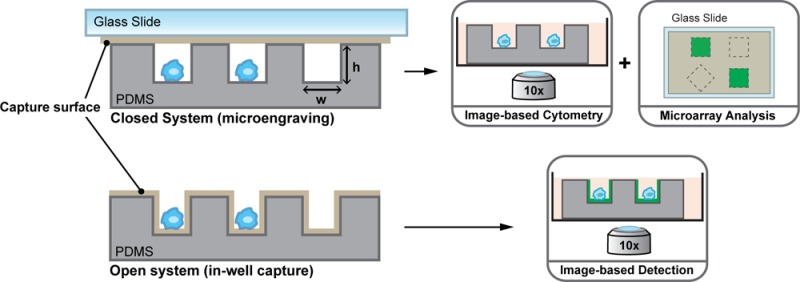
Conventional methods for analyzing populations of cells, such as Enzyme Linked Immunospot (ELISpot) and flow cytometry, provide single-cell resolution of secreted proteins, but they are often destructive in nature. Single-cell analysis based on arrays of nanowells offers several advantages over traditional assays, including control of mechanical and environmental cues,14,15 the ability to follow cellular responses over long time scales (days),16 and the ability to recover cells of interest,6,17 among others. To date, two distinct approaches to capture and quantify proteins secreted from cells isolated on arrays of nanowells have emerged. Microengraving involves a configuration wherein the wells are closed (for short intervals, ∼1–3 h) with a glass slide supporting immobilized capture antibodies (Figure 1A).6−8,14 Other reported approaches have used open arrays of wells modified with capture antibodies either on the bottoms of the wells5 or on the planar surfaces between wells10 to bind proteins released from cells over hours to days of incubation (see Figure 1B, as well as Figure S1 in the Supporting Information). Capture of analytes at or near the surface of the nanowells relies on principles similar to ELISpot, with the added advantage that isolating cells in individual wells allows the analysis of both secreted proteins and cell cytometry, as well as subsequent recovery of specific cells by microscopy and manipulation.
Figure 1.

Schematic of (A) microengraving and (B) in-well capture assays for single-cell analysis of secreted proteins. Arrays of nanowells allow selective capture of analytes at the surface modified with capture antibody or ligand. The spatial addresses of the wells allow identification of specific cells secreting proteins of interest, and their subsequent recovery. In panel A, microengraving involves a closed configuration wherein the wells are sealed with a solid substrate such as a glass slide coated with an affinity ligand that captures secreted protein. The solid substrate is removed and further processed using fluorescently labeled antibodies. Panel B illustrates in-well capture of secreted proteins from single cells. The secreted protein is captured on the surface of the wells coated with affinity ligands. All steps from incubation with fluorescent detection antibodies and washing are performed on the device containing the cells. In panel (C), the scale for the simulations in this work is expressed in units of pmol/m2, which is convenient to generalize secretion of any protein with different molecular weights. For convenience, we have included the scales representing concentrations in units of fg/μg2 for the case of antibodies (∼150 kDa) and cytokines (∼15 kDa).
While both configurations have demonstrated useful measurements of secretion from single cells, relatively little consideration has been given to how the different underlying physical principles for each may govern their operational range, sensitivity, and suitability for various applications. We have previously considered the time-dependent mass transfer of proteins secreted from cells during microengraving, as well as their accumulation on a substrate by affinity binding, using numerical simulations.6 Here, we have expanded our computational analysis to examine further both open and closed configurations of arrays of nanowells for capturing secreted proteins. We considered how operating parameters such as incubation time, ligand affinity, rates of secretion, and spatial geometries impacted the time required to reach steady-state conditions and the overall efficiency (defined by the maximum total quantities of proteins captured). Based on these computational analyses, we demonstrate a modified approach for improved open-well capture of secreted proteins that offers improved capture efficiencies. Together, these data and analyses provide a conceptual framework and predicted theoretical efficiency for the design and use of arrays of nanowells as a bioanalytical tool for detecting single-cell protein secretion.
Materials and Methods
Computational Analysis of Analyte Transport and Binding
Cells were assumed to secrete proteins (e.g., cytokine or antibody) at a constant rate:
| 1 |
where Ntotal is the total number of molecules, κ is a constant that describes the rate of secretion, and t is the time secreted proteins diffuse radially into the media. We note that the rate of secretion for a live cell likely fluctuates over time, depending on extrinsic factors, such as the quality of its environment, as well as intrinsic factors, including its secretory capacity, state of activation, and stage in the cell cycle.18 Nonetheless, assuming a constant rate in this model is useful for understanding the relationship between the quantities of protein secreted and that captured on the surface, and evaluating the upper bounds of analyte binding over the course of an experiment.
The transport of analyte in the media was described by a simple diffusion model:
| 2 |
where C is the analyte concentration and D is the diffusion coefficient.
The capture surface was assumed to be coated uniformly with antibodies against the analyte (either cytokines or antibodies) and the binding reaction described by a reversible equilibrium process:
| 3 |
where B is the concentration of free binding sites at the surface and Cs is the concentration of analyte–antibody complex at the surface. The parameters kon and koff are the association and dissociation constants, respectively. Nonuniformity of the capture reagent at the molecular scale could affect these predictions, although we note that the imaging systems typically employed for assessing the capture of analytes experimentally average over length scales many-fold larger (approximately square micrometers). Other gross artifacts related to the manufacturing and preparation of the devices that could affect the interfacial uniformity such as bubbles, protein aggregations, and topographical imperfections are outside the scope of this analysis.
The walls of the device were modeled as impermeable to the proteins captured. Polydimethylsiloxane (PDMS) is permeable to certain gases, such as oxygen,19 but is impermeable to proteins and hydrophilic small molecules.20 Permeability is also generally reduced upon plasma treatment used to create hydroxyl groups, making the exposed surface hydrophilic.19 Nonspecific binding to the surface of the wells was considered negligible (Kd in the millimolar range) compared to specific capture (Kd ≈ 10–0.1 nM). Typically, the capture surface is blocked with BSA before adding the suspension of cells, and the media used for cell culture contains bovine serum (typically 10%). In this configuration, the total concentration of proteins (predominantly serum albumin) greatly exceeds that of specific analytes. Based on their abundance and expected equilibrium partitioning at the interfaces, these other proteins would be expected to represent the most prevalent ones adsorbed nonspecifically on the surfaces of the well.
The reaction and diffusion at the surface is described by a first-order Langmuir adsorption model, where the diffusive flux (−n⃗·(−D∇C)) is balanced against the reaction rate.
| 4 |
where B0 is the initial concentration of immobilized capture ligand or antibodies (i.e., binding sites) and (B0–Cs) describes the density of available binding sites. Since the capture antibodies are immobilized on the surface, the diffusion term (Ds) becomes equal to zero. Thus, the equation describing the boundary conditions is given by
| 5 |
For these simulations, we assumed that the binding of antibody and analyte occurs at a 1:1 stoichiometry. Furthermore, we assumed that the initial concentration (at t = 0) of free analyte (C) and bound analyte (Cs) are negligible (Cs = 0, C = 0). The boundary condition for the edges of the medium above the wells was C = 0, assuming that the concentration far from a secreting cell is negligible. All simulations were performed using COMSOL Multiphysics 4.3. The range of values for all parameters used for the simulations was taken from the literature and summarized in Table 1. The scale used in these simulations to represent the amount of bound proteins is expressed in units of pmol/m2; however, we have included a scale comparing the values in terms of pmol/m2 to fg/μm2 in Figure 1C to simplify any conversion to mass of protein bound to the surface.
Table 1. Values of Parameters Used in the Finite-Element Analysis.
| parameter | value |
|---|---|
| cell diameter | 10 μm |
| density of total binding sites, B0 | 1 × 10–9 mol/m2 (ref (21)) |
| dissociation binding constant, Kd = koff/kon | |
| #1 | 10 nM (kon = 1 × 105 1/(M s), koff = 1 × 10–3 s–1) |
| #2 | 0.1 nM (kon = 1 × 106 1/(M s), koff = 1 × 10–4 s–1) |
| diffusion coefficient, D | 3 × 10–11 m2/s (ref (22)) |
Preparation of Arrays of Nanowells
Arrays of nanowells were manufactured by injecting polydimethylsiloxane (PDMS, Dow Corning) prepared at a 10:1 base/catalyst weight ratio into a custom-built mold encasing a silicon master micropatterned with SU-8 by photolithography. Arrays of nanowells were cured at 70 °C for 4 h. Each array comprised 72 × 24 blocks, each containing a 7 × 7 (50 μm × 50 μm × 50 μm) subarray of nanowells with a center-to-center distance of 100 μm, a 12 × 12 (30 μm × 30 μm × 30 μm) subarray with a center-to-center distance of 60 μm, or a 24 × 24 (15 μm × 15 μm × 15 μm) subarray with a center-to-center distance of 30 μm. The elastomeric arrays adhered directly to a 3 in. × 1 in. glass slide forming a 1-mm-thick layer. The PDMS device was plasma oxidized for 90–120 s using a plasma cleaner (Harrick Plasma Model PDC-001, 115 V) before adsorption of the antigen.
Cell Culture
A hybridoma cell line (No. 099-01, obtained from the Statens Serum Institut) secreting anti-ovalbumin (anti-OVA) IgG was cultured in DMEM (10% FBS, 100 U/mL penicillin and 100 mg/mL streptomycin), as described previously.8 Cells were split every 2–3 days under sterile conditions. The cells were centrifuged at 1500 rpm and resuspended in fresh media to remove excess secreted antibodies (that accumulate over days in culture) before each experiment. The devices were placed in a four-well slide processing dish (Nunc, Thermo Scientific) and rinsed with media. After aspirating the media, ∼300 μL of the cells (at 106 cells/mL in media) were deposited on the array by slowly pipetting the volume over the entire area of the device that had been previously blocked with bovine serum albumin (BSA) (see below). Cells were allowed to settle for 5 min; excess cells were removed by gently aspirating and replacing media.
In-Well Capture and Detection of Secreted Proteins from Single Cells
Chicken ovalbumin (Pierce, Thermo Scientific) was immobilized on the surface of the arrays by incubation overnight with a concentration of 500 μg/mL in phosphate-buffered saline (PBS) and then blocked with 1 mg/mL PBS/BSA for 30 min before adding cells. After the cells were seeded into the array and incubated for 2 h, the cells were rinsed with media and incubated with fluorescently labeled (Alexa Fluor 488) goat anti-mouse IgG (H+L) (Molecular Probes, Life Technologies) at a concentration of 3 μg/mL and calcein violet (Molecular Probes, Life Technologies) to label live cells in PBS containing 1 mg/mL BSA for 30 min at room temperature. The cells were rinsed first with PBS, then with media, and imaged. The elastomeric substrates were covered with a No. 2 Lifter slip and imaged using an automated, inverted epifluorescence microscope (Axio Observer, 10×/0.3 NA objective; Carl Zeiss, Jena, Germany) with an EM-CCD camera (ImagEM; Hamamatsu Photonics, Hamamatsu, Japan).
Microengraving
Epoxy-functionalized glass slides (Super epoxy 2, Arrayit Corporation) were coated with 100 μg/mL ovalbumin in PBS overnight at 4 °C and then rinsed with PBS and blocked with 1 mg/mL BSA/PBS for 30 min. After incubation of the cell-loaded array, the ovalbumin-coated glass slide was then placed onto the loaded array for printing. The microarray and glass slide were held together by compression in a hybridization chamber (Agilent Technologies, G2534A) and incubated for 2 h at 37 °C with 5% CO2 as described previously.6 The glass slide was then separated from the array and placed in PBS. After microengraving, slides were incubated for 30 min with blocking buffer (PBS, 3% w/v milk powder and 0.05% (v/v) Tween-20), washed with PBST (PBS + 0.05% v/v Tween-20), and then incubated with fluorescent antibodies (Alexa Fluor 488 goat anti-mouse IgG) at 2 μg/mL for 45 min at 25 °C. The slides were washed with PBST and PBS, rinsed briefly with water, and dried with a N2 stream. Slides were scanned using a Genepix 4200AL microarray scanner (Molecular Devices). The median fluorescence intensity of each element was analyzed using Genepix Pro. Data extracted from both on-chip cytometry and printed proteins were matched in Microsoft Excel using unique identifiers assigned to each well within the array. The dataset was filtered to include wells containing only single cells.
On-Chip Image-Based Cytometry before Microengraving
Before imaging, cells were stained with calcein violet (Invitrogen). The cell-loaded arrays of nanowells were mounted face-up on the microscope with a coverslip placed on top of the array. Images were acquired on an automated inverted epifluorescence microscope (Carl Zeiss). Transmitted light and epifluoresence micrographs were collected block by block. The resulting collection of images was analyzed using a custom program to determine the number of cells present in each well and the mean fluorescent intensity of each label. Only viable cells (with a calcein fluorescent intensity of more than three standard deviations above background) were considered for the analysis.
Results
Theoretical Analysis of Surface Capture of Proteins on Open Arrays of Nanowells
Previous reports for the capture of secreted proteins in an open-well configuration have established the utility of this format for detecting single-cell secretions. It remains unclear, however, how the single-cell rates of secretion, the equilibrium binding constant of the capture antibody, the dimensions of the wells, and the spatial distribution of the affinity ligands (capture antibody or ligand) in the wells affect the total amount of captured analyte and the time scales required to reach steady state.
We first simulated the steady-state spatial distribution of antibodies secreted from cells as a function of the spatial distributions of the capture ligands based on published configurations.5,8,10 For these analyses, we modeled plasma cells secreting antibodies; the specific productivities of these cells range from 500 molecules/s to 2000 molecules/s.8,23 In the open-well configuration, secreted proteins are free to diffuse into the surrounding media or adjacent wells as well as bind to the affinity ligands supported on the substrate. Comparing two reported configurations for open-well arrays,5,10 we found that the interfacial concentration of captured proteins at steady state was higher when the basal surface of the wells was modified with affinity ligands than when only the planar surfaces between wells were coated (see Figure S1 in the Supporting Information). The local proximity of the cell to the affinity ligands positioned in-well and the radial diffusion of the analyte into the bulk media influence these calculated differences.
Reported approaches for in-well detection have also used patterned hydrogels containing polyethylene glycol on the side walls to minimize nonspecific capture of the secreted proteins.5 Our initial simulations suggested that uniform coating of the capture ligand on all sides of the wells should enhance the retention of secreted proteins in the well, in addition to allowing a simple method of fabrication. We examined this configuration further by calculating the interfacial concentrations of captured protein when the nanowells were uniformly coated with affinity ligands. In this model, we tested the effects of both the size and spacing of wells and the affinity of the binding interactions (Kd = 10 nM and Kd = 0.1 nM) (see Figure 2). These simulations showed 4-fold enhanced retention of analytes (on entire available binding area) and 2-fold for high affinity ligand, compared to arrays where only the top planar surfaces support moderate affinity ligands (see Figure S1 in the Supporting Information). The majority of the captured analytes were also spatially confined to the well, increasing the interfacial concentration. Decreasing the sizing of the wells from 50 μm to 15 μm in width and depth only increased the concentration of captured protein in wells by 2.7-fold. Importantly, the interfacial concentrations of protein in empty wells adjacent to ones with highly active secreting cells remained more than an order of magnitude lower than that in the well with the secreting cell.
Figure 2.
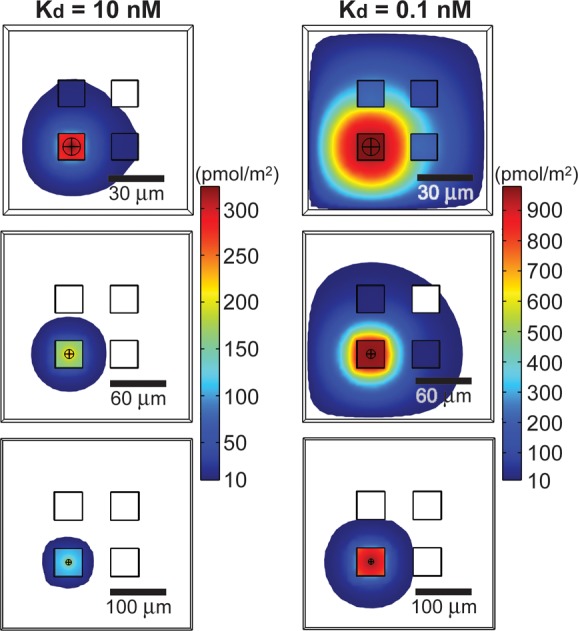
Theoretical surface distribution of bound analyte from single-cell secretions captured in-well. Finite-element simulations of secreted proteins captured on the surface of the nanowells as a function of the volume of wells and affinity of the capture ligand when proteins are secreted at a rate of 1000 molecules/s.
Comparative Analysis of Efficiency of In-Well Capture and Microengraving
Activated B cells (plasma cells and plasmablasts) as well as optimized cell lines used in biomanufacturing (e.g., CHO cells) secrete proteins at high rates, but the range of specific productivities for these types of cells are typically 1–2 orders of magnitude higher than that for other proteins secreted by primary cells (such as cytokines and chemokines, ∼5–50 molecules/s).6 We calculated how the rates of single-cell secretion impacted the time required to reach steady-state conditions and the total amounts of secreted product captured in both open-well and closed-well (microengraving) formats (see Figure 3). These results show that the open-well arrangement reaches steady state within 1 h, regardless of the rate of secretion for moderate affinity ligands, and that the maximum interfacial concentrations of captured protein varies strongly with the rates of secretion. For higher affinity ligands, however, the time required to reach steady state increases.
Figure 3.

Time-dependent interfacial concentrations of proteins captured from a single cell in open and closed configurations. Plots depict the average surface concentration of secreted analyte bound to the capture surface for both microengraving and in-well capture, as a function of the rates of secretion and dimensions of the nanowells. The equilibrium association-binding constant used for these plots are (A) Kd = 10 nM and (B) Kd = 0.1 nM.
In contrast, microengraving operates in a nonequilibrium state.6 The interfacial concentrations increase linearly over a somewhat wider range of times (0–4+ h, depending on the size of the well and rates of secretion), and reach higher values than those possible for the open-well format. Furthermore, at steady state, the interfacial concentrations are independent of the rates of secretion (see Figure S2A in the Supporting Information), although the time required to reach saturation does depend on the rate of secretion. However, the differences between the two configurations (open and closed wells) diminish significantly, as the rates of secretion increase and the affinity of the capture reagent increases (from Kd = 10 nM to Kd = 0.1 nM) (see Figure 3, as well as Figure S2B in the Supporting Information).
In these simulations, we have assumed an ideal position of the cell at the center of the basal surface of the wells. We had previously considered how alternate locations of the cell within the well (for example, a corner) could introduce variation to the captured analytes by microengraving and found no significant difference.6 Here, we also examined whether or not the positioning of the cell could introduce variations in captured amounts using in-well detection (open system). In this configuration, we found there was also no significant effects on the total average concentration of captured analyte over the capture surface of the well when a cell was positioned in the corner of a well (see Figure S3 in the Supporting Information). However, we note that minute gradients in concentration can form (for in-well detection) in the proximity of the cells, forming a minor halo effect (as seen in Figure 4). In addition, the effects of nonspecific binding have been found to be negligible (see Figure S4 in the Supporting Information).
Figure 4.

In-well capture of antigen-specific antibodies from single cells. Transmitted light and fluorescent composite micrographs of live cells (red) labeled with calcein violet and the captured secreted anti-OVA antibody (green) detected by Alexa 488 anti-mouse IgG in nanowells uniformly coated with ovalbumin. Scale bar = 30 μm.
Detection of Secreted Antigen-Specific Antibodies Inside Nanowells
Our numerical simulations indicated that microengraving offers enhanced sensitivity to the detection of secreted proteins across a wide range of single-cell rates, but that in-well detection should provide biased detection of cells with high rates of secretion. Such bias would highlight cells with high specific productivities—a criterion that should facilitate the selection of both hybridomas producing antigen-specific antibodies and cell lines for bioprocess development.13 We sought to demonstrate that a device containing arrays of nanowells is experimentally amenable for the efficient detection of secreted proteins when the surfaces of the wells are uniformly coated with capture antibodies (as depicted in Figure 1B). We used a mouse hybridoma line (099-01) that produces monoclonal antibodies specific for chicken ovalbumin. Ovalbumin was first adsorbed onto the surfaces of an array of nanowells. Then cells were deposited onto the array from a suspension in a manner that favored single-cell occupancies in the wells. The loaded array was incubated for 2 h at 37 °C. A fluorescently labeled anti-mouse IgG antibody was then added and further incubated with the cells for 30 min at room temperature. The array was rinsed gently to avoid washing away or repositioning cells. This method allowed the enumeration of both the number of viable cells in each well and the detection of captured ovalbumin-specific antibodies by automated epifluorescence microscopy (EM) (see Figure 4). The cells remained viable during the course of the experiment, including the labeling and washing steps at room temperature. We found that the incubation of 2 h yielded detectable quantities of captured antibodies, consistent with the time predicted from the numerical simulations to reach equilibrium in the open-well format. Also consistent with the models, the fluorescent signal predominantly localized to the bottom surface and sidewalls of the nanowells, even though the entire surface of the array bore antigen. This localization within the wells created sharp, clear patterns of fluorescence, which facilitated simple image analysis to establish the spatial address of wells of interest. The observed patterns of captured antibodies also matched those predicted for antibodies with high affinities in the numerical simulations; the affinity of the anti-OVA antibody is 0.11 nM, as determined by surface plasmon resonance.11
We then compared the dynamic range of signal for captured proteins in-well to that determined by microengraving (see Figure 5). Microengraving provided fine resolution of the variations in the rates of secretion, with a 3-fold greater dynamic range, compared to in-well detection. Both measures were performed using 16-bit detectors and imaging parameters individually optimized to maximize the signals detected. However, we note that, because of constraints of the thickness of the arrays used, the systems upon which the data were collected were different—in-well detection used an EM-CCD and xenon lamp for epifluorescent microscopy while the microengraved array was imaged with a photomultiplier tube (PMT) and laser excitation in a microarray scanner. Nevertheless, both methods demonstrate an ability to enumerate cells with the highest specific productivities, and the results for each method agree well with the predictions from the numerical simulations. Together, these results support the computational framework developed here to facilitate the design and understanding of the specific configuration of assays in nanowells for in-well capture and establishes the utility of uniform coatings of capture ligands for in-well detection of single-cell secreted proteins.
Figure 5.

Experimental comparison of in-well capture and microengraving. Intensity distribution of Alexa 488 anti-mouse IgG used to detect secreted ovalbumin specific antibodies from the hybridoma cells. (A) Intensity distribution from detected surface captured analyte in the in-well capture configuration. (B) Intensity distribution obtained by microengraving. The blue histogram represents background florescence obtained from wells with no cells; the black histogram shows the mean fluorescent intensities quantified for wells with single cells (total fluorescence per unit area). Scale bar = 30 μm.
Discussion
In this work, we developed a set of guidelines to aid in the design of experiments that involve the use of arrays of nanowells to quantify and analyze proteins secreted from single cells. We analyzed the transport and binding dynamics of analytes secreted from single cells, using an array of nanowells for both in-well capture and microengraving. Based on these analyses, we presented a modified method for in-well detection wherein the entire surface of the array is coated with capture ligands to detect single-cell secretions. As expected, the two most important parameters influencing the efficiency of detecting secreted analytes are the dissociation binding constant (Kd) and the rates of secretion for the cells. The affinity constant is particularly important, because the use of suboptimal capture antibodies or antigens could lead to false negatives.
On one hand, the use of nanowells for in-well capture with a uniformly coated surface to detect secreted proteins offers flexibility and ease of use. This configuration eliminates complex fabrication schemes5,10 and is compatible with a range of molding methods and materials (e.g., polystyrene) that are more conducive for mass production than arrays formed with silicone elastomers. In addition, contrary to other approaches,10 analytes bind to the sidewalls to create sharp and well-defined spatial patterns that simplify automated image analysis and concentrate signal to facilitate the detection of positive wells and high-throughput data extraction. The single substrate employed for in-well detection further simplifies post-assay processing and data integration and, thus, reduces the total time required for the process. One disadvantage evident from the numerical simulations is that the maximum signal in open-well configurations is strongly dependent on the rates of secretion, as a result of persistent diffusion of material out of the wells. The constraints imposed by thermodynamics and mass transport make the method less useful for quantitative measures. These features together suggest that the open-well configuration is best-suited for rapid screening of highly productive cells (e.g., antibody-secreting cells, manufacturing cell lines), and biases toward high secretors and high affinity interactions.
On the other hand, microengraving provides a complementary method for single-cell analysis of secreted proteins. The temporary confinement of the volume into which proteins are secreted enhances the sensitivity of the method for low rates of secretion, and makes the measurements more robust across a range of dissociation constants for the capture ligand, compared to open-well configurations. Microengraving also establishes an operating condition that expands the dynamic range of measurements, facilitating quantification of the amounts of secreted protein. The increase in sensitivity and quantitative potential compensates for the additional time required for processing and data integration. These features make the method well-suited for assessing heterogeneous populations of secreting cells, such as T cells.86,11,14
One possible perturbation introduced in a closed system is that secretions from the cells could be affected by the local concentration of analyte or other soluble factors. Sealed wells, as in the case of microengraving, would also retain higher concentrations of secreted protein relative to open wells. This effect could induce signaling mechanisms for feedback (such as autocrine signaling) that may change cellular processes (including secretion) over time, and could, therefore, affect the total amount of proteins available for capture in open versus closed wells as a result of biological feedback. Nevertheless, this phenomena will not likely be a concern for the screening of antibody-secreting cells or when local concentrations of ligands vastly exceed the numbers of available receptors on the surface of a single cell.
Conclusions
The computational analyses presented here should aid the design of bioanalytical assays that rely on arrays of nanowells for measuring secreted proteins from single cells. The established framework addresses the thermodynamics and mass transport of the two common configurations used in practice. Our results should provide practical guidance on the operating range and limitations of each configuration, their expected sensitivities and optimal incubation times based on rates of secretion and affinity of the capture reagent, as well as the costs of materials and labor required, with respect to the objectives of the assay.
Acknowledgments
This work was supported by the NIH/NIAID (No. 5P01AI045757) and by the Koch Institute Support (core) (Grant No. P30-CA14051) from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy And Infectious Diseases or the National Institutes of Health. J.C.L. is a Camille Dreyfus Teacher-Scholar. The authors dedicate this paper to the memory of Officer Sean Collier, for his caring service to the MIT community and for his sacrifice.
Supporting Information Available
Figure S1 compares the theoretical surface capture of secreted antibody (from single cells) in the configuration developed in this report and the configuration previously published. Figure S2 summarizes the theoretical maximum binding of analytes to the capture surface in an open system (in-well capture) or closed system (microengraving) configuration and the half-time to reach steady-state. Figure S3 analyzes the effects of deviation from centralized positioning of the cell in the wells. Figure S4 investigates the effects of nonspecific binding on the capture of analytes. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): J.C.L. is a founder and shareholder of Enumeral Biomedical.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Guldevall K.; Vanherberghen B.; Frisk T.; Hurtig J.; Christakou A. E.; Manneberg O.; Lindström S.; Andersson-Svahn H.; Wiklund M.; Önfelt B. PLoS One 2010, 5, e15453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadarajan N.; Julg B.; Yamanaka Y. J.; Chen H.; Ogunniyi A. O.; McAndrew E.; Porter L. C.; Piechocka-Trocha A.; Hill B. J.; Douek D. C.; Pereyra F.; Walker B. D.; Love J. C. J. Clin. Invest. 2011, 121, 4322–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lecault V.; VanInsberghe M.; Sekulovic S.; Knapp D. J. H. F.; Wohrer S.; Bowden W.; Viel F.; McLaughlin T.; Jarandehei A.; Miller M.; Falconnet D.; White A. K.; Kent D. G.; Copley M. R.; Taghipour F.; Eaves C. J.; Humphries R. K.; Piret J. M.; Hansen C. L. Nat. Methods 2011, 8, 581–586. [DOI] [PubMed] [Google Scholar]; b Lindström S.; Eriksson M.; Vazin T.; Sandberg J.; Lundeberg J.; Frisén J.; Andersson-Svahn H. PLoS One 2009, 4, e6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Konry T.; Dominguez-Villar M.; Baecher-Allan C.; Hafler D. A.; Yarmush M. L. Biosens. Bioelectron. 2011, 26, 2707–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ma C.; Fan R.; Ahmad H.; Shi Q.; Comin-Anduix B.; Chodon T.; Koya R. C.; Liu C.-C.; Kwong G. A.; Radu C. G.; Ribas A.; Heath J. R. Nature Medicine 2011, 17, 738–743. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Love K. R.; Politano T. J.; Panagiotou V.; Jiang B.; Stadheim T. A.; Love J. C. PLoS One 2012, 7, e37915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H.; Stybayeva G.; Silangcruz J.; Yan J.; Ramanculov E.; Dandekar S.; George M. D.; Revzin A. Anal. Chem. 2009, 81, 8150–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q.; Bradshaw E. M.; Nilsson B.; Hafler D. A.; Love J. C. Lab Chip 2010, 10, 1391–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunniyi A. O.; Story C. M.; Papa E.; Guillen E.; Love J. C. Nat. Protoc. 2009, 4, 767–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love J. C.; Ronan J. L.; Grotenbreg G. M.; van der Veen A. G.; Ploegh H. L. Nat. Biotechnol. 2006, 24, 703–707. [DOI] [PubMed] [Google Scholar]
- Bradshaw E. M.; Kent S. C.; Tripuraneni V.; Orban T.; Ploegh H. L.; Hafler D. A.; Love J. C. Clin. Immunol. 2008, 129, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin A.; Ozawa T.; Tajiri K.; Obata T.; Kondo S.; Kinoshita K.; Kadowaki S.; Takahashi K.; Sugiyama T.; Kishi H.; Muraguchi A. Nat. Med. 2009, 15, 1088–1092. [DOI] [PubMed] [Google Scholar]
- Story C. M.; Papa E.; Hu C.-C. A.; Ronan J. L.; Herlihy K.; Ploegh H. L.; Love J. C. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 17902–17907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Park S.; Kim W.; Kim Y.; Son Y. D.; Lee S.-C.; Kim E.; Kim S. H.; Kim J. H.; Kim H.-S. Anal. Chem. 2010, 82, 5830–5837. [DOI] [PubMed] [Google Scholar]; b Panagiotou V.; Love K. R.; Jiang B.; Nett J.; Stadheim T.; Love J. C. Appl. Environ. Microbiol. 2011, 77, 3154–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.; Han J.; Kim W.; Lee G. M.; Kim H.-S. J. Biotechnol. 2011, 156, 197–202. [DOI] [PubMed] [Google Scholar]
- Torres A. J.; Contento R. L.; Gordo S.; Wucherpfennig K. W.; Love J. C. Lab Chip 2013, 13, 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasson-Ochsner M.; Romano G.; Hakanson M.; Smith M. L.; Leckband D. E.; Textor M.; Reimhult E. Lab Chip 2011, 11, 2876–2883. [DOI] [PubMed] [Google Scholar]
- Zaretsky I.; Polonsky M.; Shifrut E.; Reich-Zeliger S.; Antebi Y.; Aidelberg G.; Waysbort N.; Friedman N. Lab Chip 2012, 12, 5007–5015. [DOI] [PubMed] [Google Scholar]
- Choi J. H.; Ogunniyi A. O.; Du M.; Du M.; Kretschmann M.; Eberhardt J.; Love J. C. Biotechnol. Prog. 2010, 26, 888–895. [DOI] [PubMed] [Google Scholar]
- a Love K. R.; Politano T. J.; Panagiotou V.; Jiang B.; Stadheim T. A.; Love J. C. PLoS One 2012, 7, e37915. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bird J. J.; Brown D. R.; Mullen A. C.; Moskowitz N. H.; Mahowald M. A.; Sider J. R.; Gajewski T. F.; Wang C.-R.; Reiner S. L. Immunity 1998, 9, 229–237. [DOI] [PubMed] [Google Scholar]
- Markov D.; Lillie E.; Garbett S.; McCawley L. Biomed. Microdevices 2013, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Douville N.; Takayama S.; El Sayed M. Ann. Biomed. Eng. 2012, 40, 1862–1873. [DOI] [PubMed] [Google Scholar]
- Liao W.; Wei F.; Qian M. X.; Zhao X. S. Sens. Actuators, B 2004, 101, 361–367. [Google Scholar]
- Pokrić B.; Pučar Z. Anal. Biochem. 1979, 93, 103–114. [PubMed] [Google Scholar]
- a Charlet M.; Kromenaker S. J.; Srienc F. Biotechnol. Bioeng. 1995, 47, 535–540. [DOI] [PubMed] [Google Scholar]; b Henn A. D.; Rebhahn J.; Brown M. A.; Murphy A. J.; Coca M. N.; Hyrien O.; Pellegrin T.; Mosmann T.; Zand M. S. J. Immunol. 2009, 183, 3177–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.