

Abstract
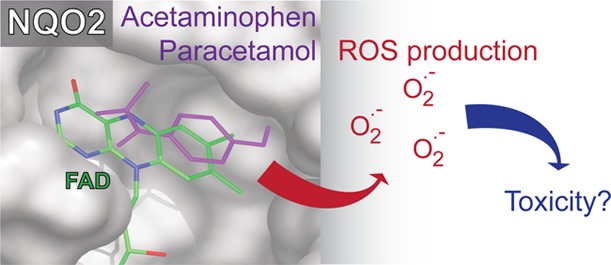
The analgesic and antipyretic compound acetaminophen (paracetamol) is one of the most used drugs worldwide. Acetaminophen overdose is also the most common cause for acute liver toxicity. Here we show that acetaminophen and many structurally related compounds bind quinone reductase 2 (NQO2) in vitro and in live cells, establishing NQO2 as a novel off-target. NQO2 modulates the levels of acetaminophen derived reactive oxygen species, more specifically superoxide anions, in cultured cells. In humans, NQO2 is highly expressed in liver and kidney, the main sites of acetaminophen toxicity. We suggest that NQO2 mediated superoxide production may function as a novel mechanism augmenting acetaminophen toxicity.
Keywords: acetaminophen, paracetamol, NQO2, quinone reductase 2, superoxide, CETSA, drug target
Introduction
Acetaminophen—also known as paracetamol, Tylenol, and acetyl-p-aminophenol (APAP)—is possibly the most used drug in the world, with around 50 million weekly users in the US alone.1 Originally cyclooxygenase (COX) enzymes, the classical target of nonsteroidal anti-inflammatory drugs, were considered the “on-target” for acetaminophen. However, the evidence for this remains controversial (reviewed in ref (2)), and other proteins, such as transient receptor potential cation channel, member A1 (TRPA1), have been recently suggested to mediate the therapeutic effects via acetaminophen metabolites.3 Direct targets of acetaminophen have remained elusive as research has focused on downstream events following acetaminophen metabolism.4
Although acetaminophen is safe at low doses, overdose is the leading cause of acute liver failure.2,5 This toxicity is considered a very complicated and incompletely understood process that takes place mainly in the liver and kidneys but involves several cell types, including hepatocytes and cells of the immune system (reviewed in refs (4−6)). At the molecular level the two main factors in acetaminophen toxicity are formation of a highly reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI) and production of reactive oxygen species (ROS), especially superoxide anions.7−11 These events are considered separate, although they both center on mitochondria.4 The reactive oxygen species and NAPQI are largely counteracted by glutathione in the early stages of toxicity.4,5 However, after glutathione depletion, NAPQI forms protein adducts and damages mitochondria, which are considered as the main source of superoxide in later stages of toxicity.4,9,10,12,13 Formation of NAPQI is essential for toxicity,14,15 but it alone may be insufficient for causing necrotic cell death.16,17
Production of ROS can activate c-Jun N-terminal kinase (JNK) and transient receptor potential melanostatine 2 (TRPM2) Ca2+ channels leading into further mitochondrial damage and amplified ROS formation.4,6,16,18 Changes in cytosolic Ca2+ levels have been observed early in hepatotoxicity,11,13 and TRPM2 channels have been recently shown to mediate acetaminophen hepatotoxicity.16 It is believed that an initial oxidative stress may be needed for the activation of JNK and Ca2+ channels.4,6 However, NAPQI mediated mitochondrial toxicity and ROS formation should only take place after glutathione depletion, and oxidative stress, which is commonly measured by indirect indicators of ROS, is not considered to take place early in toxicity.7,9 Interestingly, recent advances in live oxidative stress monitoring in vivo have observed excess ROS formation almost immediately after acetaminophen overdose.19 This early ROS formation was only seen with high acetaminophen doses. Importantly, the molecular source and cell type of origin for the initial ROS remains unclear.4
Here we show that the cytosolic protein quinone reductase 2 (NQO2) is a novel off-target for acetaminophen and many acetaminophen-like compounds. NQO2, which is highly expressed in human liver and kidneys, mediates acetaminophen induced superoxide production in cultured cells.
Experimental Section
Small Molecules and NQO Proteins
Imatinib mesylate, NMEH (1-methyl-1,4-dihydronicotinamide) and MnTBAP chloride were from Santa Cruz Biotechnology. All other compounds, including acetaminophen (meets USP testing specifications, 98.0–101.0%) and its analogues, were from Sigma-Aldrich unless stated otherwise. Acetaminophen and its analogues were diluted in DMSO, which alone served as a vehicle in all experiments, except when indicated otherwise. Recombinant DT diaphorase (NQO1) and NQO2 (both human enzymes) were from Sigma-Aldrich. For thermal shift assays involving APAP analogues, a His6-tagged NQO2 (human) expressed in Escherichia coli BL21(DE3) was used.
Acetaminophen Target Affinity Purification
Acetaminophen affinity column was prepared by coupling 5 mM 4-acetamidothiophenol (Sigma-Aldrich) to SulfoLink coupling gel (Pierce) in 10 mM Tris-HCl, 5 mM EDTA, 25 mM TCEP, pH 8.5. A control column was prepared by omitting 4-acetamidothiophenol. The column was blocked with 20 mM β-mercaptoethanol. One gram of mouse liver was homogenized in 10 mL of PBS containing 1% Triton X-100, 1 mM DTT, and 1× proteinase inhibitor cocktail (Sigma) and batch purified using 1 mL of affinity matrix. Columns were washed with lysis buffer and eluted with 20 mM acetaminophen. Eluted proteins were concentrated by trichloroacetic acid/acetone precipitation, separated on 4–12% SDS–PAGE, and stained with colloidal Coomassie followed by mass spectrometry identification.
In Vitro Thermal Shift Assays
Thermal shift assays were performed as described.20 Briefly, 5 μM NQO1 or NQO2, 5× SYPRO orange (Sigma-Aldrich), and indicated chemicals were mixed in buffer (10 mM Hepes, 150 mM NaCl, pH 7.5) on ice with final sample volume of 50 μL. DMSO was used as solvent for all chemicals with final DMSO concentration of 1% (w/v). Sample temperature was increased 1 °C/min, and fluorescence (λex = 470 nm; λem = 520 nm) was measured using Eppendorf Mastercycler ep realplex2 thermal cycler.
Cell Culture Based Experiments
Cell culture, counting, and fluorescence measurements were done essentially as in ref (21). HeLa cells were cultured in high glucose DMEM supplemented with 10% FBS (Sigma-Aldrich), 1% l-glutamine, and 1% Pen Strep. All experiments were done before cells reached confluency. Cell counting and fluorescence measurements were done using an Accuri C6 cytometer (Becton-Dickinson) so that only cells of viable size were included in the analysis. For measuring superoxide levels, MitoSOX Red (Life Technologies) was added to 5 μM final concentration for 30 min, after which cells were washed twice with PBS and analyzed with a flow cytometer. For measuring Ca2+ levels, Fluo-3 (Sigma-Aldrich) was added to 5 μM final concentration for 30 min followed by an additional 30 min with 1 mM probenecid (Sigma-Aldrich), after which cells were collected and analyzed as with MitoSOX measurements. Cell viability was assessed by counting cell number and measuring membrane integrity as descripted below for CETSA experiments.
RNAi was performed by reverse-transfecting with 40 nM siRNA with HiPerfect (Qiagen). NQO2 (cat. no. HSC.RNAI.N000904.12.1 and HSC.RNAI.N000904.12.7) and NC1 negative control siRNAs were from Integrated DNA Technologies (IDT). For overexpressions human codon optimized NQO2 (GeneArt) was Gateway cloned into pDEST40 vector containing 3xV5 tag in C terminus. Plasmids (3 μg of plasmid per well on a 12-well plate) were transfected with FuGENE HD (Promega).
For Western blots, antibodies were used at their recommended concentrations and detected using infrared-dye conjugated secondary antibodies and LICOR Odyssey detection system. Antibodies used were GAPDH (#5174) from Cell Signaling Technology, SOD1 (HPA001401) from Sigma-Aldrich, and NQO2 (NBP1-31563) from Novus Biologicals. All cell culture experiments were done before cells reached confluency.
Cellular Thermal Shift Assay (CETSA)
CETSA was performed as in ref (22) with minor modifications and with addition of a loading control. HeLa cells were trypsinized, washed with PBS, and suspended in PBS supplemented with protease inhibitor cocktail (Sigma-Aldrich). The cell suspension (5000 cells/μL) was treated with compounds in DMSO (final concentration not exceeding 0.5% (w/v)) for 1 h at 37 °C with gentle mixing. Each sample (70 μL) was then heated at the indicated temperature for 3 min using an Eppendorf Thermomixer with mixing (500 rpm) and lysed with three freeze–thaw cycles using dry ice and a 42 °C water bath. Cell lysates were centrifuged at 16000g for 15 min at 4 °C, and analyzed by Western blotting. For quantitation, band intensities were normalized to the mean of the three lowest temperature bands, in which protein levels stayed constant, and then NQO2 bands were normalized to SOD1 bands used as loading control. ITDRFCETSA experiments were done using constant temperature of 72 °C. Band intensities were normalized to the highest concentration and SOD1 levels.
For analysis of cellular membrane integrity and cell viability, cells were heated as the CETSA samples, followed by 10 min incubation with 30 μg/mL propidium iodide and analysis by flow cytometry.
Analysis of NQO2 Binding Site
Acetaminophen binding to NQO2 structure (PDB: 1ZX1) was modeled with DockingServer (www.dockingserver.com) using default parameters of the program. Acetaminophen binding to the NQO2 active site was measured using electronic absorption spectroscopy essentially as described.23 Briefly, 2 mg/mL His6-NQO2 was mixed with 0.5 mM acetaminophen or DMSO in 25 mM Hepes, 150 mM NaCl, pH 7.5, and absorption spectra were measured between 250 and 550 nm. The absorption spectrum of free FAD (100 μM) with 5 mM acetaminophen or DMSO was measured as a control.
Enzyme Assays
NQO1 and NQO2 inhibition was measured by mixing 50 μL of 50 mM Hepes-KOH, pH 7.4, with 0.01% Tween20, 0.18 mg/mL BSA, and 1 μM FAD containing 100 ng of recombinant NQO1 or NQO2 (Sigma-Aldrich) and mixing with 50 μL of the same buffer with test compounds. Reactions were initiated by adding 50 μL of assay buffer containing 50 mM Hepes-KOH, pH 7.4, with 0.01% Tween20, 0.18 mg/mL BSA, 1 μM FAD, and 500 μM NADH (for NQO1) or 500 μM NMEH (for NQO2) as cosubstrates as well as 600 μM MTT and 300 μM menadione. The absorbance of the samples was measured at 595 nm.
Substrate assays were performed similarly to inhibition assays except omitting the menadione substrate. For kinetic assays acetaminophen and menadione were diluted directly into the reaction mix in order to omit DMSO as DMSO alone yields some background activity. Absorbance was measured every 30 s after the reaction was started and background correction was made using a sample without NQO2. All samples were measured simultaneously in a 96-well assay format.
NQO2 Expression in Human Tissues
Human Normal Tissue Blot II was obtained from ProSci Incorporated (Poway, CA) and stained with NQO2 antibody. Each lane contains same amount of total protein (15 μg). NQO2 mRNA expression levels are from Illumina Body Map RNA-Seq data (E-MTAB-513). An independent human skeletal muscle sample was purchased from Abcam (ab29330).
Statistical Analysis
Western blot band intensities were analyzed with ImageJ. Correlation values for Figure 1c were calculated using logarithmic two parameter nonlinear regression equation in SigmaPlot. Sigmoidal curves were fitted to CETSA and ITDRFCESTA data using SigmaPlot. For kinetic analysis, ligand binding curves and confidence intervals were fitted and Vmax, Km, and regression correlation (R2) were analyzed using SigmaPlot and its enzyme kinetics application. Statistical significances were evaluated by ANOVA and two tailed t test with Bonferroni correction (n.s. = nonsignificant, * = p < 0.05, ** = p < 0.01, *** = p < 0.001 in all figures and panels). All data with error bars depict mean and standard deviation.
Figure 1.

Acetaminophen binds to NQO2. (a) Structures of acetaminophen and thioacetaminophen coupled to affinity matrix. (b) SDS–PAGE separation of protein eluates from thioacetaminophen (+) and control (−) columns. NQO2 detection from Western blot of the eluates is shown at bottom. (c) Thermal shift assays of APAP and AMAP with NQO2 and APAP with NQO1. See panel d for controls. (d) NQO2 thermal shift assay of acetaminophen regioisomers (2 mM), nitroacetanilide (2 mM), and NQO2 inhibitors quercetin (1 μM) and resveratrol (10 μM). AOAP is acetyl-o-aminophenol. Dicoumarol (10 μM) control for NQO1 is shown (N = 3–4 in panels c and d).
Results
Acetaminophen Binds NQO2 in Vitro
We investigated potential direct targets of acetaminophen by affinity purification from a mouse liver lysate. To simplify the coupling chemistry, we attached thioacetaminophen to sulforeactive affinity matrix via thioether bond as chemical properties of ethers and thioethers are very similar (Figure 1a). Proteins were eluted with free acetaminophen. Despite high background in control and thioacetaminophen columns, a protein of ∼25 kDa was specifically observed in the thioacetaminophen column eluate (Figure 1b). Mass spectrometry unambiguously identified this protein with 92% sequence coverage as quinone reductase 2 (Supporting Information Figure S1), formally known as NAD(P)H dehydrogenase, quinone 2 (NQO2), which is a cytoplasmic protein also known as quinone reductase 2 (QR2). NQO2/QR2 should not be confused with the mitochondrial NQO2 (formally known as NDUFV2) involved in oxidative phosphorylation. The binding of NQO2 to thioparacetamol column was confirmed by Western blotting of the eluates (Figure 1b, bottom).
Relatively little is known about NQO2, and the endogenous substrates of NQO2 have remained largely elusive, although dopamine has been suggested.24 NQO2 is known to be important in drug metabolism,25 and NQO2 has been associated with numerous disorders, including cancer and neurodegenerative diseases.26 Exogenous substrates of NQO2 include menadione, and several inhibitors have been described, for example, quercetin, resveratrol, and imatinib/Gleevec26−29 (see Supporting Information Figure S2 for structures). Despite its name, NQO2 uses nonphosphorylated dihydronicotinamides and not NAD(P)H as its cosubstrate30 and NQO2 substrate specificity is not limited to quinones.25,26 NQO2 mediated metabolism can also generate free radicals when appropriate substrates are available.31
Inverse pharmacological approaches have identified NQO2 as a binding partner for many interesting compounds such as resveratrol, chloroquine, and melatonin.28,32,33 To validate acetaminophen binding to NQO2 in vitro we performed thermal shift assays using recombinant NQO2.20 Human NQO2 was used in this and all subsequent assays. Acetaminophen and its regioisomer 3-acetaminophenol (AMAP) both stabilized NQO2 against thermal denaturation in a concentration dependent manner (Figure 1c). Acetaminophen interaction was selective for NQO2 as no binding was observed with the closely related NQO1 (DT-diaphorase, Figure 1c), while a known inhibitor of NQO1, dicoumarol, significantly stabilized this enzyme (Figure 1d). NQO2 inhibitors quercetin and resveratrol also caused substantial thermal stabilization of NQO2 (Figure 1d). Analysis of all regioisomers of acetaminophen indicated that the hydroxyl group position is not critical for NQO2 binding (Figure 1d and Supporting Information Table S1).
Optimized Cellular Thermal Shift Assay Reveals NQO2-Acetaminophen Interaction in Live Cells
It is well-known that in vitro assays represent cellular binding poorly, as drug concentration and target engagement are dependent on ADME properties (absorption, distribution, metabolism, and excretion). To better reflect physiological binding conditions inside cells, we performed cellular thermal shift assays (CETSA) between acetaminophen and NQO2 in live HeLa cells (Figure 2a).22 We improved the original method by including superoxide dismutase 1 (SOD1) as a control protein. SOD1 is a thermostable protein,34 and we did not observe any significant reduction in SOD1 levels with CETSA treatment temperatures up to 80 °C (Figure 2b). We also validated that none of the compounds in our assay affected SOD1 levels (Figure 2c). To validate that the observed bindings take place inside intact cells, we measured cell membrane permeability using cell impermeable propidium iodide staining. Although membrane integrity was lost in the highest temperatures used, in temperatures where NQO2 became destabilized membranes remained intact (Figure 2d). We conclude that the binding observed is taking place in intact cells and not just cell lysates.
Figure 2.

Cellular thermal shift assays of acetaminophen binding to NQO2. (a) Schematic presentation of CETSA. The interaction between drug and target protein increases the thermal stability of the protein, which can be measured by separating and quantifying nonaggregated levels of the target protein at different temperatures. (b) SOD1 levels in HeLa cells treated as for CETSA experiments. No significant changes in protein levels were seen in the used temperatures. (c) SOD1 protein levels in cellular thermal shift samples. SOD1 levels did not change significantly when HeLa cells were treated as in CETSA (left panel) or ITDRFCETSA (right panel) experiments. For the ITDRFCETSA experiment the second highest concentration of each chemical was used, as samples reached maximal NQO2 intensity at this concentration. GAPDH was used as a loading control. (d) HeLa cell count and membrane integrity after CETSA treatment. HeLa cells were treated as for CETSA, after which cells were treated with propidium iodide for 15 min before analyzing membrane integrity and cell count using flow cytometer. Values are normalized to samples treated at 37 °C. (e) Cellular thermal shift assay of NQO2. APAP (10 mM), AMAP (10 mM), and quercetin (100 μM) were incubated with HeLa cells for 1 h. Representative NQO2 Western blots of each sample are shown. NQO2 levels were normalized to SOD1 levels used as loading control (not shown). Statistical significances between DMSO and APAP are indicated. Note that the membrane integrity is not maintained at the highest temperatures (panel d), but NQO2 binding is observed already before this, indicating binding in intact cells. APAP and AMAP did not differ significantly. (f) Isothermal (at 72 °C) dose response fingerprints in HeLa cells after 1 h exposure to APAP, nitroacetanilide, resveratrol, and menadione. Representative NQO2 Western blots of each sample are shown. Statistical significance between APAP and nitroacetanilide with 0.5 mM concentration is indicated (N = 3–4 in all panels).
Consistent with the in vitro thermal shift assays, treatment with acetaminophen, AMAP, and quercetin stabilized NQO2 in live HeLa cells (Figure 2e). To more quantitatively understand the NQO2 binding affinity in living cells, we performed isothermal dose response fingerprint analysis based CETSA (ITDRFCETSA).22 This assay indicated that the cellular binding affinity of acetaminophen was ∼500 times weaker than that of resveratrol and approximately 1 mM extracellular concentration is required to observe binding under our experimental conditions (Figure 2f).
Acetaminophen Binds NQO2 near the Active Site
We next investigated acetaminophen binding to NQO2 in more detail. The binding pocket of NQO2 is hydrophobic, and known inhibitors of NQO2 have planar aromatic ring systems (Supporting Information Figure S2), which bind adjacent to the isoalloxazine ring of flavin adenine mononucleotide (FAD) and are coordinated by aromatic amino acid residues in the catalytic pocket.28In silico docking to NQO2 suggested a similar mode of binding for acetaminophen (Figures 3a and 3b). To validate this prediction, we utilized electronic absorption spectroscopy, which has been used previously to assess NQO2 composition and drug binding.23,29 The absorption spectrum of NQO2 bound flavin (absorbance in the 350–500 nm range) was affected by acetaminophen and its regioisomers (Figure 3c and Supporting Information Figure S3) in a similar manner to that observed with imatinib binding at the active site.23 We observed no effect of acetaminophen on free FAD (Figure 3c, bottom right). The spectroscopy data indicates that acetaminophen binds very close to FAD in the active site. Further supporting this, thermal shifts caused by quercetin and menadione, which both bind to the NQO2 active site, were not additive with acetaminophen (Figure 3d). These data suggest that acetaminophen binds NQO2 near the active site similarly to known inhibitors, substrates, and cosubstrates. To conclusively confirm the binding orientation of acetaminophen, cocrystallization of acetaminophen with NQO2 would be required.
Figure 3.

Acetaminophen binds to NQO2 active site. (a) In silico docking analysis of APAP to NQO2 (PDB: 1ZX1) indicates binding at the active site adjacent to the isoalloxazine ring of flavin adenine mononucleotide (FAD). (b) Suggested interacting amino acids in NQO2 based on in silico docking assays. (c) Electronic absorption spectroscopy of NQO2 with 0.5 mM APAP (red) and DMSO (blue). Inset on right top corner shows difference spectrum, and inset on right bottom shows spectra with free FAD. (d) NQO2 thermal shifts of APAP (10 mM) with known NQO2 active site binding molecules menadione (50 μM) and quercetin (1 μM) (N = 4).
Acetaminophen Appears To Act as a Weak NQO2 Substrate
We next tested if acetaminophen inhibits NQO2. However, NQO2 activity was not affected by acetaminophen or AMAP (up to 10 mM), whereas known inhibitors of NQO2 potently inhibited NQO2 activity in an in vitro enzyme assay (Figure 4a and Supporting Information Figure S4a). Similarly, acetaminophen displayed no inhibitory effect on NQO1 (Supporting Information Figure S4b). We next considered the possibility that acetaminophen might act as a substrate or cosubstrate. No activity was observed when the assay cosubstrate NMEH was replaced by acetaminophen, indicating that acetaminophen is not a cosubstrate (Supporting Information Figure S4c). Instead, NQO2 could utilize acetaminophen and AMAP as weak substrates (Figure 4b). This activity was abrogated by quercetin (Figure 4b), and no substrate activity was seen with NQO1 (Supporting Information Figure S4d), indicating a NQO2-dependent reaction. Kinetic substrate analysis validated acetaminophen acting as a weak NQO2 substrate with Km = 417 ± 10 μM compared to Km = 4.3 ± 0.1 μM for menadione (Figure 3c). As the weak substrate activity of acetaminophen could be potentially explained by synthesis byproducts and/or degradation products, we tested if simple quinones and hydroquinones might explain this activity. However, 4-benzonquinone and 4-hydroquinone displayed no such activity in the NQO2 assay (Supporting Information Figure S4e). Acetaminophen seems to therefore function as a novel, albeit poor, NQO2 substrate, although it is not clear what NQO2 modifies acetaminophen to.
Figure 4.

Acetaminophen functions as a weak substrate for NQO2. (a) NQO2 in vitro inhibition assay. NQO2 was incubated in the presence of compounds at the concentrations indicated and NQO2 activity measured with a colorimetric assay. (b) NQO2 substrate assay. Substrate activities of menadione (25 μM), APAP (2.5 mM), AMAP (2.5 mM), and control (DMSO) are shown. All substrate activities were abolished by addition of quercetin (10 μM). (c) Kinetic analysis of NQO2 substrate activity with menadione (left) and acetaminophen (right). Red lines represent 95% confidence intervals of the fitted ligand binding curves (black). NQO2 activity was measured with increasing substrate concentrations at fixed enzyme and cosubstrate (500 μM NMEH) concentrations. Michaelis constants and maximum reaction rates are displayed with standard errors. Note that acetaminophen and menadione were dissolved in the assay buffer in order to omit DMSO (N = 3 in all panels).
Library of Acetaminophen-like Molecules Displays a Wide Range of NQO2 Interactions and Correlating Levels of Superoxide Production
Acetaminophen and AMAP were not obvious candidates for possible NQO2 substrates based on previously known substrate structures. We therefore extended our in vitro thermal shift and substrate assays by screening 22 compounds with structural similarity to acetaminophen (Supporting Information Figure S2). NQO2 could bind and utilize many acetaminophen analogues as substrates (Supporting Information Table S1), and there was a weak correlation between the binding and substrate assays (Figure 5a, Supporting Information Table S1). Analogues with acidic side chains display no NQO2 binding or substrate activity. In contrast, nitroacetanilide had high binding affinity in vitro (Figure 1d), and it displayed stronger NQO2 binding than acetaminophen in ITDRFCETSA (Figure 2e).
Figure 5.

Acetaminophen-like compounds display wide range of NQO2 substrate activity in vitro and correlating superoxide production in cultured HeLa cells. (a) Comparison of NQO2 thermal shifts and NQO2 substrate activities for acetaminophen analogues (2 mM). Structures of some of the analogues are shown for comparison. (b) Specificity of MitoSOX measurement. HeLa cells were treated with APAP (5 mM) for 2.5 h followed by 0.5 h treatment with superoxide scavenger, MnTBAP (200 μM). (c) Comparison of APAP and NAPQI mediated superoxide production. HeLa cells were treated with APAP (2 mM) or NAPQI (20 μM) for 8 h. Note that only a small fraction of APAP is metabolized to NAPQI, and higher NAPQI concentrations are lethal to cells. (d) Effect of P450 inhibition on APAP mediated superoxide production. HeLa cells were treated with P450 inhibitor disulfiram (2 μM) for 1 h followed by APAP treatment (5 mM) for 2 h. Data normalized to controls without APAP treatment for clarity. (e) Comparison of NQO2 substrate activity and cellular superoxide production by acetaminophen and its analogues. For MitoSOX measurements HeLa cells were treated with 2 mM chemicals for 1 h. See Supporting Information Figure S1 and Table S1 for structures and data, respectively (N = 3 in all panels).
NQO2 has been shown to be capable of producing ROS,31 and acetaminophen overdose causes extensive oxidative stress. Most studies on acetaminophen induced ROS are done in vivo and have therefore relied on indirect measurements of ROS, like nitrosylation of tyrosine residues and lipid peroxidations, which are only observable after ROS mediated damage accumulates. To measure if acetaminophen interaction with NQO2 could be involved in superoxide production, we used MitoSOX fluorescence probe, which can react with both intra- and extramitochondrial ROS and is most sensitive toward superoxide. Importantly, acetaminophen induced MitoSOX signal was fully reversed with the addition of superoxide scavenger MnTBAP (Figure 5b). We used HeLa cells as a cell culture model for NQO2 mediated ROS production as HeLa cells express NQO2 and have relatively low baseline ROS levels (data not shown). Furthermore, we did not observe superoxide production in HeLa cells after treatment with NAPQI, and acetaminophen induced superoxide levels were unaltered when P450 enzymes required for NAPQI formation were inhibited with disulfiram35 (Figures 5c and 5d), indicating that the ROS formation by acetaminophen is largely separate from NAPQI mediated damage in this cell culture model. Furthermore, to focus on direct effects of acetaminophen, we used DMSO, which inhibits acetaminophen metabolism and NAPQI formation,4 as a solvent for acetaminophen. In summary, the HeLa cell model is thus ideal to confirm binding and to separate ROS effects from NAPQI mediated toxic effects.
Acetaminophen and its analogues displayed large differences in superoxide production, and there was a positive correlation (R2 = 0.499) between superoxide production in HeLa cells and NQO2 substrate activity in vitro (Figure 5e, Supporting Information Table S1). A notable exception was AMAP, which did not increase superoxide levels although it clearly binds NQO2 in cells (see Figure 2e). It has been suggested that reactive metabolites of AMAP and APAP differ in their ability to diffuse across mitochondrial membranes and arylate mitochondrial matrix proteins.13 This data suggests that NQO2 may be involved in acetaminophen induced superoxide production, although other mechanisms must also exist to explain the difference between AMAP and APAP.
NQO2 Is Highly Expressed in the Sites of Acetaminophen Toxicity
We next wanted to understand if there are biological consequences for the NQO2 interaction. As relatively little is known about NQO2, we examined the NQO2 protein and mRNA expression levels in a panel of normal human tissues. Although NQO2 was present in all tested tissues, the levels were clearly highest in liver and kidney (Figure 6), the main sites of acetaminophen toxicity. These findings are consistent with previous observations in rat.36 We also observed unreported NQO2 variants of approximately 35 kDa in the skeletal muscle. However, these NQO2 variants may have been partially donor specific as a similar NQO2 pattern was not seen in an independent human skeletal muscle sample (Supporting Information Figure S5).
Figure 6.
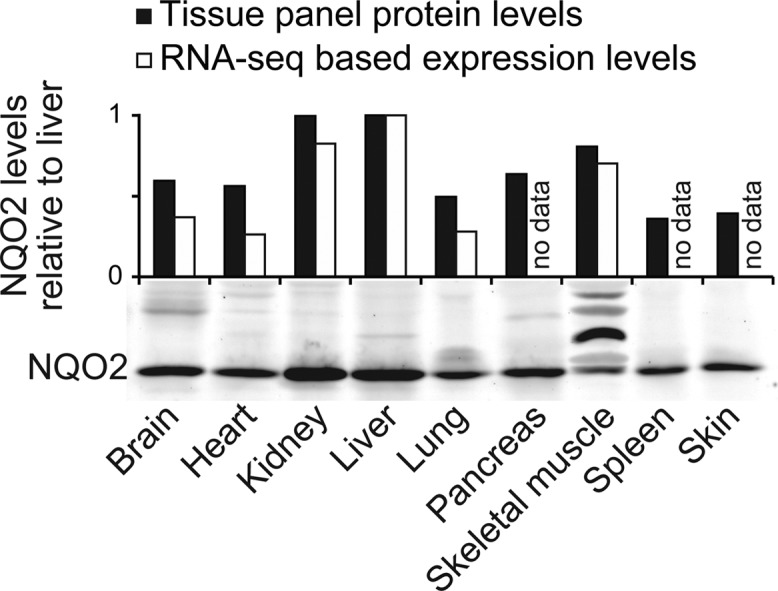
NQO2 protein and mRNA levels in normal human tissues. Tissue Western blot displays unknown NQO2 variants in skeletal muscle. For quantifications, mRNA and protein levels were normalized to NQO2 in liver. See also Supporting Information Figure S3.
NQO2 Levels Modulate Acetaminophen Induced Superoxide Production and Ca2+ Levels
The correlation between NQO2 substrate activity and superoxide production with the acetaminophen-like compounds suggested that NQO2 might be involved in acetaminophen mediated ROS production. To examine this we inhibited NQO2 with quercetin, resveratrol, and imatinib. Acetaminophen induced superoxide levels were significantly lower in the presence of all these inhibitors in HeLa cells (Figure 7a, left). Although these NQO2 inhibitors have off-target effects and possible antioxidant effects, it seems more likely that these inhibitors reduce superoxide through their common target, NQO2. To confirm this, we inhibited NQO2 with two independent siRNAs and similarly observed partial reduction in superoxide production by acetaminophen (Figure 7b, middle). Knock-down efficiency of NQO2 was approximately 50%, as measured by Western blotting (Figure 7b, left), matching the reduction in superoxide levels. Overexpression of NQO2 in HeLa cells resulted in a significant increase in superoxide production by acetaminophen (Figure 7c, middle), and this roughly correlated with the level of NQO2 overexpression (Figure 7c, left).
Figure 7.

NQO2 is involved in acetaminophen induced superoxide production. (a) APAP induced superoxide production (left) and intracellular Ca2+ levels (right) after chemical inhibition of NQO2. HeLa cells were treated with control (DMSO), quercetin (10 μM), resveratrol (10 μM), or imatinib (20 μM) for 1 h followed by 4 h treatment with vehicle (DMSO), APAP (5 mM), or AMAP (5 mM). (b) APAP induced superoxide production after NQO2 RNAi. HeLa cells were treated with control or NQO2 siRNA for 48 h followed by 4 h treatment with vehicle (DMSO), APAP (5 mM), or AMAP (5 mM). (c) APAP induced superoxide production after NQO2 overexpression. HeLa cells were transfected with empty vector or NQO2, incubated for 36 h followed by 5 h treatment with vehicle (DMSO) or APAP (5 mM). In panels b and c representative Western blots of NQO2 levels are on the left, superoxide levels in the middle, and intracellular Ca2+ levels on the right. Data is normalized to each control for clarity (N = 3–4 in all panels).
Changes in both cytosolic and mitochondrial Ca2+ levels are considered as one of the earliest signatures of acetaminophen hepatotoxicity.11,13 As ROS levels may impact Ca2+ homeostasis, we measured if NQO2 has an effect on acetaminophen modulated Ca2+ homeostasis, as measured by the Ca2+ sensitive fluorescence probe Fluo-3. Inhibition of NQO2 reduced the changes in Ca2+ levels caused by acetaminophen (Figures 7a and 7b, right). Although HeLa cells cannot be considered as a suitable model for acetaminophen toxicity, both genetic and chemical inhibition of NQO2 in HeLa cells resulted in a modest protection from high doses of acetaminophen (Supporting Information Figure S6). Altogether, this data indicates that NQO2 activity can modulate superoxide production and changes in Ca2+ levels induced by acetaminophen in HeLa cell culture.
Discussion
Very few direct targets for acetaminophen are known.4 We have identified NQO2 as an off-target for acetaminophen with a role in superoxide production and modulation of Ca2+ levels in cultured HeLa cells. While we have comprehensively validated the binding of acetaminophen to NQO2 with several independent methods in vitro, in mouse liver lysate, and in cultured HeLa cells, all of the data indicates that this is a relatively weak interaction and the in vivo significance of this binding will require further studies. Although in our experiments acetaminophen did not inhibit NQO2 in vitro, our results are consistent with previous findings that acetaminophen protects cells from menadione toxicity,37 possibly through NQO2 substrate competition. Instead of NQO2 inhibition, we observed that acetaminophen acts as a weak NQO2 substrate. The substrate activity of acetaminophen was approximately 100-fold less than that of menadione, which is a highly active NQO2 substrate.26 The thermal shift assay similarly suggested that acetaminophen binding is >100-fold weaker than that of menadione. Menadione toxicity requires both NQO1 and NQO2 catalyzed metabolism, and in mice, the LD50 for menadione is 13 mg/kg, but this depends on the cosubstrate availability.38 For comparison, acetaminophen has an intraperitoneal LD50 ≈ 500 mg/kg. Thus, the difference in substrate activity is slightly higher than the difference in the in vivo toxicity, which may be because of assay conditions and due to the well-known routes of acetaminophen toxicity.
Whereas most NQO2 binding compounds are inhibitors of this enzyme, our observations suggesting that acetaminophen acts as a weak NQO2 substrate propose a new metabolic route for acetaminophen. Although it is theoretically possible that NQO2 catalyzes a reaction between acetaminophen and NAPQI,8,26 whether and exactly how NQO2 is involved in acetaminophen metabolism in vivo remains unknown. Interestingly, if NQO2 has a role in acetaminophen metabolism and toxicity in vivo, the presence of NQO2 cosubstrates may represent a source of variation in the individual responses to acetaminophen.
We observed NQO2 mediated ROS production in HeLa cells. Although superoxide is a very reactive molecule as such, the cytotoxic effects of superoxide are attenuated through the action of superoxide dismutases and only excessive amounts overwhelming the capacity of the dismutases will result in oxidative damage. Such cytotoxic concentrations are achieved as a result of acetaminophen overdose.7−11 Although the MitoSOX probe does not react exclusively with superoxide, the acetaminophen induced ROS could be rescued with a superoxide scavenger, suggesting that acetaminophen produces mainly superoxide in our model. This superoxide production was also seen with many acetaminophen-like compounds that display NQO2 substrate activity in vitro, supporting the conclusion that NQO2 is a cellular source for acetaminophen induced ROS. Furthermore, our screen of acetaminophen-like compounds, some of which are also fragments of more specific drugs,39 provides a basis for structure–activity studies to better understand NQO2 function.
The low affinity binding between NQO2 and acetaminophen suggests that NQO2 does not play an active role in the therapeutic effects of acetaminophen. However, the pharmacokinetics of acetaminophen result in very high plasma concentrations, especially when compared to other known NQO2 targeting chemicals, like resveratrol.2 With ITDRFCETSA we detect an interaction between acetaminophen and NQO2 with approximately 1 mM extracellular concentrations of the drug, which is in agreement with the in vitro substrate assay with a Km value of ∼0.4 mM. Acetaminophen plasma concentrations can reach up to 10 mM in overdose patients,40 and, at least in rodents, acetaminophen plasma and tissue concentrations are known to be essentially the same.41 We also observed that NQO2 is predominantly present in liver and kidney, the tissues where acetaminophen is known to cause toxicity. It seems therefore possible that NQO2 may have a role in acetaminophen induced superoxide production also in vivo.
High levels of oxidant stress occur after most of the acetaminophen has been metabolized in vivo, NAPQI mediated damage and JNK kinase mediated amplification of ROS being key events. Interestingly, JNK activation is considered to require an initial ROS stimulus, although the source of this stimulus in acetaminophen toxicity is unknown.2,4 In NQO2 knockout mouse keratinocytes JNK activation is abolished.42 We propose that acetaminophen may act in part through NQO2 in the initial phase of toxicity, where increases in ROS and cytosolic Ca2+ levels concomitant with glutathione depletion are observed.13,19 If so, NQO2 mediated ROS production would not necessarily be the main cause of toxicity but rather a damage augmenting mechanism. This theory does not contradict the current model of acetaminophen toxicity where inhibition of NAPQI formation protects from acetaminophen overdose,14 and N-acetyl cysteine, which is a nonspecific ROS scavenger, reverses oxidative stress and toxicity.10 Consistent with our observations, NQO2 inhibitors, resveratrol and quercetin, have also been shown to alleviate acetaminophen toxicity in mice.43,44 However, additional studies are required to fully validate NQO2 inhibition as the protective mechanism instead of antioxidant properties or inhibition of NAPQI formation.4 Conclusive in vivo validation that NQO2 participates in ROS production in early acetaminophen toxicity would require live monitoring of ROS19 in NQO2 knockout animal model, which is beyond the scope of this work.
Altogether, our findings broaden the spectrum of known acetaminophen targets and suggest a potential new mechanism through which acetaminophen can mediate its biological effects.
Acknowledgments
We thank A. McLeod and K. Woods for technical assistance; I. Gilbert and K. Woods for discussions; A. McLeod, H. Pessa, and E. Sheils for critical reading of the manuscript; and College of Life Sciences Fingerprints facility for mass spectrometry analysis. This study was funded by Scottish Universities Life Sciences Alliance (SULSA) and the Wellcome Trust Career Development Fellowship to M.B. (Grant 089999).
Glossary
Abbreviations Used
- AMAP
acetyl-m-aminophenol
- AOAP
acetyl-o-aminophenol
- APAP
acetyl-p-aminophenol (acetaminophen)
- CETSA
cellular thermal shift assay
- ITDRFCETSA
isothermal dose response fingerprint analysis based CETSA
- DMSO
dimethyl sulfoxide
- FAD
flavin adenine dinucleotide
- MnTBAP
manganese(III) tetrakis(4-benzoic acid)porphyrin
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NADH
nicotinamide adenine dinucleotide (reduced form)
- NMEH
1-methyl-1,4-dihydronicotinamide
- NQO1
DT-diaphorase
- NQO2
NAD(P)H dehydrogenase, quinone 2
- ROS
reactive oxygen species
Supporting Information Available
Six additional figures with control data and compound structures, figure legends, and one table with acetaminophen-like compound measurement data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Both authors contributed equally to all aspects of the study.
The authors declare no competing financial interest.
Supplementary Material
References
- Moynihan R. FDA fails to reduce accessibility of paracetamol despite 450 deaths a year. BMJ 2002, 3257366678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham G. G.; Davies M. J.; Day R. O.; Mohamudally A.; Scott K. F. The modern pharmacology of paracetamol: therapeutic actions, mechanism of action, metabolism, toxicity and recent pharmacological findings. Inflammopharmacology 2013, 213201–32. [DOI] [PubMed] [Google Scholar]
- Andersson D. A.; Gentry C.; Alenmyr L.; Killander D.; Lewis S. E.; Andersson A.; Bucher B.; Galzi J. L.; Sterner O.; Bevan S.; Hogestatt E. D.; Zygmunt P. M. TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Delta(9)-tetrahydrocannabiorcol. Nat. Commun. 2011, 2, 551. [DOI] [PubMed] [Google Scholar]
- McGill M. R.; Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm. Res. 2013, 3092174–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessems J. G.; Vermeulen N. P. Paracetamol (acetaminophen)-induced toxicity: molecular and biochemical mechanisms, analogues and protective approaches. Crit. Rev. Toxicol. 2001, 31155–138. [DOI] [PubMed] [Google Scholar]
- James L. P.; Mayeux P. R.; Hinson J. A. Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos. 2003, 31121499–506. [DOI] [PubMed] [Google Scholar]
- Knight T. R.; Kurtz A.; Bajt M. L.; Hinson J. A.; Jaeschke H. Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicol. Sci. 2001, 622212–20. [DOI] [PubMed] [Google Scholar]
- Dahlin D. C.; Miwa G. T.; Lu A. Y.; Nelson S. D. N-acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc. Natl. Acad. Sci. U.S.A. 1984, 8151327–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James L. P.; McCullough S. S.; Knight T. R.; Jaeschke H.; Hinson J. A. Acetaminophen toxicity in mice lacking NADPH oxidase activity: role of peroxynitrite formation and mitochondrial oxidant stress. Free Radical Res. 2003, 37121289–97. [DOI] [PubMed] [Google Scholar]
- Bajt M. L.; Knight T. R.; Lemasters J. J.; Jaeschke H. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: protection by N-acetyl cysteine. Toxicol. Sci. 2004, 802343–9. [DOI] [PubMed] [Google Scholar]
- Moore M.; Thor H.; Moore G.; Nelson S.; Moldeus P.; Orrenius S. The Toxicity of Acetaminophen and N-Acetyl-Para-Benzoquinone Imine in Isolated Hepatocytes Is Associated with Thiol Depletion and Increased Cytosolic Ca-2+. J. Biol. Chem. 1985, 260243035–3040. [PubMed] [Google Scholar]
- Cover C.; Mansouri A.; Knight T. R.; Bajt M. L.; Lemasters J. J.; Pessayre D.; Jaeschke H. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J. Pharmacol. Exp. Ther. 2005, 3152879–87. [DOI] [PubMed] [Google Scholar]
- Tirmenstein M. A.; Nelson S. D. Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3′-hydroxyacetanilide, in mouse liver. J. Biol. Chem. 1989, 264179814–9. [PubMed] [Google Scholar]
- Cheung C.; Yu A. M.; Ward J. M.; Krausz K. W.; Akiyama T. E.; Feigenbaum L.; Gonzalez F. J. The cyp2e1-humanized transgenic mouse: role of cyp2e1 in acetaminophen hepatotoxicity. Drug Metab. Dispos. 2005, 333449–57. [DOI] [PubMed] [Google Scholar]
- Cohen S. D.; Khairallah E. A. Selective protein arylation and acetaminophen-induced hepatotoxicity. Drug Metab. Rev. 1997, 291–259–77. [DOI] [PubMed] [Google Scholar]
- Kheradpezhouh E.; Ma L.; Morphett A.; Barritt G. J.; Rychkov G. Y. TRPM2 channels mediate acetaminophen-induced liver damage. Proc. Natl. Acad. Sci. U.S.A. 2014, 11183176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukkanen M. O.; Leppanen P.; Turunen P.; Tuomisto T.; Naarala J.; Yla-Herttuala S. EC-SOD gene therapy reduces paracetamol-induced liver damage in mice. J. Gene Med. 2001, 34321–5. [DOI] [PubMed] [Google Scholar]
- Jaeschke H.; Bajt M. L. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol. Sci. 2006, 89131–41. [DOI] [PubMed] [Google Scholar]
- Shuhendler A. J.; Pu K.; Cui L.; Uetrecht J. P.; Rao J. Real-time imaging of oxidative and nitrosative stress in the liver of live animals for drug-toxicity testing. Nat. Biotechnol. 2014, 32, 373–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 292212–21. [DOI] [PubMed] [Google Scholar]
- Miettinen T. P.; Pessa H. K.; Caldez M. J.; Fuhrer T.; Diril M. K.; Sauer U.; Kaldis P.; Bjorklund M. Identification of transcriptional and metabolic programs related to Mammalian cell size. Curr. Biol. 2014, 246598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Molina D.; Jafari R.; Ignatushchenko M.; Seki T.; Larsson E. A.; Dan C.; Sreekumar L.; Cao Y.; Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341614184–7. [DOI] [PubMed] [Google Scholar]
- Winger J. A.; Hantschel O.; Superti-Furga G.; Kuriyan J. The structure of the leukemia drug imatinib bound to human quinone reductase 2 (NQO2). BMC Struct. Biol. 2009, 9, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Buryanovskyy L.; Zhang Z. Quinone reductase 2 is a catechol quinone reductase. J. Biol. Chem. 2008, 2833523829–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli C. M.; Tran N.; Knox R.; Jaiswal A. K. NRH:quinone oxidoreductase 2 (NQO2) catalyzes metabolic activation of quinones and anti-tumor drugs. Biochem. Pharmacol. 2006, 723366–76. [DOI] [PubMed] [Google Scholar]
- Vella F.; Ferry G.; Delagrange P.; Boutin J. A. NRH:quinone reductase 2: an enzyme of surprises and mysteries. Biochem. Pharmacol. 2005, 711–21–12. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Eberhard D.; Abraham Y.; Bastuck S.; Boesche M.; Hobson S.; Mathieson T.; Perrin J.; Raida M.; Rau C.; Reader V.; Sweetman G.; Bauer A.; Bouwmeester T.; Hopf C.; Kruse U.; Neubauer G.; Ramsden N.; Rick J.; Kuster B.; Drewes G. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007, 2591035–44. [DOI] [PubMed] [Google Scholar]
- Buryanovskyy L.; Fu Y.; Boyd M.; Ma Y.; Hsieh T. C.; Wu J. M.; Zhang Z. Crystal structure of quinone reductase 2 in complex with resveratrol. Biochemistry 2004, 433611417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine M.; Marcheteau E.; Delagrange P.; Ferry G.; Boutin J. A. Characterization of cofactors, substrates and inhibitor binding to flavoenzyme quinone reductase 2 by automated supramolecular nano-electrospray ionization mass spectrometry. Int. J. Mass Spectrom. 2012, 312, 87–96. [Google Scholar]
- Knox R. J.; Jenkins T. C.; Hobbs S. M.; Chen S.; Melton R. G.; Burke P. J. Bioactivation of 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB 1954) by human NAD(P)H quinone oxidoreductase 2: a novel co-substrate-mediated antitumor prodrug therapy. Cancer Res. 2000, 60154179–86. [PubMed] [Google Scholar]
- Reybier K.; Perio P.; Ferry G.; Bouajila J.; Delagrange P.; Boutin J. A.; Nepveu F. Insights into the redox cycle of human quinone reductase 2. Free Radical Res. 2011, 45101184–95. [DOI] [PubMed] [Google Scholar]
- Nosjean O.; Ferro M.; Coge F.; Beauverger P.; Henlin J. M.; Lefoulon F.; Fauchere J. L.; Delagrange P.; Canet E.; Boutin J. A. Identification of the melatonin-binding site MT3 as the quinone reductase 2. J. Biol. Chem. 2000, 2754031311–7. [DOI] [PubMed] [Google Scholar]
- Graves P. R.; Kwiek J. J.; Fadden P.; Ray R.; Hardeman K.; Coley A. M.; Foley M.; Haystead T. A. Discovery of novel targets of quinoline drugs in the human purine binding proteome. Mol. Pharmacol. 2002, 6261364–72. [DOI] [PubMed] [Google Scholar]
- Forman H. J.; Fridovich I. On the stability of bovine superoxide dismutase. The effects of metals. J. Biol. Chem. 1973, 24882645–9. [PubMed] [Google Scholar]
- Manyike P. T.; Kharasch E. D.; Kalhorn T. F.; Slattery J. T. Contribution of CYP2E1 and CYP3A to acetaminophen reactive metabolite formation. Clin. Pharmacol. Ther. 2000, 673275–82. [DOI] [PubMed] [Google Scholar]
- Liao S.; Williams-Ashman H. G. Enzymatic Oxidation of Some Non-Phosphorylated Derivatives of Dihydronicotinamide. Biochem. Biophys. Res. Commun. 1961, 43208–13. [DOI] [PubMed] [Google Scholar]
- Tripathy D.; Grammas P. Acetaminophen protects brain endothelial cells against oxidative stress. Microvasc. Res. 2009, 773289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long D. J. 2nd; Iskander K.; Gaikwad A.; Arin M.; Roop D. R.; Knox R.; Barrios R.; Jaiswal A. K. Disruption of dihydronicotinamide riboside:quinone oxidoreductase 2 (NQO2) leads to myeloid hyperplasia of bone marrow and decreased sensitivity to menadione toxicity. J. Biol. Chem. 2002, 2774846131–9. [DOI] [PubMed] [Google Scholar]
- Chung C. W.; Dean A. W.; Woolven J. M.; Bamborough P. Fragment-based discovery of bromodomain inhibitors part 1: inhibitor binding modes and implications for lead discovery. J. Med. Chem. 2012, 552576–86. [DOI] [PubMed] [Google Scholar]
- Shah A. D.; Wood D. M.; Dargan P. I. Understanding lactic acidosis in paracetamol (acetaminophen) poisoning. Br. J. Clin. Pharmacol. 2011, 71120–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister C. F.; McLean A. E. Inhibition of DNA synthesis by paracetamol in different tissues of the rat in vivo. Toxicology 1997, 1161–349–57. [DOI] [PubMed] [Google Scholar]
- Ahn K. S.; Gong X.; Sethi G.; Chaturvedi M. M.; Jaiswal A. K.; Aggarwal B. B. Deficiency of NRH:quinone oxidoreductase 2 differentially regulates TNF signaling in keratinocytes: up-regulation of apoptosis correlates with down-regulation of cell survival kinases. Cancer Res. 2007, 672010004–11. [DOI] [PubMed] [Google Scholar]
- Sener G.; Toklu H. Z.; Sehirli A. O.; Velioglu-Ogunc A.; Cetinel S.; Gedik N. Protective effects of resveratrol against acetaminophen-induced toxicity in mice. Hepatol. Res. 2006, 35162–8. [DOI] [PubMed] [Google Scholar]
- Janbaz K. H.; Saeed S. A.; Gilani A. H. Studies on the protective effects of caffeic acid and quercetin on chemical-induced hepatotoxicity in rodents. Phytomedicine 2004, 115424–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.