

Abstract
Ameloblastoma is a benign but locally infiltrative odontogenic neoplasm. Although ameloblastomas rarely metastasise, recurrences together with radical surgery often result in facial deformity and significant morbidity. Development of non-invasive therapies has been precluded by a lack of understanding of the molecular background of ameloblastoma pathogenesis. When addressing the role of ERBB receptors as potential new targets for ameloblastoma, we discovered significant EGFR over-expression in clinical samples using real-time RT–PCR, but observed variable sensitivity of novel primary ameloblastoma cells to EGFR-targeted drugs in vitro. In the quest for mutations downstream of EGFR that could explain this apparent discrepancy, Sanger sequencing revealed an oncogenic BRAF V600E mutation in the cell line resistant to EGFR inhibition. Further analysis of the clinical samples by Sanger sequencing and BRAF V600E-specific immunohistochemistry demonstrated a high frequency of BRAF V600E mutations (15 of 24 samples, 63%). These data provide novel insight into the poorly understood molecular pathogenesis of ameloblastoma and offer a rationale to test drugs targeting EGFR or mutant BRAF as novel therapies for ameloblastoma.
Keywords: ameloblastoma, BRAF, EGFR, odontogenic tumour, oncogenic mutation, targeted therapy
Introduction
Ameloblastoma is a slow-growing but locally infiltrative odontogenic neoplasm of the jaws 1,2, whose aetiology and pathogenesis are not understood. Due to their often complex growth pattern, most ameloblastomas are treated by surgical resection, often resulting in facial deformity and significant morbidity. More conservative approaches tend to result in recurrence and further surgery. Targeted therapy could potentially eliminate the need for extensive and/or repeated surgery.
ERBB receptors are a receptor tyrosine kinase subfamily that includes the epidermal growth factor receptor (EGFR), ERBB2, ERBB3 and ERBB4. ERBB receptors are indispensable for development and frequently dysregulated in human malignancies 3. EGFR and its ligands, EGF and transforming growth factor-α (TGFA) are expressed in the odontogenic epithelium of normal developing teeth 4, and strong EGFR expression has also been detected in ameloblastoma 4–6.
Here, we analysed the expression of all ERBB receptors in clinical ameloblastoma samples, using real-time RT–PCR. We also studied the role of ERBB signalling and assessed the feasibility of ERBB-targeted therapeutics in novel primary ameloblastoma cell lines. Furthermore, we report a high frequency of oncogenic BRAF V600E mutations in clinical ameloblastoma samples and demonstrate that BRAF V600E mutation was associated with resistance to EGFR-targeted drugs in primary ameloblastoma cells.
Materials and methods
Patients and tissue specimens
Fresh frozen tumour samples from 24 conventional intra-osseous ameloblastomas (Table 1), eight sporadic keratocystic odontogenic tumours (KCOT) and six samples of normal oral mucosa (see supplementary material, Table S1) were included in the study. Two ameloblastoma samples were from the primary and recurrent tumours of the same patient (samples 17 and 18; Table 1). Ethics Committee approvals (1–11 March 2007, 0/H0703/054 and CPP53-10) and the patients' written informed consents were obtained in accordance with the Helsinki Declaration.
Table 1.
Clinical information and BRAF mutation status of the ameloblastoma patients; cases arranged as in Figure 1
| Sampleno. | Age | Sex | Ethnicity | Location | Primary/recurrent | Histology | Notes | BRAF status | BRAF V600EIHC |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 66 | M | Caucasian | Right mandible | Primary | S/MC follicular | Enucleated | V600E | Pos |
| 2 | 70 | F | Asian | Left mandible | Primary | S/MC follicular | Enucleated | V600E | Pos |
| 3 | 61 | M | Caucasian | Right posterior mandible | Primary | S/MC plexiform | Resected | WT | Neg |
| 4 | 27 | F | Black-Somali | Middle mandible | Primary | S/MC follicular | Enucleated | V600E | Pos |
| 5 | 24 | F | Black | Right mandible | Primary | S/MC plexiform | Enucleated | V600E | Pos |
| 6 | 50 | F | Black | Left mandible | Recurrence | Plexiform | Resected, tumour sample decalcified in formic acid | V600E | Neg* |
| 7 | 36 | M | Afro-Caribbean | Left mandible | Residual | S/MC follicular | Enucleated | WT | Neg |
| 8 | 47 | M | Caucasian | Mandible | Primary | S/MC follicular | Resected | V600E | Pos |
| 9 | 32 | M | North African | Right posterior mandible | 1st recurrence | S/MC plexiform | Primary tumour at age 27; enucleated | WT | NA |
| 10 | 46 | M | African | Middle anterior mandible | 3rd recurrence | S/MC follicular | Primary tumour at age 25; radical resection | V600E | NA |
| 11 | 14 | M | Black-North African | Left posterior mandible | Primary | S/MC plexiform | Left angle and ramus | V600E | Pos |
| 12 | 34 | F | Caucasian | Left posterior mandible | 1st recurrence | S/MC follicular | Resected | V600E | Pos |
| 13 | 84 | M | Caucasian | Right mandible | Primary | S/MC follicular | Resected | WT | Neg |
| 14 | 18 | F | Caucasian | Left anterior mandible | Primary | S/MC follicular | Enucleated | V600E | NA |
| 15 | 16 | M | Caucasian | Mandible | Primary | S/MC plexiform | Enucleated | V600E | Pos |
| 16 | 61 | F | NA | Right mandible | Primary | S/MC plexiform/follicular | Enucleated | WT | Neg |
| 17 | 69 | M | Caucasian | Left posterior mandible | Primary | S/MC plexiform | Primary tumour for sample 18; enucleated | WT | Neg |
| 18 | 77 | M | Caucasian | Left posterior mandible | 1st recurrence | S/MC plexiform | Recurred after 8 years, primary tumour sample 17 | WT | Neg |
| 19 | 69 | M | Caucasian | Right posterior mandible | Primary | S/MC plexiform | Patient died after operation | WT | Neg |
| 20 | 43 | M | Caucasian | Right body mandible | Primary | S/MC follicular | Enucleated | V600E | Pos |
| 21 | 44 | F | Black-African | Right posterior mandible | Primary | S/MC plexiform | No recurrence after 5 years | V600E | Pos |
| 22 | 33 | M | African | Middle anterior mandible | 3rd recurrence | S/MC follicular | Primary tumour at age 20; enucleated | V600E | Pos |
| 23 | 46 | F | Afro-Caribbean | Left body mandible | 1st recurrence | S/MC plexiform | Primary tumour at age 34; resected | V600E | NA |
| 24 | 31 | M | Black-African | Left posterior mandible | Primary | S/MC follicular | Recurred after 5 years, no real-time RT–PCR data available | WT | Neg |
Decalcification of ameloblastoma tumour samples using formic acid may hinder the BRAF V600E immunohistochemistry.
S/MC, solid/multicystic; WT, wild-type; NA, data/sample not available; IHC, immunohistochemistry.
Real-time RT–PCR
RNA isolation and real-time RT–PCR (TaqMan, Applied Biosystems) analyses were carried out as previously described 7,8. The amount of tumour tissue in the samples was >90%. Primer and probe sequences have been described previously 8 or are described in Table S2 (see supplementary material). One ameloblastoma sample (patient sample 24) was not included in the analysis, due to insufficient RNA concentration.
Establishment of primary ameloblastoma cells and ameloblastoma fibroblasts
Fresh samples from ameloblastoma tumours (AB10, patient 3; ABSV, patient 12; see Table 1) were cut into small pieces (approximately 1 × 1 mm) and placed in T25 cell culture flasks with 1 ml CnT-24 medium (CELLnTEC), supplemented with Pen/Strep/Amphotericin B Solution (CELLnTEC). Outgrowing ameloblastoma cells were harvested at confluence and maintained in the CnT-24 medium. Primary ameloblastoma fibroblast (AF) cultures were established from an ameloblastoma sample (tumour not analysed in this study) as above, using Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum (FCS), 50 U/ml penicillin and 50 µg/ml streptomycin as the culture medium.
Antibodies and inhibitors
The EGFR antibodies cetuximab and panitumumab, as well as the ERBB2 antibody trastuzumab, were purchased from Turku University Hospital pharmacy. The EGFR tyrosine kinase inhibitors gefitinib and erlotinib were purchased from Santa Cruz Biotechnology. EGFR/ERBB4 inhibitor AG1478, PI3K inhibitor LY294002 and MEK inhibitor PD98059 were purchased from Calbiochem (Merck Millipore).
MTT cell viability assay
AB10 and ABSV cells were plated onto 96-well plates in triplicate, at a density of 3500 cells/well, in CnT-24 Oral Epithelium Medium, with or without inhibitors. After 72 h, the number of viable cells was estimated using the MTT assay (CellTiter 96 non-radioactive cell proliferation assay; Promega) according to the manufacturer's guidelines.
Western blotting
Cells growing on six-well plates were treated overnight with EGFR, ERBB2, AKT or MEK inhibitors or for 10 min with EGF (R&D). Samples of cell lysates (25 µg total protein) were analysed by western blotting with antibodies against phospho-EGFR, phospho-ERK, phospho-AKT and ERK (cat. nos 2220, 9101, 9271 and 9102, Cell Signaling Technology) and EGFR and AKT (sc-03 and sc-1618, Santa Cruz Biotechnology).
Targeted cDNA sequencing
Sequences coding for EGFR kinase domain and KRAS, NRAS, HRAS or BRAF genes were PCR-amplified and purified using NucleoSpin Gel and PCR Clean-up kit (Macheney-Nagel). Both strands of amplified fragments were Sanger-sequenced for recurrent mutations (kinase domain for EGFR, codons 12, 13 and 61 for RAS genes, codon 600 for BRAF). Primers used in cDNA sequencing are described in Table S2 (see supplementary material).
BRAF V600E immunohistochemistry
Immunohistochemical staining was performed using BRAF V600E mutation-specific antibody VE1 (Spring Biosciences), as previously described 9.
Statistical analyses
Real-time RT–PCR data were analysed by one-way ANOVA, using the Games–Howell post hoc test. MTT cell viability assays were analysed by t-test. To analyse the relationships between BRAF mutation status and clinical patient data, Fisher's exact test was used. Association of BRAF mutation status with EGFR expression (high or low; above or below median expression, respectively) was analysed using Fisher's exact test. Statistical analyses were carried out using SPSS statistics v 20 (IBM).
Results
EGFR and ERBB4 are over-expressed in ameloblastoma
A real-time RT–PCR analysis of 23 solid/multicystic ameloblastomas (patient samples 1–23; Table 1) was performed to study the expression of ERBB receptors. Eight KCOTs and six normal oral mucosa samples were included in the analysis as controls (see supplementary material, Table S1). EGFR and ERBB4 were specifically over-expressed in ameloblastoma when compared to normal samples (EGFR, p = 0.003; ERBB4, p = 0.01) or to KCOT (EGFR, p = 0.001; ERBB4, p < 0.001) (Figure 1A, D). EGFR over-expression is in accordance with previous studies reporting high EGFR protein levels in ameloblastoma 4–6. The predominantly expressed ERBB4 receptor isoforms in ameloblastoma were the JM-a isoforms (see supplementary material, Figure S1). For ERBB2, no statistically significant differences were observed (Figure 1B). ERBB3 was significantly more highly expressed in KCOT than in ameloblastoma (p = 0.011) (Figure 1C).
Figure 1.
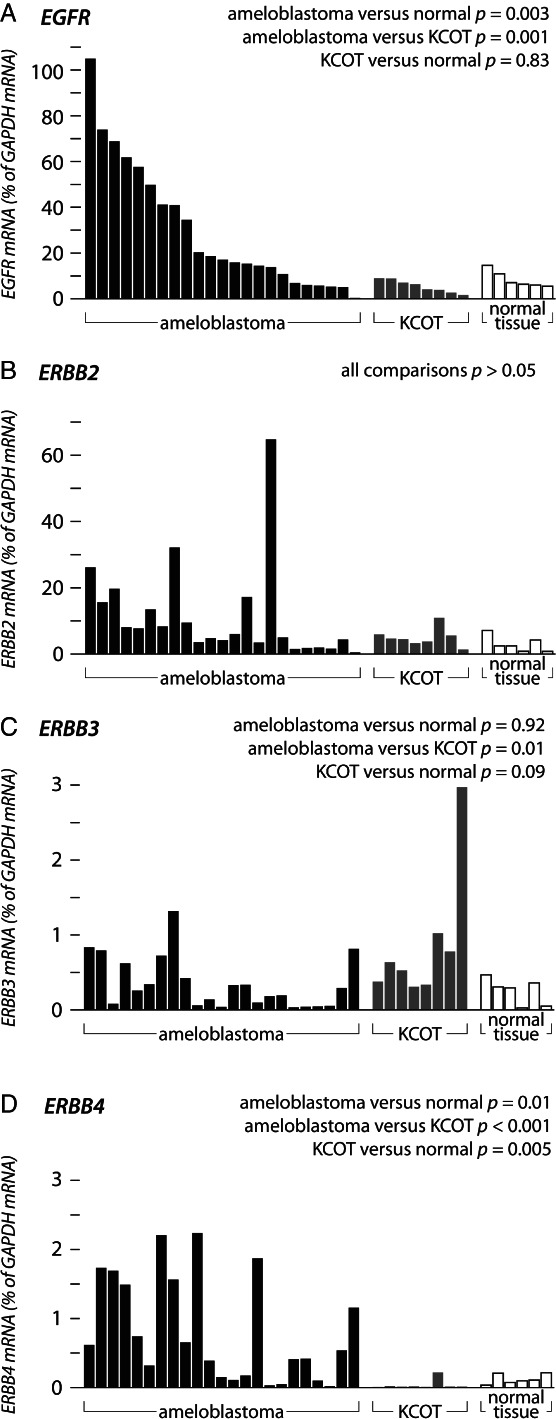
Real-time RT–PCR analysis of ERBB receptor expression in ameloblastoma, keratocystic odontogenic tumour (KCOT) and normal oral mucosa. Twenty-three ameloblastomas, eight KCOTs and six normal samples were analysed for EGFR (A), ERBB2 (B), ERBB3 (C) or ERBB4 (D) expression.
Establishment of ameloblastoma cell lines
To address the function of ERBB receptors in ameloblastoma, two non-immortalized primary ameloblastoma cell lines, AB10 and ABSV, were established from patient samples 3 and 12, respectively (Table 1). A primary fibroblast cell line (ameloblastoma fibroblasts, AFs) was also established (from a tumour not analysed in this study). AB10 and ABSV cells were morphologically identical and formed an epithelial-like monolayer very similar to those of two previously published ameloblastoma cell lines 10,11, whereas ameloblastoma fibroblasts demonstrated a typical spindle-shaped fibroblastic morphology (Figure 2A). The ameloblastoma cells expressed high levels of epithelial markers KRT14 (keratin 14), KRT19 (keratin 19) and CDH1 (E-cadherin) (Figure 2B), whereas the expression of mesenchymal markers CDH2 (N-cadherin) and VIM (vimentin) was almost undetectable (Figure 2B). The ERBB receptor expression pattern was similar in both ameloblastoma cell lines (Figure 2D) and corresponded to that observed in the ameloblastoma tumour samples (Figure 1). However, neither of the cell lines expressed detectable levels of ERBB4, although ERBB4 was expressed in the original tumour from which the AB10 cell line was established. This suggests that ERBB4 expression was lost during cell line establishment.
Figure 2.
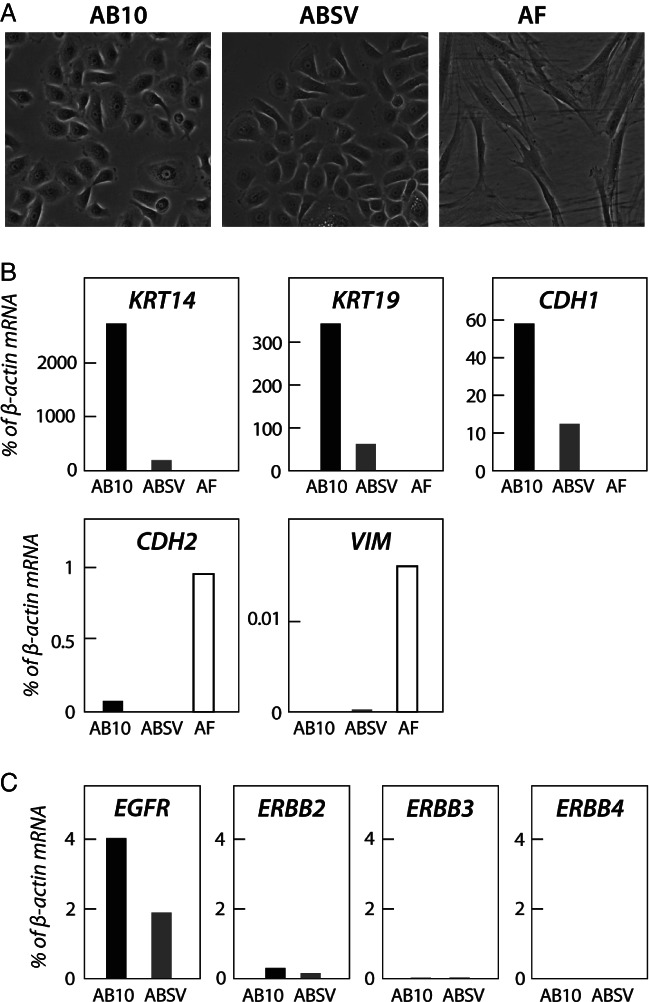
Characterization of established primary ameloblastoma tumour cell lines. (A) Established AB10, ABSV and ameloblastoma fibroblast cultures were grown on six-well plates and photographed using ×200 magnification. (B) The cell lines were analysed for the expression of epithelial markers KRT14 (keratin 14), KRT19 (keratin 19) and CDH1 (E-cadherin) and mesenchymal markers CDH2 (N-cadherin) and VIM (vimentin), using real-time RT–PCR. (C) AB10 and ABSV cell lines were analysed for EGFR, ERBB2, ERBB3 and ERBB4 expression, using real-time RT–PCR.
EGFR inhibition in ameloblastoma cells
The effect of EGFR inhibition on the proliferation of primary ameloblastoma cells was analysed by MTT cell viability assays. In AB10 cells, 72 h of treatment with the EGFR antibodies cetuximab and panitumumab already promoted a significant, dose-dependent reduction in proliferation at a concentration of 0.1 µg/ml (p < 0.001) (Figure 3A). Consistently, the EGFR tyrosine kinase inhibitors (TKI) erlotinib, gefitinib and AG1478 already significantly suppressed AB10 cell growth at the concentration of 0.01 μm (p < 0.001, p = 0.004, and p < 0.001, respectively). However, similar treatment with the ERBB2-antibody trastuzumab did not demonstrate a dose-dependent effect (Figure 3A). Treating AB10 cells with EGFR-targeted antibodies or TKIs abolished EGFR phosphorylation and silenced the RAS–RAF–MAPK and PI3K–AKT signalling pathways downstream of EGFR, as seen by a reduction of ERK and AKT phosphorylation after treatment (Figure 3B). Consistent with the proliferation assays, ERBB2 inhibition had no major effect on the phosphorylation of ERK or AKT. When tested on the ABSV cells, however, neither panitumumab nor cetuximab had a significant effect on proliferation (Figure 3A). The EGFR TKIs reduced proliferation of ABSV cells by 50%, but the effect was achieved only with the highest concentration of 10 μm. Indeed, there was a clear, statistically significant difference in the sensitivity of AB10 and ABSV cells lines to EGFR inhibition (Figure 3A).
Figure 3.

Effect of EGFR inhibition on ameloblastoma cells. (A) AB10 and ABSV cells were plated on 96-well plates and incubated for 72 h, with or without the indicated concentrations of EGFR monoclonal antibodies cetuximab or panitumumab, ERBB2 monoclonal antibody trastuzumab or EGFR tyrosine kinase inhibitors erlotinib, gefitinib or AG1478. Cell viability after incubation was estimated using the MTT assay. *Significant differences between the proliferation of AB10 and ABSV cells (*p < 0.05, **p < 0.01, ***p < 0.001; Student's t-test). (B) AB10 cells were grown on six-well plates and treated overnight with cetuximab (10 µg/ml), panitumumab (10 µg/ml), trastuzumab (10 µg/ml), erlotinib (10 μm), AG1478 (10 μm), PI3-K inhibitor LY294002 (10 μm) or MEK inhibitor PD98059 (10 μm) or for 10 min with EGF (50 ng/µl). After treatment, phosphorylation of EGFR, AKT and ERK was analysed by western blotting, using phospho-specific antibodies (n.s., non-specific band). Total EGFR, AKT and ERK levels were determined using anti-EGFR, anti-AKT and anti-ERK antibodies. (C) The coding region of the BRAF gene was PCR-amplified from AB10 and ABSV cell cDNA, followed by Sanger sequencing of both strands of a 320 bp amplicon flanking the V600 codon of BRAF. The arrow indicates the mutated nucleotide responsible for the V600E substitution.
BRAF mutation is associated with resistance to EGFR-targeted drugs in primary ameloblastoma cells
Specific genetic markers predict the response to EGFR-targeted cancer drugs in the clinic and in vitro 12–20. To test whether the differences observed in the responses to EGFR-targeted drugs in ameloblastoma cells were due to genetic factors, cDNA sequencing was performed to reveal mutations in the EGFR kinase domain or in the RAS–RAF–MAPK signalling pathway genes KRAS, NRAS, HRAS and BRAF. No mutations were observed in the EGFR kinase domain or in the RAS genes in either of the two cell lines. However, the unresponsive ABSV cell line harboured a BRAF V600E mutation (Figure 3C), caused by a point mutation 1799T > A. These findings demonstrate that the resistance of the ABSV cell line to EGFR-targeted drugs was associated with an activating mutation in a downstream signalling pathway.
BRAF mutations are frequent in ameloblastoma
Twenty-four ameloblastoma samples were analysed for BRAF mutations by cDNA sequencing. BRAF V600E-specific immunohistochemistry was also performed for cases where appropriate tumour sections were available. Of 24 tumours, 15 (63%) were observed to harbour the BRAF V600E (1799T > A) mutation (Figure 4, Table 1). In most cases, as in the ABSV cell line (Figure 3C), the mutation appeared heterozygous (Figure 4A). However, as the sequencing was performed from cDNA and the involvement of normal tissue could not be totally excluded, gene-level zygosity of the mutation could not be comprehensively addressed in the tumour samples. The mutations were found in patients with different ethnicities, including individuals of Caucasian (n = 6), Black-African (n = 7), Afro-Caribbean (n = 1) and Asian (n = 1) origin, with no significant association to specific ethnicity. BRAF mutation status also did not correlate with the sex or age of the patients, tumour histology, tumour recurrence or EGFR expression level (p > 0.1). As expected, the original tumour from which the AB10 cell line was established (patient sample 3; Table 1) was negative for the mutation and the tumour from which the ABSV line was established (patient sample 12; Table 1) was positive. These findings suggest the presence of a high-frequency oncogenic mutation in clinical ameloblastoma.
Figure 4.
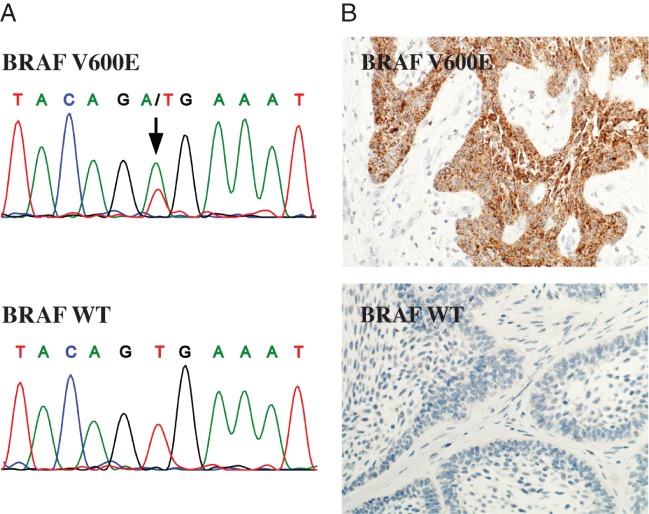
Analysis of BRAF V600E mutations in ameloblastoma tumour samples. (A) The coding region of the BRAF gene was PCR-amplified from tumour cDNA, followed by Sanger sequencing of both strands of a 320 bp region flanking the V600 codon of BRAF; arrow indicates the mutated nucleotide responsible for the V600E substitution. (B) Immunohistochemistry was performed for tumour sections using BRAF V600E-specific monoclonal antibody. Matching electropherograms and images from BRAF V600E immunohistochemistry are shown for BRAF V600E-positive (top, patient sample 20) and negative (bottom, patient sample 24) tumours.
Discussion
The pathogenesis of ameloblastoma has remained elusive, despite considerable efforts to characterize the underlying molecular mechanisms. Here, the ERBB signalling in ameloblastoma was studied using clinical ameloblastoma samples as well as novel ameloblastoma cell lines. Real-time RT–PCR analysis of ERBB expression demonstrated a statistically significant over-expression of EGFR and ERBB4 in ameloblastomas (n = 23) when compared to clinical samples of KCOT (n = 8) or to normal mucosa (n = 6). Pharmacological inhibition of EGFR suppressed the proliferation of one primary ameloblastoma cell line (AB10) but was ineffective for the other (ABSV). In the quest for factors underlying this difference, a BRAF V600E mutation was found in the resistant ABSV cell line. These observations demonstrate that, similar to KRAS mutations in colorectal cancer 12,17,18,20, the BRAF V600E mutation is associated with resistance to EGFR-targeted drugs in ameloblastoma cells. BRAF V600E mutations were found in 63% (15 of 24) of the ameloblastoma samples. The high incidence of BRAF mutations represents the first indication of a high-frequency oncogenic mutation in ameloblastoma. Given the high response rate of BRAF V600E-positive metastatic melanoma to mutant BRAF-targeted inhibition by vemurafenib 21 or dabrafenib 22, our observations also offer a rationale for future testing of BRAF-targeted therapeutics for ameloblastoma.
The presence of activating BRAF mutations suggests the involvement of a hyperactive RAS–RAF–MAPK pathway in the pathogenesis of ameloblastomas. The observed dependency of AB10 cells on EGFR signalling supports this hypothesis, as the RAS–RAF–MAPK pathway is well known to operate downstream of EGFR 23, and as ERK phosphorylation was completely abolished in response to EGFR inhibition in these cells. Interestingly, transgenic mice carrying the vHa-RAS oncogene, a constitutively active homologue of the human HRAS, develop odontogenic tumours that closely resemble human ameloblastoma 24,25.
Taken together, our results indicate that a hyperactive RAS–RAF–MAPK pathway is closely associated with ameloblastoma pathogenesis, either through EGFR-mediated signalling or through frequent activating mutations in the BRAF gene. Thus, our observations offer for the first time a rationale for designing targeted therapies in ameloblastoma.
Acknowledgments
The skilful technical assistance of Marja Uola and Mariia Valkama (Institute of Dentistry, University of Turku), Tiia Heinonen (Turku Centre for Biotechnology), Minna Santanen (Department of Medical Biochemistry and Genetics, University of Turku) and Maria Tuominen (MediCity Research Laboratories, University of Turku) is gratefully acknowledged. The work was financially supported by the Academy of Finland (Grant No. 137845), Finnish Cancer Organizations, the Sigrid Jusélius Foundation, Turku University Central Hospital, the Mariza and Reino Salonen Foundation and the French Ministry of Health (Grant No. AOM09186). Seven patient tissue samples and data were provided by Guy's and St Thomas's Head and Neck Biobank, which is supported by the UK Department of Health via the National Institute for Health Research (NIHR), a comprehensive Biomedical Research Centre award and Guy's and St Thomas's NHS Foundation Trust.
Author contributions
KK, KE and KH designed the study; KH, PRM, BR and JK provided the clinical ameloblastoma samples; KH and PRM diagnosed the tumours; JC and PRM established the AB10 cell line; AR performed BRAF V600E immunohistochemistry; KK conducted all the remaining experimental work; and KK, KE and KH prepared the manuscript.
Supporting Information
The following supplementary material may be found in the online version of this article:
Real-time RT–PCR analysis of the expression of ERBB4 receptor isoforms in ameloblastoma, keratocystic odontogenic tumour (KCOT) and normal oral mucosa.
Clinical information for KCOT patients and normal sample donors.
Primer and probe sequences used in real-time RT–PCR and targeted cDNA sequencing.
References
- 1.Gardner DG, Heikinheimo K, Shear M. Pathology and genetics of the head and neck tumours: ameloblastoma. In: Barnes L, Eveson JW, Reichart P, Sidransky D, et al., editors. Word Health Organization Classification of Tumours. Lyon: IARC Press; 2005. pp. 296–300. [Google Scholar]
- 2.Mendenhall WM, Werning JW, Fernandes R, et al. Ameloblastoma. Am J Clin Oncol. 2007;30::645–648. doi: 10.1097/COC.0b013e3181573e59. [DOI] [PubMed] [Google Scholar]
- 3.Holbro T, Hynes NE. ErbB receptors: directing key signalling networks throughout life. Annu Rev Pharmacol Toxicol. 2004;44::195–217. doi: 10.1146/annurev.pharmtox.44.101802.121440. [DOI] [PubMed] [Google Scholar]
- 4.Heikinheimo K, Voutilainen R, Happonen RP, et al. EGF receptor and its ligands, EGF and TGFα, in developing and neoplastic human odontogenic tissues. Int J Dev Biol. 1993;37::387–396. [PubMed] [Google Scholar]
- 5.Ueno S, Miyagawa T. Immunohistochemical investigation of epidermal growth factor receptor expression in ameloblastomas. J Pathol. 1994;173::33–38. doi: 10.1002/path.1711730106. [DOI] [PubMed] [Google Scholar]
- 6.Vered M, Shohat I, Buchner A. Epidermal growth factor receptor expression in ameloblastoma. Oral Oncol. 2003;39:138–143. doi: 10.1016/s1368-8375(02)00034-9. [DOI] [PubMed] [Google Scholar]
- 7.Heikinheimo K, Jee KJ, Niini T, et al. Gene expression profiling of ameloblastoma and human tooth germ by means of a cDNA microarray. J Dent Res. 2002;81::525–530. doi: 10.1177/154405910208100805. [DOI] [PubMed] [Google Scholar]
- 8.Junttila TT, Laato M, Vahlberg T, et al. Identification of patients with transitional cell carcinoma of the bladder overexpressing ErbB2, ErbB3, or specific ErbB4 isoforms: real-time reverse transcription–PCR analysis in estimation of ErbB receptor status from cancer patients. Clin Cancer Res. 2003;9::5346–5357. [PubMed] [Google Scholar]
- 9.Capper D, Preusser M, Habel A, et al. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol. 2011;122::11–19. doi: 10.1007/s00401-011-0841-z. [DOI] [PubMed] [Google Scholar]
- 10.Harada H, Mitsuyasu T, Nakamura N, et al. Establishment of ameloblastoma cell line, AM-1. J Oral Pathol Med. 1998;27::207–212. doi: 10.1111/j.1600-0714.1998.tb01943.x. [DOI] [PubMed] [Google Scholar]
- 11.Kibe T, Fuchigami T, Kishida M, et al. A novel ameloblastoma cell line (AM-3) secretes MMP-9 in response to Wnt-3a and induces osteoclastogenesis. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115::780–788. doi: 10.1016/j.oooo.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26::1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 13.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350::2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 14.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from 'never smokers' and are associated with sensitivity of tumours to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101::13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paez JG, Jänne P. Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304::1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 16.Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2::e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lièvre A, Bachet J-B, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66::3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 18.Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signalling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67::2643–2648. doi: 10.1158/0008-5472.CAN-06-4158. [DOI] [PubMed] [Google Scholar]
- 19.Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26::5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 20.De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS,BRAF,NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11::753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 21.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364::2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380::358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 23.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2::127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 24.Cardiff RD, Leder A, Kuo A, et al. Multiple tumour types appear in a transgenic mouse with the ras oncogene. Am J Pathol. 1993;142::1199–1207. [PMC free article] [PubMed] [Google Scholar]
- 25.Dodds A, Cannon R, Suggs C, et al. mRNA expression and phenotype of odontogenic tumours in the v-Ha-ras transgenic mouse. Arch Oral Biol. 2003;48::843–850. doi: 10.1016/s0003-9969(03)00178-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Real-time RT–PCR analysis of the expression of ERBB4 receptor isoforms in ameloblastoma, keratocystic odontogenic tumour (KCOT) and normal oral mucosa.
Clinical information for KCOT patients and normal sample donors.
Primer and probe sequences used in real-time RT–PCR and targeted cDNA sequencing.