

Abstract
Pancreatic ductal adenocarcinoma (PDA) is one of the most lethal malignancies characterized by an intense tumor stroma with hypoperfused regions, a significant inflammatory response and pronounced therapy resistance. New therapeutic agents are urgently needed. The plant-derived agent triptolide also known as “thunder god vine” has a long history in traditional Chinese medicine for treatment of rheumatoid arthritis and cancer and is now in a clinical phase II trial for establishing the efficacy against a placebo. The authors mimicked the situation in patient tumors by induction of hypoxia in experimental models of pancreatic cancer stem cells (CSCs) and evaluated the therapeutic effect of triptolide. Hypoxia led to induction of colony and spheroid formation, aldehyde dehydrogenase 1 (ALDH1) and NF-κB activity, migratory potential and a switch in morphology to a fibroblastoid phenotype, as well as stem cell- and epithelial–mesenchymal transition-associated protein expression. Triptolide efficiently inhibited hypoxia-induced transcriptional signaling and downregulated epithelial–mesenchymal transition (EMT) and CSC features in established highly malignant cell lines, whereas sensitive cancer cells or nonmalignant cells were less affected. In vivo triptolide inhibited tumor take and tumor growth. In primary CSCs isolated from patient tumors, triptolide downregulated markers of CSCs, proliferation and mesenchymal cells along with upregulation of markers for apoptosis and epithelial cells. This study is the first to show that triptolide reverses EMT and CSC characteristics and therefore may be superior to current chemotherapeutics for treatment of PDA.
What's new?
Current treatment for pancreatic cancer does not directly target tumor hypoxia, a major mediator of aggressive growth, early metastasis, and therapy resistance. The plant-derived agent triptolide has a long history of use in rheumatoid arthritis and cancer in traditional Chinese medicine and has been shown to have potent therapeutic properties in a variety of studies. Here, the authors show for the first time that triptolide effectively inhibits hypoxia-induced signaling, leading to downregulation of NF-κB activity, epithelial-mesenchymal transition, and stem cell-like features. Triptolide may therefore be superior to current chemotherapeutics for treatment of pancreatic cancer.
Keywords: pancreatic cancer, novel antitumor agents, hypoxia, epithelial–mesenchymal transition, cancer stem cells
Pancreatic ductal adenocarcinoma (PDA) is one of the most aggressive malignancies and is usually diagnosed in an advanced state, with extensive local invasion, early systemic dissemination and marked resistance to chemotherapy and radiotherapy.1 PDA is characterized by a pronounced hypoxic tumor microenvironment,2 resulting in high expression of the hypoxia marker HIF-1α in patient tissue, which is a predictor of poor clinical outcome.3 In experimental studies, hypoxia is a marker for aggressive growth and spontaneous metastasis formation in pancreatic cancer xenografts,4 suggesting a role for hypoxia in enrichment of cancer cells with stem cell characteristics.
According to the theory, cancer stem cells (CSCs) survive hypoxia and conventional cytotoxic therapy due to defense and survival mechanisms.5 The small CSC population within the tumor mass is believed to possess self-renewal potential and the ability to differentiate, thereby generating a heterogeneous cell population of the originating tumor.6–8 In addition, CSCs are proposed to mediate uncontrolled growth, therapy resistance, invasion and metastasis.9 Markers for CSCs have been identified in PDA and involve typical surface marker expression of CD44+/CD24+/ESA+, CD133+/CXCR4+ and c-Met+.10–12 Recent reports have indicated that the emergence of CSCs occurs in part because of epithelial–mesenchymal transition (EMT).13
EMT can be activated by hypoxia-induced HIF-1 signaling,14 followed by a cellular switch from epithelial to mesenchymal properties.15 This is accompanied by reduced intercellular adhesion and formation of migratory cells with invasive properties.16 During EMT, the mesenchymal marker Vimentin is upregulated, and the epithelial marker and transmembrane adhesion molecule E-cadherin is downregulated. E-cadherin is transcriptionally repressed by Twist, Snail and Slug and causes adherens junction breakdown in concert with other signaling events.13 The involvement of EMT in tumor metastasis in vivo has been controversial for a long time17 because human carcinoma metastasis lacks a mesenchymal phenotype and presents with an epithelial morphology.18 Therefore, it has been proposed that invading tumor cells undergo mesenchymal–epithelial transition to form metastases with an epithelial phenotype.19 A recent article confirmed this hypothesis and showed in vivo the requirement of “reversible EMT” in tumor metastasis.20 Recent data have demonstrated that EMT is involved in generating cells with stem cell properties.21
Furthermore, hypoxia leads to activation of the transcription factor NF-κB and its translocation to the nucleus, where it binds to Iκ-specific promoter regions of many genes.22,23 The functions of NF-κB are diverse and include regulation of cell proliferation, resistance to apoptosis, EMT, metastasis and inflammation-induced cancer development and progression.24–26 Recent studies have indicated a role for NF-κB activation in providing signals that maintain mammary CSCs.27 Our data have demonstrated that constitutively enhanced NF-κB binding of the subunits c-Rel and Rel A confers CSC features in highly aggressive PDA cells.28,29
Traditional Chinese medicine (TCM) provides a rich source of anti-inflammatory agents with NF-κB inhibitory and anticarcinogenic activities. The herb Tripterygium wilfordii Hook f, known as the “thunder god vine” in China, has a long history in the treatment of rheumatoid arthritis and cancer.30 The major active substance in this herb is triptolide, a diterpenoid triepoxide, which is currently being evaluated in a clinical phase I trial for screening of safety (reviewed in Ref.31). Several experimental studies have explained the anti-inflammatory, proapoptotic and tumor-repressing effects of triptolide by inhibition of NFAT, proteasome activity, topoisomerase, heat-shock response and NF-κB signaling (reviewed in Ref.31). Whether triptolide might overcome hypoxia-induced NF-κB activity, EMT and CSC characteristics in PDA is unknown thus far, although these features may be the prerequisite for therapeutic long-term responses.
In our study, we demonstrate that hypoxia induces CSC characteristics and NF-κB c-Rel-dependent EMT. Downregulation of NF-κB by triptolide inhibited migration, self-renewal activity, stem cell-related signaling, tumor take and growth of established pancreatic cancer cells. Most notably, triptolide induced apoptosis and inhibited proliferation along with downregulation of CSC and EMT markers in spheroidal CSC-enriched cultures selected from patient tumors.
Material and Methods
Tumor cell lines
BxPc-3, MIA-PaCa2 and AsPC-1 pancreatic cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA) and authenticated throughout the culture by the typical morphology. To maintain authenticity of the cell lines, frozen stocks were prepared from initial stocks, and every 3 months, a new frozen stock was used for the experiments. Mycoplasma-negative cultures were ensured by monthly testing. Cells were cultured in DMEM (PAA, Pasching, Austria) supplemented with 10% heat-inactivated FCS (Sigma, Deisenhoffen, Germany) and 25 mmol/l HEPES (PAA).
Selection of CSC-enriched spheroidal cells from patient tumors by subtransplantation
A surgical nondiagnostic specimen was mechanically minced, and 2 × 107 cells in matrigel were transplanted into the flanks of 6-week-old NMRI (nu/nu) female mice. After development of a tumor, the xenograft was resected, minced and subtransplanted to new mice. Subtransplantation was repeated until a stably growing xenograft line after Passage 3 was obtained. Pancreatic cancer spheres were generated as recently described32 and used for experiments lasting between 7 and 30 days in culture. Patient material was obtained under the approval of the ethical committee of the University of Heidelberg after written informed consent of patients. The diagnoses were established by conventional clinical and histological criteria according to the World Health Organization. All surgical resections were indicated by principles and practice of oncological therapy.
Treatment of cells
For induction of hypoxia, 80% confluent cells were added to a hypoxia chamber (self-made), which was flushed using a gas mixture of 1% O2, 5% CO2 and 94% N2 (Grandpair, Heidelberg, Germany) for ∼4 min. Cells were incubated in the hypoxic environment for 24, 48 or 72 hr at 37°C. If the incubation period exceeded 24 hr, the chamber was refilled with the gas mixture every 24 hr to ensure constant oxygen concentrations. Triptolide (PG-490) was obtained from Sigma-Aldrich (St. Louis, MO). The purity of triptolide was ≥98% (HPLC), and a 10 mM stock solution was prepared in DMSO.
Viability assay
Viability was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as described previously.29
Colony-forming assay
Cells cultured under normoxia or hypoxia for 24 hr were seeded in complete medium in six-well tissue culture plates (TPP), and colony-forming assays were performed as described previously.29
Spheroid assay
For formation of spheroids, cells were cultured in NeuroCult NS-A basal serum-free medium (human; StemCell Technologies, Vancouver, Canada) supplemented with 2 μg/ml heparin (StemCell Technologies), 20 ng/ml hEGF (R&D Systems, Wiesbaden-Nordenstadt, Germany), 10 ng/ml hFGF-b (PeproTech, Hamburg, Germany) and NeuroCult NS-A Proliferation Supplements (StemCell Technologies). Cells were seeded at low densities (5 × 102 to 2 × 103 cells per milliliter) in 12-well low-adhesion plates (1 milliliter per well). For quantification of the percentage of spheroid-forming cells, cells were seeded at one cell per well in 96-well plates. Wells with more than one cell were excluded from evaluation.
Aldehyde dehydrogenase 1 activity
ALDEFLUOR substrate (5 µl; Aldagen, Durham, NC) was added to 1 × 106 treated tumor cells in 500 µl of assay buffer and incubated for 60 min at 37°C. Pretreatment with the aldehyde dehydrogenase 1 (ALDH1) inhibitor diethylaminobenzaldehyde was used as a negative control.
Scratch assay
Tumor cells (6 × 105) were seeded in six-well plates and grown to confluence overnight. A line was then scraped within confluent cells using the fine end of 10-µl pipette tips (Time 0). Cells were exposed to hypoxia for 24 hr. Images of migrating cells were sequentially acquired at 6, 12 and 24 hr.
Western blot analysis
Total cell lysates, whole-cell protein extracts were prepared by a standard protocol, and proteins were detected by Western blot analysis as described previously.29 Briefly, cells were washed twice in ice-cold PBS and were lysed in three packed cell volumes of buffer containing Tris (30 mM, pH 7.4), NaCl (150 mM), 0.5% Triton X-100, 0.5% Na-Desoxycholate, 1 µl/ml dithiothreitol (DTT) and protease inhibitor cocktails (Sigma-Aldrich). After incubation on ice for 5 min, lysed cells were centrifuged at 14,000 rpm for 20 min to pellet cellular debris. Cellular proteins (40–60 μg) were separated on 8%, 10% or 12% SDS-PAGE gel, and the proteins were transferred to the Immobilon®-P Transfer membranes (Millipore, Billerica, MA). The following antibodies were used: mouse mAbs against human Twist2, Vimentin, Notch4 (Abcam, Cambridge, UK), HIF-1α (R&D Systems, Abingdon, UK) and β-Actin (Sigma-Aldrich) and rabbit polyclonal Abs against human Snail, Slug, Jagged1 (Abcam), E-cadherin, Sox2, Nanog (Cell Signaling, Danvers, MA), c-Rel and Rel-A (Santa Cruz Biotechnology, Santa Cruz, CA).
Gel retardation analysis of NF-κB binding
Nuclear protein extracts were prepared using NE-PER® Nuclear and Cytoplasmic Extraction Reagents, and the bandshift reaction was performed using the Light Shift® Chemiluminescent EMSA Kit according to the manufacturer's instructions (Thermo Scientific). The following biotin 3′ end-labeled oligonucleotides or unlabeled oligonucleotides (MWG Biotech) were used: NF-κB-antisense: 5-GCC TGG GAA AGT CCC CTC AA-3′ and NF-κB-sense: 5′-TTG AGG GGA CTT TCC CAG GC-3′.33
Downregulation of c-Rel by siRNA transfection
Nonsense or c-Rel siRNA oligonucleotides were obtained from Santa Cruz Biotechnology (Heidelberg, Germany). One day before transfection, 2 × 105 cells per well were seeded in six-well plates. The medium was replaced with the siRNA transfection reagent, and the transfection of cells was performed as recommended by the manufacturer. The cells were analyzed 72 hr after transfection.
Immunohistochemistry and immunofluorescence staining
Endogenous biotin was blocked using the Avidin/Biotin blocking kit (Vector, Burlingame, CA) according to the instructions of the manufacturer. Endogenous peroxidase was quenched by 0.3% in methanol. Primary antibodies were rabbit polyclonal Abs against human CA IX (Santa Cruz Biotechnology), c-Met (Abcam), E-cadherin (Cell Signaling), Ki67 (Thermo Scientific, Rockford, IL) and cleaved fragment of activated human caspase-3 (R&D Systems, Abingdon, UK) and mouse mAb against CD133 (Abcam). Biotinylated goat anti-rabbit or anti-mouse IgG (Vector) was used as a secondary Ab. The signal was amplified using the ABC Elite kit (Vector). AEC (3-Amino-9-ethylcarbazole) was used as a chromogen. Samples were counterstained with hematoxylin (Mayer) and mounted on Pro Tags Aqua mount (Quartett, Berlin, Germany). Omission of the primary Ab served as a negative control. The signal was detected at 400× magnification using a Leica DMRB fluorescence microscope (Leica, Wetzlar, Germany). Immunofluorescence staining was performed in established cell lines growing in chambers of the Nunc® Lab-Tek Chamber Slide™ system (Sigma-Aldrich). According to a standard protocol, ice-cold acetone served as fixative. Primary antibodies were rabbit polyclonal Abs against human Ki67 (Thermo Scientific, Rockford, IL) and Slug (Abcam) and mouse Abs against cytokeratin 19 (Abcam) and Vimentin (Abcam). Nuclei were stained with DAPI (4,6-diamidino-2′-phenylindol, 1 µg/ml). Goat anti-rabbit Alexa Fluor 488 IgG and goat anti-mouse Alexa Fluor 594 (Invitrogen, Camarillo, CA) were used as secondary Abs.
Images of representative fields were captured using a SPOT™ FLEX 15.2 64Mp shifting pixel digital color camera (Diagnostic Instruments) and analyzed with SPOT Basic/Advanced 4.6 software.
Human pluripotent stem cell antibody array
Nitrocellulose membranes to which capture antibodies have been spotted, and reagents for detection were obtained as a kit from R&D Systems® (Wiesbaden, Germany). According to the manufacturer's instructions, protein extracts were prepared and incubated with the nitrocellulose membranes, followed by detection of specific protein binding with biotinylated secondary antibodies using streptavidin-HRP and chemiluminescence detection reagents.
Transplantation of tumor cells to the chorioallantoic membrane of fertilized chicken eggs
This assay was performed as described recently34 with modifications. Briefly, fertilized white Leghorn chicken eggs (Geflügelzucht Hockenberger, Eppingen, Germany) were incubated at a humidity of 45–55% at 37.8°C in digital motor breeders Type 168/D (Siepmann GmbH, Herdecke, Germany). At Day 4 of embryonic development, 2–3 ml of albumen was removed with a syringe, thus allowing detachment of the embryo. Thereafter, a small window was cut into the eggshell, and the window was sealed with tape. At Day 9 of embryonic development, small handmade rings from Thermanox™ cover discs (Thermo Scientific, Schwerte, Germany) were placed on the chorioallantoic membrane (CAM), and 5 × 105 of pretreated tumor cells in matrigel were deposited into the rings. Alternatively, untreated tumor cells were transplanted to eggs with viable embryos at Day 9 of embryonic development followed by ovo treatment at Days 12 and 15 of embryonic development. For treatment, a Whatman filter paper was placed to the CAM directly adjacent to the plastic ring in which the tumor xenograft was growing and dropping 10 µl PBS to eggs of the CO and 10 µl triptolide (50 nM) to the triptolide group. Tumor take and tumor growth were evaluated at Day 17. All embryos that died before Day 17 were excluded from further analyses. At this time point, tumors of the control group were resected followed by evaluation of morphology by H&E staining and detection of human cells by labeling Ki67-positive cells by immunohistochemistry and double-immunofluorescence against human Ki67 and cytokeratin 19. Tumor take was calculated by the following formula: N1 × 100/N2 (N1 = number of embryos with tumor; N2 = number of live embryos). Tumor volumes were determined after resection of tumor xenografts by the following formula: Volume = 4/3 × π × r3 (r = 1/2 × square root of diameter 1 × diameter 2).34 Tumor tissue was stored in dry ice and embedded in Tissue Tek O.C.T. Compound (Sakura, Zoeterwoude, NL) for further analyses.
Extraction of genomic DNA and human Alu PCR amplification
Genomic DNA was isolated from fresh tissue derived from the CAM surrounding the plastic ring in which the tumor cells were xenografted to chicken eggs and from liver and lung tissue of the chicken embryo using the DNeasy Blood & Tissue Kit (Quiagen). Polymerase chain reaction was performed in a thermocycler (Biozym, Oldendorf, Germany) using 5 ng genomic DNA and FastStart PCR reagents from Roche Applied Science (Mannheim, Germany). Specific primer sequences for human Alu DNA, which is not expressed in chicken, were as follows: Alu-sense: 5′-GTA AGA GTT CCG TAA CAG GAC AGC T-3′ and Alu-antisense: 5′-CCC CAC CCT AGG AGA ACT TCT CTT T-3′. The PCR conditions were 60 sec at 94°C, 120 sec at 58°C and 120 sec at 72°C and 40 amplification cycles. About 20 µl of the PCR reaction was separated by electrophoresis on 4% agarose gels and visualized by ethidium bromide staining and UV transillumination.
Statistical analysis
Experiments with established cell lines were performed three times, and the quantitative data are presented as the mean ± SD. An exception is the human pluripotent stem cell antibody array, which was performed once in duplicate due to high costs of this experiment. However, the array data were confirmed by Western blot analysis. The experiments with primary spheroidal cells isolated from patient tumor-derived xenograft lines on mice were performed twice in triplicates. The significance of data was analyzed using Student's t-test. P < 0.05 was considered to be statistically significant.
Results
Hypoxia increases self-renewal potential and migratory activity
To evaluate the effect of hypoxia on progression of pancreatic cancer, we used three established human PDA cell lines. According to the degree of differentiation of the primary tumor, mutations in K-ras and/or p53, in vitro morphology, colony- and spheroid-forming capacity, ALDH1 activity, tumorigenicity in mice, histology of xenografts and expression of E-cadherin and Vimentin, we classified MIA-PaCa2 and AsPC-1 as CSChigh and BxPc-3 as CSClow (Supporting Information Table S1). Hypoxia was induced by incubation of the cells in a gas mixture of 1% O2, 5% CO2 and 94% N2, which resulted in fast upregulation of HIF-1α and its target gene VEGF within 2 hr as we showed recently,35 demonstrating activation of hypoxia-induced signaling. To evaluate the effect of hypoxia on CSC characteristics, we examined self-renewal potential by analyzing ALDH1 activity and colony- and spheroid-forming capacity. A high colony-formation potential of the aggressive MIA-PaCa2 and AsPC-1 cells was observed under normoxia and was further increased by hypoxia. In contrast, the less aggressive BxPc-3 cells had only minimal colony-forming potential under normoxia, a process that was induced by hypoxia (Fig. 1a). This finding was associated with significant upregulation of ALDH1 in BxPc-3 cells from 5 to 15% by hypoxia, whereas the already constitutively enhanced ALDH1 levels in MIA-PaCa2 and AsPC-1 cells under normoxia were not further increased significantly by hypoxia (Fig. 1b). Likewise, culture of the cells in hypoxia for 24 hr led to enhanced spheroid formation 7 days later, which was, however, not significant after this first round of hypoxia (Fig. 1c). To increase the effect, viable cells from the first-generation spheroids were isolated and treated again with hypoxia, followed by anchorage-independent growth for spheroid formation. This resulted in significant enhanced spheroid formation in the second-generation spheroids. For analysis of hypoxia-induced stem cell signaling, we examined protein expression in BxPc-3 cells before and after induction of hypoxia using a stem cell antibody array. This array contained 15 protein markers for detection of pluripotency and tumorigenesis. Hypoxia led to upregulation of many of these markers with the most pronounced effects in SOX2 (essential for self-renewal and pluripotency), FOXA2 (involved in differentiation of pancreas and tumorigenesis) and Snail (involved in downregulation of E-cadherin and induction of EMT). Because Notch antibodies were not included in the protein array, we examined the expression of Notch signaling by Western blot analysis. Twenty-four hours after hypoxia without an additional reoxygenation phase, Notch1, its ligand Jagged1 and Notch4 were upregulated compared to normoxia (Fig. 1d, left). Upregulation of SOX2 and Nanog, as demonstrated by Western blot analysis, confirmed the results of the protein array. Although at 24 hr, hypoxia had no effect on E-cadherin protein levels in protein array analysis (Fig. 1d), almost complete downregulation of E-cadherin protein expression was observed by Western blot analysis 48 and 72 hr after hypoxia without an additional reoxygenation phase in BxPc-3 and AsPC-1 cells, whereas MIA-PaCa2 cells do not constitutively express E-cadherin (Fig. 2a). The result we had already confirmed in our recent publications, that is, the absence of E-cadherin expression in MIA-PaCa2 cells35,36 and also others found that E-cadherin expression is absent in MIA-PaCa2 cells.37 In parallel, the EMT markers Slug, Snail, Twist2 and Vimentin were upregulated, with the most pronounced effects in BxPc-3 cells. In contrast, the expression of Snail and Twist2 was already constitutively enhanced under normoxia in the two more aggressive cell lines MIA-PaCa2 and AsPC-1. Hypoxia-induced signaling changed the morphology to a fibroblastoid, spindle-shaped phenotype as detected by microscopy (Fig. 2b) and enhanced the migratory activity as measured by a scratch assay (Fig. 2c).
Figure 1.

Hypoxia enhances stemness in pancreatic cancer cells. (a) BxPc-3, MIA-PaCa2 and AsPC-1 cells were seeded in six-well plates and cultured under normoxic (N) or hypoxic (H) conditions. Twenty-four hours later, cells were trypsinized and replated in medium containing 10% FCS at different cell numbers (BxPc-3: 600, 1,000 or 2,000 cells per well; MIA-PaCa2: 200, 400 or 600 cells per well; AsPC-1: 200 or 400 cells per well). Cells were grown without a medium change for 2 weeks, followed by evaluation of fixed and Coomassie-stained colonies consisting of at least 50 cells. The plating efficiency as a percent was calculated using the following formula: 100 × number of colonies/number of seeded cells. (b) The cells were cultured under normoxic or hypoxic conditions, and the ALDH1 activity was analyzed by flow cytometry 24 hr later. The percentage of ALDH1-positive cells is presented. (c) The cells were cultured for 24 hr under normoxia or hypoxia and then seeded at clonal density in low-adhesion plates. The spheroids were grown until Day 7 and photographed at ×100 magnification or quantified as described in the “Material and Methods” section (first generation). Thereafter, the first-generation spheroids were dissociated to single cells, and equal numbers of live cells were cultured under normoxia or hypoxia for 24 hr. Subsequently, the cells were plated at clonal density, resulting in second-generation spheroids. The number of second-generation spheroids was quantified 7 days after plating by counting viable spheroids per well. The number of surviving cells in the normoxia control was set to 100%. (d) BxPc-3 cells were cultured under normoxia or hypoxia. Twenty-four hours later, protein extracts were prepared and incubated with the nitrocellulose membranes of an antibody array kit for detection of human pluripotent stem cell markers. The binding of proteins to antibodies spotted to the membrane was detected using biotinylated secondary antibodies, streptavidin-HRP and chemiluminescence. The pixel density was quantified using ImageJ software. Left panel: BxPc-3 cells were cultured 24 hr under normoxia or hypoxia, and the protein expression of Notch1, Jagged1, Notch4, SOX2 and Nanog was evaluated by Western blot analysis. *p < 0.05, **p < 0.01.
Figure 2.

Hypoxia induces mesenchymal features. (a) Protein extracts were prepared from cells cultured under normoxia or after 24, 48 or 72 hr under hypoxia. The protein expression of E-cadherin, Vimentin, Slug, Snail and Twist2 was analyzed by Western blot analysis by the use of 40–60 μg protein extract per well. The expression of β-actin was evaluated to ensure equal conditions. (b) Cells were cultured under normoxic (N) or hypoxic (H) conditions, and 24 hr later, morphological changes to more spindle-shaped cells (arrows) were evaluated with a Nikon Eclipse TS100 microscope at ×200 magnification. (c) Cells were cultured to 90% confluence, and the cell layer was scratched with the tip of a yellow pipette, followed by culturing cells under normoxia or hypoxia. During this time, closure of the wounded region was evaluated 0, 12 and 24 hr after scratching by microscopy at ×100 magnification.
siRNA-mediated inhibition of c-Rel prevents hypoxia-induced NF-κB and EMT
Hypoxia-induced EMT was associated with increased binding of NF-κB to its consensus promoter site as shown by gel retardation assays (Fig. 3a). A control EMSA experiment using the house-keeping gene AP-1 ensured functionality of our nuclear extracts (data not shown). Because we know from our previous studies that the highly transactivation-competent NF-κB subunit c-Rel is a key player in mediating pancreatic CSC features,29 we transfected BxPc-3, MIA-PaCa2 and AsPC-1 cells with specific c-Rel siRNA using liposomes. This procedure led to strong downregulation of c-Rel expression in all cell lines compared to the nonsense control siRNA (Fig. 3b). Downregulation of c-Rel was associated with inhibition of hypoxia-induced migration (Fig. 3c) and inhibition of Slug and Vimentin expression, as measured by double immunofluorescence staining (Fig. 3d). In addition, Twist2 expression was inhibited in BxPc-3 and AsPC-1 cells, whereas the constitutively enhanced Twist2 expression in MIA-PaCa2 cells was not affected by the siRNA-mediated downregulation of c-Rel. These results suggest that progression of pancreatic cancer may be targeted by therapeutic downregulation of NF-κB signaling.
Figure 3.

NF-κB is involved in hypoxia-induced progression. (a) Cells were incubated under normoxic (N) or hypoxic (H) conditions, followed by isolation of nuclear protein extracts 24 hr later. DNA binding was analyzed by EMSA using a biotin-labeled oligonucleotide probe for the NF-κB promoter consensus sequence. Specific (NF-κB) and unspecific shifts (UNSP) are marked. Competition with a tenfold excess of unlabeled oligonucleotide (10×) served as a control for specificity of binding. (b) Cells were transfected with siRNA oligonucleotides encoding a nonsense (NS) sequence or a specific sequence for inhibition of c-Rel (sicRel). Three days later, proteins were prepared, and the expression of c-Rel was evaluated by Western blot analysis. β-Actin served as a control for equal loading. The bar indicates 50 µm. (c) Three days after transfection with nonsense or c-Rel siRNA, the confluent cell layers were scratched, and then, they were cultured under hypoxic conditions. Closure of the wounded region was monitored 6 and 24 hr after scratching by microscopy and documented by photographs. The bar indicates 100 µm. (d) Likewise, the expression of EMT-related proteins Slug and Vimentin was detected by double immunofluorescence staining of cells exposed to hypoxia for 48 hr. DAPI counterstaining was used for detection of nuclei. Mouse anti-CD44 mAb/Alexa Fluor 594 IgG (red) and rabbit polyclonal anti-CA IX/Alexa Fluor 488 IgG (green) were used as secondary antibodies. Tissue sections were analyzed under ×400 magnification using a Leica DMRB fluorescence microscope. The bar indicates 25 µm. (e) Western blot analysis of Twist2 expression was performed using protein extracts derived from siRNA-transfected cells cultured under normoxia or hypoxia for 3 days.
Triptolide inhibits hypoxia-induced NF-κB activity, EMT and CSC features
Because we found that hypoxia-induced EMT is associated with increased NF-κB activity in our system (compare Fig. 3), which was also recently found by others,22,23 we evaluated the NF-κB inhibitor triptolide for therapeutic targeting. Triptolide is a diterpenoid triepoxide (Fig. 4a), with strong anti-inflammatory and NF-κB-inhibitory activity (reviewed in Ref.31). Treatment of BxPc-3, AsPC-1 and MIA-PaCa2 cells with triptolide for 24 hr potently downregulated hypoxia-induced NF-κB binding activity as examined by EMSA (Fig. 4b). This was accompanied by inhibition of protein expression of HIF-1α, the NF-κB subunits c-Rel and Rel-A and the EMT-related protein Twist2 within 24 hr of treatment, as examined by Western blot analysis (Fig. 4c). In line with the repressed NF-κB and EMT signaling, hypoxia-induced migratory activity was inhibited by coincubation with triptolide for 24 hr (Fig. 4d). Importantly, within 24 hr of treatment with triptolide, cancer cells remained viable (Fig. 4e), suggesting that the inhibition of migration was not due to the death of cells. The viability was reduced within the first 48 hr after treatment of cells with triptolide (Fig. 4e). There were no significant differences in triptolide-reduced viability between cells cultured under normoxia or hypoxia; however, the effects were most pronounced in the highly aggressive MIA-PaCa2 and AsPC-1 cells. In contrast, the less malignant BxPc-3 cells or nonmalignant pancreatic ductal cells or mesenchymal stem cells (MSCs) were less sensitive to triptolide (Fig. 4e), suggesting predominant targeting of highly malignant cells and, thus, low side effects in patients. This assumption was underlined by the inhibition of hypoxia-induced colony formation (Fig. 5a), ALDH1 activity (Fig. 5b) and spheroid-forming capacity, which was even more pronounced in second-generation spheroids (Fig. 5c). These results indicate the potent inhibition of self-renewal activity of CSC features by triptolide. Because BxPc-3 cells possess only weak self-renewal potential, we analyzed the effect of triptolide on stem cell signaling in BxPc-3 cells by a pluripotency and tumorigenesis-specific stem cell antibody array (compare Fig. 1d). After treatment of BxPc-3 cells with hypoxia alone or in the presence of triptolide, triptolide clearly reversed hypoxia-induced upregulation of many markers involved in pluripotency and tumorigenesis with the most pronounced effects in SOX2 and FOXA2 (Fig. 5d). Downregulation of SOX2 and Nanog was confirmed by Western blot analysis, which also revealed downregulation of Notch1 and its ligand Jagged1 (the latter two genes were not represented in the stem cell array).
Figure 4.
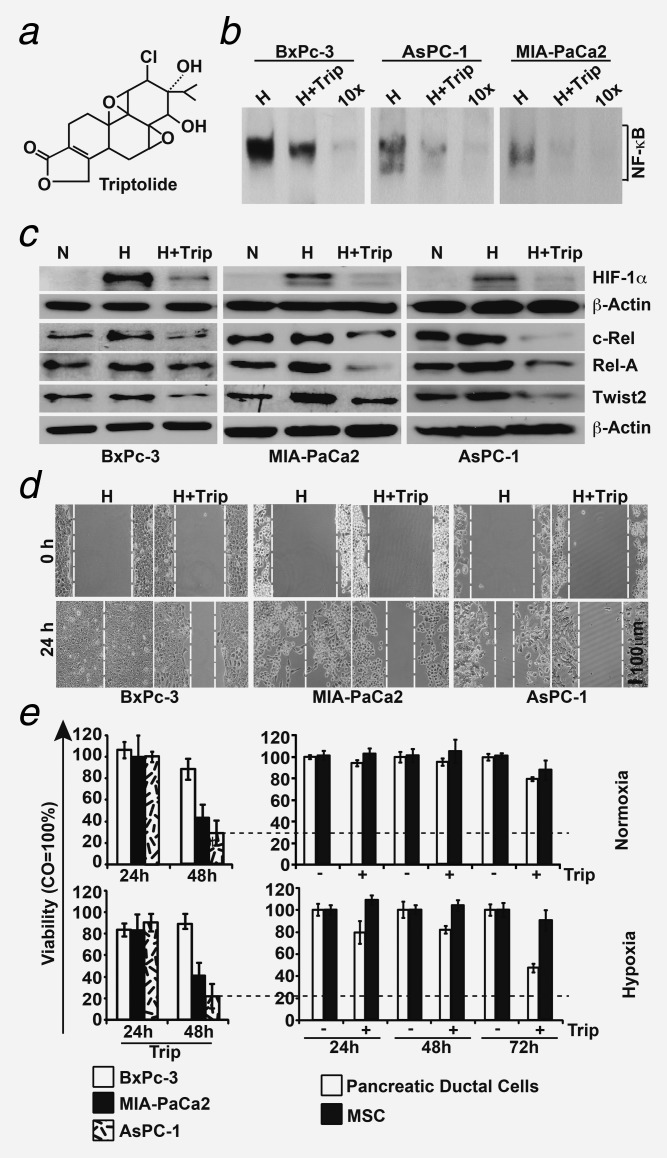
Triptolide inhibits hypoxia-induced NF-κB and migration. (a) The chemical structure of the diterpenoid triepoxide triptolide. (b) Cells were incubated under hypoxic conditions alone (CO) or combined with triptolide (Trip, 20 nM). Nuclear protein extracts were harvested 24 hr later, and DNA binding was analyzed by EMSA using a biotin-labeled oligonucleotide probe for the NF-κB promoter consensus sequence. Specific (NF-κB) and unspecific shifts (UNSP) are marked. Competition with a tenfold excess of unlabeled oligonucleotide (10×) served as a control for specificity of binding. (c) Cells were cultured under normoxia (N), hypoxia (H) or hypoxia plus triptolide (H+Trip) for 24 hr. Protein extracts were isolated, and the expression of HIF-1α, c-Rel, Rel-A and Twist2 was examined by Western blot analysis. (d) Cells were grown to 90% confluence, followed by scratching and incubation under hypoxia (H) or hypoxia plus triptolide (H+Trip). Additionally, migration was examined as described in Figure 3c. The bar indicates 100 µm. (e) MTT assay of malignant (BxPc-3, MIA-PaCa2 and AsPC-1) and nonmalignant (pancreatic ductal cells and MSC) cells grown under normoxia or hypoxia in the presence or absence of triptolide (20 nM) at the time points indicated.
Figure 5.

Triptolide downregulates cancer stem cell features. (a) The colony-forming capacity of cells grown for 24 hr under hypoxia alone or in the presence of triptolide (20 nM) was analyzed as described in Figure 1a. (b) The ALDH1 activity of cells treated as described above was measured as described in Figure 1b. (c) Spheroid formation was detected in MIA-PaCa2 and AsPC-1 cells treated as described above. The number of second-generation spheroids was quantified 3 days after plating by counting viable spheroids per well. Photographs of second-generation spheroids (left) and evaluation of the percentage of first- and second-generation spheroids (right) are shown. The bar indicates 100 µm. (d) Binding of proteins to antibodies spotted on the membrane of a Human Pluripotent Stem Cell Array was detected in BxPc-3 cells treated as described above and in Figure 1d. (e) Notch1, Jagged1, SOX2 and Nanog protein expression was detected by Western blot analysis. *p < 0.05, **p < 0.01.
Furthermore, we evaluated the effect of triptolide on tumor engraftment (tumor take) and growth in vivo using xenotransplantation to the CAM of fertilized chicken eggs. This model closely resembles the xenograft model in immunodeficient mice because the immune system of the chicken embryo is not fully developed,38 allowing the growth of three-dimensional tumors with a blood supply from the chicken embryo (Fig. 6a). Most importantly, the CAM contains no nerves, and transplantation is therefore not painful to the embryo. The size of the resected tumors can be determined using calipers (Fig. 6b), and human tumor cells grow embedded in stroma formed by chicken cells (Fig. 6c). Staining of a tumor section with the human-specific proliferation marker Ki67 indicates that the large dark-brown labeled cells are of human origin, and the smaller, nonlabeled stroma-embedded cells between the human cell islets are chicken cells (Fig. 6d). BxPc-3, MIA-PaCa2 and AsPC-1 cells were treated with hypoxia in the presence or absence of triptolide in vitro, followed by transplantation of viable cells 24 hr later (Fig. 6e). Although the percentage of tumor take of cells treated with hypoxia alone was 100%, 77% and 100% in BxPc-3, AsPC-1 and MIA-Paca2, respectively, triptolide cotreatment strongly diminished tumor take to 50%, 15% and 33%, respectively. In addition, the tumors that started to grow on eggs demonstrated a strongly reduced tumor volume in the triptolide treatment groups. These results suggest that triptolide targets CSCs, due to inhibition of tumor take, which is an essential feature of CSCs.
Figure 6.
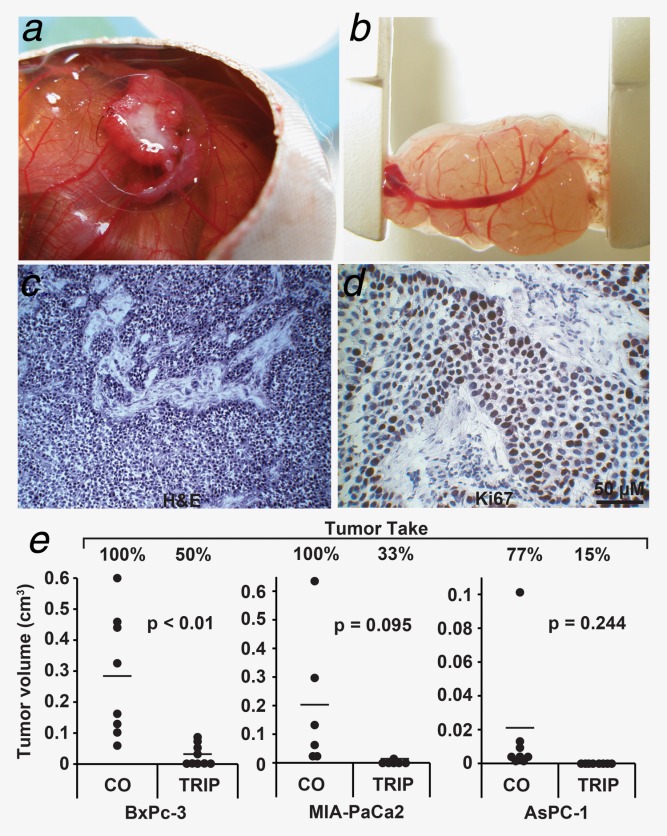
Triptolide inhibits tumor take and reduces tumor volume. (a) The three-dimensional growth of BxPc-3 cells transplanted on the CAM of fertilized chicken eggs was photographed at Day 17 of embryonic development. (b) The xenograft tumor was resected at Day 17, and the size was measured using calipers. (c) H&E staining of the frozen tumor section (×100 magnification). (d) Immunohistochemistry staining of the frozen tumor section with the human proliferation marker Ki67 (×400 magnification, brown). The bar indicates 50 μm. (e) BxPc-3, MIA-PaCa2 and AsPC-1 cells were grown under hypoxia alone or in the presence of triptolide (Trip, 20 nM) in vitro. Twenty-four hours later, 5 × 105 cells were transplanted on the CAM of fertilized chicken eggs. At Day 17 of embryonic development, tumor take was determined as the percentage of grown tumors of the 15 transplanted per group. The size of each grown tumor was determined using calipers, and the tumor volumes are presented as black dots. If no tumor grew, the volume was set to 0. The bar indicates the mean of the tumor volume in each treatment group. *p < 0.05, **p < 0.01.
Finally, we examined the effect of triptolide on tumor growth under more clinically relevant conditions transplanted BxPc-3 cells first to the CAM followed by in ovo treatment after development of a tumor. Because of the rapid three-dimensional growth of xenograft tumors (Figs. 7a–7d), the blood supply by vessels was limited, as we detected by staining of the tumor tissue with the human markers for cytokeratin 19 and the Ki67, which detects a high proliferation rate (Fig. 7e). However, the human tissue was supplied by only one chicken cell-derived blood vessel at the margin of the tumor, as detected by DAPI staining (Fig. 7f). As a consequence, hypoxia occurred in the tissue, as concluded from a positive staining of control tumor tissue with the hypoxia marker CA IX, which was accompanied by enhanced expression of the CSC marker c-Met, the proliferation marker Ki67 and the EMT marker Vimentin along with basal expression of E-cadherin and the cleaved fragment of active caspase-3 (Fig. 7i). In contrast, in ovo treatment with triptolide prevented the expression of CA IX, c-Met, Ki67 and Vimentin and enhanced the expression of E-cadherin and the cleaved fragment of active caspase-3, as shown by representative stainings and a diagram, which shows the number of average positive cells. These expression data were confirmed by the measurement of tumor volume, which was 0.42 cm3 in the control group but 0.079 cm3 in the triptolide group (Figs. 7a–7d and 7g). In addition, we also examined invasion and metastasis of human tumor cells by replication of human Alu sequences from genomic DNA of chicken tissue by PCR. Although five of the five examined CAM pieces adjacent to the tumor xenograft were positive in the control groups, suggesting invasion, triptolide totally prevented this (Fig. 7h). In contrast, Alu sequences were not detectable in the liver and lung of the chicken embryo (Supporting Information Fig. S1), suggesting that metastasis of the less malignant BxPc3 cells did not occur within the short time of growth in fertilized chicken eggs. Most importantly, side effects in chicken embryos were not observed, because the weight of chicken embryos was the same after treatment and necrosis in liver sections was not detected (data not shown). These results demonstrate that triptolide inhibits tumor engraftment, tumor growth, invasion, expression of markers for CSCs and EMT and induces E-cadherin expression and apoptosis in vivo.
Figure 7.

Triptolide inhibits tumor growth, invasion, CSC-like feature and EMT in vivo. Untreated BxPc-3 cells were transplanted on the CAM of fertilized chicken eggs at Day 9 of embryonic development followed by in ovo treatment as described in the “Material and Methods” section at Days 12 and 15 with triptolide (+Trip, 50 nM) or PBS (CO). At Day 17 of embryonic development, the tumors of (a and b) untreated or (c and d) triptolide-treated eggs were photographed before and after resection. At the right lower edge of the eggs, the filter papers to which PBS or triptolide were dropped are visible. The bar indicates 0.25 cm. (e) Double immunofluorescence staining of a frozen tumor section (×400 magnification) with the human markers for cytokeratin 19 (red) and Ki67 for proliferation (green). The bar indicates 50 μm. (f) Nuclei of the tumor section were stained with DAPI (blue). The arrows mark a blood vessel formed by chicken cells, which are not positive for the human markers cytokeratin 19 and Ki67. (g) Diagram in which tumor volumes of all examined xenografts and the means are shown. (h) Tissue from the CAM surrounding the plastic ring in which the tumor xenografts grow were excised from five eggs per group, followed by isolation of genomic DNA. Human Alu sequences were amplified using 5 ng genomic DNA as template. Double-distilled water served as negative control (Neg.), and genomic DNA isolated from tumor xenografts served as positive control (Tumor). The DNA marker is shown in the first lane. (i) Frozen tumor sections were analyzed by immunohistochemistry for the expression of CA IX, c-Met, Ki67, cleaved fragment of active caspase-3, E-cadherin and Vimentin. Representative photographs for each group under ×400 magnification are shown. The arrows mark positive cells, which appear red to dark-red. The bar indicates 50 μm. The number of positive cells was quantified in ten vision fields under ×400 magnification, and the means ± SD are shown. *p < 0.05, **p < 0.01.
Triptolide inhibits hypoxia-induced CSC marker expression in primary spheroidal cultures
To further highlight these findings in a patient-related model, we transplanted freshly resected PDA tissue subcutaneously in mice, followed by serial subtransplantation. With increasing passage number, the xenograft tumors started to grow faster, as evident from the enhanced doubling time and latency (Fig. 8a). After each subtransplantation up to Passage 7, the morphology of frozen tumor sections was examined, and the findings demonstrated that the typical duct-like structures and pronounced stroma of PDA were preserved (Fig. 8b). In parallel, the CSC marker c-Met was enriched from 4% in the primary patient tumor to 43% in xenografts of Passage 7. Tumor cells were isolated from xenografts (T29) and cultured in vitro as anchorage-independent spheroidal colonies (Fig. 8c), which led to further enrichment of c-Met+ cells to 64% (Fig. 8d). Culture of these spheres under normoxia or hypoxia for 48 hr in the presence or absence of triptolide resulted in marked reduction of viable spheroids in both triptolide groups as evaluated by microscopy (Fig. 8c). Immunohistochemistry revealed that hypoxia enhanced the percentage of hypoxia marker CA IX from 5.2 to 32%, of c-Met+ cells from 64 to 75%, of CD133+ cells from 74 to 85%, of Ki67+ cells from 23 to 36%, of caspase-3+ cells from 9 to 20% and of Vimentin+ cells from 85 to 91% along with downregulation the percentage of E-cadherin+ cells from 16 to 10%. Similar experiments with two primary spheroidal cultures derived from two different patient tumors xenografted to mice (T18 and T30) were performed and confirm the above results (Supporting Information Fig. S2), demonstrating that triptolide significantly reverses hypoxia-induced progression markers even in primary CSC-enriched spheroidal cultures.
Figure 8.

Triptolide prevents growth of patient-selected and CSC-enriched spheroids. (a) Tumor mush isolated from a surgical nondiagnostic specimen (T29) from a patient with PDA was transplanted subcutaneously in the flanks of immunodeficient 6-week-old NMRI (nu/nu) female mice (P1), followed by subtransplantation (P2 and P3) until a stably growing xenograft line developed after Passage 3. The time until a first tumor was visible after transplantation (Latency) and the tumor doubling times are shown. (b) The morphology of the primary patient tumor and tissue thereof transplanted to mice (xenograft tumors) in Passages 1, 3 and 7 (P1, P3 and P7) was detected by Trichrome staining of frozen tissue, followed by microscopy. Likewise, the tissue was stained using a c-Met antibody, and the percentage of positive cells (brown) was analyzed by counting ten vision fields under ×400 magnification. The bar indicates 50 µm. (c) Anchorage-independent spheroidal cultures were established from xenografts and cultivated under normoxia (N) or hypoxia (H) alone or combined with triptolide (Trip, 20 ng) for 48 hr. Photographs were then taken. The bar indicates 50 µm. (d) Alternatively, spheroidal cultures treated as described above were cytospinned to glass slides, and the expression of hypoxia marker (CA IX), tumor stem cell markers (c-Met and CD133), cell proliferation (Ki67), apoptosis marker (caspase-3) and EMT-related markers (E-cadherin and Vimentin) was examined by immunohistochemistry. Representative pictures of positive-labeled cells (red, arrow) under normoxia are shown. The bar indicates 20 µm. The number of positive cells was quantified in ten vision fields under ×400 magnification, and the means ± SD are shown. **p < 0.01.
Discussion
The current treatment options for PDA provide a 5-year survival rate of only 5%. To date, efforts to identify additional effective therapies have had limited success. Despite the low response rate, modest overall survival benefit and rapid development of resistance, gemcitabine has been adopted as the standard therapy for advanced pancreatic cancer.39 The newer drug combination FOLFIRINOX extends life by only 4 months compared to gemcitabine alone40 but causes severe side effects. Both, gemcitabine and FOLFIRINOX do not directly target the pronounced tumor hypoxia, a major mediator for aggressive growth, early metastasis and therapy resistance of PDA. In our study, we show that hypoxia-induced EMT and CSC features can be effectively inhibited by triptolide, an anti-inflammatory agent used in TCM.
Induction of CSC-like features and EMT by hypoxia
Our results demonstrate that hypoxia activates self-renewal potential, migration and morphological changes, as well as stem cell- and EMT-associated signaling in established PDA cell lines. Hypoxia conditions were according to our recent studies35,41 in which we used a hypoxia chamber flushed with a gas mixture of 1% O2, 5% CO2 and 94% N2 from an air liquid tank for 4 min. The chamber was sealed and kept at 37%. We did not examine more severe hypoxia, because low concentrations of oxygen are not easy to control especially at long-time treatment as used in our study. However, we ensured a constant level of 1% O2 by the use of a so-called Blood Gas Analyzer. This O2 concentration was sufficient to induce a hypoxic environment, as we concluded from enhanced expression of the hypoxia marker HIF-1α We found hypoxia-induced activation of the CSC markers Notch1, Jagged1, Notch4, c-Met and CD133 and the embryonic stem cell markers Nanog, SOX2, FOXA2, SOX17 and PDX-1. This result corresponds to recent findings of HIF-mediated induction of OCT4, Nanog, SOX2, KLF4 and cMyc in cancer cell lines and glioma-derived CD133-negative cells.42 A recent study in pancreatic cancer cells has demonstrated that the expression of FOXA2 neutralizes several EMT-related effects.37 However, FOXA2 upregulation in our system was accompanied by an epithelial-to-mesenchymal switch, downregulation of E-cadherin and upregulation of Vimentin, Slug, Snail and Twist2. An explanation for this discrepancy is provided by a model that suggests dynamic interaction among epithelial, self-renewal and mesenchymal programs in CSCs.43 Our findings are clinically relevant because intratumoral hypoxia and subsequent induction of EMT and selection of tumor cells with CSC features may occur frequently in patient tumors due to the extremely dense tumor stroma and unique hypovascular nature of PDA (reviewed in Ref.44). This concept matches recent experience with antiangiogenic therapy. Blocking of tumor blood vessel formation increases intratumoral hypoxia, and this process stimulates the CSC population as demonstrated by a previous study using experimental models of breast cancer.45 Accordingly, Louie et al.46 selected a stem-like breast cancer cell subpopulation with an EMT-like phenotype by repeated cycles of hypoxia/reoxygenation. Moreover, gene expression patterns in human cancers indicate that de-differentiated cancer cells demonstrate a combined EMT/stem cell-like phenotype because Twist1 coinduces EMT and stemness properties (reviewed in Ref.47). Twist2 signaling may be involved in linking EMT to CSCs because its overexpression resulted in morphological transformation, EMT signaling, colony-forming abilities and an enhanced number of stem-like cells in a breast cancer model.48
NF-κB as a mediator of hypoxia-induced CSC and EMT features
Our data show hypoxia-mediated upregulation of Twist2 in BxPc-3 and AsPC-1 cells, whereas MIA-PaCa2 cells exhibit constitutively enhanced Twist2 expression under normoxia. siRNA-mediated inhibition of NF-κB c-Rel downregulated the expression of hypoxia-induced Twist2, inhibited migratory activity and downregulated the expression of Slug and Vimentin. Correspondingly, pancreatic CSC-enriched cells were resensitized to cancer therapy by siRNA-mediated downregulation of c-Rel as shown in one of our earlier studies.29 Therefore, we assume that NF-κB may be a master inducer of both EMT and CSC features. Our suggestion is underscored in the study by Jiang et al.49 These authors described that during neoplastic transformation, human keratinocyte cells undergo EMT and then acquire a malignant CSC-like phenotype. Longer times for transformation increased activation of IKKβ, IκBα and NF-κB Rel-A and enhanced the expression of Snail; stem cell markers were also detected.49 Inhibition of Rel-A blocked EMT, acquisition of a CSC-like phenotype, and neoplastic transformation. Likewise, in the established gemcitabine-resistant PDA cell line SW1990, siRNA-mediated knockdown of Rel-A inhibited the ability of gemcitabine to increase the proportion of CSC and EMT markers.
Therapeutic potential of triptolide
The purity of triptolide that we used was ≥98% (HPLC). This high purity makes it unlikely that some of the observed effects are due to impurities of the substance. Although triptolide is the main active ingredient in extracts from the Chinese herb Tripterygium wilfordii Hook f, 124 other sesquiterpene derivatives have been reported to be present in Tripterygium, as extracts were often prepared in different ways (e.g., with different solvents), and they have different constituents and biological effects.31 However, this concern does not apply to our study as we used highly purified triptolide. We have identified triptolide as an effective inhibitor of hypoxia-induced transcriptional signaling that blocks HIF-1α expression and NF-κB binding activity, as well as downregulates c-Rel, Rel-A and Twist2 expression, consistent with the finding that the twist gene promoters are under the control of NF-κB and contain functional κB binding elements (reviewed in Ref.50). Additionally, we found that triptolide caused impairment of the migration, colony- and spheroid-forming ability and ALDH1 activity and repression of hypoxia-induced stem cell signaling. We did not observe cytotoxicity by triptolide 24 hr after incubation. Cytotoxicity occurred during the first 48 hr after incubation with the highest levels in CSC-enriched cell lines. In contrast, triptolide was less toxic in gemcitabine-sensitive BxPc-3 cells or nonmalignant MSCs and immortalized pancreatic ductal cells. Indeed, recent data demonstrate that triptolide generally takes ∼24 hr or more to induce apoptosis or cytotoxicity in cancer cells, much longer than the time of several hours that other cytotoxic agents require.31 Notably, although highly malignant cells do not die within 24 hr, triptolide potently inhibited hypoxia-mediated EMT and CSC signaling 24 hr after triptolide treatment. This assumption is underscored by our finding of the completely blocked in vivo tumor take of the highly malignant MIA-PaCa2 and AsPC-1 cells 24 hr after treatment with triptolide. In contrast, the tumor take of the less aggressive BxPc-3 cells was only partially blocked. Therefore, it is tempting to speculate that triptolide predominantly targets CSCs, while leaving more differentiated tumor cells or nonmalignant cells unaffected. Such a model would explain why cytotoxic effects occurred rather late and why triptolide can circumvent tumor drug resistance.31 The idea of specific targeting of CSC by triptolide is emphasized by the finding that no overt signs of toxicity occurred when mice received the triptolide derivative minnelide for 385 days at a concentration of 0.43 mg/kg.51 However, minnelide and triptolide significantly decreased tumor weight, tumor volume and tumor spread in orthotopic MIA-PaCa2 xenografts at the same concentration, and tumors did not recur after termination of treatment. In contrast, the first-line chemotherapeutic agent in pancreatic cancer, gemcitabine, at a concentration of 100 mg/kg bodyweight, did not lead to a significant decrease in tumor size versus the control.51 This promising in vivo effect of triptolide in mice was confirmed in a small clinical study where 45 leukemia patients received 30–40 μg triptolide per kilogram of body weight daily in China (reviewed in Ref.31). Complete remission was observed in 18 patients, and partial remission was observed in six patients. The efficacy of triptolide was superior to that of adriamycin and aclacinomycin. Toxicities were manifested as phlebitis and gastrointestinal abnormalities. No heart, liver and kidney toxicities were observed (reviewed in Ref.31).
Regarding inhibition of metastasis by triptolide in mouse models, we believe that downregulated expression of Notch1 and its ligand Jagged1 may be involved, as demonstrated in our study. This would further support Notch1's ability to inhibit invasion by inactivation of NF-κB, VEGF and MMP-9 in pancreatic cancer,52 implying a positive feedback loop between hypoxia-induced transcriptional signaling and target gene expression that leads to EMT signaling and CSC features.
Undoubtedly, further studies will be needed to determine the precise mechanisms by which triptolide potently eliminates highly malignant pancreatic cancer cells while sparing more differentiated cells. However, our data provide strong evidence for effective inhibition of hypoxia-induced EMT and CSC signaling by triptolide in pancreatic cancer.
Acknowledgments
The authors thank Dr. N. Giese for organization of the tissue service at their clinic and Sonja Bauer for technical assistance.
Author Contributions
I.H., L.L. and A.S.: concept and design; L.L., S.L., N.B., E.A., C.N., J.M. and J.G.: development of methodology; L.L., S.L., N.B., E.A., C.N., J.M. and J.G.: acquisition of data; L.L. and I.H.: analysis and interpretation of data; I.H. and L.L.: writing, review and/or revision of the manuscript and J.W. and P.S.: administrative, technical or material support.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- 1.Gukovskaya AS, Pandol SJ. Cell death pathways in pancreatitis and pancreatic cancer. Pancreatology. 2004;4:567–86. doi: 10.1159/000082182. [DOI] [PubMed] [Google Scholar]
- 2.Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58:1408–16. [PubMed] [Google Scholar]
- 3.Hoffmann AC, Mori R, Vallbohmer D. High expression of HIF1α is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA, VEGF, and bFGF. Neoplasia. 2008;10:674–9. doi: 10.1593/neo.08292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang Q, Jurisica I, Do T. Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res. 2011;71:3110–20. doi: 10.1158/0008-5472.CAN-10-4049. [DOI] [PubMed] [Google Scholar]
- 5.Rasheed ZA, Kowalski J, Smith BD. Concise review: emerging concepts in clinical targeting of cancer stem cells. Stem Cells. 2011;29:883–7. doi: 10.1002/stem.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lapidot T, Sirard C, Vormoor J. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 7.Al-Hajj M, Wicha MS, Benito-Hernandez A. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh SK, Hawkins C, Clarke ID. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 9.Simeone DM. Pancreatic cancer stem cells: implications for the treatment of pancreatic cancer. Clin Cancer Res. 2008;14:5646–8. doi: 10.1158/1078-0432.CCR-08-0584. [DOI] [PubMed] [Google Scholar]
- 10.Li C, Heidt DG, Dalerba P. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 11.Hermann PC, Huber SL, Herrler T. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 12.Li C, Wu JJ, Hynes M. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology. 2011;141:2218–27. doi: 10.1053/j.gastro.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–51. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higgins DF, Kimura K, Bernhardt WM. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–20. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thiery JP. Epithelial–mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–6. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Huber MA, Beug H, Wirth T. Epithelial–mesenchymal transition: NF-κB takes center stage. Cell Cycle. 2004;3:1477–80. doi: 10.4161/cc.3.12.1280. [DOI] [PubMed] [Google Scholar]
- 17.Ledford H. Cancer theory faces doubts. Nature. 2011;472:273. doi: 10.1038/472273a. [DOI] [PubMed] [Google Scholar]
- 18.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 19.Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 20.Tsai JH, Donaher JL, Murphy DA. Spatiotemporal regulation of epithelial–mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725–36. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mani SA, Guo W, Liao MJ. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koong AC, Chen EY, Mivechi NF. Hypoxic activation of nuclear factor-κB is mediated by a Ras and Raf signaling pathway and does not involve MAP kinase (ERK1 or ERK2) Cancer Res. 1994;54:5273–9. [PubMed] [Google Scholar]
- 23.Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 24.Pikarsky E, Porat RM, Stein I. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 25.Karin M, Greten FR. NF-κB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 26.Maier HJ, Schmidt-Strassburger U, Huber MA. NF-κB promotes epithelial–mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010;295:214–28. doi: 10.1016/j.canlet.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Cao YX, Luo JL, Karin M. IκB kinase α kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc Natl Acad Sci USA. 2007;104:15852–7. doi: 10.1073/pnas.0706728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. 2011;121:3804–9. doi: 10.1172/JCI57099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kallifatidis G, Rausch V, Baumann B. Sulforaphane targets pancreatic tumour-initiating cells by NF-κB-induced antiapoptotic signalling. Gut. 2009;58:949–63. doi: 10.1136/gut.2008.149039. [DOI] [PubMed] [Google Scholar]
- 30.Brinker AM, Ma J, Lipsky PE. Medicinal chemistry and pharmacology of genus Tripterygium (Celastraceae) Phytochemistry. 2007;68:732–66. doi: 10.1016/j.phytochem.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou ZL, Yang YX, Ding J. Triptolide: structural modifications, structure–activity relationships, bioactivities, clinical development and mechanisms. Nat Prod Rep. 2012;29:457–75. doi: 10.1039/c2np00088a. [DOI] [PubMed] [Google Scholar]
- 32.Lonardo E, Hermann PC, Mueller MT. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. 2011;9:433–46. doi: 10.1016/j.stem.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Klapproth K, Sander S, Marinkovic D. The IKK2/NF-{κ}B pathway suppresses MYC-induced lymphomagenesis. Blood. 2009;114:2448–58. doi: 10.1182/blood-2008-09-181008. [DOI] [PubMed] [Google Scholar]
- 34.Balke M, Neumann A, Szuhai K. A short-term in vivo model for giant cell tumor of bone. BMC Cancer. 2011;11:241. doi: 10.1186/1471-2407-11-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salnikov AV, Liu L, Platen M. Hypoxia induces EMT in low and highly aggressive pancreatic tumor cells but only cells with cancer stem cell characteristics acquire pronounced migratory potential. PLoS One. 2012;7:e46391. doi: 10.1371/journal.pone.0046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou W, Kallifatidis G, Baumann B. Dietary polyphenol quercetin targets pancreatic cancer stem cells. Int J Oncol. 2010;37:551–61. doi: 10.3892/ijo_00000704. [DOI] [PubMed] [Google Scholar]
- 37.Song Y, Washington MK, Crawford HC. Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal transition in pancreatic cancer. Cancer Res. 2010;70:2115–25. doi: 10.1158/0008-5472.CAN-09-2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janse EM, Jeurissen SH. Ontogeny and function of two non-lymphoid cell populations in the chicken embryo. Immunobiology. 1991;182:472–81. doi: 10.1016/s0171-2985(11)80211-1. [DOI] [PubMed] [Google Scholar]
- 39.Oberstein PE, Saif MW. Vol. 12. USA: Highlights from the 2011 ASCO Gastrointestinal Cancers Symposium San Francisco, CA; 2011. First-line treatment for advanced pancreatic cancer; pp. 96–100. [PubMed] [Google Scholar]
- 40.Conroy T, Desseigne F, Ychou M. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 41.Rausch V, Liu L, Apel A. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J Pathol. 2012;227:325–35. doi: 10.1002/path.3994. [DOI] [PubMed] [Google Scholar]
- 42.Mathieu J, Zhang Z, Zhou W. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71:4640–52. doi: 10.1158/0008-5472.CAN-10-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Celia-Terrassa T, Meca-Cortes O, Mateo F. Epithelial–mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest. 2012;122:1849–68. doi: 10.1172/JCI59218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tuveson DA, Neoptolemos JP. Understanding metastasis in pancreatic cancer: a call for new clinical approaches. Cell. 2012;148:21–3. doi: 10.1016/j.cell.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 45.Conley SJ, Gheordunescu E, Kakarala P. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. 2012;109:2784–9. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Louie E, Nik S, Chen JS. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. Breast Cancer Res. 2010;12:R94. doi: 10.1186/bcr2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brabletz T. EMT and MET in metastasis: where are the cancer stem cells? Cancer Cell. 2012;22:699–701. doi: 10.1016/j.ccr.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 48.Fang X, Cai Y, Liu J. Twist2 contributes to breast cancer progression by promoting an epithelial–mesenchymal transition and cancer stem-like cell self-renewal. Oncogene. 2011;30:4707–20. doi: 10.1038/onc.2011.181. [DOI] [PubMed] [Google Scholar]
- 49.Jiang R, Li Y, Xu Y. EMT and CSC-like properties mediated by the IKKβ/IκBα/RelA signal pathway via the transcriptional regulator, Snail, are involved in the arsenite-induced neoplastic transformation of human keratinocytes. Arch Toxicol. 2013;87:991–1000. doi: 10.1007/s00204-012-0933-0. [DOI] [PubMed] [Google Scholar]
- 50.Sosic D, Olson EN. A new twist on twist—modulation of the NF-κB pathway. Cell Cycle. 2003;2:76–8. [PubMed] [Google Scholar]
- 51.Chugh R, Sangwan V, Patil SP, et al. A preclinical evaluation of Minnelide as a therapeutic agent against pancreatic c. Sci Transl Med. 2012;4:156ra39. doi: 10.1126/scitranslmed.3004334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Z, Banerjee S, Li Y. Down-regulation of notch-1 inhibits invasion by inactivation of nuclear factor-κB, vascular endothelial growth factor, and matrix metalloproteinase-9 in pancreatic cancer cells. Cancer Res. 2006;66:2778–84. doi: 10.1158/0008-5472.CAN-05-4281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information