

Abstract

CXCL12 binds to CXCR4, promoting both chemotaxis of lymphocytes and metastasis of cancer cells. We previously identified small molecule ligands that bind CXCL12 and block CXCR4-mediated chemotaxis. We now report a 1.9 Å resolution X-ray structure of CXCL12 bound by such a molecule at a site normally bound by sY21 of CXCR4. The complex structure reveals binding hot spots for future inhibitor design and suggests a new approach to targeting CXCL12–CXCR4 signaling in drug discovery.
Introduction
Chemokines are small but potent chemotactic cytokines (8–14 kDa). To date, about 50 chemokines have been identified and divided into four distinct families (C, CC, CXC, and CX3C) based on the arrangement of conserved cysteines in the N-terminus.1−3 These secreted proteins orchestrate homing of cells toward areas of high chemokine concentration through binding and activation of their cognate GPCRs (G-protein coupled receptors) on the surface of cells. Processes such as cell trafficking and adhesion greatly depend on the chemokine–receptor signaling axis.3−5
CXCL12 (stromal-cell-derived factor-1, SDF-1a) is a CXC-type chemokine that binds to the CXCR4 and CXCR7 receptors attracting receptor-containing cells toward areas of elevated CXCL12 levels. Extracellular matrix glycosaminoglycans (GAGs) also bind CXCL12 and maintain a chemotactic concentration gradient.6 CXCL12 is constitutively expressed and essential during embryonic development but afterward functions mainly in inflammatory response, immune surveillance, and tissue homeostasis. This is done through trafficking of lymphocytes to where they are needed such as the lymph nodes, lung, and bone.7,8
Metastatic cancer cells exploit the same mechanism as lymphocytes by upregulating the expression of chemokine receptors.2,3,9 CXCR4, for example, is overexpressed in over 23 human cancers, allowing tumor cells to migrate to organs that produce CXCL12, leading to the formation of secondary colonies.9,10 Because metastasis contributes the most to cancer mortality rates, preventing the migration of tumor cells is of paramount medical importance.11 As a result, novel inhibitors of the CXCR4–CXCL12 signaling axis have been under active development as potential cancer therapeutics.12,13 Such efforts have mainly focused on the orthosteric site of CXCR4, a deep transmembrane pocket suitable for the binding of small molecule antagonists.14 For example, AMD3100 (Plerixafor), a CXCR4 antagonist, has been approved in promoting hematopoietic stem cell mobilization from the bone marrow to the blood in treating multiple myeloma and non-Hodgkin’s lymphoma.15 However, recent studies also suggest that neutralizing chemokines may prove to be a successful approach to cancer therapy as well.16−18 NOX-A12, an RNA oligonucleotide in l-configuration that binds CXCL12 and blocks GAG binding, is thought to increase the susceptibility of chronic lymphocytic leukemia cells to chemotherapy by interfering with chemokine-mediated cell motility.18
CXCR4 has been previously described to rest in a constitutuve dimeric form, independent of ligand binding.19 CXCL12 then binds and activates CXCR4 in a two-step/two-site process (Figure 1).20 First, CXCL12 is recognized by the extracellular N-terminal domain of the receptor (site 1 binding) (Figure 1B).21 Following recognition, the flexible N-terminus of CXCL12 docks into the receptor (site 2 binding) (Figure 1C), leading to receptor internalization and downstream signaling such as calcium influx and chemotaxis.
Figure 1.

Monomeric representation of CXCR4 bound by CXCL12 through a two-step/two-site process. (A) CXCR4 has a flexible extracellular N-terminal domain. (B) In step-1/site-1, CXCL12 recognizes and binds the N-terminal domain of CXCR4 aided by sulfotyrosine recognition. (C) In step-2/site-2, the flexible N-terminal domain of CXCL12 docks into CXCR4 causing activation. Multiple lines of evidence suggest that CXCR4 can form dimers, but there is no evidence to suggest that the site 1 interface would be altered by a change in oligomeric state of the receptor.
As with other chemokine receptors, the CXCR4 N-terminus is post-translationally sulfated at one or more tyrosines,22 including Y7, Y12, and Y21, which increases its affinity for CXCL12. Sulfation at Y21 (sY21) not only contributes the most to enhancing binding affinity but also has the largest effect on downstream signaling.23−25 Structures of locked CXCL12 dimers, in complex with sulfated (only at Y21 or triply sulfated at Y7, Y12, and Y21) CXCR41–38, identified discrete binding pockets for each sulfotyrosine,23 suggesting potential target sites for which small molecule ligands can be engineered. Thus, as molecular details of the CXCL12–CXCR4 interface emerge, structure-based inhibition of CXCL12 becomes a practical albeit challenging approach.
Previously, our in silico screening using DOCK 3.5.54 and the ZINC small molecule database identified ZINC 310454 as a novel small molecule ligand against the sY21-binding site.26 Weak binding to the sY21 site and inhibition of CXCL12–CXCR4 interactions were confirmed by NMR perturbation studies and by CXCL12-mediated Ca2+-flux assays using THP-1 cells, respectively. Subsequent analysis of ZINC 310454, fragment-based design and SAR optimization coupled with a bioisostere approach led to the design and synthesis of tetrazole derivatives, including compound 1. Like the original hits from docking, these compounds bind to CXCL12 with μM affinities. Compound 1 was synthesized by substitution of the carboxyl group with a tetrazole from the meta- to the para- position and substitution of the thioureido linker with the urea (Figure 2A).27 NMR chemical shift mapping suggested that compound 1 also binds to the sY21 site, and molecular docking was used to estimate the binding pose (Figure 2B).
Figure 2.
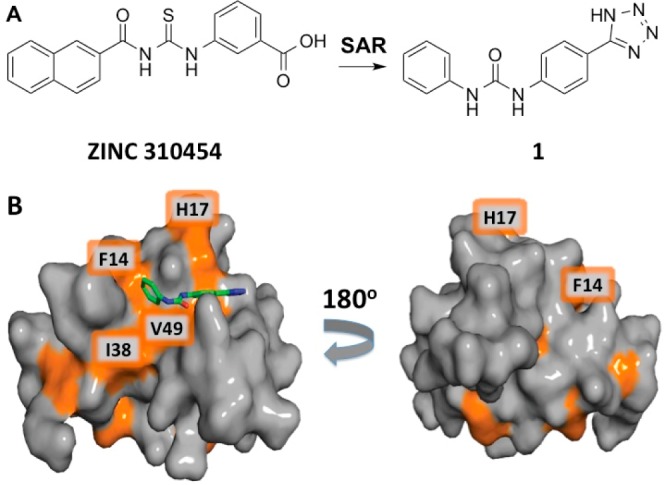
A ZINC 310454 derivative binds in the sY21 binding pocket as predicted by in silico docking. (A) Fragment-based SAR analysis of ZINC 310454 led to the design and synthesis of compound 1. (B) Significant chemical shift perturbations in the presence of 1600 μM compound 1 map to the predicted binding pocket on CXCL12 (PDB: 2K05). The residues most perturbed are colored in orange.
To date, apart from one previous study identifying chalcone-4 as a neutralizing ligand for CXCL12,17 our virtual screening hits and the tetrazole derivatives are the only reported CXCL12-specific small molecule inhibitors, yet the structure of an inhibitor bound to CXCL12 was still lacking. Besides the NMR CXCL12–CXCR41–38 structure (PDB ID: 2K05), the only structure of a CXCL12–ligand complex is the crystal structure of CXCL12 bound by a heparin disaccharide (PDB ID: 2NWG),28 a fragment of the natural ligand that binds at the same pocket as sY12 of CXCR4. Here we present an X-ray crystallographic complex structure of CXCL12 bound to compound 1 at the sY21-binding site, the first of CXCL12 with a novel small molecule ligand. The crystal structure identifies residues that mediate ligand binding and elucidates unoccupied hotspots that can be utilized in future optimization efforts.
Results and Discussion
Compound 1 Occupies the sY21 Binding Site of CXCL12
The complex crystal was obtained by soaking compound 1 into apo CXCL12 crystals. In previous experiments, we noticed that DMSO can nonspecifically bind to the CXCL12 surfaces of interest and result in a noisy electron density map. As a result, no DMSO was used in our soaking experiment and the compound was dissolved to saturation directly in the stabilization buffer. The resulting complex structure was solved at 1.9 Å resolution and has a primitive orthorhombic space group (P212121) (Table 1) with a dimer in each asymmetric unit (Figure 3). The overall structure of the CXCL12 dimer is nearly identical to that of the apo protein (PDB ID: 2J7Z) with an RMSD of 0.238 Å aligning 774 atoms. The only significant backbone movements between the complex structure and the previously determined apo structure are observed in the flexible N-terminus of each monomer.
Table 1. Data Collection and Refinement Statistics.
| Data Collection | |
|---|---|
| space group | P212121 |
| cell dimensions | |
| a, b, c (Å) | 36.93, 57.71, 72.53 |
| α, β, γ, (deg) | 90, 90, 90 |
| wavelength (Å) | 1.5418 |
| resolution range (Å) | 20.00–1.90 |
| I/σI | 21.77(3.25)a |
| completeness | 99.8% |
| Rmerge | 0.061 (0.626)a |
| redundancy | 6.89 (6.85)a |
| Refinement | |
| no. of reflections used | 12099 |
| Rfactor | 0.1863 |
| Rfree | 0.2347 |
| no. of atoms | 1178 |
| protein | 1107 |
| ligand | 21 |
| water | 40 |
| B-factors (Å2) | |
| protein | 59.3 |
| ligand | 78.9 |
| water | 55.0 |
| root-mean-square deviations | |
| bond lengths (Å) | 0.015 |
| bond angles (deg) | 1.802 |
Numbers in parentheses are for the highest resolution shell.
Figure 3.

Complex crystallographic structure of CXCL12 dimer (ribbon and surface model) with compound 1 (yellow) bound to the sY21-binding site.
The unbiased Fo – Fc density unambiguously identifies the binding mode of compound 1 (Figure 4A), residing in the pocket that normally interacts with sY21 and adjacent residues, as seen in the NMR complex structure of CXCL12 bound by a CXCR4 sulfated N-terminal peptide (Figure 4C). Compound 1 was observed only in one of the two monomers in the asymmetric unit because the corresponding binding pocket in the other monomer was partially blocked by crystal packing.
Figure 4.
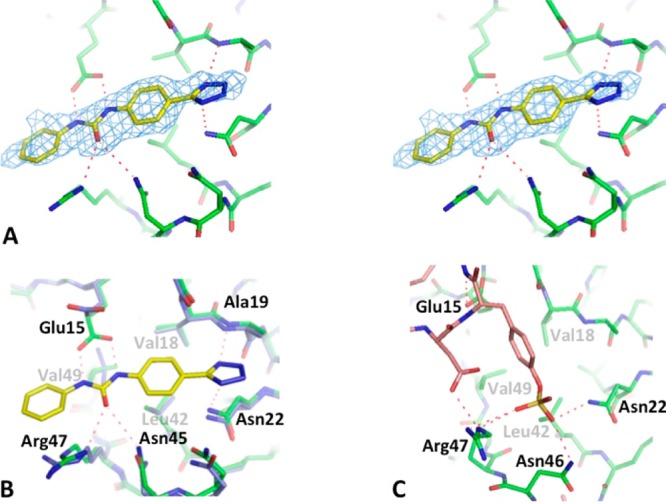
Complex crystallographic structure and characterization of the sY21 binding site. (A) Stereo image depiction of the unbiased Fo−Fc map (at 3σ) of compound 1 bound to the sY21-binding site of CXCL12. (B) Complex crystal structure of CXCL12 (green) bound by compound 1 (yellow) superimposed to the apo crystal structure (PDB ID: 2J7Z) (cyan) shows conformational changes induced upon binding. (C) NMR complex structure of CXCL12 bound to the D20-sY21-D22 segment of CXCR41–38 (PDB ID: 2K05) outlines the sY21-binding site as well as the possible hydrogen bond interactions.
A number of protein main chain and side chain functional groups are involved in polar and nonpolar interactions with the small molecule ligand. Ala19N and Asn22Nδ2 are in close contact (∼3.0 Å) to the tetrazole group, with the former establishing a favorable hydrogen bond and the latter an NH−π interaction (Figure 4B). The urea linker is within hydrogen-bonding distance to Glu15, Asn45, and Arg47. Interestingly, compared to the apo structure (PDB ID: 2J7Z), Glu15 side chain swings into the sY21-binding site and forms an ideal hydrogen-bonding network with compound 1, with the two nitrogen atoms of the urea linker serving as hydrogen donors to the two oxygen atoms of the glutamate side chain (Figure 4B). Furthermore, compared to the published apo structure, Arg47 undergoes conformational changes in order to form a hydrogen bond with the carbonyl oxygen of the urea linker while partially stacking against the terminal benzene ring of compound 1 (Figure 4B). The backbone atoms of residues 43–45 also move closer into the sY21-binding pocket by 0.5–0.8 Å, allowing a closer contact between Asn45 and the urea linker.
In addition to polar interactions, compound 1 establishes extensive hydrophobic interactions with Val18, Leu42, and Val49 (Figure 4B). The proximal benzene ring nestles in the hydrophobic pocket formed by Val18 and Leu42, making many nonpolar contacts. The distal benzene ring is also within van der Waals contact distance with Val49δ1, although the interactions with Val49 can be further optimized. In fact, most of the terminal benzene ring seems to suspend in solution past the pocket, suggesting that this part of the ligand can be further optimized to enhance binding to the chemokine. It also highlights the rigidity of the urea linker, which is further immobilized by the multiple hydrogen bonds as described above. These observations suggest additional carbon atoms may be introduced into the linker to allow adequate flexibility in future lead compounds, which we are currently investigating.
We have previously shown that compound 1 is a fragment that binds CXCL12.27 NMR perturbation studies have placed the compound in the sY21-binding site, but its binding pose was largely unknown (Figure 2B). Molecular docking successfully predicted the correct orientation of the compound and assisted us with previous optimization efforts. Docking however was unable to identify all the possible hydrogen bonds because it did not predict the protein conformational changes induced upon binding such as changes in Glu15 and Arg47. The complex crystal structure, therefore, has not only confirmed binding but has shed light on crucial interactions for the first time, previously overlooked through other complementary methods such as NMR and molecular docking.
As the sY21-binding site also resides close to the crystal-packing interface, the binding of compound 1 has caused additional conformational changes in residues from neighboring molecules in the crystal lattice. An alternative conformation is observed for His17 from a symmetry-related molecule in order to relieve a possible steric clash with the tetrazole group by the original conformation. Arg8 from another adjacent molecule also becomes less ordered. We attempted to investigate whether crystal packing may influence the binding pose of compound 1 by using crystals of R8A, H17A, and R8A/H17A mutants for the soaking experiments. Unfortunately, compound 1 was not observed in these crystals, possibly because the positive charges of these two residues can nonspecifically increase the chances of incorporating the negatively charged compound 1 into the binding pocket, an unintended but useful effect considering the challenges faced in crystallizing complexes with weak ligands. It should also be noted that His17 in monomer 1 is part of the sY21-binding site and may directly contribute to ligand binding through electrostatic interactions.
Comparison with sY21 Binding in CXCR4 Interaction
The binding of compound 1 and the CXCR41–38 peptide to the same sY21-binding site (PDB ID: 2K05) (Figure 4B,C) share many similarities yet demonstrate key differences. Both compound 1 and sY21 are negatively charged at physiological pH and interact with many of the same protein residues in a series of hydrogen bonds, including Glu15, Asn22, and Arg47. These interactions highlight the overall positively charged and highly polar environment of the binding site. However, the sulfate group of sY21 and the tetrazole group of compound 1 reside in different areas of the binding pocket, with the former hydrogen bonding with Asn22, Asn46, and Arg47 and the latter with Asn22 and the backbone N atom of Ala19. The conformations of Glu15 and Arg47 also differ in the two complexes.
The most significant difference, however, lies in the more extensive hydrophobic interactions observed in the complex with compound 1. The aromatic ring of sY21 is placed perpendicularly and relatively superficially on the protein surface in a corner of the pocket formed by Val18, Leu42, and Val49, while the benzene ring of compound 1 lays flat on the center of this small hydrophobic cavity. This increases hydrophobic contact in compound 1 binding.
Some of the differences we have observed between the two complexes undoubtedly come from the structural ambiguity in NMR structures, particularly in terms of side chain conformations, as well as from the structural biases imposed by crystal packing, especially the lack of protein flexibility. However, although CXCL12 exhibits much flexibility in solution, its core domain including the majority of the sY21-binding site displays much less variation among different NMR conformations as well as various crystal structures. This is particularly true for the hydrophobic cavity and the peptide segment spanning His17 to Asn22. The most flexible region appears to come from the three asparagine residues 44–46. Asn45 or Asn46 each makes one hydrogen bond with the ligand in the respective complex. Although certain interactions involving these residues may have been missed in the NMR or crystal structures due to the limitations of each technique, it is unlikely that such experimental caveats have drastically distorted the ligand-binding mode.
Comparison with Heparin Binding
CXCL12 was previously crystallized in complex with an unsaturated heparin disaccharide, revealing structural details of the interactions between glycosaminoglycans and CXCL12.28 Two copies of the disaccharide were observed in the crystal structure. Only one of them, residing at the dimer interface, was deemed biologically relevant, whereas the other one was most likely a crystallization artifact. The heparin-binding site at the dimer interface is characterized by a high concentration of positively charged residues, including His25, Lys27, and Arg41 from both CXCL12 monomers (Figure 5A). It is not surprising that the disaccharide, with four negatively charged functional groups, binds to this area. Most of the intermolecular interactions are mediated through a series of hydrogen bonds involving the charged moieties exposed to the solvent. Only one nonpolar contact is observed between the disaccharide and a hydrophobic residue, Leu29 (Figure 5A). These observations suggest that the binding surface for the heparin molecule may be less suitable for engineering small molecule ligands compared with the sY21-binding site. The fact that we observed nonspecific binding of DMSO to the sY21-binding site in our control crystals also highlights the features of that binding surface suitable for small molecule interactions.
Figure 5.

Heparin/sY12 vs compound 1 binding. (A) CXCL12–heparin sulfate crystal structure (PDB ID: 2NWG) positions haparin sulfate (yellow) in the region where sY12 of CXCR41–38 normally binds. (B) CXCL12–compound 1 crystal structure with heparin sulfate (yellow) and sY12 (purple) superimposed in sY12 site suggests that compounds specific to both sites could potentially be linked together via linkers.
Insights into Structure-Based Inhibitor Design
The current complex structure provides important information for structure-based inhibitor design. It highlights the urea linker as an optimal anchor in hydrogen bonding interactions; the rotation of Glu15, along with the arrangement of Asn45 and Arg47, creates an ideal hydrogen-bonding network with the urea linker of compound 1. This also suggests that amide linkers are not as efficient as urea linkers because an amide linker can only form one hydrogen bond with Glu15 compared to the two hydrogen bonds that the urea linker is forming in the crystal structure. This difference has been exemplified through our docking studies that appropriately position amide-linker derivatives in the sY21-binding site, showing that only one hydrogen bond can form between the amide and Glu15.
The crystal structure not only identifies the important interactions between compound 1 and CXCL12 but also elucidates unoccupied potential hot spots for ligand binding. The terminal benzene extends into a cleft that contains both polar and nonpolar residues, characteristics that can be exploited in optimization efforts (Figure 5B). Particularly, this cleft contains several nonpolar residues including Pro10, Leu29, and Val39 (Figure 5A). It resides on the edge of the heparin-binding site and interacts with sY12 in the complex structure with the CXCR41–38 peptide (Figure 5B). Whereas heparin fails to fully utilize the nonpolar binding surface, more hydrophobic interactions are observed between sY12 and these protein side chains. Consequently, in order to leverage these binding hot spots in our ongoing project of inhibitor design efforts, extra carbon atoms were added after the urea linker of the current inhibitor scaffold for increased flexibility. A docking pose prediction of compound 2, which differs from compound 1 through an additional two-carbon linker after the urea, suggests that a more flexible linker may increase hydrophobic interactions (Figure 6A). This hypothesis is supported by NMR that shows compound 2 inducing chemical shift perturbation of Val39, suggesting the terminal benzene may better interact with the cleft. Additionally, in a chemotaxis assay with THP-1 cells, 25 μM of compound 1 causes a ∼20% decrease in cell migration, while 25 μM of the more flexible compound 2 causes a ∼40% decrease in cell migration, further supporting the potential benefits of linker flexibility (Figure 6B).
Figure 6.

Docking pose and chemotaxis inhibition. (A) Compound 2 is based on the compound 1 scaffold but contains an additional two-carbon linker past the urea. Docking pose prediction suggests the flexible linker may help increase hydrophobic interactions with the cleft above Val39. (B) Chemotaxis assay in THP-1 cells demontrates that 25 μM compound 1 reduces cell migration by ∼20%, while 25 μM compound 2 reduces cell migration by ∼40%.
Although the sY12-binding site appears to be a more challenging target by itself, these observations suggest that small molecule ligands can be designed to span from the sY21-binding site into the sY12-binding site or even further into the heparin binding site. This would further improve the affinity and specificity of the novel ligand, increasing its utility in disrupting the CXCL12–CXCR4 signaling axis and establishing a strategy for chemokine targeted therapies across the entire chemokine family.
Conclusion
The CXCL12–CXCR4 signaling axis has been a key target for drug discovery due to its involvement in various diseases, particularly cancer. Previous efforts focused on CXCR4 solely because chemokines had been deemed “undruggable” due to their small size and shallow surfaces. However, as most drug discovery efforts against CXCR4 have failed in clinical trials due to toxicity issues, the need for alternative approaches has become apparent. We have previously demonstrated that a structure-based approach can successfully identify compounds that bind to the sY21-binding site on CXCL12. Even though binding of inhibitors was confirmed, details of the binding interactions were lacking. Our new complex structure not only validates the binding of ligands to the protein but also offers invaluable information to guide further optimization efforts through a rational design approach. Furthermore, because sulfotyrosine recognition by chemokines is predicted to contribute to most chemokine–receptor interactions, successfully targeting such sites could potentially extend to targeting other chemokines as well.29
Methods
Purity
CXCL12WT was produced and purified as previously described.30 The purity of the protein and compounds 1 and 2 (Supporting Information) was determined to be >95% by SDS-PAGE and HPLC, respectively.
NMR Spectroscopy
NMR data was collected on a Bruker Avance 600 MHz spectrometer equipped with a TCI cryoprobe at 298 K. Lyophilized [U-15N]-CXCL12WT was reconstituted in a 25 mM deuterated MES, 10% (v/v) D2O, 0.02% (w/v) NaN3 buffer, pH 6.8. NMR samples of 50 μM CXCL12WT and 0–1600 μM compound 1 were made with a LEAP PAL robot, where each sample contained 2.0% deuterated DMSO. A Bruker SampleJet was used for automated sample handling and 1H–15N heteronuclear multiple quantum coherence (HMQC) spectra were collected for each titration point. Spectral data was processed with in house scripts, and chemical shift changes were tracked using CARA software.31 Total 1H and 15N chemical shift perturbations were calculated as previously shown.32
Crystallization
CXCL12 was concentrated to 8 mg mL–1 for crystallization trials. The Qiagen JCSG suites were screened using the Phoenix Microdispenser, and hits were optimized and tested for space group determination. Hits that led to crystals with a trigonal space group (e.g., PDB ID: 3GV3) were unsuccessful in binding compound 1 because the sY21-binding site was completely blocked by crystal packing residues. Hundreds of small crystals appeared in a condition containing 3.5 AmSO4, 1%MPD, and 0.1 M MES sodium salt, pH 6.5. These were optimized (2 M AmSO4, 2% MPD, and 0.1 M MES sodium salt, pH 6.5) to consistently produce a few and large single crystals. Surprisingly, two forms of crystals appeared in this condition. One form belonged to the ineffectual trigonal space group, while the other belonged to an orthorhombic space group (e.g., PDB ID: 2J7Z).
Because crystals belonging to two different space groups would form in the same crystallization drop, seeding was used to induce crystallization of the orthorhombic space group instead of the trigonal space group form. Crystals belonging to the P212121 space group would grow to their full size in 3–4 days, measure up to 400 μm in each dimension, and diffract up to 1.8 Å.
Attempts to grow complex CXCL12 crystals in the presence of compound 1 were unsuccessful. We therefore used a soaking method. Because compound 1 was insoluble at high concentrations, we resorted to introducing it in its solid powder form at various amounts directly on the crystal drop. Soaking was also performed for only 30 min because compound 1 would quickly degrade the crystals upon introduction. Crystals were then flash-frozen in 2.2 M AmSO4, 25% glycerol, 2% MPD, and 0.1 M MES sodium salt, pH 6.5. Data sets were collected for many crystals in the search for the best candidate because solubility issues with compound 1 produced inconsistent results with compound density while damaging a lot of the crystals.
Data Collection and Processing
Data was collected at the Moffitt Cancer Center home source with a Rigaku Raxis detector. Data was processed with XDS,33 and the structure was solved through molecular replacement (using PDB ID: 2J7Z) with MOLREP34 and refinement with Refmac535 and PDB_REDO.36 Figures were made using PYMOL (www.pymol.org). The structure has been deposited into the Protein Data Bank with accession code 4UAI.
Molecular Docking
DOCK 3.5.5437 was used to dock compound 2 into an ensemble of 21 protein conformations (20 NMR conformations from PDB ID 2K05 and the current X-ray structure). The best pose was chosen based on complementarity.
THP-1 Chemotaxis
Compound dilutions were prepared in 100% DMSO and added to aliquots of a RPMI 1640, 25 mM HEPES, and 0.2% (w/v) BSA buffer and were centrifuged at 15000 rpm for 20 min at 4 °C. A 250 μL aliquot of 25 μM compound 1 or 2 along with 30 nM CXCL12WT was added to the lower well of a Corning HTS Transwell 96-well plate. THP-1 cells were washed twice in buffer, and 6 × 105 cells were added to the upper well. DMSO was held constant between upper and lower wells. Plates were incubated for 2 h at 5% CO2 and 37 °C, and migrated cells in the lower well were counted on a BD LSR II flow cytometer. Bar graph is the mean ± SEM of four replicates on two separate plates (n = 8). Significance was determined by a two-tailed, unpaired Student’s t test.
Acknowledgments
This work was supported by the National Institutes of Health under award nos. GM097381 and AI058072 to B.F.V. and CA173056 to R.L. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We would like to thank Andreas Becker, from the Moffitt Cancer Center Structural Biology Core Facility, for assistance with X-ray data collection.
Glossary
Abbreviations Used
- CXCL12
CXC chemokine ligand 12
- CXCR4
CXC chemokine receptor 4
- CXCR7
CXC chemokine receptor 7
Supporting Information Available
Synthesis and characterization of compounds 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The crystal structure of CXCL12 in complex with inhibitor has been deposited into the Protein Data Bank with accession code 4UAI.
Author Present Address
∥ For J.J.Z.: Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, 240 Longwood Avenue, Boston, Massachusetts 02115, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zlotnik A.; Yoshie O. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali S.; Lazennec G. Chemokines: novel targets for breast cancer metastasis. Cancer Metastasis Rev. 2007, 26, 401–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona S. M.; Garcia J. A.; Cardona A. E. The fine balance of chemokines during disease: trafficking, inflammation, and homeostasis. Methods Mol. Biol. 2013, 1013, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D.; Zlotnik A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [DOI] [PubMed] [Google Scholar]
- Comerford I.; McColl S. R. Mini-review series: focus on chemokines. Immunol. Cell Biol. 2011, 89, 183–184. [DOI] [PubMed] [Google Scholar]
- Amara A.; Lorthioir O.; Valenzuela A.; Magerus A.; Thelen M.; Montes M.; Virelizier J. L.; Delepierre M.; Baleux F.; Lortat-Jacob H.; Arenzana-Seisdedos F. Stromal cell-derived factor-1alpha associates with heparan sulfates through the first beta-strand of the chemokine. J. Biol. Chem. 1999, 274, 23916–23925. [DOI] [PubMed] [Google Scholar]
- Karin N. The multiple faces of CXCL12 (SDF-1alpha) in the regulation of immunity during health and disease. J. Leukocyte Biol. 2010, 88, 463–473. [DOI] [PubMed] [Google Scholar]
- Nagasawa T. CXC chemokine ligand 12 (CXCL12) and its receptor CXCR4. J. Mol. Med. (Berlin, Ger.) 2014, 92, 433–439. [DOI] [PubMed] [Google Scholar]
- Cojoc M.; Peitzsch C.; Trautmann F.; Polishchuk L.; Telegeev G. D.; Dubrovska A. Emerging targets in cancer management: role of the CXCL12/CXCR4 axis. OncoTargets Ther. 2013, 6, 1347–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Cheng G.; Hao M.; Zheng J.; Zhou X.; Zhang J.; Taichman R. S.; Pienta K. J.; Wang J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakinuma T.; Hwang S. T. Chemokines, chemokine receptors, and cancer metastasis. J. Leukocyte Biol. 2006, 79, 639–651. [DOI] [PubMed] [Google Scholar]
- Choi W. T.; Duggineni S.; Xu Y.; Huang Z.; An J. Drug discovery research targeting the CXC chemokine receptor 4 (CXCR4). J. Med. Chem. 2012, 55, 977–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachelerie F.; Ben-Baruch A.; Burkhardt A. M.; Combadiere C.; Farber J. M.; Graham G. J.; Horuk R.; Sparre-Ulrich A. H.; Locati M.; Luster A. D.; Mantovani A.; Matsushima K.; Murphy P. M.; Nibbs R.; Nomiyama H.; Power C. A.; Proudfoot A. E.; Rosenkilde M. M.; Rot A.; Sozzani S.; Thelen M.; Yoshie O.; Zlotnik A. International Union of Basic and Clinical Pharmacology [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev. 2014, 66, 1–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Chien E. Y.; Mol C. D.; Fenalti G.; Liu W.; Katritch V.; Abagyan R.; Brooun A.; Wells P.; Bi F. C.; Hamel D. J.; Kuhn P.; Handel T. M.; Cherezov V.; Stevens R. C. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPersio J. F.; Uy G. L.; Yasothan U.; Kirkpatrick P. Plerixafor. Nature Rev. Drug Discovery 2009, 8, 105–106. [DOI] [PubMed] [Google Scholar]
- Galzi J. L.; Hachet-Haas M.; Bonnet D.; Daubeuf F.; Lecat S.; Hibert M.; Haiech J.; Frossard N. Neutralizing endogenous chemokines with small molecules. Principles and potential therapeutic applications. Pharmacol. Ther. 2010, 126, 39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachet-Haas M.; Balabanian K.; Rohmer F.; Pons F.; Franchet C.; Lecat S.; Chow K. Y.; Dagher R.; Gizzi P.; Didier B.; Lagane B.; Kellenberger E.; Bonnet D.; Baleux F.; Haiech J.; Parmentier M.; Frossard N.; Arenzana-Seisdedos F.; Hibert M.; Galzi J. L. Small neutralizing molecules to inhibit actions of the chemokine CXCL12. J. Biol. Chem. 2008, 283, 23189–23199. [DOI] [PubMed] [Google Scholar]
- Hoellenriegel J.; Zboralski D.; Maasch C.; Rosin N. Y.; Wierda W. G.; Keating M. J.; Kruschinski A.; Burger J. A. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood 2013, 123, 1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock G. J.; Farzan M.; Sodroski J. Ligand-independent dimerization of CXCR4, a principal HIV-1 coreceptor. J. Biol. Chem. 2003, 278, 3378–3385. [DOI] [PubMed] [Google Scholar]
- Crump M. P.; Gong J. H.; Loetscher P.; Rajarathnam K.; Amara A.; Arenzana-Seisdedos F.; Virelizier J. L.; Baggiolini M.; Sykes B. D.; Clark-Lewis I. Solution structure and basis for functional activity of stromal cell-derived factor-1: dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997, 16, 6996–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuku Y.; Yoshiura C.; Ueda T.; Terasawa H.; Hirai T.; Tominaga S.; Hirose M.; Maeda Y.; Takahashi H.; Terashima Y.; Matsushima K.; Shimada I. Structural basis of the interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G-protein-coupled receptor CXCR4. J. Biol. Chem. 2009, 284, 35240–35250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe H.; Farzan M. Chapter 7 Tyrosine sulfation of HIV-1 coreceptors and other chemokine receptors. Methods Enzymol. 2009, 461, 147–170. [DOI] [PubMed] [Google Scholar]
- Veldkamp C. T.; Seibert C.; Peterson F. C.; De la Cruz N. B.; Haugner J. C. III; Basnet H.; Sakmar T. P.; Volkman B. F. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci. Signaling 2008, 1, ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone M. J.; Chuang S.; Hou X.; Shoham M.; Zhu J. Z. Tyrosine sulfation: an increasingly recognised post-translational modification of secreted proteins. New Biotechnol. 2009, 25, 299–317. [DOI] [PubMed] [Google Scholar]
- Veldkamp C. T.; Seibert C.; Peterson F. C.; Sakmar T. P.; Volkman B. F. Recognition of a CXCR4 sulfotyrosine by the chemokine stromal cell-derived factor-1alpha (SDF-1alpha/CXCL12). J. Mol. Biol. 2006, 359, 1400–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldkamp C. T.; Ziarek J. J.; Peterson F. C.; Chen Y.; Volkman B. F. Targeting SDF-1/CXCL12 with a ligand that prevents activation of CXCR4 through structure-based drug design. J. Am. Chem. Soc. 2010, 132, 7242–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziarek J. J.; Liu Y.; Smith E.; Zhang G.; Peterson F. C.; Chen J.; Yu Y.; Chen Y.; Volkman B. F.; Li R. Fragment-based optimization of small molecule CXCL12 inhibitors for antagonizing the CXCL12/CXCR4 interaction. Curr. Top Med. Chem. 2012, 12, 2727–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy J. W.; Cho Y.; Sachpatzidis A.; Fan C.; Hodsdon M. E.; Lolis E. Structural and functional basis of CXCL12 (stromal cell-derived factor-1 alpha) binding to heparin. J. Biol. Chem. 2007, 282, 10018–10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziarek J. J.; Heroux M. S.; Veldkamp C. T.; Peterson F. C.; Volkman B. F. Sulfotyrosine recognition as marker for druggable sites in the extracellular space. Int. J. Mol. Sci. 2011, 12, 3740–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekoshi T.; Ziarek J. J.; Volkman B. F.; Hwang S. T. A locked, dimeric CXCL12 variant effectively inhibits pulmonary metastasis of CXCR4-expressing melanoma cells due to enhanced serum stability. Mol. Cancer Ther. 2012, 11, 2516–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R. L. J.The Computer Aided Resonance Assignment/Tutorial; Cantina Verlag: Goldau, Switzerland, 2004.
- Ziarek J. J.; Getschman A. E.; Butler S. J.; Taleski D.; Stephens B.; Kufareva I.; Handel T. M.; Payne R. J.; Volkman B. F. Sulfopeptide probes of the CXCR4/CXCL12 interface reveal oligomer-specific contacts and chemokine allostery. ACS Chem. Biol. 2013, 8, 1955–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 22–25. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten R. P.; Joosten K.; Cohen S. X.; Vriend G.; Perrakis A. Automatic rebuilding and optimization of crystallographic structures in the Protein Data Bank. Bioinformatics 2011, 27, 3392–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Shoichet B. K. Molecular docking and ligand specificity in fragment-based inhibitor discovery. Nature Chem. Biol. 2009, 5, 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.