

Abstract

We report a series of 4-sulfamoylphenyl-ω-aminoalkyl ethers as carbonic anhydrase (CA, EC 4.2.1.1) inhibitors. The structure–activity relationship was drawn for the inhibition of four physiologically relevant isoforms: hCA I, II, IX, and XII. Many of these compounds were highly effective, low nanomolar inhibitors of all CA isoforms, whereas several isoform-selective were also identified. X-ray crystal structures of two new sulfonamides bound to the physiologically dominant CA II isoform showed the tails of these derivatives bound within the hydrophobic half of the enzyme active site through van der Waals contacts with Val135, Leu198, Leu204, Trp209, Pro201, and Pro202 amino acids. One of the highly water-soluble compound (as trifluoroacetate salt) showed effective IOP lowering properties in an animal model of glaucoma. Several fluorescent sulfonamides incorporating either the fluorescein-thiourea (7a–c) or tetramethylrhodamine-thiourea (9a,b) moieties were also obtained and showed interesting CA inhibitory properties for the tumor-associated isoforms CA IX and XII.
Introduction
Carbon dioxide (CO2) is a very stable form of carbon, the central element of life on this planet and one of the simplest molecules that was probably highly abundant in the primeval earth atmosphere. This gas reacts with water, leading to H2CO3, which is an unstable compound that is spontaneously transformed into bicarbonate and protons. However, the reaction between CO2 and water is particularly slow at pH values of 7.5 or lower, which is usually the physiologic pH value in many tissues and organisms.1−3 Carbon dioxide hydration becomes, on the other hand, very effective at higher pH values, being instantaneous at pH > 12.1−3 Moreover CO2 is an important molecule in all life processes, being generated in high amounts in most organisms.3−7 To catalyze its rapid transformation into bicarbonate, catalysts evolved in all life kingdoms, that is, the enzymes known as carbonic anhydrases (CAs, EC 4.2.1.1).1−7 Six genetically diverse such enzyme families are presently known—the α-, β-, γ-, δ-, ζ-, and η-CAs6−8—with the last class discovered quite recently.9
CAs not only face the conversion of the high amounts of CO2 formed in the metabolic processes, transforming it in bicarbonate and protons, but they also manage the acid–base equilibria connected to this reaction. In fact, the products formed in the catalyzed reaction are either ions with strong buffering activity (bicarbonate) or hydrated protons (H+ ions). The regulation of pH is a highly important process in all life forms, since many biochemical reactions are tightly regulated by it.1−3 This is probably the reason why so many genetic CA families are presently known so that in some organisms, a multitude of different CA families with many isoforms have been described, each with specialized functions.6−9 The necessity of a tight/precise pH regulation may thus explain why most organisms investigated so far contain multiple CA isoforms, although they differ significantly by their catalytic activity, susceptibility to various classes of inhibitors, subcellular localization, and many other such features.1−3,6−9 For example, in humans, 15 different CA isoforms, all belonging to the α-class, have been described.1−3
Most mammals (including humans) possess two blood isoforms, denominated CA I and CAII, with a total concentration of these proteins as high as 0.2 mM.10 However, the catalytic activity of the human (h) isoform hCA I is much lower compared with that of hCA II, and in addition, hCA I is also inhibited by the chloride and bicarbonate present in the plasma, leaving a lot of questions regarding the physiologic function of this isoform.10,11 On the other hand, the high activity isoform hCA II (also known as the “rapid” blood enzyme, to distinguish it from the “slow” one, hCA I) is involved in the secretion of electrolytes in a multitude of tissues, such as the bicarbonate-rich aqueous humor in the anterior chamber of the eyes, and the cerebrospinal fluid, but also in pH and CO2 homeostasis all over the body, as mentioned above.11−13 Other functions include urine formation and bicarbonate reabsorption in the kidney tubules; biosynthetic reactions, such as gluconeogenesis, lipogenesis, and ureagenesis; bone resorption and calcification; and probably many other less well understood physiological/pathological processes.11−15 Indeed, a dysregulation of the activity of these isoforms in one or more tissues has important pathologic consequences, such as glaucoma, when excessive aqueous humor is secreted within the eye, with the subsequent increase in the intraocular pressure (IOP) and edema, when not enough fluids are secreted/eliminated in the urine, leading to fluid accumulation in the body, processes in which CA II together with several other isoforms such as CA IV, XII, and XIV, are involved in the kidneys, epilepsy (the involvement of CA II and other brain CA isoforms in this disease is poorly understood and certainly not irrelevant), and some forms of cancer, in which CA II was observed to be overexpressed, alone or together with other isoforms such as CA IX and XII.11−15 CA II is also involved in other pathologies, such as acute mountain sickness and apparently, atherosclerosis and osteoporosis.16
Primary sulfonamides constitute the main class of CA inhibitors (CAIs), with a number of such derivatives in clinical use for decades, mainly as antiglaucoma agents, diuretics, antiepileptics, or antiobesity drugs.1c,1d,11−16 Recently, some sulfonamides with CA inhibitory properties entered phase I clinical trials as antitumor/antimetastatic agents targeting hypoxic tumors in which two CA isoforms, CA IX and XII, are overexpressed.1c,1d,17 The search for sulfonamide CAIs with various potentials in therapeutics is a dynamic research field, with many new classes being reported constantly and investigated in detail for inhibitory effects against mammalian and nonmammalian CAs.1,17,18 Here, we report a class of 4-sulfamoylphenyl-ω-aminoalkyl ethers, a poorly investigated chemotype in the CAIs landscape, with interesting properties as antiglaucoma agents as well as for the design of fluorescent enzyme inhibitors with potential use for imaging CAs in various tissues.
Results and Discussion
Chemistry and Drug Design
Benzenesulfonamides constitute a highly investigated class of CAIs,19a with most such compounds reported so far being derivatives of sulfanilamide, homosulfanilamide, or 4-aminoethylbenzenesulfonamide. Derivatization of the primary aliphatic/aromatic amino group from these compounds by its transformation into carboxamides, secondary sulfonamides, ureas, thioureas, or by reaction with pyrylium salts has led to a considerable number of new derivatives that showed excellent inhibitory properties against CA isoforms of medicinal interest, such as CA II, IV, VA/VB, IX, or XII.16−18,19a This is generally known as the tail approach for designing CAIs.18c Surprisingly, very few benzenesulfonamides incorporating ether or thioether moieties have been reported so far. In fact, only one paper, by Vernier et al.,19b considered these chemotypes for the design of sulfonamide-based CAIs. In a very interesting study, these authors reported compounds of the type Ar-X-Ar′-SO2NH2, where X was O or S, and Ar, Ar′ aromatic/heterocyclic six-membered rings, which showed highly effective inhibitory properties against CA isoforms involved in important physiologic processes, such as CA II and IV.19b These derivatives showed improved water solubility compared with structurally similar sulfanilamide derivatives, possessed low nanomolar inhibitory action against CA II, and were shown to penetrate eye tissues, arriving at the ciliary processes where the enzyme is present within the eye, and participating in aqueous humor secretion.19b Unfortunately no in vivo antiglaucoma studies have been performed with those compounds that possessed physicochemical properties appropriate for an antiglaucoma drug candidate.
Considering these facts, we decided to explore the synthesis and properties of ethers incorporating the benzenesulfonamide “head” and aliphatic ether moieties of the type H2N–(CH2)n–O–C6H4–SO2NH2. As mentioned above, such ethers were not investigated as CAIs until now, and considering the aromatic derivatives reported by the Pfizer group,19b the presence of aliphatic, amino moieties should also promote the water solubility of the compounds. In fact, a considerable pharmacologic problem of the first generation CAIs, such as acetazolamide, AAZ; methazolamide, MZA; or dichlorophenamide, DCP, was their poor water solubility. Only the second generation drugs, such as dorzolamide, DRZ; and brinzolamide, BRZ, have an improved water solubility because these two topically acting antiglaucoma drugs are administered as hydrochloride salts (both weak amines).

Thus, we designed the following strategy for obtaining the 4-sulfamoylphenyl ω-aminoalkyl ethers reported in this paper (Scheme 1). Reaction of ω-amino-alcohols 1a–e (n = 2–6) with tert-butyloxycarbonyl anhydride afforded the Boc-protected amines 2a–e,20 which by Mitsunobu reaction with 4-hydroxybenzenesulfonamide 3 led to the Boc-protected derivatives 4a–e.21 Removal of the protecting group in the presence of trifluoroacetic acid (TFA) afforded the trifluoroacetate salts of the 4-sulfamoylphenyl ω-aminoalkyl ethers 5a–e (Scheme 1). The alkyl chain present in the new derivatives ranged between 2 and 6 carbon atoms to investigate the influence of the spacer length for the enzyme inhibitory properties of the new derivatives.
Scheme 1. Preparation of the Sulfonamides 4, 5, 7, and 9 Reported in This Paper.

Another aspect in the design of CAIs is related to the use of such compounds as diagnostic tools, for example, for the imaging of tumors in which some CA isoforms are overexpressed. We have reported, for example, fluorescein-based sulfonamides (obtained again from sulfanilamide, homosulfanilamide or 4-aminoethylbenzenesulfonamide, which were reacted with fluorescein isothiocyanate)22 that were essential for demonstrating the role of CA IX/XII in the acidification of the extracellular tumor milieu and also in the proof-of-concept studies regarding the druggability of these novel antitumor targets.23 However, like most aromatic thioureas, the fluorescent sulfonamides reported earlier showed a rather low water solubility, which may be a limiting factor for some of their applications. Thus, we report here novel derivatives that were prepared by reaction of the 4-sulfamoylphenyl ω-aminoalkyl ethers 5a–e with fluorescein isothiocyanate 6 or [9-(2-carboxy-4-isothiocyanato-phenyl)-6-dimethylaminoxanthen-3-ylidene]-dimethylammonium chloride 8, leading to the novel fluorescent compounds of types 7a–c and 9a,b, respectively (Scheme 1). The last compounds (9a,b) incorporate a fluorophore that was not investigated earlier for its interaction with CAs.
Carbonic Anhydrase Inhibition
Inhibition data against four physiologically significant CA isoforms, that is, h (human) hCA I, II, IX, and XII, are shown in Table 1. The following structure–activity relationship (SAR) can be drawn from the data of Table 1:
Table 1. hCA I, II, IX, and XII Inhibition Data of the Newly Synthesized Sulfonamides 4a–9b and Acetazolamide AAZ as Standard, by the Stopped Flow CO2 Hydrase Assay24.
|
Ki (nM)a |
||||
|---|---|---|---|---|
| compd | hCA I | hCA II | hCA IX | hCA XII |
| 4a | 5.3 | 5.0 | 8.3 | 7.2 |
| 4b | 6.6 | 5.1 | 5.8 | 6.5 |
| 4c | 41.2 | 5.7 | 7.7 | 6.6 |
| 4d | 7.9 | 5.5 | 7.1 | 5.7 |
| 4e | 6.1 | 5.2 | 6.9 | 6.4 |
| 5a | 649 | 66.5 | 8.7 | 88.5 |
| 5b | 452 | 36.6 | 17.9 | 9.6 |
| 5c | 286 | 8.9 | 32.6 | 7.5 |
| 5d | 52.5 | 8.8 | 6.6 | 7.3 |
| 5e | 63.0 | 3.9 | 6.5 | 6.5 |
| 7a | 18.0 | 5.0 | 8.5 | 8.6 |
| 7b | 17.1 | 4.6 | 7.2 | 5.7 |
| 7c | 90.9 | 3.9 | 8.8 | 7.1 |
| 9a | 826 | 215 | 9.6 | 609 |
| 9b | 151 | 43.3 | 7.9 | 36.7 |
| AAZ | 250 | 12.1 | 25.0 | 5.7 |
Mean from three different assays; errors are in the range of ±10% of the reported value.
(i) The slow human isoform hCA I effectively inhibited by some of the sulfonamides investigated here, such as 4a–4e and 7a, 7b, which showed KI values in the low nanomolar range (5.3–41.2 nM), whereas other derivatives (e.g., 5d, 5e, and 7c) were medium potency inhibitors, with KI’s of 52.5–90.9 nM. Like acetazolamide AAZ, some of the new compounds, among which 5a–5c and 9a, 9b were less effective hCA I inhibitors, with KI’s of 151–826 nM. Thus, the best hCA I inhibitors were the Boc-protected derivatives 4, which showed a rather compact behavior of very effective inhibitor, except for the compound with the 4-carbon-atoms linker (4c) which was less effective compared with its congeners 4a, b, d, and e. The deprotected amines 5 were less effective as hCA I inhibitors compared with the corresponding Boc derivatives (Table 1). Among the fluorescent CAIs reported here, the fluorescein-containing compounds 7a and 7b were effective hCA I inhibitors, whereas the tetramethylrhodamine derivatives 9a and 9b were much less effective as hCA I inhibitors compared with the fluorescein derivatives mentioned above.
(ii) The physiologically dominant hCA II very effectively inhibited by most sulfonamides reported here. Indeed, just four compounds (5a and 5b) as well as 9a,b showed medium potency activity, with KI’s of 36.6–215 nM. The remaining sulfonamides showed very effective hCA II inhibitory properties, with KI’s ranging between 3.9 and 8.9 nM (Table 1) and are thus more effective than the clinically used drug acetazolamide AAZ. The SAR is rather clear-cut: the five BOC-protected derivatives 4a–4e showed a very compact behavior with basically no variation of the inhibitory power, with the length of the linker from 2 to 6 CH2 moieties. However, the situation is changed for the amines 5a–5e, which on one hand were weaker hCA II inhibitors compared with the corresponding Boc-protected derivatives and on the other hand showed an increase in the inhibitory power with an increase in the linker chain from 2 to 6 CH2 moieties. Indeed, between compounds 5a and 5e, there is a 17-fold difference in the inhibitory activity against this isoform. As for hCA I inhibition, again, the fluorescein-tailed sulfonamides 7a–7c were much more inhibitory compared with the tetramethylrhodamine derivatives 9a and 9b.
(iii) The tumor-associated, transmembrane isoform hCA IX was very well inhibited by all derivatives reported here, with KI’s of 5.8–32.6 nM. The SAR is almost impossible to delineate because all these compounds show excellent inhibitory activity. For example, the Boc-protected derivatives 4a–e have a minimal variation of the inhibition constants, ranging between 5.8 and 8.3 nM. This variation is slightly higher for the amines 5 (between 6.5 and 32.6 nM) and is again almost absent for the fluorescent sulfonamides 7 and 9. It is interesting to note that for this isoform, both the fluorescein and the tetramethylrhodamine derivatives were equally effective as CAIs.
(iv) The other transmembrane isoform investigated here, hCA XII, was also effectively inhibited by most of the new sulfonamides reported in this paper. Two compounds, 5a and 9b, were medium potency inhibitors (KI’s of 36.7–88.5 nM), and one (9a) was an ineffective inhibitor (KI of 609 nM). The remaining sulfonamides investigated here showed excellent hCA XII inhibitory activity, with inhibition constants ranging between 5.4 and 9.6 nM, again with no obvious SAR to be discussed (Table 1).
(v) Although most of these sulfonamides were effective CAIs against all four isoforms investigated here, several interesting selectivity cases were observed: for example, 9a is a hCA IX-selective sulfonamide inhibitor, with a KI of 9.6 nM against the tumor-associated isoform and >215 nM against hCA I, II, and XII (Table 1). Compound 5b effectively inhibits the two transmembrane isoforms (KI’s of 9.6–17.9 nM); it is a much less effective inhibitor of the two cytosolic isoforms hCA I and II (KI’s of 36.6–452 nM).
X-ray Crystallography
To rationalize some of the inhibition data presented above, two of the novel sulfonamides reported here, 4c (incorporating the Boc-aminobutyl moiety) and 5c (incorporating the 4-aminobutyl fragment) were cocrystallized with hCA II, and their crystal structures were resolved at a high resolution (Table 2). Both inhibitors were observed bound within the enzyme active site, coordinating to the Zn(II) ion by means of the deprotonated nitrogen of the sulfonamide moiety (Figures 1 and 2), like all other sulfonamide or sulfamates investigated so far by means of this technique.1,25,26 The phenyl ring and the rather long, hydrophobic alkyl tails of both inhibitors were observed to interact only with residues of the hydrophobic half of the hCA II active site (as shown in Figures 1–3), such as Val121, Phe131, Leu198, Pro201, and Pro202. The tail of compound 5c extends farther out into the enzyme’s hydrophobic cleft, allowing it to form more stabilizing interactions with amino acids, such as Pro201 and Leu204; however, the shorter tail of compound 4c is unable to perform such interactions (Figure 3).
Table 2. Crystallographic Statistics for the hCA II Adducts of 4c and 5ca.
| hCA II-4c | hCA II-5c | |
|---|---|---|
| PDB ID | 4RFC | 4RFD |
| space group | P21 | P21 |
| unit-cell parameters (Å, deg) | a = 42.4, b = 41.3, c = 71.7, β = 104.1 | a = 42.5, b = 41.3, c = 72.1, β = 104.3 |
| resolution (Å) | 1.80 (1.86–1.80) | 1.63 (1.69–1.63) |
| total no. reflections | 71 626 | 105 407 |
| individual reflections | 22 386 | 30 415 |
| redundancy | 3.2 | 3.5 |
| completeness | 98.8 (99.5) | 99.7 (97.9) |
| Rsymb | 0.163 | 0.086 |
| Rcryst/Rfreec | 0.224/0.260 | 0.178/0.206 |
| rmsd for bond lengths/angles (Å, deg) | 0.006/1.10 | 0.010/1.29 |
| av B-factors (Å2) main/side/ligand | 12.3/16.7/ | 5.3/9.1/ |
| no. protein atoms | 2086 | 2114 |
| no. water molecules | 54 | 127 |
| Ramachandran statistics most favored and additional/generously allowed | 89.4/10.5/0.5 | 87.6/11.5/0.9 |
Values in parentheses represent highest resolution bin.
Rsym = (∑|I - ⟨I⟩|/∑ ⟨I⟩).
Rcryst = (∑|F0 – Fc|/∑ |F0|). Rfree is calculated in the same way as Rcryst except it is for data omitted from refinement (5% of reflections for all data sets).
Figure 1.
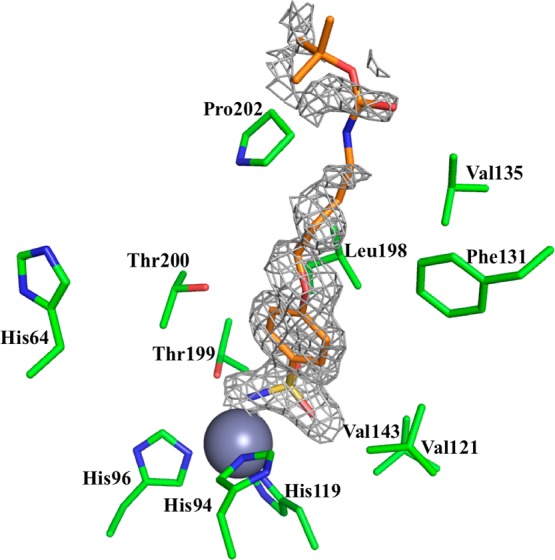
Electron density of compound 4c bound within the hCA II active site. The Zn(II) ion (gray sphere) and its three His ligands (His94, 96, and 119) as well as other residues involved in the catalytic cycle or binding with inhibitors are shown. The 2F0 – Fc electron density is represented by a 0.6σ-weighted gray mesh. Because of the less-ordered electron density for the tail region of 4c, its map was contoured at a lower sigma level compared with compound 5c.
Figure 2.
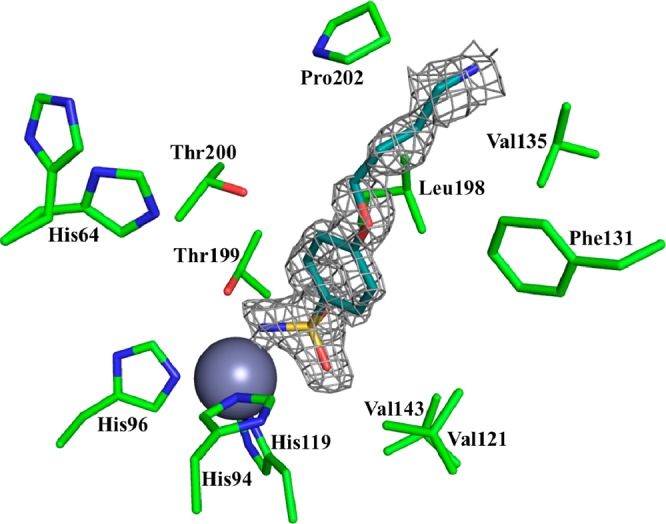
Electron density of compound 5c bound within the hCA II active site. The Zn(II) ion (gray sphere) and its three His ligands (His94, 96, and 119) as well as other residues involved in the catalytic cycle or binding with inhibitors are shown. The 2F0 – Fc electron density is represented by a 1.0σ-weighted gray mesh.
Figure 3.
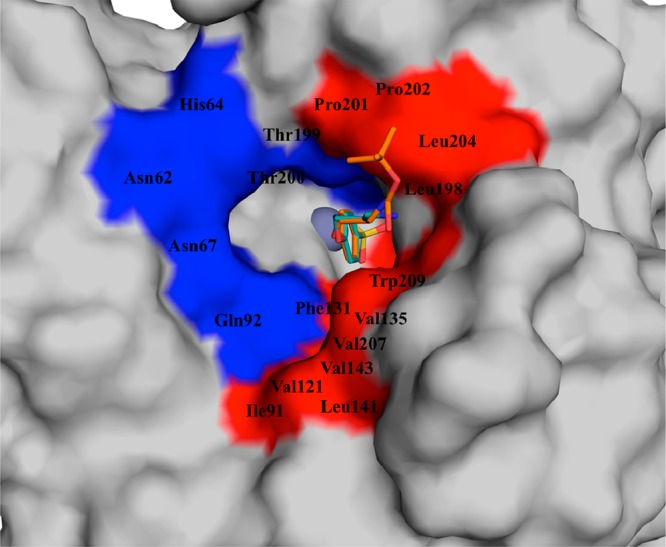
Overlay of sulfonamides 4c (orange) and 5c (teal) bound within the active site of hCA II. Hydrophobic region, red; hydrophilic region, blue; zinc ion, gray sphere at the bottom of the activity.
It should be mentioned that for the Boc-protected derivative 4c, the electron density of the tail region was not completely defined (Figure 1), probably because of its high flexibility and disorder when complexed to the enzyme. In contrast, for the amine 5c, all atoms from the tail region had the electron density well-defined, proving that this region is less disordered compared with the Boc-protected compound 4c (Figure 2). To account for this disorder and to ensure the ligand was built into the density correctly, the map for compound 4c was contoured at a lower sigma level (0.6) than that of 5c (1.0). The fact that the tails of these compounds lie only in the hydrophobic cleft of the hCA II active site is a noteworthy finding, because we showed in earlier papers25,26 that the active site region (hydrophobic versus hydrophilic halves) in which a sulfonamide binds is indicative of its isoform-selectivity properties. In fact, we observed earlier that compounds that have their tails lying only in the hydrophobic half generally do not possess isoform-selective inhibitory properties, and this is also confirmed by the present findings (although neither 4c nor 5c is highly effective as hCA I inhibitors, Table 1). In fact, these two compounds inhibit hCA II, IX, and XII in rather similar ranges and are effective inhibitors against these three isoforms. Because our interest was to obtain compound with antiglaucoma activity, for which both hCA II and XII27 effective inhibitory properties are desired, we consider the present observations of real interest.
Antiglaucoma Activity
Both the Boc-protected and the amino derivative sulfonamides reported here showed excellent water solubility and could be formulated as 2% eye drops at the neutral pH value (dorzolamide, DRZ, the clinically used drug is a hydrochloride salt with a pH of the eye drops of 5.5 which produces eye irritation and stinging as side effects).11 We have investigated the intraocular pressure (IOP) lowering properties of some of these compounds, more precisely, 4c and 5c (for which the X-ray in adduct with hCA II was reported; see the Discussion section, above), in an animal model of glaucoma.28 Indeed, both compounds were low nanomolar inhibitors of isoforms hCA II (responsible for aqueous humor secretion) and hCA XII (isoform that is overexpressed in the eyes of glaucomatous patients).27 As seen from the data of Figure 4, the Boc-protected derivative 4c showed a small decrease of IOP (of 1–2.5 mmHg) when given topically to the eye of the animals, whereas the free amine 5c (as a trifluoroacetate salt) was more effective, with an IOP decrease of 4.4 mmHg at 2 h postadministration, more effective than DRZ, the standard drug, which caused an IOP drop of 4 mmHg at 60 min postadministration. Another notable difference between 5c and DRZ was the fact that the new compound investigated here had a prolonged efficacy compared to DRZ, for which after 4 h no IOP decrease was seen. In contrast, 5c showed efficacy even after 4 h postadministration, with an IOP drop of 3 mmHg at that time point. It should be mentioned that the animal model employed here is of normotensive rabbits,28 and this is why the absolute IOP drops are not very high, but the advantage of this model is that the measurements can be done rapidly and are highly reproducible.14a
Figure 4.
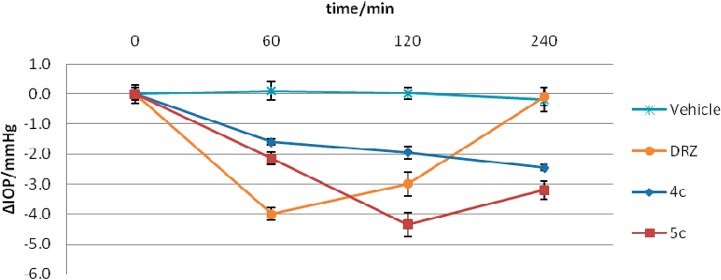
Drop of intraocular pressure (ΔIOP, mmHg) versus time (min) in hypertonic saline-induced ocular hypertension in rabbits, treated with 50 μL of 2% solution of compounds 4c, 5c, and DRZ as the standard drug and vehicle. Errors were within 10–15% of the reported IOP values (from three different measurements for each of the four animals in the study group) and were statistically significant (p = 0.045 by the Student’s t test).
Conclusions
We report a series of new 4-sulfamoylphenyl-ω-aminoalkyl ethers that have been prepared by Mitsunobu reaction. Interesting SAR has been observed for the inhibition of four physiologically relevant CA isoforms: hCA I, II, IX, and XII. Many of the new compounds were highly effective inhibitors of all these isoforms, in the low nanomolar range, with few isoform-selective compounds also identified. These findings have been rationalized by resolving the X-ray crystal structures of two of the new sulfonamides. The tails of these derivatives were observed bound only in the hydrophobic half of the enzyme active site, making van der Waals contacts with amino acids such as Val135, Leu198, Leu204, Trp209, Pro201. and Pro202. One of the compounds incorporating a free amine moiety, which was highly water-soluble as a trifluoroacetate salt, also showed effective IOP lowering properties in an animal model of glaucoma. Several fluorescent sulfonamides have also been reported that incorporate either fluorescein–thiourea or tetramethylrhodamine–thiourea moieties, which also effectively inhibited some CA isoforms investigated here.
Experimental Protocols
Chemistry
Anhydrous solvents and all reagents were purchased from Sigma-Aldrich, Alfa Aesar, and TCI. All reactions involving air- or moisture-sensitive compounds were performed under a nitrogen atmosphere using dried glassware and syringe techniques to transfer solutions. Nuclear magnetic resonance (1H NMR, 13C NMR, DEPT-135, DEPT-90, HSQC, HMBC) spectra were recorded using a Bruker Advance III 400 MHz spectrometer in DMSO-d6. Chemical shifts are reported in parts per million (ppm), and the coupling constants (J) are expressed in Hertz (Hz). Splitting patterns are designated as follows: s, singlet; d, doublet; sept, septet; t, triplet; q, quadruplet; m, multiplet; brs, broad singlet; dd, double of doubles, appt, apparent triplet, appq, apparent quartet. The assignment of exchangeable protons (OH and NH) was confirmed by the addition of D2O. Analytical thin-layer chromatography (TLC) was carried out on Merck silica gel F-254 plates. Flash chromatography purifications were performed on Merck Silica gel 60 (230–400 mesh ASTM) as the stationary phase and ethyl acetate/n-hexane were used as eluents. Melting points (mp) were measured in open capillary tubes with a Gallenkamp MPD350.BM3.5 apparatus and are uncorrected. All compounds reported here were >95% HPLC pure.
General Procedure for the Synthesis of O-Alkylbenzenesulfonamides 5a–e via Mitsunobu coupling
a. General Procedure for Boc Protection
Aminoalcohol 1a–e (1.0 equiv) was dissolved in dichloromethane (DCM) and treated with a 1 M NaOH aqueous solution or diisopropyl ethylamine (DIPEA) (1.0 equiv), then di-tert-butyl dicarbonate (1.0 equiv) was added, and the mixture was vigorously stirred O.N. until consumption of starting materials (TLC monitoring). The reaction was quenched with a 1 M hydrochloric acid aqueous solution, neutralized with NaHCO3 aqueous solution, and extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with H2O (3 × 20 mL), dried over Na2SO4, filtered, and concentrated under vacuo to give the titled product.
b. General Procedure for Mitsunobu Coupling
Boc-aminoalchol 2a–e (1.0 equiv) was dissolved in dry THF and transferred to a two neck-flask via cannula, followed by addition of Ph3P (1.0 equiv) and 4-hydroxybenzenesulfonamide (1.0 equiv). Then the solution was cooled to 0 °C, and diisopropyl azodicarboxylate (DIAD) (1.1 equiv) was added dropwise. The reaction was warmed to r.t. and stirred at the same temperature until the starting materials were consumed (TLC monitoring), quenched with slush, and extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with H2O (3 × 20 mL), dried over Na2SO4, filtered, and concentrated under vacuo to give a residue that was purified by silica gel column chromatography followed by crystallization when necessary.
c. General Procedure for Boc Deprotection
Compounds 4a–e (1.0 equiv) was dissolved in DCM or 1,4-dioxane and treated with TFA. The reaction was stirred at r.t. until the starting material was consumed (TLC monitoring). The solvent was removed under vacuo, and the obtained residue was crystallized from IPA or triturated from diethyl ether to obtain the titled compound as a white solid.
Synthesis of tert-Butyl 2-Hydroxyethylcarbamate 2a
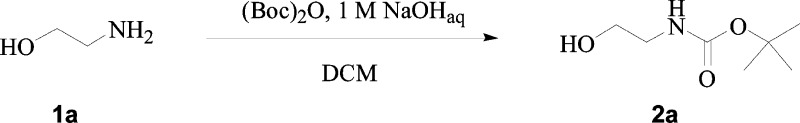
Ethanolamine 1a (1.0 g, 1.0 equiv) was dissolved in DCM (16.5 mL) and treated with a 1 M aqueous solution of NaOH (1.0 equiv), and then di-tert-butyl dicarbonate (1.0 equiv) was added. The reaction mixture was treated according to the general procedure a, previously reported, to give the titled compound 2a as a colorless liquid, which was used as it is.
tert-Butyl 2-Hydroxyethylcarbamate 2a:
66% yield; silica gel TLC Rf 0.18 (ethyl acetate/n-hexane 50% v/v); δH (400 MHz, DMSO-d6) 1.41 (9H, s), 3.0 (2H, q, J 6.0), 3.38 (2H, t, J, 6.0), 4.6 (1H, t, J 6.0, exchange with D2O, OH), 6.71 (1H, brt, exchange with D2O, NH); δC (100 MHz, DMSO-d6) 29.2, 43.6, 61.0, 78.4, 156.6. Experimental data are in agreement with reported data.29
Synthesis of tert-Butyl 3-Hydroxypropylcarbamate 2b

3-Amino-1-propanol 1b (0.98 g, 1.0 equiv) was dissolved in DCM (13 mL) and treated with a 1 M aqueous solution of NaOH (1.0 equiv), and then di-tert-butyl dicarbonate (1.0 equiv) was added. The reaction mixture was treated according to the general procedure a, previously reported, to give the titled compound 2b as a colorless liquid, which was used as it is.
tert-Butyl 3-Hydroxypropylcarbamate 2b:
80% yield; silica gel TLC Rf 0.18 (ethyl acetate/n-hexane 50% v/v); δH (400 MHz, DMSO-d6) 1.41 (9H, s), 1.55 (2H, pent, J 6.4), 3.0 (2H, q, J 6.4), 3.42 (2H, q, J, 6.0), 4.4 (1H, t, J 6.0, exchange with D2O, OH), 6.76 (1H, brt, exchange with D2O, NH); δC (100 MHz, DMSO-d6) 29.2, 33.7, 38.1, 60.7, 78.3, 156.5. Experimental data are in agreement with reported data.30
Synthesis of tert-Butyl 4-Hydroxybutylcarbamate 2c

4-Amino-1-butanol 1c (1.45 g, 1.0 equiv) was dissolved in DCM (16 mL) and treated with DIPEA (1.0 equiv) and then di-tert-butyl dicarbonate (1.0 equiv). The reaction mixture was treated according to the general procedure a, previously reported, to give the titled compound 2c as a yellow liquid, which was used as it is.
tert-Butyl 4-Hydroxybutylcarbamate 2c:
70% yield; silica gel TLC Rf 0.16 (ethyl acetate/n-hexane 50% v/v); δH (400 MHz, DMSO-d6) 1.41 (13H, s), 2.93 (2H, m), 3.40 (2H, m), 4.39 (1H, t, J, 5.0, exchange with D2O, OH), 6.80 (1H, t, J 6.0, exchange with D2O, NH); δC (100 MHz, DMSO-d6) 27.2, 29.2, 30.8, 40.7, 61.4, 78.2, 156.5. Experimental data are in agreement with reported data.31
Synthesis of tert-Butyl 5-Hydroxypentylcarbamate 2d
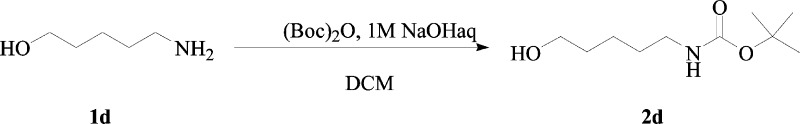
5-Amino-1-pentanol 1d (1.5 g, 1.0 equiv) was dissolved in DCM (14.5 mL) and treated with a 1 M aqueous solution of NaOH (1.0 equiv) and then di-tert-butyl dicarbonate (1.0 equiv). The reaction mixture was treated according to the general procedure a, previously reported, to give the titled compound 2d as a yellow liquid, which was used as it is.
tert-Butyl 5-Hydroxypentylcarbamate 2d:
77% yield; silica gel TLC Rf 0.3 (ethyl acetate/n-hexane 60% v/v); δH (400 MHz, DMSO-d6) 1.22–1.32 (2H, m), 1.34–1.48 (13H, m) 2.92 (2H, q, J 6.4), 3.39 (2H, m), 4.36 (1H, t, J 5.2, exchange with D2O, OH), 6.79 (1H, brt, exchange with D2O, NH); δC (100 MHz, DMSO-d6) 23.8, 29.2, 30.3, 33.1, 40.8, 61.6, 78.2, 156.5. Experimental data are in agreement with reported data.30b
tert-Butyl 6-Hydroxyhexylcarbamate 2e

6-Amino-1-hexanol 1e (0.1 g, 1 equiv) was dissolved in DCM (8.5 mL) and treated with a DIPEA (1.0 equiv) and di-tert-butyl dicarbonate (1.0 equiv). The reaction mixture was treated according to the general procedure a, previously reported, to give the titled compound 2e as a colorless oil, which was used as it is.
tert-Butyl 6-Hydroxyhexylcarbamate 2e:
81% yield; silica gel TLC Rf 0.2 (ethyl acetate/n-hexane 50% v/v); δH (400 MHz, DMSO-d6) 1.23–1.34 (4H, m), 1.34–1.48 (13H, m), 2.89 (2H, q, J 6.7), 3.36 (2H, q, J 5.2), 4.3 (1H, t, J 5.2, exchange with D2O, OH), 6.76 (1H, brt, exchange with D2O, NH); δC (100 MHz, DMSO-d6) 26.2, 27.1, 29.2, 30.5, 33.4, 40.7, 61.6, 78.2, 156.5. Experimental data are in agreement with reported data.32
Synthesis of tert-Butyl 2-(4-Sulfamoylphenoxy)-ethylcarbamate 4a

tert-Butyl 2-hydroxyethylcarbamate 2a (1.0 g, 1.0 equiv) was dissolved in dry THF (9.5 mL) and was treated with Ph3P (1.0 equiv), 4-hydroxybenzenesulfonamide 3 (1.0 equiv), and DIAD (1.1 equiv) according to the general procedure b, previously reported. The reaction was stirred at r.t. until starting materials were consumed (TLC monitoring). The reaction was quenched with slush and extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with H2O (3 × 20 mL), dried over Na2SO4, filtered, and concentrated under vacuo, and the obtained residue was purified by silica gel column chromatography eluting with ethyl acetate/n-hexane 60% v/v, followed by crystallization in EtOH/H2O mixture to afford the titled compound 4a as a white solid.
tert-Butyl 2-(4-Sulfamoylphenoxy)-ethylcarbamate 4a:
40% yield, silica gel TLC Rf 0.4 (ethyl acetate/n-hexane 60% v/v); mp 148–149 °C; δH (400 MHz, DMSO-d6) 1.42 (9H, s), 3.33 (2H, t, J 5.6), 4.07 (2H, t, J, 5.6), 7.07 (1H, brt, exchange with D2O, NH), 7.1 (2H, d, J 8.8), 7.24 (2H, s, exchange with D2O, SO2NH2), 7.77 (2H, d, J 8.8); δC (100 MHz, DMSO-d6) 29.4, 40.3, 68.0, 79.2, 115.7, 128.9, 137.2, 156.9, 162.0. Elemental analysis: calcd C 49.35, H 6.37, N 8.85, S 10.14; found C 49.43, H 6.03, N 8.68, S 9.97; m/z (ESI negative) 315.6 [M – H]−.
Synthesis of tert-Butyl 3-(4-Sulfamoylphenoxy)-propylcarbamate 4b

tert-Butyl 3-hydroxypropylcarbamate 2b (0.88 g, 1.0 equiv) was dissolved in dry THF (4.7 mL) and was treated with Ph3P (1.0 equiv), 4-hydroxybenzenesulfonamide 3 (1.0 equiv), and DIAD (1.1 equiv) according to the general procedure b, previously reported. The reaction was stirred at r.t. until starting materials were consumed (TLC monitoring). The reaction was quenched with slush and extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with H2O (3 × 20 mL), dried over Na2SO4, filtered, and concentrated under vacuo, and the obtained residue was purified by silica gel column chromatography eluting with ethyl acetate/n-hexane 55% v/v, followed by crystallization in IPA to afford the titled compound 4b as a white solid.
tert-Butyl 3-(4-sulfamoylphenoxy)-propylcarbamate 4b:
21% yield, silica gel TLC Rf 0.35 (ethyl acetate/n-hexane 55% v/v); mp 134–135 °C; δH (400 MHz, DMSO-d6) 1.41 (9H, s), 1.88 (2H, pent, J 6.4), 3.10 (2h, q, J 6.4), 4.08 (2H, t, J, 6.4), 6.95 (1H, t, J 6.4, exchange with D2O, NH), 7.09 (2H, d, J 8.8), 7.23 (2H, s, exchange with D2O, SO2NH2), 7.77 (2H, d, J 8.8); δC (100 MHz, DMSO-d6) 29.2, 30.0, 37.7, 66.6, 78.5, 115.3, 128.6, 137.0, 156.6, 161.9. Elemental analysis: calcd C 50.89, H 6.71, N 8.48, S 9.70; found C 51.09, H 7.01, N 8.64, S 9.54; m/z (ESI negative) 329.40 [M – H]−.
Synthesis of tert-Butyl 4-(4-Sulfamoylphenoxy)-butylcarbamate 4c

tert-Butyl 4-hydroxybutylcarbamate 2c (1.11 g, 1.0 equiv) was dissolved in dry THF (10.0 mL) and was treated with Ph3P (1.0 equiv), 4-hydroxybenzenesulfonamide 3 (1.0 equiv), and DIAD (1.1 equiv) according to the general procedure b, previously reported. The reaction was stirred at r.t. until starting materials were consumed (TLC monitoring). The reaction was quenched with slush and extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with H2O (3 × 20 mL), dried over Na2SO4, filtered, and concentrated under vacuo, and the obtained residue was purified by silica gel column chromatography eluting with 60% v/v ethyl acetate/n-hexane to afford the titled compound 4c as a white solid.
tert-Butyl 4-(4-Sulfamoylphenoxy)-butylcarbamate 4c:
22% yield, silica gel TLC Rf 0.40 (ethyl acetate/n-hexane 60% v/v); mp 93–94 °C; δH (400 MHz, DMSO-d6) 1.41 (9H, s), 1.56 (2H, m), 1.74 (2H, m), 3.10 (2h, q, J 6.5), 4.08 (2H, t, J 6.5), 6.95 (1H, t, J 5.2, exchange with D2O, NH), 7.09 (2H, d, J 8.8), 7.23 (2H, s, exchange with D2O, SO2NH2), 7.77 (2H, d, J 8.8); δC (100 MHz, DMSO-d6) 26.9, 27.0, 29.2, 30.2, 68.6, 78.4, 115.4, 128.6, 137.0, 156.6, 162.0; Elemental analysis: calcd C 52.31, H 7.02, N 8.13, S 9.31; found C 52.65, H 6.76, N 8.01, S 8.91; m/z (ESI negative) 343.17 [M – H]−.
Synthesis of tert-Butyl 5-(4-Sulfamoylphenoxy)-pentylcarbamate 4d

tert-Butyl 5-hydroxypentylcarbamate 2d (1.2 g, 1.0 equiv) was dissolved in dry THF (9.0 mL) and was treated with Ph3P (1.0 equiv), 4-hydroxybenzenesulfonamide 3 (1.0 equiv), and DIAD (1.1 equiv) according to the general procedure b, previously reported. The reaction was stirred at r.t. until starting materials were consumed (TLC monitoring). The reaction was quenched with slush and extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with H2O (3 × 20 mL), dried over Na2SO4, filtered, and concentrated under vacuo, and the obtained residue was purified by silica gel column chromatography, eluting with 60% v/v ethyl acetate/n-hexane to afford the titled compound 4d as a white solid.
tert-Butyl 5-(4-Sulfamoylphenoxy)-pentylcarbamate 4d:
27% yield, silica gel TLC Rf 0.42(ethyl acetate/n-hexane 60% v/v); mp 95–96 °C; δH (400 MHz, DMSO-d6) 1.37–1.51 (13H, m), 1.73 (2H, pent, J 6.8), 2.94 (2H, q, J 6.4), 4.04 (2H, t, J 6.4), 6.80 (1H, t, J 5.7, exchange with D2O, NH), 7.08 (2H, d, J 8.8), 7.20 (2H, s, exchange with D2O, SO2NH2), 7.75 (2H, d, J 8.8); δC (100 MHz, DMSO-d6) 23.6, 29.1, 29.2, 30.1, 40.6, 68.7, 78.2, 115.3, 128.6, 136.9, 156.5, 162.0; Elemental analysis: calcd C 53.61, H 7.31, N 7.82, S 8.95; found C 53.50, H 7.01, N 7.87, S 8.69; m/z (ESI negative) 357.60 [M – H]−.
Synthesis of tert-Butyl 6-(4-Sulfamoylphenoxy)-hexylcarbamate 4e

tert-Butyl 6-hydroxyhexylcarbamate 2e (1.37 g, 1.0 equiv) was dissolved in dry THF (9.0 mL) and was treated with Ph3P (1.0 equiv), 4-hydroxybenzenesulfonamide 3 (1.0 equiv), and DIAD (1.1 equiv) according to the general procedure b, previously reported. The reaction was stirred at r.t. until starting materials were consumed (TLC monitoring). The reaction was quenched with slush and extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with H2O (3 × 20 mL), dried over Na2SO4, filtered, and concentrated under vacuo, and the obtained residue was purified by silica gel column chromatography, eluting with 50% v/v ethyl acetate/n-hexane to afford the titled compound 4e as a white solid.
tert-Butyl 6-(4-Sulfamoylphenoxy)-hexylcarbamate 4e:
42% yield; silica gel TLC Rf 0.35 (ethyl acetate/n-hexane 50% v/v); 101–102 °C; δH (400 MHz, DMSO-d6) 1.41 (17H, m), 1.75 (2H, q, J 8.0), 2.94 (2H, q, J 8.0), 4.08 (1H, t, J 5.2, exchange with D2O, OH), 6.80 (1H, brt, exchange with D2O, NH), 7.11 (2H, d, J 8), 7.23 (2H, s, exchange with D2O, SO2NH2), 7.77 (2H, d, J 8); δC (100 MHz, DMSO-d6) 23.6, 24.8, 26.4, 29.2, 30.1, 39.5, 68.3, 78.2, 115.4, 128.7, 136.9, 156.3, 162.1. Elemental analysis: calcd C 54.82, H 7.58, N 7.52, S 8.61; found C 54.66, H 7.58, N 7.36, S 8.56; m/z (ESI negative) 371.25 [M – H]−.
Synthesis of 2-(4-Sulfamoylphenoxy)-ethylammonium Trifluoroacetate Salt 5a

TFA (7.0 equiv) was added to a stirring mixture of tert-butyl 2-(4-sulfamoylphenoxy)-ethylcarbamate 4a (0.5 g, 1.0 equiv) in 10 mL of DCM. The reaction was stirred at r.t. according to the general procedure c, previously reported until starting material was consumed (TLC monitoring). The solvents were evaporated under vacuo, and the obtained residue was triturated with diethyl ether and dried under vacuo to afford the titled compound 5a as a white solid.
2-(4-Sulfamoylphenoxy)-ethylammonium Trifluoroacetate Salt 5a:
88% yield, mp 142–143 °C; δH (400 MHz, DMSO-d6) 3.30 (2H, t, J 5.2), 4.27 (2H, t, J 5.2), 7.17 (2H, d, J, 8.8), 7.29 (2H, s, exchange with D2O, SO2NH2), 7.81 (2H, d, J 8.8), 8.02 (3H, brt, exchange with D2O, NH3); δC (100 MHz, DMSO-d6) 39.2, 65.7, 115.7, 118.2 (d, J1C–F 299), 128.7, 137.8, 159.4 (q, J2C–F 31), 161.2; δF (376 MHz, DMSO-d6) −73.5 (3F, s). Elemental analysis: calcd C 36.37, H 3.97, N 8.48, S 9.71; found C 36.76, H 3.72, N 8.08, S 10.02; m/z (ESI positive) 217.08 [M – CF3COO–]+.
Synthesis of 3-(4-Sulfamoylphenoxy)-propylammonium Trifluoroacetate Salt 5b.

tert-Butyl 3-(4-sulfamoylphenoxy)-propylcarbamate 4b (0.8 g, 1.0 equiv) was dissolved in 1,4-dioxane (10.0 mL), followed by addition of TFA (50.0 equiv). The reaction was treated according to the general procedure c, previously reported (TLC monitoring). The solvents were evaporated under vacuo, and the obtained residue was triturated with diethyl ether and dried under vacuo to afford the titled compound 5b as a white solid.
3-(4-Sulfamoylphenoxy)-propylammonium Trifluoroacetate Salt 5b:
91% yield, mp 129–130 °C; δH (400 MHz, DMSO-d6) 2.05 (2H, pent, J 6.4), 3.00 (2H, m,), 4.17 (2H, t, J 6.0), 7.12 (2H, d, J, 8.8), 7.26 (2H, s, exchange with D2O, SO2NH2), 7.78 (3H, brt, exchange with D2O, −NH3), 7.79 (2H, d, J 8.8); δC (100 MHz, DMSO-d6) 27.6, 37.1, 66.0, 115.43, 118.2 (d, J1C–F 297), 128.6, 137.3, 159.4 (q, J2C–F 31) 161.6; δF (376 MHz, DMSO-d6) −73.64 (3F, s), Elemental analysis: calcd C 38.37, H 4.39, N 8.14, S 9.31; found C 38.42, H 4.60, N 7.95, S 9.66; m/z (ESI positive) 231.30 [M – CF3COO–]+.
Synthesis of 4-(4-Sulfamoylphenoxy)-butylammonium Trifluoroacetate Salt 5c

tert-Butyl 4-(4-sulfamoylphenoxy)-butylcarbamate 4c (0.38 g, 1.0 equiv) was dissolved in DCM (5.0 mL), followed by addition of TFA (12.0 equiv). The reaction was treated according to the general procedure c previously reported (TLC monitoring). The solvents were evaporated under vacuo, and the obtained residue was crystallized from IPA to afford the titled compound 5c as a white solid.
4-(4-Sulfamoyl-phenoxy)-butylammonium Trifluoroacetate Salt 5c:
50% yield; mp 110–111 °C; δH (400 MHz, DMSO-d6) 1.74 (2H, m), 1.83 (2H, m), 2.90 (2H, t, J 7.4), 4.11 (2H, t, J 6.0), 7.10 (2H, d, J 8.0), 7.25 (2H, s, exchange with D2O, SO2NH2), 7.79 (5H, m, Ar–H, NH3+); δC (100 MHz, DMSO-d6) 24.8, 26.4, 39.5, 68.3, 115.4, 118.2 (d, J1C–F 298) 128.6, 137.1, 159.15 (q, J2C–F 31), 161.8; δF (376 MHz, DMSO-d6) −73.4 (3F, s). Elemental analysis: calcd C 40.22, H 4.78, N 7.82, S 8.95; found C 40.30, H 4.63, N 7.72, S 9.56; m/z (ESI positive) 245.17 [M – CF3COO–]+.
Synthesis of 5-(4-Sulfamoylphenoxy)-pentylammonium Trifluoroacetate Salt 5d

tert-Butyl 5-(4-sulfamoylphenoxy)-pentylcarbamate 4d (0.46 g, 1.0 equiv) was dissolved in DCM (5.0 mL), followed by addition of TFA (7.0 equiv). The reaction was treated according to the general procedure c, previously reported (TLC monitoring). The solvents were evaporated under vacuo, and the obtained residue was triturated from diethyl ether to afford the titled compound 5d as a white solid.
5-(4-Sulfamoylphenoxy)-pentylammonium Trifluoroacetate Salt 5d:
90% yield; mp 121–122 °C; δH (400 MHz, DMSO-d6) 1.50 (2H, m), 1.63 (2H, m), 1.79 (2H, m), 2.85 (2H, m), 4.09 (2H, t, J 6.0), 7.10 (2H, d, J 9.0), 7.24 (2H, s, exchange with D2O, SO2NH2), 7.71 (3H, brt, exchange with D2O, NH3+), 7.77 (2H, d, J 9.0); δC (100 MHz, DMSO-d6) 23.4, 27.6; 28.9, 39.6, 68.6, 115.3, 118.1 (d, J1C–F 298), 128.6, 137.0, 159.1 (q, J2C–F 31), 161.9; δF (376 MHz, DMSO-d6) −73.5 (3F, s). Elemental analysis: calcd C 41.93, H 5.14, N 7.52, S 8.61; found C 41.73, H 5.23, N 7.25, S 8.34; m/z (ESI positive) 259.17 [M-CF3COO–]+.
Synthesis of 6-(4-Sulfamoylphenoxy)-hexylammonium Trifluoroacetate Salt 5e

tert-Butyl 6-(4-sulfamoylphenoxy)-hexylcarbamate 4e (0.10 g, 1.0 equiv) was dissolved in DCM (1.8 mL), followed by addition of TFA (5.0 equiv). The reaction was treated according to the general procedure c, previously reported (TLC monitoring). The solvents were evaporated under vacuo, and the obtained residue was crystallized from IPA to afford the titled compound 5e as a white solid.
6-(4-Sulfamoylphenoxy)-hexylammonium Trifluoroacetate Salt 5e:
30% yield; mp 122–123 °C; δH (400 MHz, DMSO-d6) 1.45 (4H, m), 1.59 (2H, pent, J 7.5), 1.76 (2H, pent, J 7.5), 2.82 (2H, t, J 7.5), 4.08 (2H, t, J 6.5), 7.11 (2H, d, J 9.0), 7.25 (2H, brs, exchange with D2O, SO2NH2), 7.63 (3H, brt, exchange with D2O, NH3+), 7.77 (2H, d, J 9.0); δC (100 MHz, DMSO-d6) 25.9, 26.4, 27.9, 30.7, 39.7, 68.7, 115.4, 118.1 (d, J1C–F 298), 128.6, 137.0, 159.3 (q, J2C–F 31), 162.0; δF (376 MHz, DMSO-d6) −73.5 (1F, s). Elemental analysis: calcd C 43.52, H 5.48, N 7.25, S 8.30; found C 43.19, H 5.24, N 6.95, S 8.16; m/z (ESI positive) 273.40 [M – CF3COO–]+.
General Procedure for Synthesis of Fluorescent Tagged Sulfonamides 7a–c
The O-alkylbenzenesulfonamide salt 5 (1.0 equiv) and 2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)-5-isothiocyanatobenzoic acid 6 (1.0 equiv) were poured into a two-neck flask, and dry DMA (1.0 mL) was added, followed by addition of TEA (1.5 equiv). The reaction was stirred at r.t. until starting materials were consumed (TLC monitoring). It was then quenched with slush and a 6 M aqueous hydrochloric acid solution and then extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with H2O (3 × 20 mL), dried over Na2SO4, filtered, and concentrated in vacuo to afford the titled compound 7 as an orange powder.
Synthesis of 2-(6-Hydroxy-3-oxo-3H-xanthen-9-yl)-5-{3-[2-(4-sulfamoylphenoxy)-ethyl]-thioureido}-benzoic Acid 7a

2-(4-Sulfamoylphenoxy)-ethylammonium trifluoroacetate salt 5a (50 mg, 1.0 equiv) and 2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)-5-isothiocyanatobenzoic acid 6 (1.0 equiv) were treated according to the general procedure previously reported afford the titled compound 7a as an orange powder.
2-(6-Hydroxy-3-oxo-3H-xanthen-9-yl)-5-{3-[2-(4-sulfamoylphenoxy)-ethyl]-thioureido}-benzoic Acid 7a
76% yield; silica gel TLC Rf 0.10 (MeOH/DCM 10% v/v); mp 172–173 °C (dec.); δH (400 MHz, DMSO-d6) 3.98 (2H, brt), 4.32 (2H, t, J 5.2), 6.61–6.66 (4H, m), 6.71 (2H, d, J 2.0), 7.23 (5H, m, 3H Ar–H, 2H exchange with D2O, SO2NH2), 7.76 (1H, exchange with D2O, NH), 7.80 (2H Ar–H, d, J 8.8), 8.28 (1H, s), 8.35 (1H, exchange with D2O, NH), 10.11 (1H, exchange with D2O, NH), 10.15 (2H, exchange with D2O, OH); δC (100 MHz, DMSO-d6) 44.0, 67.1, 84.1, 103.2, 110.7, 113.6, 115.5, 117.6, 125.1, 127.6, 128.7, 130.0, 130.6, 137.4, 142.1, 148.3, 152.9, 160.5, 161.7, 169.5, 181.8 (C=S). Elemental analysis: calcd C 57.51, H 3.83, N 6.94, S 10.59; found C 57.56, H 4.03, N 6.68, S 10.84; m/z (ESI positive) 606.50 [M + H]+.
Synthesis of 2-(6-Hydroxy-3-oxo-3H-xanthen-9-yl)-5-{3-[3-(4-sulfamoylphenoxy)-propyl]-thioureido}-benzoic Acid 7b

3-(4-Sulfamoylphenoxy)-propylammonium trifluoroacetate salt 5b (50 mg, 1.0 equiv) and 2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)-5-isothiocyanatobenzoic acid 6 (1.0 equiv) were treated according to the general procedure e, previously reported, to afford the titled compound 7b as an orange powder.
2-(6-Hydroxy-3-oxo-3H-xanthen-9-yl)-5-{3-[3-(4-sulfamoylphenoxy)-propyl]-thioureido}-benzoic Acid 7b
83% yield; silica gel TLC Rf 0.40 (MeOH/DCM 20% v/v); mp 185–186 °C (dec); δH (400 MHz, DMSO-d6) 2.11 (2H, t, J 6.4), 3.72 (2H, brt), 4.18 (2H, t; J 5.8), 6.62 (4H, m), 6.71 (2H, s), 7.15 (2H, d, J 8.8), 7.23 (2H, s, exchange with D2O, SO2NH2), 7.79 (2H, d, J 8.8), 8.26 (1H, exchange with D2O, NH), 10.00 (1H, exchange with D2O, NH), 10.15 (2H, exchange with D2O, OH); δC (100 MHz, DMSO-d6) 29.0, 41.9, 66.9, 85.2, 103.3, 110.8, 113.7, 115.5, 117.7, 125.1, 127.6, 128.7, 130.1, 130.6, 137.2, 142.4, 148.0, 153.0, 160.6, 162.0, 169.5, 181.6 (C=S); m/z (ESI positive) 621.0 [M + 2H]2+.
Synthesis of 2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)-5-{3-[4-(4-sulfamoylphenoxy)-butyl]-thioureido}-benzoic Acid 7c

4-(4-Sulfamoylphenoxy)-butylammonium trifluoroacetate salt 5c (50 mg, 1.0 equiv)and 2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)-5-isothiocyanatobenzoic acid 6 (1.0 equiv) were treated according to the general procedure e, previously reported, to afford the titled compound 7c as an orange powder.
2-(6-Hydroxy-3-oxo-3H-xanthen-9-yl)-5-{3-[4-(4-sulfamoylphenoxy)-butyl]-thioureido}-benzoic Acid 7c
79% yield; silica gel TLC Rf 0.70 (MeOH/DCM 20% v/v); mp 200–201 °C (dec); δH (400 MHz, DMSO-d6) 1.77–1.87 (4H, m), 3.64 (2H, brs), 4.14 (2H, t, J 6.1), 6.60–6.66 (4H, m), 6.71 (2H, d, J 2.2), 7.13 (2H, d, J 8.8), 7.22 (3H, d, J 8.3, Ar–H, 2H exchange with D2O, SO2NH2), 7.77 (2H, d, J 8.8), 8.30 (2H, s, 1H exchange with D2O, NH), 10.09 (3H, brs, exchange with D2O, 2xOH, NH); δC (100 MHz, DMSO-d6) 26.4, 26.9, 45.4, 68.5, 85.3, 103.2, 110.0, 113.7, 115.4, 117.1, 126.0, 128.1, 128.7, 129.8, 130.0, 137.1, 141.9, 148.9, 153.1, 160.8, 161.9, 168.6, 182.6 (C=S); m/z (ESI positive) 634.5 [M + H]+.
General Procedure for Synthesis of Florescent Tagged O-Alkyl Benzenesulfonamides 9a,b
Compound 5 (1.0 equiv) and [9-(2-carboxy-4-isothiocyanatophenyl)-6-dimethylaminoxanthen-3-ylidene]dimethylammonium chloride 8 (1.0 equiv) were poured into a two-neck flask, and dry DMA (1.0 mL) was added, followed by addition of TEA (1.5 equiv). The reaction was stirred at r.t. until starting materials were consumed (TLC monitoring), and then it was quenched with slush and a 6 M aqueous hydrochloric acid solution. The precipitate formed was centrifuged, collected, dried under vacuo, washed with diethyl ether (3 × 10 mL), and dried under vacuo to afford the titled compound 9 as a red powder.
Synthesis of 9-(2-Carboxy-4-{3-[2-(4-sulfamoylphenoxy)ethyl]thioureido}phenyl)-6-dimethylaminoxanthen-3-ylidene]-dimethylammonium Chloride 9a

2-(4-Sulfamoyl-phenoxy)ethylammonium trifluoroacetate salt 5a (6.9 mg, 1.0 equiv) and [9-(2-carboxy-4-isothiocyanatophenyl)-6-dimethylaminoxanthen-3-ylidene]-dimethylammonium chloride 8 (1.0 equiv) were treated according to the previously reported procedure to afford the titled compound 9a as a red powder.
9-(2-Carboxy-4-{3-[2-(4-sulfamoylphenoxy)-ethyl]-thioureido}-phenyl)-6-dimethylaminoxanthen-3-ylidene]-dimethylammonium Chloride 9a
70% yield; silica gel TLC Rf 0.40 (MeOH/DCM 20% v/v); mp 177–178 °C (dec); δH (400 MHz, DMSO-d6) 3.30 (12H, s), 3.99 (2H, brt), 4.33 (2H, t, J 5.4), 7.00 (4H, m), 7.14 (3H, m), 7.21 (2H, d, J 8.8), 7.26 (2H, s, exchange with D2O, SO2NH2), 7.41 (1H, m), 7.72 (1H, d, J 8.8), 7.81 (2H, d, J 8.8), 8.46 (2H, brs, 1H, exchange with D2O, NH), 10.38 (1H, exchange with D2O, NH); δC (100 MHz, DMSO-d6) 35.1, 41.5, 43.9, 67.1, 97.2, 114.1, 115.3, 122.6, 124.5, 125.9, 128.7, 131.7, 137.5, 142.6, 157.6, 160.2, 161.4, 161.6, 166.8, 167.1 (C=O), 181.5 (C=S); m/z (ESI positive) 660.50 [M – Cl]+.
Synthesis of 9-(2-Carboxy-4-{3-[3-(4-sulfamoyl-phenoxy)-propyl]-thioureido}-phenyl)-6-dimethylaminoxanthen-3-ylidene]-dimethylammonium Chloride 9b

3-(4-Sulfamoylphenoxy)-propylammonium trifluoroacetate salt 5b (7.2 mg, 1.0 equiv) and [9-(2-carboxy-4-isothiocyanatophenyl)-6-dimethylamino-xanthen-3-ylidene]-dimethylammonium chloride 8 (1.0 equiv) were treated according to the previously reported procedure to afford the titled compound 9b as a red powder.
9-(2-Carboxy-4-{3-[3-(4-sulfamoyl-phenoxy)-propyl]-thioureido}-phenyl)-6-dimethylamino-xanthen-3-ylidene]-dimethylammonium Chloride 9b
74% yield; silica gel TLC Rf 0.30 (MeOH/DCM 20% v/v); mp 192–193 °C (dec); δH (400 MHz, DMSO-d6) 3.30 (12H, s), 4.13 (2H, t, J 6.2), 4.21 (2H, t, J 6.2), 6.98 (3H, m), 7.08–7.25 (8H, m), 7.40 (1H, d, J 8.8), 7.75 (1H, d, J 8.8), 7.79 (2H, d, J 8.8), 7.87 (1H, brs), 7.94 (1H, d, J 8.8), 8.58 (1H, brt, exchange with D2O, NH), 10.51 (1H, brs, exchange with D2O, NH), 10.65 (1H, s); δC (100 MHz, DMSO-d6) 28.8, 35.0, 41.4, 41.5, 66.8, 97.2, 114.2, 115.5, 122.6, 124.2, 127.0, 128.6, 131.7, 137.1, 142.6, 157.7, 160.8, 161.9, 161.6, 166.8, 167.1 (C=O), 181.5 (C=S); m/z (ESI positive) 674.42 [M – Cl]+.
X-ray Crystallography
Co-Crystallization
Two microliters of a 600 mM concentration of 4c was added to a 500 μL 1.6 M sodium citrate, 50 mM Tris–HCL pH 7.8 reservoir solution. One microliter of this reservoir solution was added to 5 μL of CA II at a final concentration of 10 mg/mL so that the final drug concentration was at 0.24 mM. Hanging drops were set up, and crystals were seen within 5 days. This was repeated for 5c.
Diffraction Data and Collection
Diffraction data for CA II-4c and CA II-5c complexes were collected on an in-house R-Axis IV+2 image plate detector using a RU-H3R rotating Cu anode (Kα = 1.5418 Å) operating at 50 kV and 22 mA. Images were collected every 1° with an exposure time of 5 min at a detector distance of 100 mm. The crystal data were integrated, merged and scaled using HKL2000.33
Structure Determination
Phasing was carried out in the PHENIX34 suite of programs using the Auto Molecular Replacement procedure to obtain the initial phases using a previously solved HCA II structure with water molecules removed (PDB code 3KS3).35 The graphics program Coot36 was used to view the electron density map, and the structure was adjusted on the basis of the calculated electron density. Topology files of the inhibitors were generated using the PRODRG37 server, and these files were used to model the drug into the density generated. Refinement was continued using PHENIX.REFINE until the Rcrys and Rfree were minimized. The geometric restraints of the final model were analyzed using PROCHECK.38 The data diffraction and final model refinement statistics are summarized in Table 2.
Normotensive Rabbit IOP Lowering Studies
Male New Zeland albino rabbits weighing 1500–2000 g were used in these studies. Animals were anaesthetized using Zoletil (tiletamine chloride + zolazepam chloride, 3 mg/kg body weight, i.m. injection) and injected with 0.05 mL hypertonic saline solution (5% in distilled water) into the vitreous of both eyes. IOP was measured by using a digital tonometer (Tomo-Pen Avia Tonometer, Reichert Inc. Depew, NY 14043, USA) prior to hypertonic saline injection (basal) at 1, 2, and 4 h after administration of the drug.14a Vehicle (phosphate buffer, pH 7.0 plus DMSO 2%) or drugs were instilled immediately after the injection of hypertonic saline into the conjunctive pocket. Eyes were randomly assigned to different groups. Four different animals were used for the tested compounds. Experiments with animals were conducted in agreement with current ethical guidelines and norms approved by the ethical committee of our university.
Acknowledgments
This research was financed by two EU grants of the seventh framework program (Metoxia and Dynano projects to AS and CTS) and the National Institutes of Health Grant CA165284 (to R.M.).
Glossary
Nonstandard Abbreviations
- CA
carbonic anhydrase
- CAI
CA inhibitor
- DCM
dichloromethane
- DIAD
diisopropyl azodicarboxylate
- DIPEA
diisopropylethylamine
- DMA
dimethylacetamide
- IOP
intraocular pressure
- IPA
propan-2-ol
- KI
inhibition constant
- SAR
structure–activity relationship
- TFA
trifluoroacetic acid
Accession Codes
4RFC and 4RFD.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- a Krishnamurthy V. M.; Kaufman G. K.; Urbach A. R.; Gitlin I.; Gudiksen K. L.; Weibel D. B.; Whitesides G. M. Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein-ligand binding. Chem. Rev. 2008, 108, 946–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Smeulders M. J.; Barends T. R.; Pol A.; Scherer A.; Zandvoort M. H.; Udvarhelyi A.; Khadem A. F.; Menzel A.; Hermans J.; Shoeman R. L.; Wessels H. J.; van den Heuvel L. P.; Russ L.; Schlichting I.; Jetten M. S.; Op den Camp H. J. Evolution of a new enzyme for carbon disulphide conversion by an acidothermophilic archaeon. Nature 2011, 478, 412–416. [DOI] [PubMed] [Google Scholar]; c Alterio V.; Di Fiore A.; D’Ambrosio K.; Supuran C. T.; De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms?. Chem. Rev. 2012, 112, 4421–4468. [DOI] [PubMed] [Google Scholar]; d Supuran C. T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery 2008, 7, 168–181. [DOI] [PubMed] [Google Scholar]
- a Aggarwal M.; Boone C. D.; Kondeti B.; McKenna R. Structural annotation of human carbonic anhydrases. J. Enzyme Inhib. Med. Chem. 2013, 28, 267–277. [DOI] [PubMed] [Google Scholar]; b Gieling R. G.; Parker C. A.; De Costa L. A.; Robertson N.; Harris A. L.; Stratford I. J.; Williams K. J. Inhibition of carbonic anhydrase activity modifies the toxicity of doxorubicin and melphalan in tumour cells in vitro. J. Enzyme Inhib. Med. Chem. 2013, 28, 360–369. [DOI] [PubMed] [Google Scholar]; c Rummer J. L.; McKenzie D. J.; Innocenti A.; Supuran C. T.; Brauner C. J. Root effect hemoglobin may have evolved to enhance general tissue oxygen delivery. Science 2013, 340, 1327–1329. [DOI] [PubMed] [Google Scholar]; d Ebbesen P.; Pettersen E. O.; Gorr T. A.; Jobst G.; Williams K.; Kienninger J.; Wenger R. H.; Pastorekova S.; Dubois L.; Lambin P.; Wouters B. G.; Supuran C. T.; Poellinger L.; Ratcliffe P.; Kanopka A.; Görlach A.; Gasmann M.; Harris A. L.; Maxwell P.; Scozzafava A. Taking advantage of tumor cell adaptations to hypoxia for developing new tumor markers and treatment strategies. J. Enzyme Inhib. Med. Chem. 2009, 24S11–39. [DOI] [PubMed] [Google Scholar]; e Supuran C. T. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnologic use for CO2 capture. J. Enzyme Inhib. Med. Chem. 2013, 28, 229–230. [DOI] [PubMed] [Google Scholar]
- a Neri D.; Supuran C. T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discovery 2011, 10, 767–777. [DOI] [PubMed] [Google Scholar]; b Smith K. S.; Jakubzick C.; Whittam T. S.; Ferry J. G. Carbonic anhydrase is an ancient enzyme widespread in prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 15184–15189. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Swietach P.; Patiar S.; Supuran C. T.; Harris A. L.; Vaughan-Jones R. D. The role of carbonic anhydrase 9 in regulating extracellular and intracellular pH in 3-D tumor-cell growths. J. Biol. Chem. 2009, 284, 20299–20310. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Supuran C. T. Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 2012, 27, 759–772. [DOI] [PubMed] [Google Scholar]
- a Xu Y.; Feng L.; Jeffrey P. D.; Shi Y.; Morel F. M. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008, 452, 56–61. [DOI] [PubMed] [Google Scholar]; b Supuran C. T. Bacterial carbonic anhydrases as drug targets: towards novel antibiotics?. Front. Pharmacol. 2011, 2, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Capasso C.; Supuran C. T. Antiinfective carbonic anhydrase inhibitors: A patent and literature review. Expert Opin. Ther. Pat. 2013, 23, 693–704. [DOI] [PubMed] [Google Scholar]; d Hirohashi N.; Alvarez L.; Shiba K.; Fujiwara E.; Iwata Y.; Mohri T.; Inaba K.; Chiba K.; Ochi H.; Supuran C. T.; Kotzur N.; Kakiuchi Y.; Kaupp U. B.; Baba S. A. Sperm from sneaker male squids exhibit chemotactic swarming to CO2. Curr. Biol. 2013, 23, 775–781. [DOI] [PubMed] [Google Scholar]
- a Cottier F.; Leewattanapasuk W.; Kemp L. R.; Murphy M.; Supuran C. T.; Kurzai O.; Mühlschlegel F. A. Carbonic anhydrase regulation and CO2 sensing in the fungal pathogen Candida glabrata involves a novel Rca1p ortholog. Bioorg. Med. Chem. 2013, 21, 1549–1554. [DOI] [PubMed] [Google Scholar]; b Schlicker C.; Hall R. A.; Vullo D.; Middelhaufe S.; Gertz M.; Supuran C. T.; Mühlschlegel F. A.; Steegborn C. Structure and inhibition of the CO2-sensing carbonic anhydrase Can2 from the pathogenic fungus Cryptococcus neoformans. J. Mol. Biol. 2009, 385, 1207–1220. [DOI] [PubMed] [Google Scholar]; c Moya A.; Tambutté S.; Bertucci A.; Tambutté E.; Lotto S.; Vullo D.; Supuran C. T.; Allemand D.; Zoccola D. Carbonic anhydrase in the scleractinian coral Stylophora pistillata: characterization, localization, and role in biomineralization. J. Biol. Chem. 2008, 283, 25475–25484. [DOI] [PubMed] [Google Scholar]
- a Capasso C.; Supuran C. T. Sulfa and trimethoprim-like drugs – antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J. Enzyme Inhib. Med. Chem. 2014, 29, 379–387. [DOI] [PubMed] [Google Scholar]; b Maresca A.; Vullo D.; Scozzafava A.; Manole G.; Supuran C. T. Inhibition of the β-class carbonic anhydrases from Mycobacterium tuberculosis with carboxylic acids. J. Enzyme Inhib. Med. Chem. 2013, 28, 392–396. [DOI] [PubMed] [Google Scholar]; c Maresca A.; Scozzafava A.; Vullo D.; Supuran C. T. Dihalogenated sulfanilamides and benzolamides are effective inhibitors of the three β-class carbonic anhydrases from Mycobacterium tuberculosis. J. Enzyme Inhib. Med. Chem. 2013, 28, 384–387. [DOI] [PubMed] [Google Scholar]; d Güzel-Akdemir Ö.; Akdemir A.; Pan P.; Vermelho A. B.; Parkkila S.; Scozzafava A.; Capasso C.; Supuran C. T. A class of sulfonamides with strong inhibitory action against the α-carbonic anhydrase from Trypanosoma cruzi. J. Med. Chem. 2013, 56, 5773–5781. [DOI] [PubMed] [Google Scholar]
- a Vullo D.; Del Prete S.; Osman S. M.; De Luca V.; Scozzafava A.; AlOthman Z.; Supuran C. T.; Capasso C. Sulfonamide inhibition studies of the γ-carbonic anhydrase from the oral pathogen Porphyromonas gingivalis. Bioorg. Med. Chem. Lett. 2014, 24, 240–244. [DOI] [PubMed] [Google Scholar]; b Vullo D.; Del Prete S.; Osman S. M.; De Luca V.; Scozzafava A.; AlOthman Z.; Supuran C. T.; Capasso C. Sulfonamide inhibition studies of the δ-carbonic anhydrase from the diatom Thalassiosira weissflogii. Bioorg. Med. Chem. Lett. 2014, 24, 275–279. [DOI] [PubMed] [Google Scholar]; c De Luca V.; Vullo D.; Scozzafava A.; Carginale V.; Rossi M.; Supuran C. T.; Capasso C. An α-carbonic anhydrase from the thermophilic bacterium Sulphurihydrogenibium azorense is the fastest enzyme known for the CO2 hydration reaction. Bioorg. Med. Chem. 2013, 1465–1469. [DOI] [PubMed] [Google Scholar]; d Supuran C. T. Carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1377–1378. [DOI] [PubMed] [Google Scholar]
- a Del Prete S.; De Luca V.; Scozzafava A.; Carginale V.; Supuran C. T.; Capasso C. Biochemical properties of a new α-carbonic anhydrase from the human pathogenic bacterium Vibrio cholerae. J. Enzyme Inhib. Med. Chem. 2014, 29, 23–27. [DOI] [PubMed] [Google Scholar]; b Maresca A.; Vullo D.; Scozzafava A.; Supuran C. T. Inhibition of the α- and β-carbonic anhydrases from the gastric pathogen Helycobacter pylori with anions. J. Enzyme Inhib. Med. Chem. 2013, 28, 388–391. [DOI] [PubMed] [Google Scholar]
- Del Prete S.; Vullo D.; Fisher G. M.; Andrews K. T.; Poulsen S. A.; Capasso C.; Supuran C. T. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum – the η-carbonic anhydrases. Bioorg. Med. Chem. Lett. 2014, 24, 4389–4396. [DOI] [PubMed] [Google Scholar]
- a Ridderstråle Y.; Fierke C. A.; Roush E. D.; Wistrand P. J. Localization of a protein inhibitor of carbonic anhydrase in pig tissues. Acta Physiol. Scand. 2002, 176, 27–31. [DOI] [PubMed] [Google Scholar]; b Durdagi S.; Vullo D.; Pan P.; Kähkönen N.; Määttä J. A.; Hytönen V. P.; Scozzafava A.; Parkkila S.; Supuran C. T. Protein–protein interactions: Inhibition of mammalian carbonic anhydrases I-XV by the murine inhibitor of carbonic anhydrase and other members of the transferrin family. J. Med. Chem. 2012, 55, 5529–5535. [DOI] [PubMed] [Google Scholar]
- a De Simone G.; Alterio V.; Supuran C. T. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin. Drug Discovery 2013, 8, 793–810. [DOI] [PubMed] [Google Scholar]; b Masini E.; Carta F.; Scozzafava A.; Supuran C. T. Antiglaucoma carbonic anhydrase inhibitors: A patent review. Expert Opin. Ther. Pat. 2013, 23, 705–716. [DOI] [PubMed] [Google Scholar]; c Supuran C. T. Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3467–3474. [DOI] [PubMed] [Google Scholar]
- a Arechederra R. L.; Waheed A.; Sly W. S.; Supuran C. T.; Minteer S. D. Effect of sulfonamides as selective carbonic anhydrase VA and VB inhibitors on mitochondrial metabolic energy conversion. Bioorg. Med. Chem. 2013, 21, 1544–1548. [DOI] [PubMed] [Google Scholar]; b Scozzafava A.; Supuran C. T.; Carta F. Antiobesity carbonic anhydrase inhibitors: A literature and patent review. Expert Opin. Ther. Pat. 2013, 23, 725–735. [DOI] [PubMed] [Google Scholar]
- a Aggarwal M.; McKenna R. Update on carbonic anhydrase inhibitors: a patent review (2008–2011). Expert Opin. Ther. Pat. 2012, 22, 903–915. [DOI] [PubMed] [Google Scholar]; b Carta F.; Supuran C. T. Diuretics with carbonic anhydrase inhibitory action: A patent and literature review (2005–2013). Expert Opin. Ther. Pat. 2013, 23, 681–691. [DOI] [PubMed] [Google Scholar]; c Thiry A.; Dognè J. M.; Supuran C. T.; Masereel B. Anticonvulsant sulfonamides/sulfamates/sulfamides with carbonic anhydrase inhibitory activity: drug design and mechanism of action. Curr. Pharm. Des. 2008, 14, 661–671. [DOI] [PubMed] [Google Scholar]; d Thiry A.; Dognè J. M.; Masereel B.; Supuran C. T. Carbonic anhydrase inhibitors as anticonvulsant agents. Curr. Top. Med. Chem. 2007, 7, 855–864. [DOI] [PubMed] [Google Scholar]
- a Fabrizi F.; Mincione F.; Somma T.; Scozzafava G.; Galassi F.; Masini E.; Impagnatiello F.; Supuran C. T. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J. Enzyme Inhib. Med. Chem. 2012, 27, 138–147. [DOI] [PubMed] [Google Scholar]; b Carta F.; Supuran C. T.; Scozzafava A. Novel therapies for glaucoma: a patent review 2007–2011. Expert Opin. Ther. Pat. 2012, 22, 79–88. [DOI] [PubMed] [Google Scholar]; c Aggarwal M.; Kondeti B.; McKenna R. Anticonvulsant/antiepileptic carbonic anhydrase inhibitors: a patent review. Expert Opin. Ther. Pat. 2013, 23, 717–724. [DOI] [PubMed] [Google Scholar]
- a Krall N.; Pretto F.; Decurtins W.; Bernardes G. J. L.; Supuran C. T.; Neri D. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew. Chem., Int. Ed. 2014, 53, 4231–4235. [DOI] [PubMed] [Google Scholar]; b Monti S. M.; Supuran C. T.; De Simone G. Anticancer carbonic anhydrase inhibitors: A patent review (2008–2013). Expert Opin. Ther. Pat. 2013, 23, 737–749. [DOI] [PubMed] [Google Scholar]; c Ward C.; Langdon S. P.; Mullen P.; Harris A. L.; Harrison D. J.; Supuran C. T.; Kunkler I. New strategies for targeting the hypoxic tumour microenvironment in breast cancer. Cancer Treat. Rev. 2013, 39, 171–179. [DOI] [PubMed] [Google Scholar]; d Touisni N.; Maresca A.; McDonald P. C.; Lou Y.; Scozzafava A.; Dedhar S.; Winum J. Y.; Supuran C. T. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J. Med. Chem. 2011, 54, 8271–8277. [DOI] [PubMed] [Google Scholar]
- Supuran C. T.; McKenna R.. Carbonic anhydrase inhibitors drug design. In Carbonic Anhydrase: Mechanism, regulation, Links to Disease, and Industrial Applications; McKenna R., Frost S., Eds.; Springer Verlag: Heidelberg, 2014; pp 291–323 (. [DOI] [PubMed] [Google Scholar]; Subcell. Biochem. 2014, 75, 291 - 323. [DOI] [PubMed]
- Pacchiano F.; Carta F.; McDonald P. C.; Lou Y.; Vullo D.; Scozzafava A.; Dedhar S.; Supuran C. T. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. [DOI] [PubMed] [Google Scholar]
- a Wilkinson B. L.; Bornaghi L. F.; Houston T. A.; Innocenti A.; Vullo D.; Supuran C. T.; Poulsen S.-A. Carbonic anhydrase inhibitors: inhibition of isozymes I, II, and IX with triazole-linked O-glycosides of benzene sulfonamides. J. Med. Chem. 2007, 50, 1651–1657. [DOI] [PubMed] [Google Scholar]; b Wilkinson B. L.; Bornaghi L. F.; Houston T. A.; Innocenti A.; Supuran C. T.; Poulsen S.-A. A novel class of carbonic anhydrase inhibitors: glycoconjugate benzene sulfonamides prepared by “click-tailing”. J. Med. Chem. 2006, 49, 6539–6548. [DOI] [PubMed] [Google Scholar]; c Scozzafava A.; Menabuoni L.; Mincione F.; Briganti F.; Mincione G.; Supuran C. T. Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: Is the tail more important than the ring?. J. Med. Chem. 1999, 42, 2641–2650. [DOI] [PubMed] [Google Scholar]; d Winum J.-Y.; Poulsen S.-A.; Supuran C. T. Therapeutic applications of glycosidic carbonic anhydrase inhibitors. Med. Res. Rev. 2009, 29, 419–435. [DOI] [PubMed] [Google Scholar]; e Vomasta D.; Innocenti A.; König B.; Supuran C. T. Carbonic anhydrase inhibitors: two-prong versus mono-prong inhibitors of isoforms I, II, IX, and XII exemplified by photochromic cis-1,2-α-dithienylethene derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 1283–1286. [DOI] [PubMed] [Google Scholar]
- a Supuran C. T.; Scozzafava A.; Casini A.. Development of sulfonamide carbonic anhydrase inhibitors (CAIs). In Carbonic anhydrase – Its Inhibitors and Activators; Supuran C.T., Scozzafava A., Conway J., Eds.; CRC Press: Boca Raton (FL), 2004; pp 67–147. [Google Scholar]; b Vernier W.; Chong W.; Rewolinski D.; Greasley S.; Pauly T.; Shaw M.; Dinh D.; Ferre R. A.; Meador J. W. 3rd; Nukui S.; Ornelas M.; Paz R. L.; Reyner E. Thioether benzenesulfonamide inhibitors of carbonic anhydrases II and IV: structure-based drug design, synthesis, and biological evaluation. Bioorg. Med. Chem. 2010, 18, 3307–3319. [DOI] [PubMed] [Google Scholar]
- a Quilico A. Sulfonation of phenolic ethers with aminosulfonic acid. Atti Accad. Naz. Lincei 1927, 512–517. [Google Scholar]; b Hsieh Y. Y.; Chue Y. C.; Huang C. H.; Yang I. T.; Chow C. T.; Kyi Z. Y. Chemotherapeutic studies on schistosomiasis. IV. Some aryl ethers of dimercaptopropanol and their antimonial mercaptides. Huaxue Xuebao 1957, 447–454. [PubMed] [Google Scholar]; c Zhuang Z. P.; Kung M. P.; Kung H. F. Synthesis of biphenyltrienes as probes for β-amyloid plaques. J. Med. Chem. 2006, 49, 2841–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kolb H. C.; Walsh J. C.; Kasi D.; Mocharla V.; Wang B.; Gangadharmath U. B.; Duclos B. A.; Chen K.; Zhang W.; Chen G.; Padgett H. C.; Karimi F.; Scott P. J. H.; Gao Z.; Liang Q.; Collier T. L.; Zhao T.; Xia C.. Development of molecular imaging probes for carbonic anhydrase IX using click chemistry, WO2008124703.; b Nicholas K. M.; Pettit R. Alkyne protecting group. Tetrahedron Lett. 1971, 37, 475–478. [Google Scholar]; c Chohan Z. H.; Scozzafava A.; Supuran C. T. Unsymmetrical 1,1′-disubstituted ferrocenes: Synthesis of Co(II), Cu(II), Ni(II) and Zn(II) chelates of ferrocenyl-1-thiadiazolo-1′-tetrazole, -1-thiadiazolo-1′-triazole and -1-tetrazolo-1′-triazole with antimicrobial activity. J. Enzyme Inhib. Med. Chem. 2002, 17, 261–266. [DOI] [PubMed] [Google Scholar]
- Cecchi A.; Hulikova A.; Pastorek J.; Pastoreková S.; Scozzafava A.; Winum J. Y.; Montero J. L.; Supuran C. T. Carbonic anhydrase inhibitors. Sulfonamides inhibit isozyme IX mediated acidification of hypoxic tumors. Fluorescent sulfonamides design as probes of membrane-bound carbonic anhydrase isozymes involvement in tumorigenesis. J. Med. Chem. 2005, 48, 4834–4841. [DOI] [PubMed] [Google Scholar]
- a Dubois L.; Douma K.; Supuran C. T.; Chiu R. K.; van Zandvoort M. A. M.; Pastoreková S.; Scozzafava A.; Wouters B. G.; Lambin P. Imaging the hypoxia surrogate marker CA IX requires expression and catalytic activity for binding fluorescent sulfonamide inhibitors. Radiother. Oncol. 2007, 83, 367–373. [DOI] [PubMed] [Google Scholar]; b Dubois L.; Lieuwes N. G.; Maresca A.; Thiry A.; Supuran C. T.; Scozzafava A.; Wouters B. G.; Lambin P. Imaging of CA IX with fluorescent labelled sulfonamides distinguishes hypoxic and (re)-oxygenated cells in a xenograft tumour model. Radiother. Oncol. 2009, 92, 423–428. [DOI] [PubMed] [Google Scholar]; c Akurathi V.; Dubois L.; Lieuwes N. G.; Chitneni S. K.; Cleynhens B. J.; Vullo D.; Supuran C. T.; Verbruggen A. M.; Lambin P.; Bormans G. M. Synthesis and biological evaluation of a 99mTc-labelled sulfonamide conjugate for in vivo visualization of carbonic anhydrase IX expression in tumor hypoxia. Nuclear Med. Biol. 2010, 37, 557–564. [DOI] [PubMed] [Google Scholar]; d Dubois L.; Peeters S.; Lieuwes N. G.; Geusens N.; Thiry A.; Carta F.; Scozzafava A.; Dogné J. M.; Supuran C. T.; Harris A. L.; Masereel B.; Lambin P. Specific inhibition of CA IX activity enhances the therapeutic effect of tumor irradiation. Radiother. Oncol. 2011, 99, 424–431. [DOI] [PubMed] [Google Scholar]; e Groves K.; Bao B.; Zhang J.; Handy E.; Kennedy P.; Cuneo G.; Supuran C. T.; Yared W.; Peterson J. D.; Rajopadhye M. Synthesis and evaluation of near-infrared fluorescent sulfonamide derivatives for imaging of hypoxia-induced carbonic anhydrase IX expression in tumors. Bioorg. Med. Chem. Lett. 2012, 22, 653–657. [DOI] [PubMed] [Google Scholar]; f Scozzafava A.; Menabuoni L.; Mincione F.; Mincione G.; Supuran C. T. Carbonic anhydrase inhibitors. Synthesis of sulfonamides incorporating DTPA tails and of their zinc complexes with powerful topical antiglaucoma properties. Bioorg. Med. Chem. Lett. 2001, 11, 575–582. [DOI] [PubMed] [Google Scholar]
- Khalifah R. G. The carbon dioxide hydration activity of carbonic anhydrase. J. Biol. Chem. 1971, 246, 2561–2573. [PubMed] [Google Scholar]
- a Avvaru B. S.; Wagner J. M.; Maresca A.; Scozzafava A.; Robbins A. H.; Supuran C. T.; McKenna R. Carbonic anhydrase inhibitors. The X-Ray crystal structure of human isoform II in adduct with an adamantyl analogue of acetazolamide resides in a new hydrophobic binding pocket. Bioorg. Med. Chem. Lett. 2010, 20, 4376–4381. [DOI] [PubMed] [Google Scholar]; b Menchise V.; De Simone G.; Alterio V.; Di Fiore A.; Pedone C.; Scozzafava A.; Supuran C. T. Carbonic anhydrase inhibitors: Stacking with Phe131 determines active site binding region of inhibitors as exemplified by the X-ray crystal structure of a membrane-impermeant antitumor sulfonamide complexed with isozyme II. J. Med. Chem. 2005, 48, 5721–5727. [DOI] [PubMed] [Google Scholar]; c Carta F.; Aggarwal M.; Maresca A.; Scozzafava A.; McKenna R.; Supuran C. T. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem. Commun. 2012, 48, 1868–1870. [DOI] [PubMed] [Google Scholar]
- a Pacchiano F.; Aggarwal M.; Avvaru B. S.; Robbins A. H.; Scozzafava A.; McKenna R.; Supuran C. T. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem. Commun. 2010, 46, 8371–8373. [DOI] [PubMed] [Google Scholar]; b Hen N.; Bialer M.; Yagen B.; Maresca A.; Aggarwal M.; Robbins A. H.; McKenna R.; Scozzafava A.; Supuran C. T. Anticonvulsant 4-aminobenzenesulfonamide derivatives with branched-alkylamide moieties: X-ray crystallography and inhibition studies of human carbonic anhydrase isoforms I, II, VII, and XIV. J. Med. Chem. 2011, 54, 3977–3981. [DOI] [PubMed] [Google Scholar]
- Liao S. Y.; Ivanov S.; Ivanova A.; Ghosh S.; Cote M. A.; Keefe K.; Coca-Prados M.; Stanbridge E. J.; Lerman M. I. Expression of cell surface transmembrane carbonic anhydrase genes CA9 and CA12 in the human eye: overexpression of CA12 (CAXII) in glaucoma. J. Med. Genet. 2003, 40, 257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss A. H. P.; Impagnatiello F.; Toris C. B.; Gale D. C.; Prasanna G.; Borghi V.; Chiroli V.; Chong W. K. M.; Carreiro S. T.; Ongini E. Ocular hypotensive activity of BOL-303259-X, a nitric oxide donating prostaglandin F2α agonist, in preclinical models. Exp. Eye Res. 2011, 93, 250–255. [DOI] [PubMed] [Google Scholar]
- Hanmei L.; Xun S.; Dong Z.; Zhirong Z. A cell-specific poly(ethylene glycol) derivative with a wheat-like structure for efficient gene delivery. Mol. Pharmaceutics 2012, 9, 2974–2985. [DOI] [PubMed] [Google Scholar]
- a Priet S.; Zlatev I.; Barvik I.; Geerts K.; Leyssen P.; Neyts J.; Dutartre H.; Canard B.; Vasseur J. J.; Morvan F.; Alvarez K. 3′-Deoxy phosphoramidate dinucleosides as improved inhibitors of hepatitis C virus subgenomic replicon and NS5B polymerase activity. J. Med. Chem. 2010, 53, 6608–6617. [DOI] [PubMed] [Google Scholar]; b Kane B. E.; Grant M. K.; El-Fakahany E. E.; Ferguson D. M. Synthesis and evaluation of xanomeline analogs – probing the wash-resistant phenomenon at the M1 muscarinic acetylcholine receptor. Bioorg. Med. Chem. 2008, 16, 1376–1392. [DOI] [PubMed] [Google Scholar]
- Simonin J.; Vernekar S. K.; Thompson A. J.; Hothersall J. D.; Connolly C. N.; Lummis S. C.; Lochner M. High-affinity fluorescent ligands for the 5-HT(3) receptor. Bioorg. Med. Chem. Lett. 2012, 22, 1151–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S.; Tanaka H.; Hioki K.; Yamada K.; Kunishima M. Labeling study of avidin by modular method for affinity labeling (MoAL). Bioorg. Med. Chem. Lett. 2010, 20, 7050–7053. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avvaru B. S.; Kim C. U.; Sippel K. H.; Gruner S. M.; Agbandje-McKenna M.; Silverman D. N.; McKenna R. A short, strong hydrogen bond in the active site of human carbonic anhydrase II. Biochemistry 2010, 49, 249–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Schüttelkopf A. W.; van Aalten D. M. F. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr., D 2004, 60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar]