

Abstract
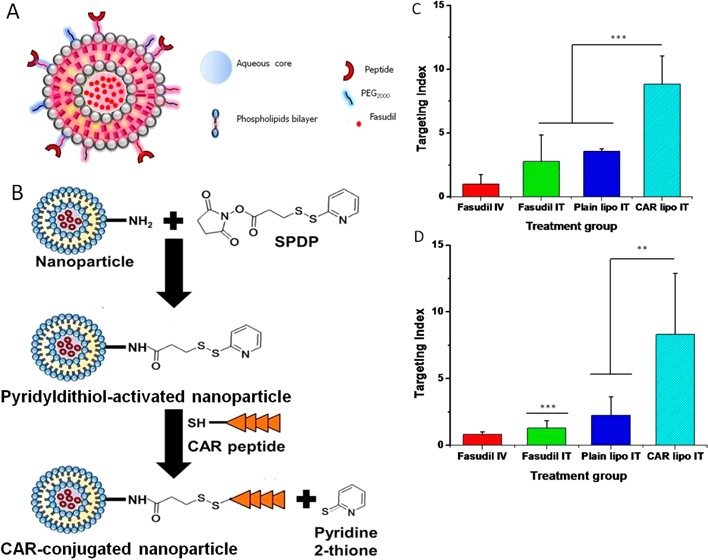
This study sought to develop a liposomal delivery system of fasudil—an investigational drug for the treatment of pulmonary arterial hypertension (PAH)—that will preferentially accumulate in the PAH lungs. Liposomal fasudil was prepared by film-hydration method, and the drug was encapsulated by active loading. The liposome surface was coated with a targeting moiety, CARSKNKDC, a cyclic peptide; the liposomes were characterized for size, polydispersity index, zeta potential, and storage and nebulization stability. The in vitro drug release profiles and uptake by TGF-β activated pulmonary arterial smooth muscle cells (PASMC) and alveolar macrophages were evaluated. The pharmacokinetics were monitored in male Sprague–Dawley rats, and the pulmonary hemodynamics were studied in acute and chronic PAH rats. The size, polydispersity index (PDI), and zeta potential of the liposomes were 206–216 nm, 0.058–0.084, and −20–42.7 mV, respectively. The formulations showed minimal changes in structural integrity when nebulized with a commercial microsprayer. The optimized formulation was stable for >4 weeks when stored at 4 °C. Fasudil was released in a continuous fashion over 120 h with a cumulative release of 76%. Peptide-linked liposomes were taken up at a higher degree by TGF-β activated PASMCs; but alveolar macrophages could not engulf peptide-coated liposomes. The formulations did not injure the lungs; the half-life of liposomal fasudil was 34-fold higher than that of plain fasudil after intravenous administration. Peptide-linked liposomal fasudil, as opposed to plain liposomes, reduced the mean pulmonary arterial pressure by 35–40%, without influencing the mean systemic arterial pressure. This study establishes that CAR-conjugated inhalable liposomal fasudil offers favorable pharmacokinetics and produces pulmonary vasculature specific dilatation.
Keywords: pulmonary arterial hypertension, CAR peptide, peptide link liposomes, inhalation delivery
1. Introduction
The goal of a targeted drug delivery system is to maximize therapeutic concentration at the disease site, overcome the chief limitation of conventional delivery strategies—off-target effects due to systemic exposure—and amplify the efficacy of a given therapeutic entity. Thus, targeted drug delivery systems use homing devices or targeting moieties to escort the drug to its site of action; the homing devices can be attached either to the drug itself or to the carrier system containing the drug. Carriers protect the drugs from rapid degradation, reduce clearance from the body, and modulate pharmacokinetics for extended drug release. Further, carriers engineered with homing devices—such as antibodies,1 fragments of antibodies,2 aptamers,3 PEGs,4 polysaccharides,5 folic acid,6 and proteins or peptides7,8—can chaperone the drug to the site of interest.
Peptide based homing devices are preferred over other targeting moieties because peptides have reduced immunogenicity, are less expensive, and are flexible for grafting onto a variety of carrier surfaces.9 Peptides can specifically bind with the receptors overexpressed at the site of action, and thus help accumulate the carrier around the diseased tissue.10 Peptides with cell penetrating property can deliver the drug or carriers to cells or cellular organelles by permeating through the cell membrane.11 CARSKNKDC, CAR, a 9 amino acid cyclic peptide, translocates various small drug molecules, therapeutic proteins, and antisense oligonucleotides into the cell cytoplasm.11 CAR also accumulates in pulmonary arterial endothelial and smooth muscle cells upon binding with cell surface heparan sulfate overexpressed in the lung vasculature of rats afflicted with pulmonary arterial hypertension (PAH), a disease of the pulmonary vasculature12,13
PAH, a debilitating disease, causes obstruction and obliteration of the small pulmonary arteries, vascular remodeling, inflammation, intimal and medial hypertrophy, wall thickening, intimal fibrosis, and plexiform lesions.14,15 Occluded blood vessels resist blood flow through the lungs and lead to heart failure and premature death.16,17 Despite some success with current PAH therapy with systemic vasodilators, PAH remains a debilitating disease with disappointing long-term survival.18 Current PAH treatment strategies suffer from limitations of short half-lives, frequent administration (9–12 times/day), and painful IV or subcutaneous injections.19 Systemic hypotension,20 impaired intrapulmonary gas exchange, reduced cardiac function, liver damage (endothelin receptor antagonist), and even death12 are major side effects of current PAH therapy.
Recently, we demonstrated that intratracheal liposomal fasudil,21 an investigational anti-PAH drug, extends the duration of pulmonary vasodilation and minimizes systemic hypotension. Thus, to direct the therapeutic cargo preferentially to the pulmonary vasculature and address the limitations of systemic exposure associated with current PAH therapy, we hypothesize that CAR-conjugated liposomal formulation of fasudil produces pulmonary vasculature-specific dilatory action in PAH lungs. We posit that liposomes will accumulate on the diseased tissue and exert prolonged local vasodilation and thereby eliminate both systemic hypotensive effects and pharmacokinetic limitations associated with conventional treatments.
To test this hypothesis, we have developed inhalable CAR-conjugated liposomal formulations of fasudil (Figure 1). The formulation was optimized for particulate properties, stability, and drug release profiles. The uptake of the optimized formulations by pulmonary arterial smooth muscle cells and alveolar macrophage was evaluated. The pharmacokinetics, safety, and pulmonary hemodynamics of the formulations were studied in either healthy or PAH rats.
Figure 1.
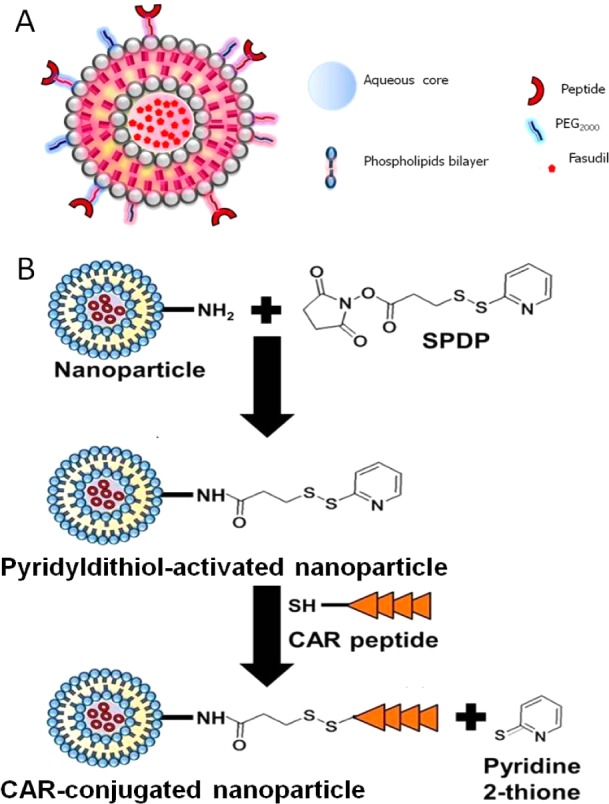
Schematic diagram of the conjugation of the liposomes with the CAR peptide (A) and reaction steps of CAR peptide conjugation with the liposomes (B).
2. Experimental Section
2.1. Materials
Lipids were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). N-Succinimidyl 1,3-(2-pyridyldithio)propionate (SPDP) was obtained from Molecular Biosciences (Boulder, CO). TGF-β was purchased from PeproTech (Rocky Hill, NJ). CAR peptide, containing an extra cysteine for conjugation through the free sulfhydryl, was obtained from Dr. Ruoslahti’s lab at the University of California in Santa Barbara. Cell medium was purchased from ATCC (Manassas, VA). The extruder and polycarbonate membranes were purchased from Avestin Inc. (Ontario, Canada). All other chemicals, unless otherwise stated, were purchased from Fisher Scientific (Pittsburgh, PA) and Sigma-Aldrich Inc. (St Louis, MO). Chemicals were of analytical grades and used without further purification.
2.2. Preparation of CAR Conjugated Liposomal Formulations of Fasudil
Liposomes were prepared using 50 μmol lipids comprising 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol, and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG2000). Various formulations were prepared by adding either 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000 (DSPE-Mal2000), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE), or 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) as the fourth lipid. The molar ratios were 70:30:5:5 (Table 1). DSPE, DSPE-Mal2000, DPPE, and DOPE were conjugated with the cysteine group of CAR peptide. Lipids were first dissolved in 2 mL of chloroform/methanol mixture at a ratio of 4:1(v/v) in a round-bottom flask, and a thin lipid film was developed under vacuum in a water bath at 45 °C using Buchi R-114 Rotavapor (Buchi Laboratories AG, Flawil, Switzerland). Lipid film was then rehydrated with 250 mM ammonium sulfate (pH 4.5). Liposomes were sonicated for 1 h and extruded sequentially at 65 °C through 400 and 200 nm polycarbonate (PC) membranes (Avanti Polar Lipids, Inc., Alabaster, Alabama). External buffer (ammonium sulfate) was then replaced with phosphate buffer saline (PBS, pH 7.4) by using a PD-10 column (Sephadex-25, GE Healthcare, Piscataway, NJ). Fasudil was encapsulated by creating ammonium sulfate gradient as reported previously.21 The liposomes were then incubated with 10 mg of fasudil after dissolving in the external buffer for 2 h at 65 °C. Unencapsulated fasudil was removed by the PD-10 column.22
Table 1. Composition and Physicochemical Characteristics of Liposomes.
| formulation | lipid composition | size (nm) | PDI | zeta potential (mV) | entrapment efficiency (%) |
|---|---|---|---|---|---|
| F-1 | DPPC:CH:DSPE-Peg-2000:DSPE-Mal | 206.167 ± 4.474 | 0.058 ± 0.007 | –42.733 ± 1.250 | 47.366 ± 3.479 |
| F-2 | DPPC:CH:DSPE-Peg-2000:DSPE | 205.500 ± 3.617 | 0.061 ± 0.005 | –33.633 ± 3.156 | 72.333 ± 6.506 |
| F-3 | DPPC:CH:DSPE-Peg-2000:DPPE | 202.267 ± 6.484 | 0.084 ± 0.002 | –35.567 ± 2.511 | 78.300 ± 5.696 |
| F-4 | DPPC:CH:DSPE-Peg-2000:DOPE | 216.833 ± 8.458 | 0.062 ± 0.009 | –20.070 ± 0.709 | 49.267 ± 5.550 |
CAR peptide was conjugated onto liposome surface by SPDP chemistry according to a previously described procedure.23 Briefly, SPDP was dissolved in 30 μL of dimethylformamide (DMF) and incubated with liposomes at room temperature for 30 min to functionalize the amine groups of the phospholipid. Excess SPDP was removed from the liposomes by ultracentrifugation and sedimented at 355000g for 1 h at 4 °C using TL-100 ultracentrifuge (Beckman, USA). Liposomes were resuspended in PBS and incubated with 1 mg of CAR peptide, dissolved in 100 μL of PBS, for 1 h at room temperature. Unconjugated peptide was further removed by ultracentrifugation as described above. Liposomes were resuspended in PBS and stored at 4 °C for further evaluation.
2.3. Physicochemical Characterization of CAR–Liposomes
CAR–liposomes were characterized for size, polydispersity index (PDI), zeta potential, and entrapment efficiency of fasudil according to our previously published method.22 To measure the size and zeta potential, 20 μL of liposomes was taken in an Eppendorf tube and diluted to 1000 μL with PBS and water. Both the size and zeta potential of the liposomes were measured by Nano ZS90 Zetasizer (Malvern Instruments Ltd., Worcestershire, U.K.). To quantify entrapped fasudil, 20 μL of liposomes was placed in an Eppendorf tube, mixed with 980 μL of methanol, sonicated for 15 min, and centrifuged at 17000g (Legend Micro 17R, Thermo Scientific) for 15 min, and the absorbance of the supernatant was measured at 320 nm in a UV spectrophotometer (HP 8453A, Olis Inc.) The fasudil encapsulation efficiency was calculated using the formula L/T × 100 (where L = the amount of fasudil incorporated into liposome, T = total amount of fasudil).
2.4. Stability Study
For in vitro stability studies, liposomes were stored at 4 °C for 28 days, and samples were periodically collected to measure liposome size and drug entrapment as describe in section 2.3. Since these formulations will be administered to rats using a MicroSprayer (PennCentury, Inc., Philadelphia, PA), we also evaluated the stability of the optimized formulation (F-3) after nebulization. For this stability study, we first measured the size, PDI, zeta potential, and entrapment efficiency of the formulations, then nebulized the formulation three times using the MicroSprayer, and collected the sample to measure particle size, PDI, zeta potential, and drug entrapment as explained above.21
2.5. In Vitro Release Study
To evaluate in vitro release profiles, we chose two optimized formulations based on their in vitro stability. This study was performed in phosphate buffer saline (PBS) at 37 °C using molecular weight cutoff (3.5K) cassettes (Slide-A-Lyzer, Thermo-Scientific, Waltham, MA). An aliquot (500 μL) of optimized liposomal formulations was placed in MW cutoff cassette placed in 50 mL of PBS in a beaker. Periodically, 1 mL of sample was collected and replaced with 1 mL of fresh PBS solution. Samples were analyzed using a UV spectrophotometer (HP 8453A, Olis Inc., Hayward, CA). The release of fasudil was calculated by dissolving the liposomes with 1% Triton X-100 at time zero. Percent fasudil released at different time points was determined by the following equation: % release = 100 × (Ft – F0)/(F100 – F0), where Ft and F0 are the concentrations at times t and 0, respectively. F100 represents 100% fasudil concentration from the liposomes.22
2.6. Uptake of CAR–Liposome by Activated Pulmonary Smooth Muscle Cells
The cellular uptake of CAR conjugated fluorescent liposomes was studied in TGF-β activated rat pulmonary arterial smooth muscle cells (PASMCs). Rat PASMCs were cultured in a 75 cm2 flask in 10% FBS, penicillin/streptomycin, and glutamine containing DMEM1/2 medium (American Type Cell Culture, Manassas, VA) at 37 °C in 5% CO2 atmosphere. The cells were seeded at a density of 5000 cells/mL on the top of a coverslip in a 12-well plate and incubated overnight. Next day, cells were activated with TGF-β (10 ng/mL) for 24 h24 and the medium was replaced with 500 μL of fresh medium. CAR conjugated fluorescent liposomes and plain fluorescent liposomes were then incubated with the cells, TGF-β activated and normal cells, for 2 h followed by fixation with 4% paraformaldehyde in PBS for 10 min and subsequent incubation with 0.1% Triton X for 40 min at room temperature. Upon treatment, cells were incubated with a blocking solution containing goat serum and Tween-20, washed with PBS three times, incubated again overnight with monoclonal anti-β-actin primary antibodies (Sigma-Aldrich, St. Louis, MO) at 4 °C, washed three times with PBS, and finally incubated with Alexa Fluor 594 goat anti-mouse IgG (Invitrogen, Grand Island, NY). Cells’ nuclei were then stained with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min and washed three times. Finally, coverslips containing the cells were mounted on the top of a glass slide, and the liposomal uptake was examined with a fluorescent microscope (IX-81, Olympus, Center Valley, PA).22
2.7. Safety of the Formulation toward Pulmonary Vasculature and Lung
2.7.1. Cell Viability Study
An MTT assay was performed to evaluate the safety of the optimized formulations upon treatment with bronchial epithelial cells (Calu-3) and PASMCs.22 Briefly, 5 × 104 PASMC and Calu-3 cells were seeded in 96-well flat bottom plates and incubated overnight for cell attachment. Next day, the medium was replaced with the fresh medium, and the cells were incubated with different concentrations of fasudil for 24 h at 37 °C. The following day, the medium was removed, cells were incubated for 4 h with MTT solution, which was later removed, 100 μL of dimethyl sulfoxide was added to dissolve the precipitate, and the absorbance was measured at 570 nm. The percent of viable cells was calculated from the following equation: % viable cells = (Atreated – AMTT)/(Acontrol – AMTT) × 100.
2.7.2. Bronchoalveolar Lavage (BAL) Study
We further tested the safety of the optimized formulations by bronchoalveolar lavage (BAL) study as reported previously.25 Saline, lipopolysaccharide (LPS), and CAR-conjugated liposomes were administered to rats by intratracheal instillation. Rats were anesthetized, 24 h after the treatment, by a cocktail of ketamine (90 mg/kg) and xylazine (10 mg/kg), and lungs were isolated and weighed. To the preweighed isolated lungs, 5 mL of ice cold saline was administered; the BAL fluid was collected and centrifuged at 500g for 10 min, and the supernatant was stored at −20 °C. The protein levels in BAL fluid were assessed by bicinchoninic assay kit (Pierce, Rockford, IL), and the concentrations of lactate dehydrogenase (LDH) and alkaline phosphatase (ALP) in BAL were measured using commercial kits (Pointe Scientific, Canton, MI).
2.8. Uptake of the Formulations by Alveolar Macrophage
In this study, we measured the extent of engulfment of the CAR–liposomes by the alveolar macrophages that line the lung epithelial surfaces. The alveolar macrophages were isolated from the pellet of the BAL fluid21 collected as described above. The fluid was centrifuged at 500g for 1 min, supernatant was removed, the pellet containing the macrophages was collected, and macrophages were seeded at a cell density of 4 × 105 cells/mL on a coverslip in 12-well plates and incubated for 1 h in Dulbecco’s PBS. The medium was removed, replaced with fresh medium, and incubated with fluorescent CAR conjugated liposomes (F-3) for an hour at 37 °C. The macrophages were then fixed with 4% paraformaldehyde in PBS and incubated with a blocking solution containing goat serum and Tween 20 in PBS followed by incubation with monoclonal anti-β-actin primary antibodies (Sigma-Aldrich, St. Louis, MO) and Alexa Fluor 594 goat anti-mouse IgG (Invitrogen, Grand Island, NY). The glass coverslips were placed on the top of fluorogel on glass slides and were sealed. The macrophages were washed three times after each step with ice cold PBS, and uptake of the liposomes was observed under a fluorescence microscope (IX-81, Olympus).
2.9. In Vivo Absorption Study
We evaluated in vivo absorption profiles of the optimized formulation in healthy male Sprague–Dawley (SD) rats weighing 250–300 g (Charles River Laboratories, Wilmington, MA) according to our published method.19 Briefly, rats were divided into three groups to receive 6 mg/kg fasudil in the following forms: (i) plain fasudil IV; (ii) plain fasudil IT, and (iii) CAR–liposomes containing fasudil IT. Prior to drug administration, rats were anesthetized by an intramuscular injection of a combination of ketamine (90 mg/kg) and xylazine (10 mg/kg). Plain drug or formulations were administered either intravenously via the penile vein or intratracheally using a small animal Microsprayer (model IA-1B; Penn Century Inc., Philadelphia, PA, USA).26 Blood samples were collected in citrated Eppendorf tubes from the tip of the tail at different time points, and immediately placed on an ice bath. To separate the plasma, blood was centrifuged at 2400g for 10 min at 4 °C and stored at −20 °C until further analysis. Fasudil concentration in the plasma was measured in high performance liquid chromatography (HPLC) with the following conditions: C18× 250 mm × 4.5 mm, particle size 5 μM; mobile phase 0.02 mM phosphate buffer:acetonitrile = 68:32; flow rate 1 mL/min; wavelength 225 nm; and injection volume 100 μL.22 An aliquot of plasma (100 μL) was placed in an Eppendorf tube, mixed with 30 μL of 3% Triton X-100 plus 170 μL of acetonitrile, vortexed for few minutes, and centrifuged at 17000g for 15 min at 4 °C, and then the supernatant was injected into the HPLC. The drug concentration was determined from the standard curve prepared with plasma as the vehicle.
All animal studies were performed in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals under a protocol approved by Texas Tech University Health Sciences Center (TTUHSC) Animal Care and Use Committee (AM-02004). Rats were housed at TTUHSC Amarillo animal facility with free access to food and water.
2.10. Hemodynamic Studies in PAH Rats
The pharmacological efficacy of the formulation was studied in two established PAH models: monocrotaline (MCT) and SUGEN 5416/hypoxia-induced rodent models of PAH. MCT PAH was induced by a subcutaneous injection of MCT (50 mg/kg) into adult Sprague–Dawley (SD) rats (weight 250 to 300 g). Injected rats were then housed for 28 days to develop PAH.21 To induce SUGEN 5416/hypoxia PAH, SUGEN 5416 (20 mg/kg) was subcutaneously administered into SD rats (weight 200 to 225 g) and the rats were kept in hypoxia (10% oxygen) (Whole Animal Hypoxia Chamber, BioSpherix, Lacona, NY) for 3 weeks.27
PAH animals were divided into four groups to receive 3 mg/kg fasudil as (a) plain fasudil IV, (b) plain fasudil IT, (c) fasudil–plain liposomes (no CAR peptide on the surface) IT, and (d) fasudil–CAR–liposomes IT. The ventral neck area of anesthetized rats was shaved, the right jugular vein and carotid artery were surgically exposed, and a polyvinyl (PV-1, Tygon, Lima, OH) catheter with a tip curved at a 60–65° angle was inserted via the right internal jugular vein and right ventricle into the pulmonary artery. This catheter was used to measure mean pulmonary arterial pressure (mPAP). Similarly, another PE-50 catheter (BD Intramedic, Sparks, MD) was inserted 3–4 cm into the carotid artery to measure mean systemic arterial pressure (mSAP). Pressure measurements were performed using Memscap SP844 physiological pressure transducers (Memscap AS, Scoppum, Norway) and bridge amplifiers. Data was acquired by a PowerLab 16/30 system using LabChart Pro 7.0 software (AD Instruments, Inc., Colorado Springs, CO).21 After recording initial pressures, the treatments, listed above, were administered and pressures were measured for 6 h. The extent of reduction of both mPAP and mSAP was calculated by considering initial pressure as 100%. Further, the lung targeting index (LTI) of the formulations was calculated from the ratio of the area above the pressure/time curve (AAC) for mPAP and mSAP using the equation LTI = AAC of mPAP/AAC of mSAP.
2.11. Data Analysis
All data are presented as mean ± SD and were analyzed by ANOVA followed by a post hoc analysis using Tukey’s comparison (GraphPad Prism, version 5.0, GraphPad Software, San Diego, CA). In statistical analysis, p value less than 0.05 was considered statistically significant. Pharmacokinetic parameters were calculated by standard noncompartmental analysis using Phoenix WinNonlin (Sunnyvale, CA).
3. Results
3.1. Physicochemical Characteristics of the Liposomes
The average hydrodynamic diameter of the liposomes ranged from 202 to 216 nm (Table 1) with PDIs ranging from 0.058 to 0.062, indicating a monodispersed colloidal system.23 The zeta potential of the liposomes was between −20 and −42.7 mV. The entrapment efficiency of the liposomes was 47–49% for formulation F-1 and F-4, respectively. However, for formulations F-2 and F-3, the entrapment efficiencies were high: 72% and 78%, respectively. Because fasudil is a weakly basic hydrophilic molecule (pKa 9.72), conventional passive loading method produced liposomes with reduced drug entrapment.21,28 Thus, we used active loading method to entrap fasudil by creating a concentration gradient with ammonium sulfate solution (250 mM) that renders the liposomal core rich in ammonium sulfate and generates extra proton in the liposomal core. Upon entering the liposomal core, uncharged fasudil becomes protonated and forms a complex with SO42–. The complex gets trapped in the aqueous core of the liposomes, which results in increased drug entrapment28 Conjugation of CAR on the liposome surface was confirmed by spectroscopic measurement of pyridine-2-thione at 343 nm, which is released upon reaction of SPDP-activated liposomal surface and the unpaired thiol of CAR (Figure 2B). No such peak was observed (Figure 2A) prior to the addition of CAR because pyridine-2-thione was not released in the absence of CAR. The extent of peptide conjugation on the liposome surface was around 36%.
Figure 2.
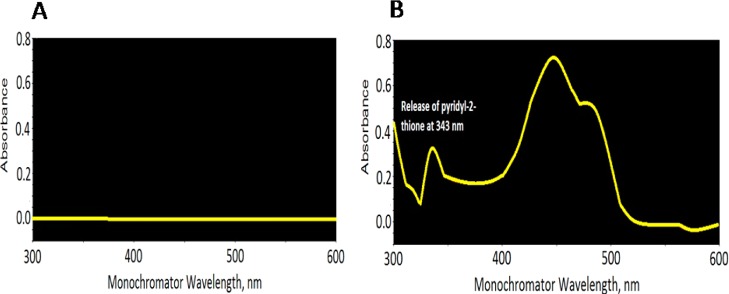
Confirmation of conjugation of the CAR peptide with the liposomes. Peak at 343 nm suggests that CAR was conjugated with liposomes. Absorbance before conjugation (A); after conjugation (B).
3.2. Stability of Liposomes
The in vitro stability of the CAR–liposomes stored at 4 °C was evaluated by measuring the size and entrapment efficiency at different time points. F-1 and F-4 containing DSPE-Mal and DOPE, respectively, were not stable for 28 days at 4 °C. With time, liposomes became larger due to aggregation of the liposomes, pointing to the liposomal instability (Figure 3A).26 However, the size of F-2 and F-3 formulations did not change during the storage period; thus no aggregation in the colloidal system was observed. Drug content of F-1 and F-4 liposomes went down upon storage, perhaps because of drug leaching from the formulations (Figure 3B), but no such reduction in the drug contents of F-2 and F-3 was observed.
Figure 3.

Stability of different formulations upon storage at 4 °C: (A) size of the lipsomes; (B) entrapment efficiency. Data represent mean ± SD, n = 3.
We have also assessed the nebulization stability of one of the formulations, F-3. For inhalation delivery, the formulation is expected to be aerosolized by means of a nebulizer. Thus, we examined whether our formulation remains stable upon nebulization. No significant changes in the size, zeta potential, and entrapment efficiency of the liposomes were observed after spraying with the microsprayer (Table 2), suggesting that the formulation F-3 can withstand the physical force applied by the nebulizers and that the formulation can be used for inhalational therapy.
Table 2. Nebulization Stability of the CAR Peptide Liposomes.
| nebulization | size (nm) | PDI | zeta potential (mV) | entrapment efficiency (%) |
|---|---|---|---|---|
| no | 207.733 ± 4.509 | 0.045 ± 0.004 | –29.603 ± 1.035 | 78.540 ± 6.741 |
| yes | 206.100 ± 4.419 | 0.088 ± 0.003 | –30.353 ± 1.027 | 78.307 ± 5.735 |
3.3. Optimized Formulations Demonstrated a Continuous Release of the Drug
Fasudil was released in a controlled fashion from the two formulations (F-2 and F-3) with a cumulative release of 60% and 76% drug, respectively (Figure 4). The difference in drug release between the two formulations may stem from the differences in the lipid composition: F-2 contained 5 mol % DSPE, but F-3 had 5 mol % DPPE. The higher phase transition temperature of DSPE might have contributed to slower release of fasudil from F-2. All in all, F-3 has higher entrapment efficiency, is stable at storage condition, and produces continuous release of the drug over 5 days. Thus, this formulation was chosen for the subsequent experiments.
Figure 4.

In vitro release profiles of fasudil-encapsulated CAR–liposomes (F-2 and F-3) in phosphate buffered saline at 37 °C. Data represent mean ± SD, n = 3.
3.4. CAR–Liposomes Were Preferentially Taken Up by PASMCs
The cellular uptake of plain liposomes and CAR–liposomes was studied in TGF-β activated PASMCs. Fluorescent microscopic images demonstrated a much higher uptake of CAR-conjugated liposomes by the TGF-β activated PASMCs compared with the uptake of plain liposomes (Figure 5). Enhanced uptake of CAR–liposomes could be due to increased expression of heparan sulfate (HS) by cells treated with TGF-β. Overexpressed HS24 enhances binding of CAR peptide on the liposome surface, which results in higher uptake of the formulation by the activated cells.
Figure 5.
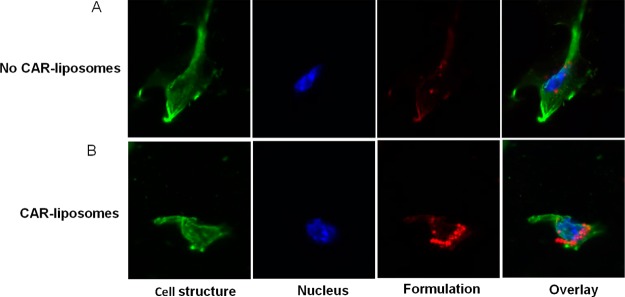
Representative fluorescence microscopic images showing the uptake of (A) liposomes without CAR peptide; (B) CAR-conjugated liposomes by the TGF-β activated pulmonary arterial smooth muscle cells. Green color represents cellular structure; blue color represents cell nucleus stained with DAPI; red color represents rhodamine B labeled liposomes; rightmost panel is the overlay.
3.5. CAR–Liposomes Escape Uptake by Alveolar Macrophages
The alveolar epithelial macrophages engulf and help remove inhaled particles. The fluorescent microscopic study demonstrated that the uptake of the CAR–liposomes by alveolar epithelial macrophages was minimal (Figure 6). True to this data, a published study also demonstrated that particles with 250 nm size range can evade macrophage uptake29 and thus can reside in the lung region for a longer time than larger particles. Further, the presence of PEG molecule on liposomes might help evade recognition by alveolar macrophages. Thus, the slower clearance of the particles might be the result of reduced uptake and poor recognition by the alveolar macrophages.21,30
Figure 6.
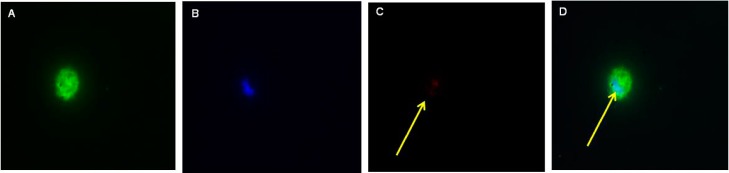
Representative fluorescence microscopic images showing macrophageal uptake of the CAR–liposomes. Green color represents cellular structure (A); blue color represents cell nucleus stained with DAPI (B); red color represents rhodamine B labeled liposomes (C); rightmost panel is the overlay (D).
3.6. CAR–Liposomes Are Safe to the Lung and Pulmonary Vasculature
We evaluated the effect of different concentrations of fasudil containing CAR–liposomes with two cell lines: PASMCs and Calu-3. Exposure to increasing concentrations of CAR–liposomes did not reduce the viability of either cell type. While SDS, the positive control, caused ∼85% cell death at a concentration of 0.1%, the cell death was only 10% (Figure 7A and Figure 7B) with CAR–liposomes incubated for 24 h at its highest concentration (250 μM). Thus, the formulations were safe for bronchial epithelial and PASMCs.
Figure 7.

Cell viability of fasudil-encapsulated CAR–liposomes in (A) lung epithelial (Calu-3) and (B) pulmonary arterial smooth muscle cells upon acute exposure. Data represent mean ± SD; n = 8.
The safety of the formulation was further evaluated in vivo following intratracheal administration. The BAL study showed that the wet-lung weight of the saline treated animal was 0.45 ± 0.01 g/100 g of rat weight; whereas the LPS treated lung weight was 0.56 ± 0.07 g/100 g. Increase in the wet lung weight suggests edema formation in the lungs after LPS treatment. Following administration of CAR–liposomes, lung weight was 0.44 ± 0.02 g/100 g, indicating no injury in the lung caused by the administration of CAR–liposomes (Figure 8A). Like saline, CAR–liposomes did not increase protein level compared with that observed in the LPS treated group (Figure 8B). Injury of the respiratory epithelial cells increases the permeability of pulmonary microvasculature, which promotes infiltration of different types of immune cells, which increases the protein concentration in BAL fluid.19 Like the protein levels, LDH and ALP levels in saline treated rats were no different from those in CAR–liposome treated rats (Figures 8C and 8D). These observations suggest that formulations would be safe for acute treatment.
Figure 8.

Effect of CAR–liposomes on the (A) wet lung weight, (B) total protein content, and (C) levels of injury marker lactate dehydrogenase (LDH), (D) alkaline phosphatase (ALP) in bronchoalveolar lavage (BAL) fluid. Data represent mean ± SD (n = 3–4). *p < 0.05, **p < 0.01, ***p < 0.001.
3.7. Inhalational Liposomes Improved the Pharmacokinetics of Fasudil
We measured the pulmonary absorption of the optimized CAR–liposomes following IT administration in healthy rats, and compared it with plain fasudil after IV and IT administration. The plasma half-life of plain fasudil after IV administration was 19 ± 4 min, which was increased by 1.8-fold upon IT administration. Intratracheal CAR–liposomes extended the plasma half-life by 34-fold compared with that of plain fasudil IV (Figure 9). The AUC of CAR–liposome was 16-fold greater than the AUC of fasudil IT (inset in Figure 9). The higher AUC of liposomal formulation can be explained by the fact that the liposomes were lysed using Triton-X to measure both the released and encapsulated drug at each time point, which represents the amount of total drug remaining in the body at the given time. In such cases, the AUC of the delivery system, depending on drug release rate, could be several-fold higher than that after IV dosing of the plain drug. Such observations have been reported previously.31 The increase in the half-life of fasudil from the liposomal formulation reflects the observation in the macrophage uptake study: reduced uptake by alveolar epithelial macrophages and slower clearance by reticular endothelial systems due to the presence of PEG on the CAR–liposome surface.32
Figure 9.

Absorption profiles of fasudil following intratracheal administration of plain fasudil and fasudil-encapsulated CAR–liposome. Data represent mean ± SD; n = 4.
3.8. Efficacy of Liposomal Fasudil in PAH Rats
We studied the pharmacological efficacy by monitoring the extent of reduction in mPAP in two rodent PAH models: MCT and SUGEN 5416/hypoxia-induced PAH. In the case of the MCT model, the average mPAP was ∼37 ± 10 mmHg 4 weeks after MCT injection. Upon treatment with fasudil IV and IT, the mPAP was reduced by 35% and 25%, respectively. However, the pressure returned to 15% of the initial value within 60 and 90 min of fasudil IV and IT administration, respectively (Figures 10A and 10B). When fasudil containing liposomes without CAR were administered, mPAP was 30% of initial and the vasodilation lasted for 150 min (Figure 10C). However, CAR–liposomes produced a greater reduction in mPAP, 40% reduction for 250 min, and the vasodilation was maintained at 20% of initial mPAP for 6 h (Figure 10D). A significant reduction in mSAP was observed for all the treatment groups except the CAR–liposome group, where little or no reduction in mSAP was observed. This could be attributed to the accumulation of the fasudil–CAR–liposomes in the lung. Thus, the released fasudil produced vasodilation in the arteries and arterioles of PAH lungs. For CAR–liposomes, a reduced amount of drug was available in the systemic circulation, thus little or no peripheral hypotension was observed.
Figure 10.

Hemodynamic efficacy of the formulations in MCT-induced PAH rats. The percent reduction of initial mean pulmonary arterial pressure (mPAP) and mean systemic arterial pressure (mSAP) upon administration of (A) plain fasudil IV; (B) plain fasudil IT; (C) plain liposome encapsulated fasudil; and (D) fasudil entrapped CAR conjugated liposomes. The rats received a single dose of 3 mg/kg plain fasudil and formulations containing an equivalent dose of the drug. Data represent mean ± SD (n = 3–6).
SUGEN 5416/hypoxia-induced PAH rats showed an initial mPAP of 45 ± 7 mmHg after 3 weeks of PAH development. Upon treatment, mPAP was reduced by 38%, 40%, and 30% by fasudil IV, fasudil IT, and fasudil entrapped plain liposomes, respectively. However, the duration of reduction in mPAP was 70, 90, and 200 min for fasudil IV (Figure 11A), fasudil IT (Figure 11B), and fasudil–plain liposomes (Figure 11C), respectively. Fasudil–CAR–liposomes produced a 30–35% reduction in mPAP for 360 min (Figure 11D). Like MCT-induced PAH model, the effect of CAR–liposomes in reducing systemic pressure was minimal. The higher extent of dilation in pulmonary arteries can be explained by the extended retention of CAR conjugated fasudil liposomes in PASMCs in the hypertensive arteries of both MCT- and SUGEN 5416/hypoxia-induced PAH lungs.12 Thus, fasudil entrapped CAR–liposomes produce pulmonary vasodilation with minimal effect on the systemic circulation.
Figure 11.

Hemodynamic efficacy of the formulations in SUGEN 5416/hypoxia-induced PAH rats. The percent reduction of initial mean pulmonary arterial pressure (mPAP) and mean systemic arterial pressure (mSAP) upon administration of (A) plain fasudil IV, (B) plain fasudil IT, (C) plain liposome encapsulated fasudil, and (D) fasudil entrapped CAR conjugated liposomes. The rats received a single dose of 3 mg/kg plain fasudil and formulations containing an equivalent dose of the drug. Data represent mean ± SD (n = 4–9).
The targeting index (TI) was determined to quantify the site-specific therapeutic action of the formulation. In the case of MCT rats, the TI values for CAR–liposomes IT (Figure 12A) were 3.2-fold and 2.5-fold greater than those of fasudil IT and fasudil containing plain liposomes IT, respectively. In the SUGEN/hypoxia model, like the MCT model, index values increased by 6.4-fold and 3.7-fold compared with plain fasudil IT and fasudil–plain liposomes, respectively (Figure 12B). In both models, higher indices were due to reduced peripheral vasodilation by CAR–liposomes, demonstrating the proof-of-principle of the proposed approach.
Figure 12.

Targeting indices of fasudil IV, fasudil IT, fasudil plain liposomes, and fasudil CAR–liposomes in MCT induced PAH rats (A) and SUGEN 5416/hypoxia induced PAH rats (B). Data represent mean ± SD (n = 3–6). **p < 0.01, ***p < 0.001.
4. Discussion
In this study, we reported a peptide-decorated inhalable formulation of fasudil for pulmonary selective vasodilation in PAH lungs. Pulmonary administration of particulate carrier-based systems does not limit the drug release only in the pulmonary region. Rather, inhaled liposomes with a size smaller than 100–200 nm can produce systemic side effects by entering the circulation.33 With this in mind, we have decorated the liposomal surface with the homing peptide, CAR. This peptide binds with cell surface heparan sulfate and accumulates within the hypertensive pulmonary arteries. CAR has also the ability to penetrate cells,34 which can facilitate particle movement from the alveolar ducts into the pulmonary arterioles, the site of action for anti-PAH drugs. Upon entering the arterioles, CAR–liposomes possibly bind with damage endothelial cells and release the drug in a slow release fashion. Particles that get into the systemic circulation may also return to the pulmonary arteries and arterioles where CAR–liposomes bind with remodeled vessels and thus release the drug at PAH afflicted vessels.
We have developed a stable formulation with favorable drug entrapment and physicochemical properties. Fasudil, a weak base, was encapsulated by active loading using high ammonium sulfate concentration gradient at 65 °C. The fluidity of the lipid membrane increases at elevated temperature, which allows fasudil to enter the aqueous core of the liposomes. The drug molecules then become confined in the liposomal aqueous core due to complexation with sulfate ion. This osmotic gradient-based confinement of the drug results in higher encapsulation upon active loading than traditional passive loading.28 The size of the liposomes prevented the uptake and clearance by alveolar macrophages, while the surface charge provided colloidal stability. The net negative charge could be attributed to the presence of zwitterionic phosphatidylcholine in the liposomes.35 Moreover, the presence of carbamate linkage in the formulations could also induce a net negative charge on the phosphate moiety at physiological pH.36 The relatively less negative charge of F-4 may have stemmed from DOPE, which is a cationic lipid.37 Nevertheless, the presence of optimal charge in most of the formulations suggests colloidal stability.
The optimized formulation showed no change in size and no aggregation of liposomes over 28 days at 4 °C. The presence of PEG chain in DSPE produces a steric hindrance around particle surface that prevents aggregation of the liposomes and yields a stable system.26 However, F-1 and F-4 showed increase in size over this storage time, which may have resulted from broken liposomes and aggregates formed in the colloidal system. Stability after nebulization established the suitability of the formulation for administration via the intratracheal route using an aerosolizer. Liposomes also showed a continuous release of the drug over 5 days, suggesting a prolonged lung specific delivery of the drug. Overall, the physicochemical properties of the liposomes demonstrated the feasibility for inhalational formulation of an anti-PAH drug.
This study also addressed whether the liposomes preferentially accumulate on proliferative smooth muscle cells in the PAH lung. The higher uptake of CAR–liposomes by TGF-β activated PASMCs indicated the target-specific binding. In vitro safety studies reflected that CAR–liposomes did not cause injury to either pulmonary arterial smooth muscle or bronchial epithelial cells. In vivo, studies did not show any evidence of acute cellular injury, as shown by no change in the lung weight, bronchoalveolar protein concentration, or LDH and ALP levels. These acute safety studies support that CAR–liposomes would not be a safety problem for inhalation therapy. However, these acute safety studies did not provide any information on whether the peptide or liposome would produce any immune reaction by the respiratory cells. Based on numerous published reports on pegylated liposomes and availability of commercial pegylated liposomal drug formulations, we anticipate that our peptide-linked liposomes would produce little or no untoward immune reaction.
The CAR–liposomes are expected to yield a long-acting formulation for the anti-PAH drug, fasudil. The reduced macrophage uptake suggested reduced clearance by alveolar macrophages and longer residence time in the lungs. The absorption profile also showed long plasma half-life of the drug when encapsulated in the liposomes as compared with the drug itself, pointing to the reduced clearance of the pegylated liposomes. The pharmacological efficacy of the peptide-conjugated liposomes encapsulating the drug exhibited a prolonged reduction in the pulmonary arterial pressure with minimal reduction of systemic blood pressure, which is reflected by a higher targeting index in the presence of homing peptide on liposome surface than plain fasudil administered intravenously and intratracheally, and liposomes without peptide. These data are consistent with the pharmacokinetic profiles of the drug in the formulation: prolonged plasma half-life as compared to the plain drug.
While this proof-of-principle study demonstrated the potential for targeted PAH treatment with reduced peripheral vasodilation and enhanced patient compliance, the translational potential of the formulation is still unknown. The mechanism of CAR-mediated pulmonary localization of the liposomal formulations must be elucidated. The stability of the liposomes was evaluated after storage for one month at 4 °C. To deliver liposomes as a nebulized solution via oral inhalation, we may have to lyophilize the liposomes to confer long-term stability and to develop a clinically acceptable and commercially viable formulation. Also we have to determine the maximal number of CAR peptide molecules that can be conjugated on the liposomal surface for maximal targeting efficiency. Mechanistic studies are required to delineate the extent of peptide accumulation in the pulmonary vasculature. Importantly, the scale-up of the formulation for clinical application would be a major hurdle to overcome before we can make any conclusions regarding the translational potential of the formulation. Thus, additional studies will dictate whether this peptide–liposome hybrid formulation has any clinical future.
5. Conclusions
This study established the feasibility of an inhalable delivery system that accumulates in the pulmonary vasculature of PAH lungs by the homing action of the surface bound peptide, CAR. The optimized formulations are taken up preferentially by the proliferating pulmonary arterial cells, are acutely safe to the lung tissue, and have a markedly improved pharmacokinetic profile. Pulmonary administration of CAR–liposomes produced a prolonged reduction in pulmonary arterial pressure without causing systemic vasodilation. Further research should explore the biochemical and cellular mechanisms and chronic effect of the formulation in PAH animals.
Acknowledgments
This work was supported in part by an American Recovery and Reinvestment Act Fund, NIH 1R15HL103431 (F.A.), and RO1 HL086680 (E.N.-G.).
Glossary
Abbreviations Used
- AAC
area above the curve
- AUC
area under the curve
- BCA
bicinchoninic acid
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco’s modified Eagle medium
- DMSO
dimethyl sulfoxide
- DPPC
1,2-dipalmitoyl-sn-glycero-3-phosphocholine
- DSPE-PEG2000
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000]
- FBS
fetal bovine serum
- HPLC
high performance liquid chromatography
- MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PAH
pulmonary arterial hypertension
- PASMC
pulmonary arterial smooth muscle cells
- PBS
phosphate buffered saline
- PDI
polydispersity index
- TEM
transmission electron microscopy
- TGF-β
transforming growth factor beta
- TI
targeting index
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Chacko A. M.; Li C.; Nayak M.; Mikitsh J. L.; Hu J.; Hou C.; Grasso L.; Nicolaides N. C.; Muzykantov V. R.; Divgi C. R.; Coukos G. Development of 124I Immuno-PET Targeting Tumor Vascular TEM1/Endosialin. J. Nucl. Med. 2014, 553500–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberger P.; Skerra A. Sortase-catalyzed in vitro functionalization of a HER2-specific recombinant Fab for tumor targeting of the plant cytotoxin gelonin. MAbs 2014, 62354–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Zhang J.; Ma Y.; Pei X.; Liu Q.; Lu B.; Jin L.; Wang J.; Liu J. A Cell-based Single-stranded DNA Aptamer Specifically Targets Gastric Cancer. Int. J. Biochem. Cell Biol. 2013, 10.1016/j.biocel.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Betancourt T.; Byrne J. D.; Sunaryo N.; Crowder S. W.; Kadapakkam M.; Patel S.; Casciato S.; Brannon-Peppas L. PEGylation strategies for active targeting of PLA/PLGA nanoparticles. J. Biomed Mater. Res. A 2009, 911263–76. [DOI] [PubMed] [Google Scholar]
- Jain A.; Mishra S. K.; Vuddanda P. R.; Singh S. K.; Singh R.; Singh S. Targeting of diacerein loaded lipid nanoparticles to intra-articular cartilage using chondroitin sulfate as homing carrier for treatment of osteoarthritis in rats. Nanomedicine 2014, 1051031–40. [DOI] [PubMed] [Google Scholar]
- Pradhan P.; Giri J.; Rieken F.; Koch C.; Mykhaylyk O.; Doblinger M.; Banerjee R.; Bahadur D.; Plank C. Targeted temperature sensitive magnetic liposomes for thermo-chemotherapy. J. Controlled Release 2010, 1421108–21. [DOI] [PubMed] [Google Scholar]
- Brooks N. A.; Pouniotis D. S.; Tang C. K.; Apostolopoulos V.; Pietersz G. A. Cell-penetrating peptides: application in vaccine delivery. Biochim. Biophys. Acta 2010, 1805125–34. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv. Mater. 2012, 24283747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladner R. C.; Sato A. K.; Gorzelany J.; de Souza M. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discovery Today 2004, 912525–9. [DOI] [PubMed] [Google Scholar]
- Molek P.; Strukelj B.; Bratkovic T. Peptide phage display as a tool for drug discovery: targeting membrane receptors. Molecules 2011, 161857–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart K. M.; Horton K. L.; Kelley S. O. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org. Biomol. Chem. 2008, 6132242–55. [DOI] [PubMed] [Google Scholar]
- Urakami T.; Jarvinen T. A.; Toba M.; Sawada J.; Ambalavanan N.; Mann D.; McMurtry I.; Oka M.; Ruoslahti E.; Komatsu M. Peptide-directed highly selective targeting of pulmonary arterial hypertension. Am. J. Pathol. 2011, 17862489–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvinen T. A.; Ruoslahti E. Target-seeking antifibrotic compound enhances wound healing and suppresses scar formation in mice. Proc. Natl. Acad. Sci. U.S.A. 2010, 1075021671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman S. A.; Zhu S.; White R. E. RhoA/Rho-kinase signaling: a therapeutic target in pulmonary hypertension. Vasc. Health Risk Manage. 2009, 5, 663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runo J. R.; Loyd J. E. Primary pulmonary hypertension. Lancet 2003, 36193681533–44. [DOI] [PubMed] [Google Scholar]
- Austin E. D.; Loyd J. E. Heritable forms of pulmonary arterial hypertension. Semin. Respir. Crit. Care Med. 2013, 345568–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeper M. M.; Rubin L. J. Update in pulmonary hypertension 2005. Am. J. Respir. Crit. Care Med. 2006, 1735499–505. [DOI] [PubMed] [Google Scholar]
- Loyd J. E.; Phillips J. A. III. Heritable Pulmonary Arterial Hypertension. GeneReviews,posted 2002, latest revision2012. [Google Scholar]
- Gupta V.; Ahsan F. Influence of PEI as a core modifying agent on PLGA microspheres of PGE(1), a pulmonary selective vasodilator. Int. J. Pharm. 2011, 4131–251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsapenko M. V.; Tsapenko A. V.; Comfere T. B.; Mour G. K.; Mankad S. V.; Gajic O. Arterial pulmonary hypertension in noncardiac intensive care unit. Vasc. Health Risk Manage. 2008, 451043–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V.; Gupta N.; Shaik I. H.; Mehvar R.; McMurtry I. F.; Oka M.; Nozik-Grayck E.; Komatsu M.; Ahsan F. Liposomal fasudil, a rho-kinase inhibitor, for prolonged pulmonary preferential vasodilation in pulmonary arterial hypertension. J. Controlled Release 2013, 1672189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahar K.; Absar S.; Patel B.; Ahsan F. Starch-coated magnetic liposomes as an inhalable carrier for accumulation of fasudil in the pulmonary vasculature. Int. J. Pharm. 2014, 4641–2185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Absar S.; Nahar K.; Kwon Y. M.; Ahsan F. Thrombus-targeted nanocarrier attenuates bleeding complications associated with conventional thrombolytic therapy. Pharm. Res. 2013, 3061663–76. [DOI] [PubMed] [Google Scholar]
- Chen J. K.; Hoshi H.; McKeehan W. L. Stimulation of human arterial smooth muscle cell chondroitin sulfate proteoglycan synthesis by transforming growth factor-beta. In Vitro Cell. Dev. Biol. 1991, 2716–12. [DOI] [PubMed] [Google Scholar]
- Hussain A.; Ahsan F. State of insulin self-association does not affect its absorption from the pulmonary route. Eur. J. Pharm. Sci. 2005, 252–3289–98. [DOI] [PubMed] [Google Scholar]
- Bai S.; Ahsan F. Inhalable liposomes of low molecular weight heparin for the treatment of venous thromboembolism. J. Pharm. Sci. 2010, 99114554–64. [DOI] [PubMed] [Google Scholar]
- Stenmark K. R.; Meyrick B.; Galie N.; Mooi W. J.; McMurtry I. F. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am. J. Physiol. 2009, 2976L1013–32. [DOI] [PubMed] [Google Scholar]
- Ishida T.; Takanashi Y.; Doi H.; Yamamoto I.; Kiwada H. Encapsulation of an antivasospastic drug, fasudil, into liposomes, and in vitro stability of the fasudil-loaded liposomes. Int. J. Pharm. 2002, 2321–259–67. [DOI] [PubMed] [Google Scholar]
- Chono S.; Tanino T.; Seki T.; Morimoto K. Uptake characteristics of liposomes by rat alveolar macrophages: influence of particle size and surface mannose modification. J. Pharm. Pharmacol. 2007, 59175–80. [DOI] [PubMed] [Google Scholar]
- Patel B.; Gupta V.; Ahsan F. PEG-PLGA based large porous particles for pulmonary delivery of a highly soluble drug, low molecular weight heparin. J. Controlled Release 2012, 1622310–20. [DOI] [PubMed] [Google Scholar]
- Kim J. Y.; Kim J. K.; Park J. S.; Byun Y.; Kim C. K. The use of PEGylated liposomes to prolong circulation lifetimes of tissue plasminogen activator. Biomaterials 2009, 30295751–6. [DOI] [PubMed] [Google Scholar]
- Lian T.; Ho R. J. Trends and developments in liposome drug delivery systems. J. Pharm. Sci. 2001, 906667–80. [DOI] [PubMed] [Google Scholar]
- Conhaim R. L.; Eaton A.; Staub N. C.; Heath T. D. Equivalent pore estimate for the alveolar-airway barrier in isolated dog lung. J. Appl. Physiol. 1988, 6431134–42. [DOI] [PubMed] [Google Scholar]
- Toba M.; Alzoubi A.; O’Neill K.; Abe K.; Urakami T.; Komatsu M.; Alvarez D.; Jarvinen T. A.; Mann D.; Ruoslahti E.; McMurtry I. F.; Oka M. A novel vascular homing peptide strategy to selectively enhance pulmonary drug efficacy in pulmonary arterial hypertension. Am. J. Pathol. 2014, 1842369–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan J. V.; Chattopadhyay S.; Ang M.; Darwitan A.; Foo S.; Zhen M.; Koo M.; Wong T. T.; Venkatraman S. S. Sustained release of an anti-glaucoma drug: demonstration of efficacy of a liposomal formulation in the rabbit eye. PLoS One 2011, 69e24513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atyabi F.; Farkhondehfai A.; Esmaeili F.; Dinarvand R. Preparation of pegylated nano-liposomal formulation containing SN-38: In vitro characterization and in vivo biodistribution in mice. Acta Pharm. 2009, 592133–44. [DOI] [PubMed] [Google Scholar]
- Even-Chen S.; Cohen R.; Barenholz Y. Factors affecting DNA binding and stability of association to cationic liposomes. Chem. Phys. Lipids 2012, 1654414–23. [DOI] [PubMed] [Google Scholar]