

Abstract

The successful search for orally active iron chelators to treat transfusional iron-overload diseases, e.g., thalassemia, is overviewed. The critical role of iron in nature as a redox engine is first described, as well as how primitive life forms and humans manage the metal. The problems that derive when iron homeostasis in humans is disrupted and the mechanism of the ensuing damage, uncontrolled Fenton chemistry, are discussed. The solution to the problem, chelator-mediated iron removal, is clear. Design options for the assembly of ligands that sequester and decorporate iron are reviewed, along with the shortcomings of the currently available therapeutics. The rationale for choosing desferrithiocin, a natural product iron chelator (a siderophore), as a platform for structure–activity relationship studies in the search for an orally active iron chelator is thoroughly developed. The study provides an excellent example of how to systematically reengineer a pharmacophore in order to overcome toxicological problems while maintaining iron clearing efficacy and has led to three ligands being evaluated in human clinical trials.
Introduction
Life without iron is virtually unknown.1 This transition metal represents 5% of the earth’s crust and serves as nature’s most efficient redox engine. Iron is a member of the third transition triad and occurs in a variety of oxidation states, ranging from −2 in the iron carbonyls to +6 in iron oxides.2 However, the oxidation states [Fe(II), 3d6; Fe(III), 3d5] and the ensuing redox couple (Chart 1) are the most relevant and represent an equilibrium that is sensitive to both pH and the nature of the ligands surrounding the metal.3
Chart 1. Electron Transport Chain Illustrating the Redox Role of Iron.

Nature has exploited Fe(II)/Fe(III), pH, and ligand sensitivity very effectively. Depending on its environment, the metal can serve either as a reducing agent or an oxidizing agent (Chart 1). Probably, iron’s most well-studied role is how it facilitates the reduction of oxygen (eq 1). Since oxygen has a triplet ground state, it is unable to react with most organic species without the presence of iron. This reduction is accomplished in a stepwise fashion by a variety of iron-containing enzymes, providing 572 kJ of energy. While the metal plays an essential role in the operation of biochemical transformations critical to life4 (Table 1), as we shall see, too much iron can be problematic, even lethal, to humans. We will briefly overview catalase,5 peroxidase,6 and aconitase,7 which play important roles in controlling iron-mediated damage and homeostasis.8
Table 1. Iron Has a Central Role in a Large Number of Biological Redox Systems.
| •Hemoglobin: oxygen delivery | •Catalase: conversion of hydrogen peroxide to water and oxygen |
| •Cytochromes: oxidations | •Peroxidases: conversion of hydrogen peroxide to water |
| •Ribonucleotide Reductase: conversion of ribonucleotides to deoxyribonucleotides | •Iron Sulfur Proteins, e.g., aconitases and rubredoxins |
| 1 |
Iron is key to the function of nearly all redox systems in both primitive and advanced life forms. Thus, the importance of iron homeostasis is obvious. The mechanisms for the uptake, distribution, and excretion of iron are highly complicated but are nevertheless fairly well understood. The process represents a fragile balance, and there are dire consequences associated with failure. Although the effects of too little iron are easily reversed by providing the metal, the impact of too much iron is far more dangerous, especially in higher life forms, e.g., humans9 (Table 2). Table 2 is divided into two kinds of iron-mediated diseases. The first six are systemic disorders, and the last four are focal. The first of the systemic diseases, primary hemochromatosis,10 is defined by an uncontrolled iron absorption problem, while the next five derive from transfusional iron overload. The last four disorders are related by virtue of a focal iron buildup.
Table 2. Iron Is Pivotal in a Number of Disease States in Both Global and Focal Iron Overloada.
| •Primary Hemochromatosis |
| •Cooley’s Anemia |
| •Aplastic Anemia |
| •Sickle Cell Disease |
| •Myelodysplasia |
| •Diamond Blackfan Anemia |
| •Parkinson’s Disease |
| •Reperfusion Injury |
| •Hemorrhagic Stroke |
| •Macular Degeneration |
Global iron overload, first six syndromes; focal iron overload, last four syndromes.
Primary hemochromatosis is a genetic disorder that is caused by the absorption of excess iron from the gastrointestinal (GI) tract. This leads to a profound buildup of the metal in the liver, pancreas, and heart, and, if left untreated, it may ultimately lead to death due to cardiac failure. However, the disease can be easily managed by therapeutic phlebotomy. On the other hand, although Cooley’s anemia,11−13 aplastic anemia,14 sickle cell disease,15 myelodysplasia,16 and Diamond Blackfan anemia17 are also characterized by iron overload, the excess iron is a consequence of the required blood transfusions.
The transfusional iron-overload diseases are probably best exemplified by Cooley’s anemia, β-thalassemia. Thalassemia is a genetic disorder that derives from β-gene mutations, leading to a reduction in hemoglobin synthesis and a disruption of the ratio of α/β chains. The consequence of this unstable ratio is that α-chain aggregates precipitate and compromise red blood cell membranes, thus shortening the life of the erythrocytes. Patients become anemic and require blood transfusions for their entire lives. Each unit of red blood cells introduces ≈250 mg of iron into a closed loop. As with primary hemochromatosis, iron-induced organ damage unfolds unless the metal is removed. Since the patients are anemic, they cannot be bled to remove the excess metal. Iron chelation therapy is the only solution.
As with global iron-overload diseases, the focal iron-overload issues, e.g., Parkinson’s disease,18 hemorrhagic stroke,19 reperfusion damage,20 and macular degeneration,21 are defined by a number of diverse scenarios with one common denominator, unmanaged iron, which is frequently referred to as the nontransferrin-bound iron (NTBI) pool.22 While the origins of iron-mediated diseases are very different, the mechanism of the iron-induced damage is virtually always the same: production of hydroxyl radicals by the metal’s reaction with hydrogen peroxide,23,24 the Fenton reaction (eq 2). The hydroxyl radical25 is a highly reactive species, often compromising everything from membrane components to DNA. The availability of any number of biological reducing agents, e.g., glutathione, ascorbate, superoxide anion, and others, that reduce Fe(III) back to Fe(II) (eq 3) serves only to exacerbate the situation.
| 2 |
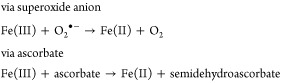 |
3 |
The Fenton reaction can be viewed as part of the body’s normal defense system. For example, when macrophages excrete peroxide,26 the peroxide is converted to hydroxyl radicals that likely serve to destroy foreign bodies, e.g., bacteria. The extent of the reaction is, of course, limited by the availability of iron. Therefore, a scenario in which there is too much iron would be problematic. Interesting examples of untoward Fenton chemistry in higher life forms are seen in reperfusion damage27 and in hemorrhagic stroke.19 Reperfusion damage derives from oxidative stress that is dependent on iron reducing hydrogen peroxide that originates from the conversion of hypoxanthine to xanthine and xanthine to uric acid.28 A molecule of hydrogen peroxide is produced each time the cycle is repeated. The physical cause of this phenomenon is usually a temporary vascular occlusion, during which time there is a buildup of xanthine and hypoxanthine. On release of the occlusion, oxygen becomes available, and there is a burst of hydrogen peroxide that can react with Fe(II) to produce hydroxyl radicals. Likewise, the mechanism behind the brain damage that occurs following a hemorrhagic stroke arises from the lysis of the red blood cells that are released during the bleed.29 Heme oxygenase liberates the iron from hemoglobin, and Fenton chemistry unfolds.29 Again, since humans have no means of eliminating the excess iron, the only way to prevent untoward iron-mediated damage is to sequester the metal and promote its excretion. A review of how primitive life forms and humans manage iron will help to circumscribe the solution to the iron-overload problem.
Iron and Primitive Life Forms: Siderophores
In the early biosphere, 3.5 billion years ago, iron existed largely as Fe(II), a highly soluble form of the metal.30 With the onset of blue–green algae and the production of oxygen generated by the ensuing photosynthesis, Fe(III) became the major oxidation state of the metal in the environment. This presented a serious access problem for microorganisms. The solubility product of ferric hydroxide31 under physiological conditions, Ksp = 10–38, translates to a free Fe(III) concentration of <10–18 M, a concentration well below that required for bacterial growth, ≈10–7 M. In order to overcome the problems associated with iron access, early prokaryotes managed to assemble and excrete iron-specific chelators to sequester the otherwise unavailable metal and render it utilizable. These ligands, siderophores32 (Figures 1, 2, 4, and 5), are highly Fe(III)-specific, with molecular weights generally between 400 and 1400. They are small molecules relative to their proteinaceous eukaryotic counterparts, e.g., transferrin,33,34 molecular weight 80 000. For example, to access iron, bacteria, e.g., Paracoccus denitrificans, a Gram-negative soil microorganism, secretes its siderophore, l-parabactin, into the environment.35 The five aromatic hydroxyls and the oxazoline ring nitrogen, shown to be the donor centers, coordinate to Fe(III).35 The siderophore forms a 1:1 metal complex (Figure 3) that eventually encounters the high-affinity receptor on the bacterial surface. The iron is released from the ligand, probably by a reductase; the free chelator dissociates to seek another Fe(III) atom, and the process begins again.
Figure 1.

Catecholamide siderophores.
Figure 2.

Hydroxamate siderophores.
Figure 4.

Retrosynthetic analysis of petrobactin.
Figure 5.

Siderophores outside of the catecholamate and hydroxamate motif: rhizobactin, rhizoferrin, pyochelin, and desferrithiocin (DFT).
Figure 3.

Putative structure of the Fe(III)/parabactin complex.
Although there are a number of notable exceptions, siderophores are most often hexacoordinate ligands, forming 1:1 complexes with Fe(III) and are largely defined by either their catechol36 (Figure 1) or hydroxamate37 (Figure 2) donors. These ligands are often predicated on polyamine38 or polyamine precursor backbones. For example, both l-parabactin35,39,40 and l-agrobactin,41,42 produced by Agrobacterium tumefaciens, are built on spermidine backbones; vibriobactin,47−49 isolated from Vibrio cholerae, is assembled on a norspermidine framework (Figure 1). Each of these chelators employs catecholamide and oxazoline phenol donors.
One of the most notable exceptions to ligands predicated on polyamine backbones is enterobactin, which is based on a macrocyclic serine system.32,43−46 This ligand was one of the very first siderophores discovered and is certainly the one most thoroughly studied. It provided investigators with a remarkable and quantitative understanding of the significance of ligand denticity and “fit” in metal binding. The advantage of a single ligand with three bidentate donors, e.g., enterobactin, is an entropic one and translates to enormous formation constants.50 The donor groups are held in position for optimal complexation with iron. The importance of the entropy issue becomes obvious when comparing the stability of the tris (2,3-dihydroxy-N,N-dimethylbenzamide) (DHBA)/Fe(III) complex with that of the enterobactin/Fe(III) complex. DHBA forms a 3:1 complex with the metal. The stepwise reactions and respective equilibrium constants are50
 |
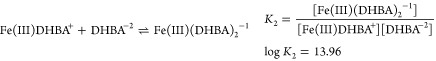 |
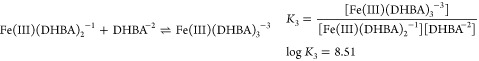 |
The stepwise equilibria for K1, K2, and K3 can be articulated in a nonstepwise format or as overall equilibrium constants.
The relationship between the stepwise and nonstepwise expressions is given as β3 = K1•K2•K3...•Kn = ∏Ki, i = 1 – n. Close attention must be given to the literature numbers so as not to confuse β with K values. The K values are useful in identifying which species are present and generally decrease as the chelator to metal ratio increases.
Thus, β3 for these reactions is calculated as β3 = K1•K2•K3 or log β3 = log K1 + log K2 + log K3 = 40.24. The equilibrium constant for the Fe(III)[enterobactin]−3 complex is50
 |
Members of the other large family of multidentate ligands, hydroxamates, are shown in Figure 2. Desferrioxamine51−55 (DFO), DFO G,56 DFO E,57 and Nannochelins A58 and B are all hexacoordinate ligands, forming 1:1 complexes with iron. It is notable that the formation constant for the Fe(III)–hydroxamate complexes is almost always significantly lower than that of their hexacoordinate catecholamide counterparts. The first three hydroxamate ligands were isolated from the same microorganism, Streptomyces pilosus. The question remains as to why one strain would produce three similar ligands. DFO, the most well known of these chelators, has served as the standard of care for patients suffering from iron-overload disorders for decades and is still widely used today.
Bisucaberin,59 isolated from the deep sea microorganism Alteromonas haloplanktis, and alcaligin,60 isolated from Alcaligenes denitrificans, form 1:1 complexes with Fe(III) under acidic conditions and 2:3 Fe2L3 complexes at and above neutral pH. Once again, as with the catecholamide chelators, almost all of the ligands depicted in Figure 2 are predicated on polyamine (cadaverine) or polyamine precursor (lysine) backbones, a relevant issue when articulating synthetic designs. This is probably best illustrated by the retrosynthetic description of the synthesis of petrobactin61−63 (Figure 4). Petrobactin is, in several ways, a very unusual chelator. It was first isolated from the oil-degrading marine microorganism Marinobacter hydrocarbonoclasticus.61 Later, the same siderophore was isolated from anthrax.63,64 The initially reported structure of petrobactin suggested that the compound was derived from citric acid, spermidine, and 2,3-dihydroxybenzoic acid (Figure 4, upper right). However, when the ligand was synthesized and the structure was verified by NMR spectra, it was discovered that petrobactin utilizes a 3,4-dihydroxybenzoic acid fragment.
Finally, Figure 5 illustrates several notable examples of ligands not presenting with either catecholamide or hydroxamate donors, i.e., not members of the two major siderophore families. Both rhizobactin,65 isolated from Rhizobium meliloti, and rhizoferrin,66 from Rhizopus microsporus, employ carboxylate and hydroxyl donors, forming 1:1 iron–ligand complexes at physiological pH. Again, both are predicated on a polyamine precursor backbone, e.g., lysine/rhizobactin or putrescine/rhizoferrin. The remaining two ligands, pyochelin,67 from Pseudomonas aeruginosa, and desferrithiocin,68 from Streptomyces pilosus, employ a thiazoline platform utilizing a phenol, a thiazoline nitrogen, and a carboxyl group as donors. The majority of our discussion will in fact focus on the latter siderophore, desferrithiocin [(S)-4,5-dihydro-2-(3-hydroxy-2-pyridinyl)-4-methyl-4-thiazolecarboxylic acid, DFT].
How Humans Manage Iron
Iron homeostasis in humans is defined by a well-controlled balance among absorption, distribution, storage, and excretion.69 The human body contains approximately 4–5 g of iron, but the metal is not evenly distributed. Nearly 60–70% is found in circulating erythrocytes, 20–30% is sequestered in the iron storage protein ferritin, and ∼10% serves as a redox component in systems such a myoglobin, cytochrome, and various enzymes (Table 1). Less than 0.1% is associated with the iron shuttle protein transferrin.
Absorption and Distribution of Iron
Iron is largely absorbed through the proximal small intestine as either inorganic iron or from heme. The metal first travels through the apical side of the enterocyte (Figure 6). The divalent metal transporter 1 (DMT1),70,71 also known as the natural resistance-associated macrophage protein 2 (NRAMP 2), or divalent cation transporter 1 (DCT1) only provides passage for Fe(II) into the enterocyte. Consequently, dietary iron, which is largely in the Fe(III) state, must first be reduced to Fe(II). A duodenal membrane-bound cytochrome b enzyme72 has been assigned this role. Precisely how heme iron, the most plentiful source of the metal, makes it into the enterocyte still remains somewhat unclear, although once inside, it is released by heme oxygenase.73,74 How iron moves within the enterocyte, again, remains undefined. However, it is understood that it exits at the basolateral side of the enterocyte, largely through the ferroportin transporter75 with the assistance of hephaestin,76,77 a transmembrane multicopper oxidase, ceruloplasmin78,79 analogue. The export of the metal from the enterocyte is now known to be controlled by hepcidin, an iron-dependent peptide hormone secreted from the liver. Hepcidin80,81 is upregulated in the liver under conditions of iron overload. This hormone, once released into the plasma, binds to ferroportin 1, causing it to be internalized and preventing iron release, thus completing the homeostatic loop for iron absorption (Figure 6).
Figure 6.

Iron absorption and processing.
Once outside of the enterocyte, Fe(III) is sequestered by apotransferrin and is shuttled as needed to a number of key locations. Diferric transferrin provides iron to the bone marrow for hematopoiesis, to the liver for storage by ferritin, and to various tissues to be incorporated by iron-dependent redox proteins. One of the critical steps in the iron cycle involves red blood cells. As described earlier, most of the body’s iron resides within the erythrocytes. The red blood cells normally die at approximately 120 days and are processed within the spleen. The released iron is picked up by apotransferrin and is moved to required sites. This iron loop is nearly closed with no mechanism to dispose of excess metal. The regulation of transferrin,82,83 the iron shuttle protein, and ferritin,84,85 the body’s iron storage protein, is highly efficient and controlled by iron levels. The fact that there is no conduit for the excretion of excess iron is the driver for the entire research area articulated in this review.
Transferrin
Transferrin is a homodimeric globular protein with a molecular weight of about 80 000. It is responsible for shuttling iron around the body and supplying it to depleted areas.82 A kind of eukaryotic siderophore, transferrin is a far larger molecule than the microbial siderophores described earlier, e.g., parabactin, molecular weight 625, for delivering iron to a prokaryote. In humans, while some transferrin synthesis occurs in the brain, testes, and mammary tissue, most of the protein is assembled in the liver. It binds two iron atoms each in a hexacoordinate array. The donors consist of two tyrosines, a histidine, and an aspartate and two oxygens from a carbonate anion. At physiological pH ∼ 7.2, the Fe(III) Kd is 10–23 M, while no significant chelation occurs at pH 4.5. Cells in need of iron effectively exploit the pH sensitivity: iron is released intracellularly within a vesicle by a pH change.83
Operationally, transferrin function is fairly well established, although precisely from where and how transferrin accesses its iron remains somewhat ill defined, thus the expression the “non-transferrin-bound iron” pool. Once iron is fixed, the complex binds to the transferrin receptor86 (TfR, Figure 7) on the cell surface. The receptor binding affinity varies as diferric transferrin ≫ monoferric transferrin > apotransferrin. The Kd of diferric transferrin, for example, is 25 nM. Because the plasma concentration of diferric transferrin is 3–6 μM, it is likely that most of the exposed receptors are saturated.87
Figure 7.

Transferrin/transferrin receptor cycle. The major steps, depicted counter clockwise, are (a) binding of Fe(III) (●) to transferrin (□, Tf), (b) binding of diferric transferrin to the transferrin receptor (TfR), (c) endocytosis by way of a clathrin-coated pit, (d) iron removal, (e) return of the apotransferrin–transferrin receptor complex to the cell surface, and (f) release of apotransferrin (ApoTf).
Once bound to the TfR at a ratio of 2:1, the megacomplex reacts with an adapter protein within a clathrin-coated pit, and the megacomplex is endocytosed. Reaching the endosome within the cell, the iron is released because of the low pH within the endosome (Figure 7). The iron is next passed through an endosomal DMT1 and picked up by other iron binding systems, e.g., heme, ferritin, and so forth. At this point, the apotransferrin receptor complex moves back to the cell surface, and the apotransferrin is released for further iron sequestration. It is notable that, generally speaking, transferrin is not a good target for iron chelation for two reasons: (1) it represents a minor fraction of the total iron pool and (2) the poor accessibility of ligands to transferrin-bound iron, derived from the fact that iron is buried within the protein; even ligands with a far higher Kf for iron than transferrin cannot access the metal.
Ferritin
The second largest pool of iron in the body is ferritin.88,89 Ferritin is a protein that stores iron in a soluble, nontoxic, readily available form and releases it in a controlled fashion. This protein consists of 24 subunits of heavy (H) and light (L) chains that are assembled into a hollow spherical shell. The H chain, believed to have ferroxidase activity, converts soluble Fe(II) entering the sphere to Fe(III) for mineralization.89 The molecular weight of ferritin, with all 24 subunits combined, is about 450 000, with 4500 stored iron atoms. Iron is released from ferritin by reduction back to Fe(II). Again, it is not a good target for chelation therapy simply because most ligands do not have access to the metal.
Control Mechanism for Iron Storage and Transport Proteins
The issue is simply how does cellular iron concentration control the storage and transport of iron. In the diseases we will discuss, when intracellular iron levels increase, ferritin levels would also be expected to increase, or, conversely, when intracellular iron levels decrease, the iron uptake apparatus should be upregulated. All of this occurs at a post-transcriptional level. For example, both transferrin receptor mRNA (mRNA)90 and ferritin mRNA91 contain an iron responsive element (IRE), to which the iron responsive protein (IRP) binds.
The IRP is an unusual bifunctional molecule. When cells are replete with iron, IRP, which contains an Fe–S cluster, exhibits aconitase activity.7 In this form, IRP cannot bind to the IRE. Under low iron conditions, apo-IRP binds very tightly to the IRE. The transferrin receptor mRNA contains five IREs in the 3′-untranslated region, while the ferritin mRNA presents with an IRE in the 5′-untranslated region. The IRE can either be an enhancer or a repressor of translation.
With ferritin, when apo-IRP is bound to the mRNA, it prevents the recruitment of the 43S preinitiation complexes. Thus, when iron levels increase, the IRP iron complex is released from the IRE, and ferritin synthesis begins. The mechanism of cellular iron control of transferrin receptor assembly is somewhat different. Apo-IRP binds to the 3′-IRE, enhancing mRNA stability and increasing translation by preventing degradation. Thus, a decrease in cellular iron upregulates the transferrin receptor mRNA. Other iron regulatory proteins, e.g., DMT192 (see above), have similar 3′-IRE control responses.
Transfusional Iron Overload
Because of space limitations, we will focus on transfusional iron-overload diseases, e.g., thalassemia,11−13 a global iron-overload problem. Nevertheless, many of the drugs that might derive from drug discovery and development efforts focused on this disease will likely be applicable to other global, e.g., sickle cell disease,15,93 and focal, e.g., hemorrhagic stroke,19,29 iron-overload issues.
There is no physiologic mechanism for the excretion of excess iron from the body other than blood loss, e.g., menstruation. Metal clearance through either the urine or the bile (feces) is virtually nonexistent. The body maintains approximately 4–5 g of iron, moving it very efficiently through a closed metabolic loop. Figure 6 best articulates the problem that needs to be overcome with global iron overload, e.g., thalassemia.
As described earlier, thalassemia is a genetic disorder that derives from a rather large number of point mutations, leading to a reduction in β-chain assembly. Often, the lack of correct equilibrium between the α- and β-chains produces unstable α-chain aggregates94 that precipitate, impacting on red blood cell membrane stability, causing cell lysis. Patients develop severe anemia and require frequent blood transfusions.
Each unit of blood contains approximately 250 mg of iron. The transfused red blood cells live for approximately 90 days and then die and are processed in the spleen. The iron released from heme is shuttled from the spleen by transferrin. The metal builds up in the liver, pancreas, and ultimately in the heart and can lead to primary hepatoma, diabetes, and heart failure. Unlike with patients with primary hemochromatosis,10 in which the excess iron may be removed by frequent phlebotomy, transfusional iron-overload patients cannot be bled. Again, the only solution is to chelate the metal and promote its excretion.
The nature of transfusional iron-overload diseases sets narrow boundary conditions on a potential chelator’s properties. Because these are genetic disorders, the patients will require lifelong exposure to the drug; this can translate to compliance issues. For example, one of the main drawbacks of desferrioxamine (DFO)53 is that it must be administered by subcutaneous (sc) infusion in the abdomen for 8–12 h a day, 5–7 days a week. Patients were often not compliant. Thus began the search for orally effective iron chelators. The ligands, then, must be highly specific for iron, should be orally active, and present with an especially clean toxicity profile.
Animal Models and Iron Clearing Efficiency
Because of the dependence of iron binding on so many different parameters, e.g., ligand denticity, stoichiometry, pH, and others, investigators developed the pM scale as a means to compare the potential effectiveness of ligands at sequestering iron.95 The pM values (−log [Fe3+]) express the amount of free iron present at equilibrium under the following conditions: a ligand concentration of 10–5 M, a total iron concentration of 10–6 M, and a pH of 7.4. The larger the pM value, the less free iron is present, and the more effective the chelator is at binding. A similar scale, referred to as iron clearing efficiency (ICE), was required to assess the performance of iron chelators in animals. The ICE, expressed as a percent, is calculated as (ligand-induced iron excretion/theoretical iron excretion) × 100.96 To illustrate, the theoretical iron excretion after the administration of 1 mmol of DFO, a hexadentate chelator that forms a 1:1 complex with Fe(III), is 1 milli-g-atom of the metal.
Two animal models are used in our laboratories to determine the ICE of the ligands, the non-iron-overloaded, bile duct-cannulated rodent97 and the iron-overloaded Cebus apella primate.98,99 The ICE of a new drug is first determined in the bile duct-cannulated rodent. This is a quick and efficient model to assess whether a chelator should be pursued further. Briefly, the rats are housed singly in metabolic cages. Bile samples are collected via a torque-transmitting tether and a fraction collector at 3 h intervals for up to 48 h. Urine samples are collected at 24 h intervals. Figure 8 illustrates the iron clearance properties of a DFT analogue given to the rodents orally (po) at a dose of 300 μmol/kg (equivalent to 100 mg/kg of DFO). With this ligand, maximal iron clearance occurs 3–6 h postdrug and has returned to baseline levels by 12–15 h postdrug. Approximately 90% of the iron was excreted in the bile, while 10% was cleared in the urine (Figure 8, bottom). The biliary ferrokinetics curve (iron excretion vs time, Figure 8, top) provides valuable information with regard to determining how frequently a drug may need to be given to allow for optimal iron clearance. If no overt toxicity was observed with the rodents,100 then the drugs were eligible to be evaluated in the iron-overloaded Cebus apella monkey model.
Figure 8.

Biliary ferrokinetics and iron excretion in non-iron-overloaded, bile duct-cannulated rats given a DFT analogue orally at a dose of 300 μmol/kg.
Briefly, the monkeys were iron-overloaded with intravenous (iv) iron dextran as previously described.101 At least 20 half-lives, 60 days, was allowed to pass before the animals were used in an iron clearance experiment. One week prior to the administration of an iron chelator, the monkeys were transferred from their usual housing to metal-free metabolic cages and were started on a low-iron liquid diet (<10 ppm Fe).100,101 Urine and stool samples were collected from the metabolic cages at 24 h intervals beginning on day −3 until day +5. The monkeys were anesthetized on day 0 and were given the chelator either po or sc. The iron content of the urine and bile/feces samples were assessed as previously described.101
Figure 9 illustrates the iron excretion induced by DFO given to the monkeys sc at a dose of 150 μmol/kg (100 mg/kg). Very little variability is noted in the predrug urine or stool samples. The DFO-induced iron excretion is observed in the urine and feces samples collected 1 day postdrug. The iron output then generally returned to baseline levels by day +2 and for the remainder of the collection period. The ICE of DFO in this group of primates was 5.1 ± 2.4%; approximately 40% of the iron was excreted in the urine.
Figure 9.

Iron excretion induced by DFO given to Cebus monkeys sc at a dose of 150 μmol/kg.
The Cebus apella primates have proven to be very effective at predicting how well a chelator will perform in humans.96,102 For example, the iron excretion induced by the administration of three chelators, DFO, 1,2-dimethyl-3-hydroxy-4-pyridone (L1), and N,N′-bis(2-hydroxybenzyl)ethylenediamine-N,N′-diacetic acid (HBED), to rodents, primates, and humans is depicted in Figure 10. DFO was given to the rats and monkeys as a sc bolus at a dose of 150 μmol/kg and to humans as an 8 h sc infusion at a dose of 92 μmol/kg.102 L1 was given po to the rats and primates at a dose of 450 μmol/kg and to humans po at a dose of about 540 μmol/kg.102 Finally, HBED was given to the rats and monkeys po at a dose of 150 μmol/kg and to humans po at doses of 103 or 206 μmol/kg.96,102 In the rats, the order of chelator efficacy was HBED > DFO > L1. However, in both the monkeys and humans, the order was DFO > L1 > HBED (Figure 10). In addition, the mode of excretion, i.e., the fraction of iron excreted in the urine and feces, is very similar when comparing the data from the primates and man. Interestingly, although HBED is ineffective when given po to the primates, it is very effective when dosed sc.103−105 However, the efficacy of HBED given sc to patients has not been determined.
Figure 10.
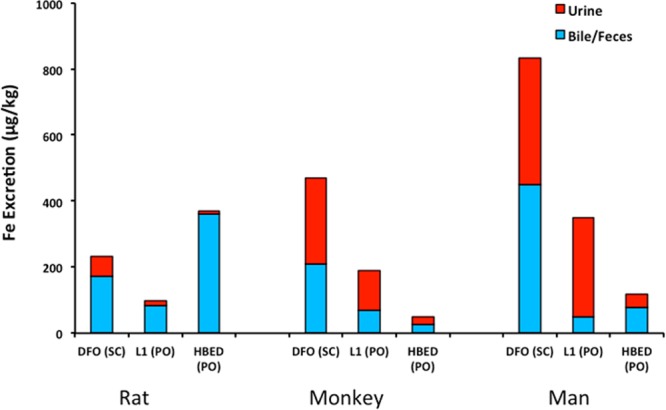
Comparison of chelator-induced iron excretion in rats, monkeys, and man. DFO was given as a sc injection in rats and primates and as an 8 h sc infusion in humans. HBED and L1 were given to all three species po.
Chelator Design Concepts
There are three potential platforms from which to choose: (1) a natural product iron chelator (a siderophore, Figures 1, 2, and 5), (2) a totally synthetic system, or (3) a hybrid, i.e., a structurally modified siderophore. The best example of a natural product iron chelator for treatment of transfusional iron overload is, of course, desferrioxamine (Figure 2).51−54 This trihydroxamate ligand, isolated from Streptomyces pilosus, forms a 1:1 hexacoordinate complex with Fe(III), Kd = 1 × 10–28 M. It was the standard of care for many years and is still in use today. While DFO is highly selective for Fe(III), it is only moderately effective in humans when given sc or iv; it is not active when given po. Its lack of oral activity is almost certainly due to its highly polar nature, as reflected in its poor lipid solubility, log Papp < −3.2.106
There is a concern about utilizing a siderophore or a siderophore analogue as a platform for developing iron decorporation therapeutics. Although siderophores are very efficient at sequestering iron, they can also promote the growth of bacterial pathogens.46,107,108 For example, desferrioxamine administration has been associated with the occurrence of Yersinia enterocolitica septicemia in humans,55 but this is uncommon. However, it has also been demonstrated that small changes in the siderophore, e.g., stereochemistry, can profoundly reduce the ligand’s capacity to promote bacterial growth. For example, when Escherichia coli was presented with enantioenterobactin,107,109 there was no growth stimulation. This was also the case with enantioparabactin.108 It is also noteworthy that when microorganisms are exposed to foreign chelators, such ligands can shut down their growth.110
Nature has, in fact, been very effective in managing the issue of catechol chelator-based siderophore stimulation of bacterial growth. Many animals, including humans, chickens, and quail, secrete siderocalins that bind catechol siderophores, thus intercepting iron meant for an invading pathogen and limiting its growth.111 In the end, it is difficult to predict if modified siderophore platforms will serve to stimulate bacteria growth. However, investigators must remain cognizant of these issues in the early development stages of chelator design strategies.
Many synthetic ligands (Figure 11) have been assembled and studied for their ability to bind iron in situ as well as for their capacity to clear iron from animals. The earlier work is probably best exemplified by Martell’s studies on polycarboxylate112−115 chelators. While the work was largely focused on the thermodynamics of iron binding, a number of therapeutically useful chelators, e.g., diethylenetriaminepentaacetic acid (DTPA)116,117 and HBED,118−120 were identified. Both of these ligands form 1:1 hexacoordinate complexes with Fe(III). Although they are moderately effective at iron decorporation from humans when given sc or iv, they are not effective when administered po. While HBED is fairly selective for Fe(III), DTPA is not. Its capacity to decorporate a variety of metals other than iron, e.g., zinc, results in a number of unacceptable side effects. Interestingly, DTPA emerged in the clinic slightly before desferrioxamine for the treatment of transfusion-related iron overload.
Figure 11.

Six synthetic chelators, four of which (Exjade, DTPA, L1, and HBED) have been used successfully in humans. CP94 represents a failed attempt to improve on the plasma residence of L1 with the idea of increasing the ICE. TREN-(Me-3,2-HOPO) articulates a successful effort to construct a hexacoordinate ligand from the bidentate L1 platform. Unfortunately, it did not perform well in animals.
Four additional synthetic ligands are depicted in Figure 11. These include three hydroxypyridones, deferiprone (L1, CP20),121−123 1,2-diethyl-3-hydroxy-4-pyridinone (CP94),124 tris[(3-hydroxy-1-methyl-2-oxo-1,2-dihydropyridine-4-carboxamido)ethyl]amine [TREN-(Me-3,2-HOPO)],125−127 and the triazole 4-[3,5-bis(2-hydroxyphenyl)-1,2,4-triazol-1-yl]benzoic acid (deferasirox, Exjade).128−130 The hydroxypyridones represent a well-articulated structure–activity relationship (SAR) study.121−124,131,132 L1 is a bidentate ligand133,134 that forms a 3:1 complex with Fe(III) at millimolar concentrations at neutral pH.133 However, a dilution effect seen in speciation studies suggests that a substantial fraction of a 2:1 Fe(III) complex (40%) exists at micromolar ligand concentrations at pH 7.135 It has been suggested that this may account for some of the ligand-induced side effects seen in patients. The 2:1 stoichiometry allows for access to the metal by biological reductants and could lead to the production of reactive oxygen species.
Nevertheless, L1 was the first orally active iron chelator136 and served as a platform for the development of more active, less toxic analogues.137 The first issue investigators focused on was improving the ligand’s iron clearing efficiency, which was moderate at best. Studies suggested that a daily dose of 75 mg/kg was required to keep patients in negative iron balance.138 This was problematic in view of the toxicity issues. The poor iron decorporation in man was attributed to the high level of the drug’s phase II metabolism in the liver. The hydroxyl group, which is essential for chelation, undergoes extensive (>85%) glucuronidation.139 Thus, the residence time for the active ligand is shortened.
In an attempt to solve this problem, investigators constructed CP94, the diethyl analogue of L1 (Figure 11).132 It had a better ICE than L1 in rodents and was 3.5 times more effective than L1 in primates.140 In addition, CP94 given to the monkeys po was 1.3 times as effective as an equimolar dose of DFO administered sc.140 This increase in ICE was attributed to less glucuronidation. In rodents, only 14% of CP94 was found to form a 3-O-glucoronide versus 44% for L1.139 Unfortunately, the increase in efficacy noted in rats and primates was not reflected in human clinical trials.141,142 A variety of other modifications on L1 have been carried out,143−145 but none have been evaluated clinically.
A further attempt to improve on the hydroxypyridone platform involved the assembly of a hexacoordinate hydroxypyridone, TREN-(Me-3,2-HOPO)125−127 (Figure 11). This hexacoordinate ligand could, in principle, alleviate the toxicity problems associated with the formation of a 2:1 Fe(III) complex that was an issue with L1. Furthermore, it could, in theory, be more selective for Fe(III). The molecule’s iron binding properties were thoroughly studied in both iron-loaded and non-iron-loaded rodents.125 When given po to iron-loaded rats at a dose of 30 μmol/kg, the ICE was 8.3 ± 2.6%. However, the same dose in the non-iron-overloaded animals was virtually ineffective.125 This was very surprising and disappointing. It seems like little else has been done with this ligand as an iron decorporating agent.
Certainly, the most well known of all of the synthetic chelators is Exjade,128−130 desferasirox, developed by Novartis (Figure 11). The ligand emerged from a well-articulated, rational drug design program including very extensive molecular modeling and screening studies. It is a tridentate ligand built on a triazole platform that is highly selective for iron and has a low affinity for trace metals, such as zinc or copper. It forms 2:1 complexes with Fe(III) and is orally active. Unfortunately, the ligand presents with several shortcomings: it does not show noninferiority to DFO and is associated with numerous side effects, including some serious renal toxicity issues.146
The last platform, hybrid ligands predicated on modified siderophores, is probably best characterized by the work on catecholamides. This received tremendous attention early on. The catecholamide bidentate fragments are found in a variety of siderophores, e.g., enterobactin, parabactin, vibriobactin, and others (Figure 1), all of which bind iron very tightly. These natural products form 1:1 hexacoordinate complexes with Fe(III) with formation constants ≈1048 M–1. While some of these siderophores do, in fact, remove iron from animals when they are administered sc, they do not work when given po. However, the catecholamide-based chelators were key in defining coordination chemistry issues surrounding ligand design concepts.
Much of the original work was dedicated to assembling ligands with catecholamide donors and the evaluation of the significance of entropy on Fe(III) binding.147−150 Unfortunately, in spite of the enormous number of catecholamide-based chelators synthesized, very little is published on their ICE in animals, and virtually nothing in humans has been published. Nevertheless, the outcome of these studies did underscore issues that are important in the design and assembly of clinically useful iron chelation agents. The significance of the geometry and denticity of ligands in the thermodynamics of iron binding was well-articulated. Assuming the same donor groups and approximate distance between them, iron binding generally followed the trend hexadentate ≫ tridentate > bidentate. Unfortunately, the thermodynamics of iron binding is almost of no value in predicting how well ligands will perform as iron decorporating agents in animals, particularly when administered orally. The formation constant serves as a kind of go/no-go gauge. If the ligands bind iron poorly, then they are not likely to work in animals. If they bind iron tightly, they at least merit animal trials. Several parameters govern the performance of chelators in animals: GI absorption of the drug, organ distribution, and plasma residence time. Once it has been shown that a chelator can remove iron from an animal when administered either po or sc, there is the entirely separate but critical issue of ligand toxicity. The modified siderophore platforms that best characterize the success of this approach are the ligands predicated on desferrithiocin (DFT).
DFT: Structure–Activity Studies
DFT is a siderophore that was isolated from Streptomyces antibioticus.68 It is a tricoordinate ligand and can be viewed as derived from the condensation of 3-hydroxypicolinic acid (1) and (S)-α-methyl cysteine (2) (Figure 12). It forms a 2:1 complex with Fe(III) with a β2 = 4 × 1029.151 On the basis of studies of the Cr(III) surrogate, two iron diastereomers, the Λ and Δ complexes, are likely formed.152 The compound was shown to be an excellent deferration agent when given po to rodents (ICE 5.5 ± 3.2%)97 and Cebus apella primates (ICE 16.1 ± 8.5%).101,153 However, it caused severe nephrotoxicity in rats.100,154 Nevertheless, the ligand’s remarkable oral activity spurred an extensive SAR study focused on the DFT platform aimed at identifying an analogue with similar iron clearing properties but without the attendant nephrotoxicity. These SAR studies have led to three different DFT analogues being evaluated in human clinical trials.
Figure 12.

A retrosynthetic overview of desferrithiocin (DFT).
Two synthetic considerations guided the first SAR study on DFT: the potential problems in accessing its unusual α-methyl cysteine fragment and the complications associated with modifying the pyridine ring. Removal of the DFT methyl group155,156 (Figure 13) provided (S)-4,5-dihydro-2-(3-hydroxy-2-pyridinyl)-4-thiazolecarboxylic acid (DMDFT, 3) and diminished the ICE in both rodents (2.4 ± 0.6%) and primates (8.0 ± 2.5%). Removal of the pyridine nitrogen, leading to (S)-4,5-dihydro-2-(2-hydroxyphenyl)-4-methyl-4-thiazolecarboxylic acid (DADFT, 4), provided a chelator with ICE values similar to the parent, 2.7 ± 0.5% (rodents) and 21.5 ± 12% (primates). Removal of both the nitrogen and the methyl, yielding (S)-4,5-dihydro-2-(2-hydroxyphenyl)-4-thiazolecarboxylic acid (DADMDFT, 5), resulted in a compound with a reduced ICE in rodents (1.4 ± 0.6%) and primates (12.4 ± 7.6%). While the three new ligands did not present with any renal toxicity, 4 and 5 did cause serious GI toxicity (Figure 14).
Figure 13.

Structure–activity relationships of the desferrithiocins and iron clearing efficiency. The dose of DFT or analogue in the rats is 150 μmol/kg; the dose in the monkeys is as shown in parentheses for each ligand. The mode of administration is shown in parentheses next to the efficiency (%, ±standard deviation). The fraction of iron excreted in the bile or stool and urine is shown in brackets.
Figure 14.

Structure–activity relationship of the DFTs and toxicity. The ligands were administered orally at a dose of 384 μmol/kg/day for up to 10 days. Note that this dose is equivalent to 100 mg/kg/day of the sodium salt of DFT.
Nevertheless, because of the profound change in toxicity profiles noted with DMDFT (3), DADFT (4), and DADMDFT (5), the ligands served as pharmacophores on which to base further SAR studies. It was anticipated that we would be able to program out the toxicity. In addition, derivatives of 4 and 5 are more synthetically accessible than those of DFT, principally because of the absence of the 3-hydroxypicolinic acid fragment (1). Thus, the systematic alteration of up to five structural parameters of the ligands were evaluated for their impact on the molecules’ toxicity and/or ICE.153,155−159 The structural alterations included (1) modifying the distance between the donor groups, (2) modifying the thiazoline ring, (3) changing the stereochemistry at C-4, (4) altering the lipophilicity of the ligand by benzfusion, or by (5) fixing small substituents to the aromatic ring.
Changing the distance separating the donor groups of DADMDFT (5) by the insertion of a methylene between the thiazoline and the aromatic rings (6) or a methylene (7) or ethylene (8) between the thiazoline and carboxylate resulted in ligands with virtually no ICE (Figure 15).157 Very little iron clearance was also seen when converting the thiazoline ring to dihydropyrrole 9, thiazole 10, thiazolidine 11, dihydroimidazole 12, dihydro-1,3-thiazine 13, or oxazolines 14 and 15 (Figure 16).158 The effect on ICE of changing the stereochemistry at C-4 of (S)-DMDFT (3), (S)-DADMDFT (5), and (S)-2-(2,4-dihydroxyphenyl)-4,5-dihydro-4-thiazolecarboxylic acid [4′-(HO)-DADMDFT, 17] to the corresponding (R) compounds 19, 18, or 16 was interesting, but it was not consistent.153,159 The most notable data set was for 3 and 4′-(HO)-DADMDFT (17): the primates had better ICE with these (S)-enantiomers than with corresponding (R)-enantiomers 19 and 16 (Figure 17).153,159 Changes in the lipophilicity of (R)- and (S)-DADMDFT and DMDFT was achieved through benzfusion.159 This manipulation had very little effect on the molecules’ ICE values, but it did serve to further underscore the idea that the (S) enantiomers perform better in the primates than their (R) counterparts (23 vs 22 and 25 vs 24) (Figure 18).159
Figure 15.

Alteration of distances between chelating centers. The rats were given the ligands po or sc at a dose of 150 μmol/kg.
Figure 16.

Thiazoline ring modifications. The rats were given the ligands po or sc at a dose of 150 μmol/kg; the dose in the monkeys is as shown in parentheses. The mode of administration is shown in parentheses next to the efficiency.
Figure 17.

Impact of C-4 stereochemistry of DMDFT, DADMDFT, and 4′-(HO)-DADMDFT on iron clearing efficiency. The rats were given the chelators po at a dose of 150 μmol/kg; the dose in the monkeys is as shown in parentheses for each ligand. The mode of administration is shown in parentheses next to the efficiency (%, ± standard deviation). The fraction of iron excreted in the bile or stool and urine is shown in brackets.
Figure 18.

Increase in lipophilicity by benzfusion. The rats were given the chelators at a dose of 150 μmol/kg; the dose in the monkeys is as shown in parentheses for each ligand. The mode of administration is shown in parentheses next to the efficiency (%, ± standard deviation). The fraction of iron excreted in the bile or stool and urine is shown in brackets.
Finally, changing the lipophilicity (log Papp) of the DADFT and DADMDFT platforms by fixing small substituents to the aromatic ring had a profound effect on ICE (Table 3).160 The log Papp data are expressed as the log of the fraction of chelator in the octanol layer; measurements were done in TRIS buffer, pH 7.4, using a shake flask direct method.161 The more negative the log Papp, the less chelator is in the octanol phase, the less lipophilic. Within a structural subtype, e.g., the 2′,4′- and 2′,3′-dihydroxy substituted ligands, there is a linear relationship between lipophilicity and ICE (Figure 19).160 It is clear that the more lipophilic chelators are more effective at removing iron. However, there exists a delicate balance among lipophilicity, ICE, and toxicity. The more highly lipophilic compounds tend to be more toxic (Table 4).162
Table 3. Desferrithiocin Analogues’ Iron Clearing Activity When Administered Orally to C. apella Primates vs the Partition Coefficients of the Compounds.

In the monkeys [n = 4 (17, 26, 27, 30), 7 (28), 5 (32), 6 (31), or 8 (29)], the dose was 150 μmol/kg. The efficiency of each compound was calculated by averaging the iron output for 4 days before the administration of the drug, subtracting these numbers from the 2 day iron clearance after the administration of the drug, and then dividing by the theoretical output; the result is expressed as a percent. The relative percentages of the iron excreted in the stool and urine are in brackets.
Data are expressed as the log of the fraction in the octanol layer (log Papp); measurements were done in TRIS buffer, pH 7.4, using a shake flask direct method.
Figure 19.

Iron-clearing efficiency (percent) in Cebus monkeys of 4′-substituted ligands 17, 26–28 (blue circles) and 3′-substituted analogues 29–32 (red squares) plotted versus the respective partition coefficients (log Papp) of the compounds. The primates were given the drugs po at a dose of 150 μmol/kg.
Table 4. Partition Coefficients and Tolerability of DFT Analogues To Examine the Relationship between Log Papp (Lipophilicity) and Ligand Toxicity.

Data are expressed as the log of the fraction in the octanol layer (log Papp); measurements were done in TRIS buffer, pH 7.4, using a shake flask direct method.
The rats were given the drugs po once daily at a dose of 384 μmol/kg/day for up to 10 days. Note that this dose is equivalent to 100 mg/kg/day of the DFT sodium salt.
The most promising finding from these studies was that ligands with small substituents, such as a hydroxyl group on the aromatic ring, with the “correct” lipophilicity can be profoundly less toxic than the parent and have excellent ICE values (Tables 3 and 4). As can be seen in Figure 20, the success of this SAR is perhaps best illustrated by the DADFT analogue (S)-2-(2,4-dihydroxyphenyl)-4,5-dihydro-4-methyl-4-thiazolecarboxylic acid [(S)-4′-(HO)-DADFT, deferitrin, 26]153 and the corresponding DADMDFT ligand (S)-2-(2,4-dihydroxyphenyl)-4,5-dihydro-4-thiazolecarboxylic acid [(S)-4′-(HO)-DADMDFT, 17].102 The kidney from the animal treated with DFT is blanched and very friable, whereas the stomach is normal (Figure 20). In contrast, the kidney from rats given DADFT (4) or DADMDFT (5) appears normal, whereas the stomach is bloated and hemorrhagic. Finally, the kidney and stomach of rats dosed with (S)-4′-(HO)-DADFT (26) or (S)-4′-(HO)-DADMDFT (17) appear normal. Thus, the renal and GI abnormalities found in rodent toxicity studies of DFT were essentially ameliorated. The gross anatomical observations were supported by histopathology. These results led to a human clinical trial with deferitrin (26).
Figure 20.

Outcome of structure–activity relationship studies on desferrithiocin. Small structural alterations can have a profound effect on renal and GI toxicity. The kidney from the animal treated with DFT is blanched and very friable, while the stomach is normal. In contrast, the kidney from rats given DADFT (4) or DADMDFT (5) appears normal, while the stomach is bloated and hemorrhagic. Finally, the kidney and stomach of rats dosed with (S)-4′-(HO)-DADFT (26) or (S)-4′-(HO)-DADMDFT (17) appear normal.
Initial clinical trials with deferitrin looked very promising.163 Chelator 26 was well-tolerated in patients at doses of 5, 10, or 15 mg/kg/day once daily (s.i.d.) for up to 12 weeks, and iron clearance levels were approaching the requisite 450 μg/kg/day.164 However, when the drug was given twice daily (b.i.d.) at a dose of 12.5 mg/kg (25 mg/kg/day), unacceptable renal toxicity was observed in three patients after only 4–5 weeks of treatment, and the study was terminated.165 The directive then became to engineer a ligand that would not cause any nephrotoxicity even when it was given b.i.d.
Reengineering (S)-4′-(HO)-DADFT (26)
The guiding tenet for this SAR, and an overview of the relationship between a ligand’s log Papp and tolerability, can be seen in Table 4. Previous studies revealed that within a family of ligands the more lipophilic compounds generally have better iron clearing efficiency. However, there also exists a second, albeit somewhat more disturbing, relationship: the greater the lipophilicity of a chelator, the more toxic it is.(166) Thus, the challenge was to design ligands that balance the lipophilicity/toxicity problem while maintaining ICE. Earlier studies with (S)-4,5-dihydro-2-(2-hydroxy-4-methoxyphenyl)-4-methyl-4-thiazolecarboxylic acid [(S)-4′-(CH3O)-DADFT, 28]166 and (S)-2-(2-hydroxy-3-methoxyphenyl)-4,5-dihydro-4-methyl-4-thiazolecarboxylic acid [(S)-3′-(CH3O)-DADFT, 32]160 indicated that these methyl ethers were ligands with excellent ICE in both rodents and primates (Table 3). However, the former ligand was too toxic.162 With these observations in hand, it was decided that these chelators would serve as platforms from which to generate more water-soluble, less lipophilic compounds with acceptable ICEs. The plan then became, in a formal sense, to anneal a 2,5,8-trioxanonyl polyether fragment to the DADFT aromatic ring methyl groups of 28 and 32, providing (S)-4,5-dihydro-2-[2-hydroxy-4-(3,6,9-trioxadecyloxy)phenyl]-4-methyl-4-thiazolecarboxylic acid [(S)-4′-(HO)-DADFT-PE, 34], a polyether analogue,162 and the corresponding 3′-polyether analogue [(S)-4,5-dihydro-2-[2-hydroxy-3-(3,6,9-trioxadecyloxy)phenyl]-4-methyl-4-thiazolecarboxylic acid [(S)-3′-(HO)-DADFT-PE, 36].167 The next step would be to determine if this functionalization was compatible with good ICE properties in rodents and primates, and reduced toxicity in rodents. As will be described below, this was a very effective strategy.
Synthesis of (S)-4′-(HO)-DADFT-PE (34) and (S)-3′-(HO)-DADFT-PE (36)
The boundary conditions for the synthetic design of the polyether ligands required that they had to be easily accessible in large quantities (Schemes 1 and 2). Deferitrin ethyl ester 37 was selectively alkylated at the 4′ postion by heating it with tosylate 38 and potassium carbonate in acetone, providing masked chelator 39. Cleavage of ethyl ester 39 in aqueous base gave 4′-polyether 34 (Scheme 1).168
Scheme 1. Synthesis of 34.

Reagents and conditions: (a) K2CO3 (2.1 equiv), acetone, 84%; (b) 50% NaOH (13 equiv), CH3OH, then 1 N HCl, rt, 16 h, 93%.
Scheme 2. Synthesis of 36.

Reagents and conditions: (a) 60% NaH (2.0 equiv), DMSO, 70%; (b) CH3OH(aq), pH 6, 70 °C, 16 h, 90%.
The less hindered phenolic group of 2,3-dihydroxybenzonitrile (40) was alkylated with tosylate 38 and sodium hydride (2 equiv) in DMSO at room temperature, generating nitrile 41 (Scheme 2). Cyclization of 41 with (S)-α-methyl cysteine (2) in aqueous CH3OH buffered at pH 6 completed the synthesis of 3′-polyether 36.168
ICE of (S)-4′-(HO)-DADFT-PE (34) and (S)-3′-(HO)-DADFT-PE (36) in Rats and Primates
When a 3,6,9-trioxadecyl group was fixed to either the 4′-(HO) of (S)-4′-(HO)-DADFT (26), providing (S)-4′-(HO)-DADFT-PE (34) (Table 5), or the 3′-(HO) of (S)-3′-(HO)-DADFT (30), providing (S)-3′-(HO)-DADFT-PE (36) (Table 5), both ligands were less lipophilic than both their parents and their corresponding O-methyl analogues, 34 vs 26 and 28; 36 vs 30 and 32.162,167,168 Thermodynamically, (S)-3′-(HO)-DADFT-PE (36) (log β2 = 33.1)169 is superior to parent molecule DFT (log β2 = 29.6)151 as an iron(III) chelator. Moreover, the concentration of free iron(III) in the presence of excess 36 (pM = 22.3) is comparable to that with desferasirox, also a tricoordinate ligand, (pM = 22.5) at a pH of 7.4.169
Table 5. Iron Clearing Activity of Desferrithiocin Analogues Administered Orally to Rodents and Primates and the Partition Coefficients of the Compounds.

Data are expressed as the log of the fraction in the octanol layer (log Papp); measurements were done in Tris buffer, pH 7.4, using a shake flask direct method.
In the rodents [n = 4 (28, 30, 36), 5 (32, 34), or 8 (26)], the rats were given a single 300 μmol/kg dose of the ligands orally by gavage. The compounds were given as their sodium salts, prepared by the addition of 1 equiv of NaOH to a suspension of the free acid in distilled water. Compound 34 was solubilized in distilled water. The efficiency of each compound was calculated by subtracting the iron excretion of control animals from the iron excretion of the treated animals. The number was then divided by the theoretical output; the result is expressed as a percent. The relative percentage of the iron excreted in the bile and urine are in brackets.
In the monkeys [n = 4 (30, 34), 5 (32), 6 (26), or 7 (28, 36)], the drugs were given po at a dose of 75 μmol/kg (36) or 150 μmol/kg (26, 28, 30, 32, 34). The compounds were solubilized in either distilled water (34), 40% Cremophor (28, 30, 32), or were given as their monosodium salts, prepared by the addition of 1 equiv of NaOH to a suspension of the free acid in distilled water (26, 36). The efficiency of each compound was calculated by averaging the iron output for 4 days before the administration of the drug, subtracting these numbers from the 2 day iron clearance after the administration of the drug, and then dividing by the theoretical output; the result is expressed as a percent. The relative percentages of the iron excreted in the stool and urine are in brackets.
The performance ratio (PR) is defined as the mean ICEprimates/ICErodents.
In the bile duct-cannulated rodent (Table 5), the iron clearing efficiency of the parent (S)-4′-(HO)-DADFT (26) was poor, 1.1 ± 0.8%. However, 4′-(HO) methylation to (S)-4′-(CH3O)-DADFT (28) had a profound effect on ICE, 6.6 ± 2.8%.162 Interestingly, the less lipophilic (S)-4′-(HO)-DADFT-PE (34)162 had an ICE similar to that of the methoxy compound 28, 5.5 ± 1.9%. The scenario with the (S)-3′-(HO)-DADFT (30) series was similar.162,167,168,170,171 The 3′-(HO) parent 30 itself performed better in rodents than its 4′-(HO) counterpart (26), 4.6 ± 0.9% vs 1.1 ± 0.8%. Again, methylation to (S)-3′-(CH3O)-DADFT (32) also increased the ICE relative to the parent, 12.4 ± 3.5% vs 4.6 ± 0.9%. Fixing a polyether to the 3′-position to produce (S)-3′-(HO)-DADFT-PE (36) led to a ligand similar to the methoxy analogue with an ICE of 10.6 ± 4.4%.167
The chelators all performed significantly better in the primates than in the rats, and the results were much more homogeneous (Table 5).167,168,170,171 The least effective ligand was the parent, (S)-4′-(HO)-DADFT (26), with an ICE of 16.8 ± 7.2%. The performance ratio (PR) of this ligand, defined as the mean ICEprimates/ICErodents, was 15.3. All of the other analogues were very effective iron decorporating agents in the primates with ICE values of around 25% and PRs ranging from 1.8 to 5.0 (Table 5). Thus, the changes in log Papp did not have near the effect on ICE in the primates as was observed in the rodents. What was most remarkable, though, was the change in the toxicity profile.
Toxicity Assessment of Deferitrin (26), (S)-4′-(HO)-DADFT-PE (34), and (S)-3′-(HO)-DADFT-PE (36) in Rodents: Renal Perfusion Studies
Recall that although deferitrin was generally well tolerated when given to patients s.i.d. at doses of 5, 10, or 15 mg/kg/day, administering the drug b.i.d. at 12.5 mg/kg/dose (25 mg/kg/day) was associated with unacceptable renal toxicity.163,165 Because of the apparent increase in renal toxicity observed in patients treated with 26 b.i.d. versus s.i.d., we elected to determine if this damage could be reproduced in the rodents.
In a preliminary dose-range finding study, male Sprague–Dawley rats were given deferitrin b.i.d. for 7 days at doses of 237, 355, or 474 μmol/kg/dose (474, 711, or 947 μmol/kg/day).162 The drug was found to cause moderate to severe vacuolization in the renal proximal tubules at all doses. It was decided that 26 would serve as a positive control and would be given to the rats s.i.d. at 474 μmol/kg/day × 7 days or b.i.d. at 237 μmol/kg/dose (474 μmol/kg/day) × 7 days. Note that this dose represents 120 mg/kg/day of 26. Additional groups of rats were given the 3′- and 4′-polyethers (36 and 34, respectively) b.i.d. at 237 μmol/kg/dose (474 μmol/kg/day) × 7 days. Aged-matched animals would serve as untreated controls.
Under light microscopy, the proximal and distal tubules of kidneys from the control animals show normal tubular architecture (Figure 21A).162 The kidneys of rodents treated with 26 s.i.d. (Figure 21B) displayed some vacuolization of the proximal tubule cells. However, the damage was much more severe when the drug was given twice daily. The kidneys of rodents treated with 26 b.i.d. at 237 μmol/kg/dose (474 μmol/kg/day) for 7 days show heavy vacuolization and thinning of the apical membranes (Figure 21C).162,168 Interestingly, and much to our surprise, besides some inclusion bodies seen when either 4′-polyether 34 or 3′-polyether 36 was given at 237 μmol/kg b.i.d. for 7 days, there was little if any damage to the proximal tubules (panels D and E of Figure 21, respectively).162,168 This allows for tremendous flexibility in dosing schedules with the polyethers.
Figure 21.

Renal perfusion. Control (A), 26 474 μmol/kg s.i.d. × 7 days (B), 26 237 μmol/kg b.i.d. × 7 days (C), 34 237 μmol/kg b.i.d. × 7 days (D), and 36 237 μmol/kg b.i.d. × 7 days (E). Magnification = 400×.
Metabolism of (S)-4′-(CH3O)-DADFT (28) and 3′- (36) and 4′-Polyethers (34)
Early metabolic studies with (S)-4′-(CH3O)-DADFT160 (28) given to rats sc at a dose of 300 μmol/kg revealed that the drug was demethylated in the liver, producing (S)-4′-(HO)-DADFT (26).160 This observation encouraged a comparative tissue distribution/metabolism study of (S)-4′-(CH3O)-DADFT (28), (S)-4′-(HO)-DADFT-PE (34), and (S)-3′-(HO)-DADFT-PE (36) (Figure 22). If, for example, 34 was converted to deferitrin (26) to any great extent, then this would preclude it being given b.i.d. The tissues that were evaluated included the liver, kidney, heart, pancreas, and plasma. The only tissue that presented with any 4′ or 3′ ether chain cleavage at the 4′ position of 34 or the 3′ position of 36 was the liver (Figure 22). At 2 h, about 30% of (S)-4′-(CH3O)-DADFT (28) has been demethylated to (S)-4′-(HO)-DADFT (26), and the metabolite remains at fairly high levels through the 8 h time point. While the polyethers 34 or 36 show some cleavage to 26 and 30, respectively, it is minor.162 At 2 h, 2% of 34 is converted to 26, and 2.6% of 36 is metabolized to 30. The metabolites are no longer detectable at the 4 and 8 h time points (Figure 22).
Figure 22.

Metabolic profiles of desazadesferrithiocin analogues 26, 28, 34, and 36 in the rodent liver. The rats (n = 3 per group) were given the drugs sc at a dose of 300 μmol/kg.
An Alternative Concept
A comparison of the iron clearing properties of 26, 34, and 36 (Tables 5 and 6) encouraged a human clinical trial on 36. The ligand is now in phase II with Shire, being assessed as its magnesium salt.172,173 Since 36 is an oil and its sodium salt is hygroscopic, the choice of a magnesium salt may have been driven by dosage form issues. It is interesting to speculate as to whether the GI and other side effects seen with the magnesium salt172,173 derive from the magnesium itself.174−176 It remains to be determined how well the magnesium salt of 36 will perform in patients.
Table 6. Iron Clearing Activity of Desferrithiocin Analogues Given Orally to Non-Iron-Overloaded, Bile Duct-Cannulated Rodents.

The compounds were given as their sodium salts, prepared by the addition of 1 equiv of NaOH to a suspension of the free acid in distilled water. Ligand 34 (300 μmol/kg) was solubilized in distilled water.
The efficiency of each compound was calculated by subtracting the iron excretion of control animals from the iron excretion of the treated animals. The number was then divided by the theoretical output; the result is expressed as a percent.
There remained two properties of (S)-3′-(HO)-DADFT-PE (36) that left room for improvement and provided the impetus for further design considerations. As stated above, the parent drug was an oil. In addition, the dose–response curve in rodents plateaued very quickly (Table 6).168 For example, when 36 was given po to the bile duct-cannulated rats at a dose of 50 μmol/kg, it caused the excretion of 0.289 ± 0.062 mg/kg of iron and had an ICE of 20.7 ± 4.4%. At a dose of 150 μmol/kg, the drug decorporated 0.782 ± 0.121 mg/kg of iron; the ICE was 18.7 ± 2.9%. Thus, tripling the dose of the drug yielded a nearly 3-fold increase in iron excretion. However, when the dose of the chelator was further increased to 300 μmol/kg, the quantity of iron excreted, 0.887 ± 0.367 mg/kg, was within error of that induced by the drug at 150 μmol/kg (p > 0.05), and the ICE was 10.6 ± 4.4% (Table 6).168,171 The deferration induced by 36 was saturable over a fairly narrow dose range.
Synthesis and Biological Evaluation of (S)-4,5-Dihydro-2-[2-hydroxy-4-(3,6-dioxaheptyloxy)phenyl]-4-methyl-4-thiazolecarboxylic Acid [(S)-4′-(HO)-DADFT-norPE]
Synthesis
The fact that the 3′-polyether 36 was an oil and its ICE properties were readily saturable seemed potentially problematic. Thus, additional SAR studies were carried out to search for a chelator that had better physiochemical properties, i.e., a solid, and retained its ICE over a wider range of doses than that of 36. The answer would come with a very simple structural modification of (S)-4′-(HO)-DADFT-PE (34): the 3,6,9-trioxadecyloxy polyether moiety was replaced with a 3,6-dioxaheptyloxy function.168,170,177 The synthesis involved a 4′-O-alkylation of (S)-4′-(HO)-DADFT ethyl ester (37) with polyether tosylate 42. The ester 43 was next cleaved in base to produce (S)-4′-(HO)-DADFT-norPE (44) (Scheme 3). Both acid 44 and ester precursor 43 were crystalline solids. X-ray crystal structures were obtained for each compound (Figures 23 and 24).170 This unequivocally verified that the structures are correct and, in particular, that the stereochemistry at C-4 was indeed (S).
Scheme 3. Synthesis of (S)-4,5-Dihydro-2-[2-hydroxy-4-(3,6-dioxaheptyloxy)phenyl]-4-methyl-4-thiazolecarboxylic Acid (44) and Its Ethyl Ester (43).

Reagents and conditions: (a) K2CO3 (1.1 equiv), acetone, reflux, 2 days, 73%; (c) 50% NaOH(aq) (13 equiv), CH3OH, 80%.
Figure 23.

X-ray of (S)-4,5-dihydro-2-[2-hydroxy-4-(3,6-dioxaheptyloxy)phenyl]-4-methyl-4-thiazolecarboxylic acid (44). Structure is drawn at 50% probability ellipsoids.
Figure 24.

X-ray of ethyl (S)-4,5-dihydro-2-[2-hydroxy-4-(3,6-dioxaheptyloxy)phenyl]-4-methyl-4-thiazolecarboxylate (43). Structure is drawn at 50% probability ellipsoids.
Chelator-Induced Iron Clearance in Rodents and Primates
(S)-4′-(HO)-DADFT-norPE acid (44) and ethyl (S)-4,5-dihydro-2-[2-hydroxy-4-(3,6-dioxaheptyloxy)phenyl]-4-methyl-4-thiazolecarboxylate [(S)-4′-(HO)-DADFT-norPE-EE, 43] were given to the rats and primates po in capsules.170 It is clear from this data that 44 has excellent ICE properties in both rodents and primates (Table 7).170 The performance ratio (PR) is 1. This PR value is relevant in the sense that if a ligand decorporates iron well in both species, then it is likely to work well in humans also.
Table 7. Iron Clearing Efficiency of Desferrithiocin Analogues Administered Orally to Rodents and Primates with the Respective Log Papp Values and Physiochemical Properties.

In the rodents [n = 3 (44), 4 (28, 34, 43, 45), 5 (DFT), 8 (26)], the drugs were given po at a dose of 150 μmol/kg (DFT) or 300 μmol/kg (26, 28, 34, 43–45). The drugs were administered in capsules (43, 44), solubilized in either 40% Cremophor RH-40/water (DFT), distilled water (34), or were given as their monosodium salts, prepared by the addition of 1 equiv of NaOH to a suspension of the free acid in distilled water (26, 28, 45). The efficiency of each compound was calculated by subtracting the iron excretion of control animals from the iron excretion of the treated animals. The number was then divided by the theoretical output; the result is expressed as a percent.
ICE is based on a 48 h sample collection period. The relative percentages of the iron excreted in the bile and urine are in brackets.
In the primates [n = 4 (DFT, 28, 34, 43, 44, in capsules, 45) or 7 (26, 44 as the monosodium salt)], the chelators were given po at a dose of 75 μmol/kg (43–45) or 150 μmol/kg (DFT, 26, 28, 34).
The drugs were administered in capsules (43, 44), solubilized in either 40% Cremophor RH-40/water (DFT, 28), distilled water (34), or were given as their monosodium salts, prepared by the addition of 1 equiv of NaOH to a suspension of the free acid in distilled water (26, 44, 45).
The efficiency was calculated by averaging the iron output for 4 days before the drug, subtracting these numbers from the 2 day iron clearance after the administration of the drug, and then dividing by the theoretical output; the result is expressed as a percent. The relative percentages of the iron excreted in the feces and urine are in brackets.
Performance ratio is defined as the mean ICEprimates/ICErodents.
Data are expressed as the log of the fraction in the octanol layer (log Papp); measurements were done in TRIS buffer, pH 7.4, using a shake flask direct method.
The biliary ferrokinetics curves in rodents (Figure 25) showcase the progress168,170 achieved with this SAR, beginning with deferitrin (26), (S)-3′-(HO)-DADFT-PE (36), and finally, (S)-4′-(HO)-DADFT-norPE (44), which is far superior to 36 and the parent 26. The norPE 44 has a protracted residence time and continues to clear iron for 48 h. Note that although the ferrokinetics curve of 44 may appear to be biphasic (Figure 25), the reason for this unusual line shape is that several animals had temporarily obstructed bile flow.170 While the concentration of iron in the bile remained the same, the bile volume, and thus overall iron excretion, decreased. Once the obstruction was resolved, bile volume and overall iron excretion normalized.
Figure 25.

Biliary ferrokinetics of DFT-related chelators 26, 36, and 44 given orally to non-iron-overloaded, bile duct-cannulated rats at a dose of 300 μmol/kg. The iron excretion (y axis) is reported as micrograms of iron per kilogram of body weight.
Dose Response of (S)-3′-(HO)-DADFT-PE (36) vs (S)-4′-(HO)-DADFT-norPE (44)
In the final analysis, a close look at how 36 compares with 44 (Table 8) underscores several points. When going from 50 to 300 μmol/kg with 36, the ICE drops by 50%. The same dosage change with 44 shows virtually no decrease in ICE.171
Table 8. Iron Clearing Efficiency of (S)-3′-(HO)-DADFT-PE (36) and (S)-4′-(HO)-DADFT-norPE (44)a.

Dose response. Over the same dose range, 50–300 μmol/kg, the former drops in ICE by 50%, whereas the latter remains constant.
Tissue Distribution/Metabolism of (S)-4′-(HO)-DADFT-norPE (44) in Rats
A comparison of the tissue distribution of deferitrin (26), (S)-3′-(HO)-DADFT-PE (36), and (S)-4′-(HO)-DADFT-norPE (44) given to rodents sc at a dose of 300 μmol/kg clearly demonstrates that 44 achieves, by far, the highest tissue levels of any of the three ligands (Figure 26).171 As with 36 and 34, we assessed the liver, kidney, pancreas, heart, and plasma of rodents treated with 44 for 26.171 This is the product resulting from cleaving the ether fragment at the 4′ position; no 26 was observed. However, we now continue to search for other potential metabolites. On the basis of studies of similar polyether fragments178−180 fixed to various drugs, cleavage of the terminal methyl group on the ether of 44 could provide an alcohol, which would likely be converted to a carboxylic acid. This would not be unexpected and deserves further consideration.
Figure 26.

A comparison of the tissue distribution of deferitrin (26), (S)-3′-(HO)-DADFT-PE (36), and (S)-4′-(HO)-DADFT-norPE (44). Rodents were given the drugs sc at 300 μmol/kg and sacrificed at 0.5, 1, 2, 4, and 8 h postexposure. The drug concentrations (y axis) are reported as nanomoles of compound per gram of wet weight of tissue or as micromolar (plasma). For all time points, n = 3.
Toxicity Assessments of (S)-4′-(HO)-DADFT-norPE (44)
Recall that the major hurdle in exploiting the DFT pharmacophore, e.g., deferitrin (26), as an orally active iron chelator was its nephrotoxicity.163,165 Iron chelator-induced proximal tubule damage is not uncommon. In fact, one of the problems associated with a currently accepted iron-overload treatment, Exjade, is proximal tubule-derived renal damage.128,146 Therefore, a series of toxicity studies focusing on 44’s impact on renal function was carried out in rats. In the initial trial, the drug was given to the animals po once daily for 10 days at a standard dose of 384 μmol/kg/day (equivalent to 100 mg/kg/day of DFT sodium salt). Additional age-matched animals served as untreated controls. All of the rats survived the exposure to the test drug.170 The animals were euthanized on day 11, 1 day after the last dose of drug. Extensive tissues were sent out for histopathological examination. No drug-related abnormalities were identified. In addition, blood was collected immediately prior to sacrifice and was submitted for a complete blood count and serum chemistries, including the determination of the animals’ kidney function, i.e., blood urea nitrogen (BUN) and serum creatinine (SCr). No drug-related abnormalities were found, and the rats’ BUN and SCr levels were within the normal range.170
Unfortunately, biomarkers, e.g., BUN and SCr levels, often do not increase until a serious loss of renal function has occurred. In the past, this could result in the performance of long-term, expensive exposure studies before there was sufficient evidence of nephrotoxicity to warrant terminating the trial. However, this problem has now been overcome with the discovery of kidney injury molecule-1 (Kim-1, rodent) and (KIM-1, human).181,182 Kim-1 is a type 1 transmembrane protein located in the epithelial cells of proximal tubules.181,182 The ectodomain of the Kim-1 proximal tubule protein is released into the urine very early after exposure to a nephrotoxic agent or ischemia; it appears far sooner than increases in BUN or SCr are detected.183,184 BioAssay Works has recently developed RenaStick, a direct lateral flow immunochromatographic assay, which allows for the rapid detection (less than 30 min) and quantitation of urinary Kim-1 excretion.185
Accordingly, the impact of ligand (S)-4′-(HO)-DADFT-norPE (44) on urinary Kim-1 excretion has been evaluated in rodents when the drug was given po s.i.d. daily for 28 days (56.9, 113.8, or 170.7 μmol/kg/day), s.i.d. for 10 days (384 μmol/kg/day), and b.i.d. at 237 μmol/kg/dose (474 μmol/kg/day) × 7 days.171 The studies were performed on rats with normal iron stores. Untreated age-matched rats were used as negative controls. Exjade was used as a positive control for the 384 μmol/kg/day × 10 day dosing regimen, while deferitrin (26) served as a positive control for the 237 μmol/kg/dose b.i.d. (474 μmol/kg/day) × 7 day drug exposure.
Very little Kim-1 was found in the urine of the age-matched negative control rats at any time (Figure 27A). However, considerable quantities of Kim-1 were found in the urine of the Exjade-treated rats (Figure 27C); the rats’ BUN and SCr were also significantly increased. In fact, none of the Exjade-treated rats survived the planned 10 day exposure to the drug.171 The b.i.d. × 7 day dosing of deferitrin (26) was also associated with an increase in urinary Kim-1 excretion (Figure 27E). Although the BUN and SCr of the deferitrin-treated rats were slightly elevated, all of the animals survived the exposure to the drug. In sharp contrast, very little Kim-1 was found at any time in the urine of any of the rats exposed to (S)-4′-(HO)-DADFT-norPE (44) (Figure 27B,D,F).171 Finally, the BUN and SCr of the 4′-norPE-treated groups of rats were virtually identical to those of the age-matched control animals. Taken together, these results have successfully demonstrated the sensitivity and usefulness of the Kim-1 assay.
Figure 27.

Urinary Kim-1 excretion, expressed as Kim-1 (ng/kg/24 h), for the following groups: (A) untreated age-matched control rats, (B) rats treated with 44 po once daily at a dose of 170.7 μmol/kg/day × 28 days, (C) rats given Exjade po once daily at a dose of 384 μmol/kg/day, (D) rats given 44 po once daily at a dose of 384 μmol/kg/day × 10 days, (E) rats given deferitrin (26) po twice daily at a dose of 237 μmol/kg/dose (474 μmol/kg/day) × 7 days, and (F) rats given 44 po twice daily at a dose of 237 μmol/kg/dose (474 μmol/kg/day) × 7 days. Note that none of the rats survived the planned 10 day exposure to Exjade. For groups A–D and F, n = 5; for group E, n = 3.
Extensive tissues from the control and 170.7 μmol/kg/day × 28 days study were assessed for any histopathology. No drug-related abnormalities were found. Note that this dose is approximately 3 times the dose necessary to excrete 450 μg Fe/kg in the primates, the suggested iron clearance required to keep a thalassemia patient in negative iron balance.164 These results have provided additional evidence for the safety and tolerability of 44.
Tissue Iron Decorporation
As described above, rodents were given acid 44 po at a dose of 384 μmol/kg/day × 10 days.170 The rats were euthanized 24 h postdrug, and the kidney, liver, pancreas, and heart were removed. The tissue samples were wet-ashed,170 and their iron levels were determined by atomic absorption spectroscopy (Figure 28). The renal iron content of rodents treated with 44 was reduced by 7.4% when the drug was administered in capsules and by 24.8% when it given as its sodium salt (Figure 28). Although the renal iron content of the latter animals was significantly less than that of the untreated controls (p < 0.001), there was not a significant difference between the capsule or sodium salt groups (p > 0.05). The reduction in liver iron was profound, >35% in both the capsule and sodium salt groups (p < 0.001). There was a significant reduction in pancreatic iron when 44 was given as its sodium salt (p < 0.05) vs the untreated controls but not when it was dosed in capsules (Figure 28). However, as with the renal iron, there was no significant difference between the capsule vs sodium salt treatment groups (p > 0.05).170 Finally, there was a significant decrease in the cardiac iron of animals treated with 4′-norPE, 6.9 and 9.9%, when 44 was given in capsules and as its sodium salt, respectively (p < 0.05).
Figure 28.

Tissue iron concentration of rats treated with 44 once daily at a dose of 384 μmol/kg/day × 10 days. The chelator was administered orally in gelatin capsules (n = 5) or by gavage as its monosodium salt (n = 10). Age-matched rats (n = 12) served as untreated controls.
Summary of (S)-4′-(HO)-DADFT-norPE (44) Advantages
The 4-norPE 44 offers a number of attractive properties.170,171,177 It is easy to synthesize and is a crystalline solid. It is a very effective iron decorporation agent that is orally active and has an ICE in rodents and primates of ≈26%. In addition, the drug maintains its ICE over a wider range of dose levels than does the 3′-PE 36 as its sodium salt. Finally, it achieves high levels in the liver, heart, and pancreas and has a large therapeutic window.171 Ligand 44 has now been licensed by Sideris as SP420 and is in a phase I clinical trial.
The Impact of Introducing Polyethers Directly into DFT on Ligand Toxicity and ICE
DFT is a very efficient iron chelator when given po.97−99 However, it is severely nephrotoxic.100 Structure–activity studies with DFT demonstrated that removal of the aromatic nitrogen to provide desazadesferrithiocin (DADFT, 4) and introduction of either a hydroxyl group or a polyether fragment onto the aromatic ring resulted in orally active iron chelators that were much less toxic than DFT or 4. These results encouraged us to determine if a comparable reduction in renal toxicity could be achieved by performing the same structural manipulations on DFT itself, i.e., fixing either a hydroxyl or polyether functionality to the DFT platform (Figure 29). Accordingly, DFT analogues 46–48 were synthesized (Schemes 4 and 5).177 The ICE and ferrokinetics of the ligands were evaluated in rats and primates; toxicity assessments were carried out in rodents. In addition, log P values were also determined.
Figure 29.

Modifications of desferrithiocin compatible with iron clearance and the absence of renal toxicity.
Scheme 4. Synthesis of 46 and 47.

Reagents and conditions: (a) 4-methoxybenzyl alcohol, 60% NaH (2.5 equiv each), DMF, 95–100 °C, 18 h, 73%. (b) TFA, pentamethylbenzene, 22 h, quantitative. (c) CH3OH, 0.1 M pH 6 buffer, NaHCO3, 73–76 °C, 45 h. (d) EtI, DIEA (1.5 equiv each), DMF, 47 h, 70%. (e) K2CO3 (1.6 equiv), acetone, reflux, 1 d, 65%. (f) 50% NaOH(aq), CH3OH, then HCl, 96% (46), 97% (47).
Scheme 5. Synthesis of 48.

Reagents and conditions: (a) K2CO3 (2 equiv), CH3CN, 68%. (b) m-CPBA, CH2Cl2. (c) Ac2O, reflux. (d) NaOH(aq), EtOH, reflux, 4 h, 87%. (e) SO3·pyridine, NEt3, DMSO, CHCl3, 16 h, 83%. (f) H2NOH·HCl, NaOAc, CH3OH, reflux, 2 h, 90%. (g) Ac2O, reflux, 94%. (h) H2, 10% Pd–C, CH3OH, 85%. (i) CH3OH, 0.1 M pH 6 buffer, NaHCO3, 75 °C, 48 h, 95%.
Synthesis of DFT Polyethers
The preparation of 5′-hydroxydesferrithiocin (46) and its 5′-norpolyether (47) (Table 9) was somewhat more complicated than that of the DADFT series. The synthesis of 46 and 47 began with 2-cyano-3,5-difluoropyridine (50), which was converted to 2-cyano-3,5-dihydroxypyridine (52) in two steps (Scheme 4). Compound 50 was reacted with 4-methoxybenzyl alcohol and K2CO3 in acetonitrile to give the protected diol 51. Removal of the 4-methoxybenzyl groups of 51 using trifluoroacetic acid (TFA)177 in pentamethylbenzene177 at room temperature provided nitrile 52. Cyclocondensation of 52 with (S)-2-methyl cysteine (2) followed by esterification of crude acid 46 with iodoethane and N,N-diisopropylethylamine produced ethyl (S)-4,5-dihydro-2-(3,5-dihydroxy-2-pyridinyl)-4-methyl-4-thiazolecarboxylate (53). Hydrolysis of ester 53 generated 46 as a solid. Alternatively, ester 53 was alkylated at the less hindered 5′ phenol177 with tosylate 42, affording the polyether ligand precursor 54. The carboxylate was unmasked under alkaline conditions to give 47, an oil.
Table 9. Iron Clearing Efficiency of DFT Analogues Administered to Rodents and Primates with the Respective Log Papp Values.

In the rodents [n = 3 (44), 4 (4 and 48), 5 (DFT, 46, 47, 49), or 8 (26)], the ligands were given po at a dose of 150 μmol/kg (DFT and 4) or 300 μmol/kg (26 and 44, 46–49). The drugs were administered in capsules (44), solubilized in 40% Cremophor RH-40/water (DFT and 4), or given as their monosodium salts, prepared by the addition of 1 equiv of NaOH to a suspension of the free acid in distilled water (26, 46–49). The efficiency of each compound was calculated by subtracting the iron excretion of control animals from the iron excretion of the treated animals. The number was then divided by the theoretical output; the result is expressed as a percent. The relative percentages of the iron excreted in the bile and urine are in brackets.
ICE is based on a 48 h sample collection period.
In the primates [n = 4 (DFT, 4, 46, and 44 in capsules and 47–49), 6 (26), or 7 (44 as the monosodium salt)], the chelators were given po at a dose of 75 μmol/kg (4, 44, 46–49) or 150 μmol/kg (DFT and 26). Ligand 48 was also given to the primates sc at a dose of 75 μmol/kg. The drugs were administered in capsules (44), solubilized in 40% Cremophor RH-40/water (DFT and 4), or given as their monosodium salts, prepared by the addition of 1 equiv of NaOH to a suspension of the free acid in distilled water (DFT, 4, 26, 44, 46–49). The efficiency was calculated by averaging the iron output for 4 days before the drug, subtracting these numbers from the 2 day iron clearance after the administration of the drug, and then dividing by the theoretical output; the result is expressed as a percent. The relative percentages of the iron excreted in the feces and urine are in brackets.
Data are expressed as the log of the fraction in the octanol layer (log Papp); measurements were done in TRIS buffer, pH 7.4, using a shake flask direct method.
The performance ratio (PR) is defined as the mean ICEprimates/ICErodents.
Synthesis of the 4′-nor polyether DFT analogue 48, an isomer of iron chelator 47 (Table 9), started with 2-methyl-3-(benzyloxy)-4-pyridone (55), available in two steps from maltol186 (Scheme 5). O-Alkylation of 55 with tosylate 42 and K2CO3 in refluxing acetonitrile187 afforded 2-methyl-3-(benzyloxy)-4-(3,6-dioxaheptyloxy)pyridine (56). The methyl group of 56 was oxidized by known methodology,186 providing aldehyde 58. Specifically, 56 was treated with 3-chloroperbenzoic acid in CH2Cl2, and the resulting N-oxide was heated at reflux in acetic anhydride. Cleavage of the acetate ester with base gave the 2-pyridinemethanol 57. Primary alcohol 57 was further oxidized to aldehyde 58 with sulfur trioxide–pyridine complex and NEt3 in DMSO and CHCl3. The oxime 59, generated under standard conditions,162 was heated at reflux with acetic anhydride, furnishing the corresponding nitrile 60. Removal of the benzyl-protecting group from 60 by hydrogenolysis (1 atm, 10% Pd–C, CH3OH) in the presence of the cyano group and pyridine ring produced 4-(3,6-dioxaheptyloxy)-3-hydroxy-2-pyridinecarbonitrile (61). Heating 61 with amino acid 2 in aqueous CH3OH buffered at pH 6 generated 48.
Biological Evaluation of DFT Polyethers
The resulting DFT ligands demonstrated a reduction in toxicity that was equivalent to that of the DADFT polyether analogues and presented with variable iron clearing properties. DFT (Table 9) and its analogues were all significantly more water-soluble (lower log Papp) than the corresponding DADFT analogues, e.g., DFT vs 4, 46 vs 26, 47 vs 44, and 48 vs 49. There was an excellent correlation between ICE and log Papp in rodents among the DFT analogues 46, 47, and 48, with the more lipophilic ligands having a greater ICE (Table 9). This trend is in keeping with previous DADFT observations in rodents that more lipophilic ligands have better ICE properties. However, in the primates, the DADFT analogues were consistently better deferration agents than the corresponding DFT ligands, although the best DFT analogue, 47, still presented with excellent ICE in both rodents and primates.
The effects of these structural modifications of DFT on its renal toxicity were assessed in rats using a urinary Kim-1 assay177 as well as by monitoring BUN and SCr. The most notable finding was that fixing a hydroxyl group or a polyether fragment to the DFT aromatic ring resulted in a profound reduction in renal toxicity as was seen after the same modification to DADFT (Figure 30). Although some nephrotoxicity was noted with both hydroxylated DADFT and DFT analogues, 26 and 46, respectively, the introduction of polyether groups into either pharmacophore resulted in ligands with little to no impact on renal function, for example, 44, 47, and 48 (Figure 30).
Figure 30.

Urinary Kim-1 excretion (y axis) is expressed as Kim-1 (ng/kg/24 h) of rats treated with DFT, DFT analogues 46–48, or DADFT analogues 26 and 44. The rodents were given the drugs po twice daily (b.i.d.) at a dose of 237 μmol/kg/dose (474 μmol/kg/day) for up to 7 days. Note that none of the rats survived the planned 7 day exposure to DFT. n = 5 for DFT, 44, and 46–48; n = 3 for ligand 26.
In summary, rather simple manipulation of the DFT aromatic ring, for example, hydroxylation, or the introduction of a polyether functionality, can have a marked effect on the ligand’s ICE and renal toxicity (Figure 30). Although the resulting DFT chelators were generally as effective in the rodents as their DADFT counterparts, they were less active in the primates.
Discussion
In the early anaerobic biosphere, iron was largely in the Fe(II) oxidation state and highly soluble.30 This changed with the production of oxygen by blue–green algae. Iron is now predominantly Fe(III) and is in the form of highly insoluble ferric hydroxide polymers.31 Microorganisms had to develop tools to solve the iron access problem. Their solution was the assembly of highly Fe(III)-specific chelators, siderophores,188 that they secreted into their environment. These ligands bound the metal and facilitated its delivery to the microorganisms via a variety of different transport and release scenarios. These chelators are relatively low molecular weight, <1200, are highly Fe(III)-specific, and offer excellent platforms for the development of therapeutically useful iron decorporation agents. Simple eukaryotes and higher life forms, e.g., humans, developed a far more sophisticated iron access, storage, and delivery system.
Homeostatic mechanisms in humans controlling iron absorption, transport, and storage to multicompartments are very tightly regulated. There is little, if any, leakage of the metal. Here lies the crux of iron-overload diseases: there is no mechanism for excretion of excess metal. The consequence of this can be lethal to patients. The only solution for transfusional iron overload is to chelate the excess metal and promote its excretion.
The boundary conditions for the design and assembly of drugs for the treatment of transfusion-mediated iron-overload disorders are very tight. The diseases are genetic, and the patients require lifelong exposure to the therapeutic. Thus, the toxicity profile of the candidate ligand must be pristine.11−13 In addition, to avoid potential patient compliance issues, the iron chelator needs to be active when given po.
There are three fundamental platforms from which to choose: a natural product chelator (a siderophore), a totally synthetic ligand, or some combination thereof. Initially, the standard of care for transfusional iron-overload treatment was DFO, a siderophore. This was followed by four notable synthetic ligands: DTPA, HBED, L1, and Exjade. Unfortunately, DFO,51−54 DTPA,116,117 and HBED118−120 are ineffective unless given parenterally. While both L1 and Exjade work when given po, their ICE values were somewhat less than hoped. In addition, although most therapies are indeed an exercise in trade offs, both drugs presented with problematic toxicity issues. Thus, the search for chelators with better ICE properties and toxicity profiles continued. This led to extensive SAR studies on the natural product iron chelator DFT.
DFT is excreted by Streptomyces antibioticus for the express purpose of acquiring Fe(III).68 Initial animal trials with DFT in rodents and primates revealed it to be an orally active, highly efficient iron chelator. However, it presented with unacceptable renal toxicity.100,154 Nevertheless, the remarkable oral bioavailability and ICE drove a very successful structure–activity adventure aimed at ameliorating DFT-induced nephrotoxicity. The initial outcome revealed that removal of the DFT aromatic nitrogen and introduction of a hydroxyl at either the aromatic 4′ or 3′ position of DADFT (4) led to ligands with remarkably reduced toxicity in rodents and good ICE in primates. In fact, deferitrin (26) was taken into human clinical trials by Genzyme.163,165 Initial results were very promising when the drug was given s.i.d. However, when the ligand was given b.i.d., patients presented with elevated BUN and SCr, and the trials were discontinued.165 The problem then became how to solve the renal toxicity problem when the chelator was administered b.i.d.
The reengineering of deferitrin (26) was predicated on the observation that both 26 and (S)-3′-(HO)-DADFT (30) were orally active iron chelators and that when the 4′-(OH) of DADFT was methylated, there was a remarkable increase in ICE and an increase in toxicity accompanying the greater lipophilicity.162,166 It was discovered that introducing the polyether fragment 3,6,9-trioxadecyl at the 4′-(OH) of 26 or the 3′-(OH) of 30, providing 4′-(HO)- and (S)-3′-(HO)-DADFT-PE (34 and 36, Table 5), respectively, led to less lipophilic, remarkably efficient iron chelators with no renal toxicity, even when the molecules were administered b.i.d.162,167,168
It was also clearly demonstrated that fixing polyether fragments to desferrithiocin itself (Figure 29 and Table 9) resulted in ligands with excellent ICE values and minimal toxicity.177 The take home message, then, was that polyether fragments can have a profound effect on a ligand’s toxicity profile. However, the (S)-4′ and (S)-3′-DADFT-PEs 34 and 36 had better ICE properties than the DFT-PE’s 47 and 48 and were good candidates for human clinical trials. A magnesium salt of 3′-polyether ligand 36 (SPD602, deferitazole magnesium) is now in phase II clinical trials with Shire.172,173 It will be interesting to learn whether using the magnesium salt of the chelator is responsible for the ligand’s observed tolerability issues, e.g., GI effects.172,173 As all of our data was derived from the corresponding acid or sodium salt, this is difficult for us to evaluate. Nevertheless, there were two properties of 3′-polyether 36 that left room for improvement. The parent acid was an oil, and the dose–response curve for 36 plateaued in rodents very quickly: doubling the dose reduced the ICE by 40%. Changing the length of the 3,6,9-trioxadecyloxy polyether fragment on (S)-4′-(HO)-DADFT-PE (34) to a 3,6-dioxaheptyloxy group provided a ligand with all of the desired properties, (S)-4′-(HO)-DADFT-norPE (44).170,171,177 It is a very effective deferration agent in both rodents and primates (ICE ∼26%), thus increasing its index of success in humans. It is a crystalline solid and performs very well in capsules in both species. Ligand 44 also has a better dose–response profile and better access to the relevant organs in rodents than 3′-polyether 36.170,171 It has now moved into human clinical trials with Sideris as SP420. These studies will ultimately reveal which ligand is the most useful. Each may offer advantages to thalassemia subpopulations.
Probably the most relevant finding in these studies, the relationship among ligand lipophilicity, iron clearing efficiency, and toxicity, will define future chelator design strategies. The concept that unfolds is one in which highly lipophilic chelators are designed for efficient oral absorption such that, once absorbed, they can be quickly metabolized to hydrophilic, likely minimally toxic, active iron decorporation agents. As the literature continues to underscore the potential application of iron chelators to a broad spectrum of diseases, there will no doubt be a rapid expansion in interest in chelation therapy. The authors and patients remain indebted to NIDDK, the University of Florida Health Center, and the involved corporate entities for their support of these studies.
Acknowledgments
The project described was supported by grant no. R37DK049108 from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases or the National Institutes of Health. We thank Elizabeth M. Nelson and Katie Ratliff-Thompson for their technical assistance and Miranda E. Price for her editorial and organizational support. We acknowledge the spectroscopy services in the Chemistry Department, University of Florida, for the mass spectrometry analyses.
Glossary
Abbreviations Used
- GI
gastrointestinal
- NTBI
nontransferrin-bound iron
- DHBA
2,3-dihydroxy-N,N-dimethylbenzamide
- DMT1
divalent metal transporter 1
- NRAMP2
natural resistance-associated macrophage protein 2
- DCT1
divalent cation transporter 1
- DFO
desferrioxamine
- TfR
transferrin receptor
- IRE
iron responsive element
- IRP
iron responsive proteins
- ICE
iron clearing efficiency
- po
orally
- sc
subcutaneously
- iv
intravenously
- SAR
structure–activity relationship
- DFT
desferrithiocin
- DMDFT
desmethyldesferrithiocin
- DADFT
desazadesferrithiocin
- DADMDFT
desazadesmethyldesferrithiocin
- s.i.d.
once daily
- b.i.d.
twice daily
- PR
performance ratio
- Kim-1
kidney injury molecule-1
- TFA
trifluoroacetic acid
- DTPA
diethylenetriaminepentaacetic acid
- BUN
blood urea nitrogen
- SCr
serum creatinine
Biographies
Raymond J. Bergeron received his undergraduate degree from Clark University and his Ph.D. in Organic Chemistry from Brandeis University. He studied under Nobel Laureate Konrad Bloch at Harvard University and next joined the chemistry faculty at the University of Maryland. In 1978, he moved to the University of Florida Department of Medicinal Chemistry. Here, he established his expertise in polyamine and iron metabolism, leading to the development of anticancer drugs and treatments for children with iron-overload disease. He has published 200 papers and a bioorganic chemistry text, edited two books, and has generated over 200 patents. Six of his discoveries have reached human clinical trials.
Jan Wiegand graduated from the University of Florida in 1988 with a B.S. degree in Agriculture.
James S. McManis received his B.A. from Franklin and Marshall College and his Ph.D. in Organic Chemistry from Cornell University. In 1983, he moved to the University of Florida, Department of Medicinal Chemistry, and is a Chemist in the research group of Dr. Bergeron in the synthesis of potential drug molecules, specifically in the areas of iron chelators and antineoplastic polyamine analogues.
Neelam Bharti finished her Ph.D. in Organic Chemistry from New Delhi in 2002 and joined Prof. Raymond J. Bergeron’s group at the University of Florida, where she was engaged in a drug development program for iron-overload and total synthesis of siderophores. She has coauthored more than 30 articles and is currently working as Chemical Sciences Librarian at the University of Florida.
The authors declare the following competing financial interest(s): Dr. Bergeron holds patents on many of the included desferrithiocin discoveries. These patents are the property of the University of Florida. Dr. Bergeron has served in the capacity of a consultant in the initial studies of the compound now held by Shire and currently serves as a consultant on the ligand held by Sideris.
After this paper was published ASAP on September 10, 2014, a correction was made to the equation for log β3 in the sixth paragraph of the section Iron and Primitive Life Forms: Siderophores. The corrected version was reposted September 26, 2014.
Funding Statement
National Institutes of Health, United States
References
- Mladenka P.; Hrdina R.; Hübl M.; Simunek T. The Fate of Iron in the Organism and Its Regulatory Pathways. Acta Med. (Hradec Kralove, Czech Repub.) 2005, 48, 127–135. [PubMed] [Google Scholar]
- Bauer I.; Knolker H.-J.. Iron Complexes in Organic Chemistry. In Iron Catalysis in Organic Chemistry; Plietker B., Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp 1–28. [Google Scholar]
- Sutton H. C.; Winterbourn C. C. On the Participation of Higher Oxidation States of Iron and Copper in Fenton Reactions. Free Radical Biol. Med. 1989, 6, 53–60. [DOI] [PubMed] [Google Scholar]
- Crichton R.Inorganic Biochemistry of Iron Metabolism: From Molecular Mechanisms to Clinical Consequences, 2nd ed.; Wiley: New York, 2001. [Google Scholar]
- Campanella L.; Capesciotti G. S.; Russo M. V.; Tomassetti M. Study of the Catalytic Mechanism of the Enzyme Catalase on Organic Hydroperoxides in Non-polar Organic Solvent. Curr. Enzyme Inhib. 2008, 4, 86–92. [Google Scholar]
- Scott M. D. H2O2 Injury in Beta Thalassemic Erythrocytes: Protective Role of Catalase and the Prooxidant Effects of GSH. Free Radical Biol. Med. 2006, 40, 1264–1272. [DOI] [PubMed] [Google Scholar]
- Tong W. H.; Rouault T. A. Metabolic Regulation of Citrate and Iron by Aconitases: Role of Iron–Sulfur Cluster Biogenesis. BioMetals 2007, 20, 549–564. [DOI] [PubMed] [Google Scholar]
- Siddique A.; Kowdley K. V. The Iron Overload Syndromes. Aliment. Pharmacol. Ther. 2012, 5, 876–893. [DOI] [PubMed] [Google Scholar]
- Brittenham G. M. Iron-Chelating Therapy for Transfusional Iron Overload. N. Engl. J. Med. 2011, 364, 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington C. A.; Kowdley K. V. Haemochromatosis. Aliment. Pharmacol. Ther. 2002, 16, 1963–1975. [DOI] [PubMed] [Google Scholar]
- Peters M.; Heijboer H.; Smiers F.; Giordano P. C. Diagnosis and Management of Thalassemia. BMJ 2012, 344, e228. [DOI] [PubMed] [Google Scholar]
- Cappellini M. D.; Cohen A. R.; Eleftheriou A.; Piga A.; Porter J.; Taher A. T.. Guidelines for the Clinical Management of Thalassemia, 2nd ed.; Thalassemia International Federation: Strovolos, Cyprus, 2008. [PubMed] [Google Scholar]
- Vichinsky E. P. Alpha Thalassemia Major—New Mutations, Intrauterine Management, and Outcomes. Hematology 2009, 35–41. [DOI] [PubMed] [Google Scholar]
- Gao C.; Li L.; Chen B.; Song H.; Cheng J.; Zhang X.; Sun Y. Clinical Outcomes of Transfusion-Associated Iron Overload in Patients with Refractive Chronic Anemia. Patient Preference Adherence 2014, 8, 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvaruso G.; Vitrano A.; Di Maggio R.; Ballas S.; Steinberg M. H.; Rigano P.; Sacco M.; Telfer P.; Renda D.; Barone R.; Maggio A.. Deferiprone versus Deferoxamine in Sickle Cell Disease: Results from a 5-Year Long-Term Italian Multi-Center Randomized Clinical Trial. Blood Cells, Mol., Dis. 2014. DOI: 10.1016/j.bcmd.2014.04.004 . [DOI] [PubMed] [Google Scholar]
- Mitchell M.; Gore S. D.; Zeidan A. M. Iron Chelation Therapy in Myelodysplastic Syndromes: Where Do We Stand?. Expert Rev. Hematol. 2013, 6, 397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garceau P.; Nguyen E. T.; Carasso S.; Ross H.; Pendergrast J.; Moravsky G.; Bruchal-Garbicz B.; Rakowski H. Quantification of Myocardial Iron Deposition By Two-Dimensional Speckle Tracking in Patients with β-Thalassemia Major and Blackfan-Diamond Anaemia. Heart 2011, 97, 388–393. [DOI] [PubMed] [Google Scholar]
- Mochizuki H.; Yasuda T. Iron Accumulation in Parkinson’s Disease. J. Neural Transm. 2012, 119, 1511–1514. [DOI] [PubMed] [Google Scholar]
- Selim M. Deferoxamine Mesylate: A New Hope for Intracerebral Hemorrhage: From Bench to Clinical Trials. Stroke 2009, 40, S90–S91. [DOI] [PubMed] [Google Scholar]
- Millań M.; Sobrino T.; Arenillas J. F.; Rodriguez-Yańez M.; García M.; Nombela F.; Castellanos M.; de la Ossa N. P.; Cuadras P.; Serena J.; Castillo J.; Dav́alos A. Biological Signatures of Brain Damage Associated with High Serum Ferritin Levels in Patients with Acute Ischemic Stroke and Thrombolytic Treatment. Dis. Markers 2008, 25, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaief J. L. Iron Induced Oxidative Damage as a Potential Factor in Age-Related Macular Degeneration: The Cogan Lecture. Invest. Ophthalmol. Visual Sci. 2006, 47, 4660–4664. [DOI] [PubMed] [Google Scholar]
- Brissot P.; Ropert M.; Le Lan C.; Loréal O. Non-Transferrin Bound Iron: A Key Role in Iron Overload and Iron Toxicity. Biochim. Biophys. Acta 2012, 1820, 403–410. [DOI] [PubMed] [Google Scholar]
- Halliwell B.Iron, Oxidative Damage, and Chelating Agents. In The Development of Iron Chelators for Clinical Use; Bergeron R. J., Brittenham G. M., Eds.; CRC Press: Boca Raton, FL, 1994; pp 33–56. [Google Scholar]
- Carr A.; Frei B. Does Vitamin C Act as a Pro-Oxidant under Physiological Conditions?. FASEB J. 1999, 13, 1007–1024. [DOI] [PubMed] [Google Scholar]
- Jomova K.; Valko M. Advances in Metal-Induced Oxidative Stress and Human Disease. Toxicology 2011, 283, 65–87. [DOI] [PubMed] [Google Scholar]
- Wilhelm J.; Vanková M.; Maxová H.; Sisková A. Hydrogen Peroxide Production by Alveolar Macrophages Is Increased and Its Concentration Is Elevated in the Breath of Rats Exposed to Hypoxia: Relationship to Lung Lipid Peroxidation. Physiol. Res. 2003, 52, 327–332. [PubMed] [Google Scholar]
- Bolli R.; Patel B. S.; Jeroudi M. O.; Li X. Y.; Triana J. F.; Lai E. K.; McCay P. B. Iron-Mediated Radical Reactions upon Reperfusion Contributes to Myocardial “Stunning”. Am. J. Physiol. 1990, 259, 1901–1911. [DOI] [PubMed] [Google Scholar]
- Kelley E. E.; Khoo N. K. H.; Hundley N. J.; Malik U. Z.; Freeman B. A.; Tarpey M. M. Hydrogen Peroxide is the Major Oxidant Product of Xanthine Oxidase. Free Radical Biol. Med. 2009, 48, 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziai W. C. Hematology and Inflammatory Signaling of Intracerebral Hemorrhage. Stroke 2013, 44, S74–S78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilbert M.; Bonnefoy V. Insight into the Evolution of the Iron Oxidation Pathways. Biochim. Biophys. Acta 2013, 1827, 161–175. [DOI] [PubMed] [Google Scholar]
- Ponka P.; Schulman H. M.; Woodworth R. C.; Richter G. W.. Iron Transport and Storage; CRC Press: Boca Raton, FL, 1990. [Google Scholar]
- Saha R.; Saha N.; Donofrio R. S.; Bestervelt L. L. Microbial Siderophores: A Mini Review. J. Basic Microbiol. 2013, 53, 303–317. [DOI] [PubMed] [Google Scholar]
- Byrne S. L.; Chasteen N. D.; Steere A. N.; Mason A. B. The Unique Kinetics of Iron Release From Transferrin: The Role of Receptor, Lobe–Lobe Interactions, and Salt at Endosomal pH. J. Mol. Biol. 2010, 396, 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkouvatsos K.; Papanikolaou G.; Pantopoulus K. Regulation of Iron Transport and the Role of Transferrin. Biochim. Biophys. Acta 2012, 1820, 188–202. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; Weimar W. R.; Dionis J. B. Demonstration of Ferric l-Parabactin-Binding Activity in the Outer Membrane of Paracoccus denitrificans. J. Bacteriol. 1988, 170, 3711–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abergel R. J.; Wilson M. K.; Arceneaux J. E. L.; Hoette T. M.; Strong R. K.; Byers B. R. Anthrax Pathogen Evades the Mammalian Immune System Through Stealth Siderophore Production. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 18499–18503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawji E.; Gledhill M.; Milton J. A.; Tarran G. A.; Ussher S.; Thompson A.; Wolff G. A.; Worsfold P. J.; Achterberg E. P. Hydroxamate Siderophores: Occurrence and Importance in the Atlantic Ocean. Environ. Sci. Technol. 2008, 42, 8675–8680. [DOI] [PubMed] [Google Scholar]
- Pegg A. E. Mammalian Polyamine Metabolism and Function. IUBMB Life 2009, 61, 880–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron R. J.; Streiff R. R.; King W.; Daniels R. D. Jr.; Wiegand J. A Comparison of the Iron-Clearing Properties of Parabactin and Desferrioxamine. Blood 1993, 82, 2552–2557. [PubMed] [Google Scholar]
- Sedlácek V.; van Spanning R. J. M.; Kucera I. Ferric Reductase A is Essential for Effective Iron Acquisition in Paracoccus denitrificans. Microbiology 2009, 155, 1294–1301. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; McManis J. S.; Dionis J.; Garlich J. R. An Efficient Total Synthesis of Agrobactin and Its Gallium(III) Chelate. J. Org. Chem. 1985, 50, 2780–2782. [Google Scholar]
- Ong S. A.; Peterson T.; Neilands J. B. Agrobactin, a Siderophore from Agrobacterium tumefaciens. J. Biol. Chem. 1979, 254, 1860–1865. [PubMed] [Google Scholar]
- Wyckoff E. E.; Mey A. R.; Payne S. M. Iron Acquisition in Vibrio cholerae. BioMetals 2007, 20, 405–416. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; Garlich J. R.; McManis J. S. Total Synthesis of Vibriobactin. Tetrahedron 1985, 41, 507–510. [Google Scholar]
- Allred B. E.; Correnti C.; Clifton M. C.; Strong R. K.; Raymond K. N. Siderocalin Outwits the Coordination Chemistry of Vibriobactin, a Siderophore of Vibrio cholera. ACS Chem. Biol. 2013, 8, 1882–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond K. N.; Dertz E. A.; Kim S. S. Enterobactin: An Archetype for Microbial Iron Transport. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3584–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hider R. C.; Silver J.; Neilands J. B.; Morrison I. E.; Rees L. V. Identification of Iron (II) Enterobactin and Its Possible Role in Escherichia coli Iron Transport. FEBS Lett. 1979, 102, 325–328. [DOI] [PubMed] [Google Scholar]
- Klebba P. E. Three Paradoxes of Ferric Enterobactin Uptake. Front. Biosci. 2003, 8, s1422–s1436. [DOI] [PubMed] [Google Scholar]
- Guterman S. K.; Morris P. M.; Tannenberg W. J. K. Feasibility of Enterochelin as an Iron-Chelating Drug: Studies with Human Serum and a Mouse Model System. Gen. Pharmacol. 1978, 9, 123–127. [DOI] [PubMed] [Google Scholar]
- Harris W. R.; Carrano C. J.; Cooper S. R.; Sofen S. R.; Avdeef A. E.; McArdle J. V.; Raymond K. N. Coordination Chemistry of Microbial Iron Transport Compounds. 19. Stability Constants and Electrochemical Behavior of Ferric Enterobactin and Model Complexes. J. Am. Chem. Soc. 1979, 101, 6097–6104. [Google Scholar]
- Bergeron R. J.; Wiegand J.; McManis J. S.; Perumal P. T. Synthesis and Biological Evaluation of Hydroxamate-Based Iron Chelators. J. Med. Chem. 1991, 34, 3182–3187. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; Pegram J. J. An Efficient Total Synthesis of Desferrioxamine B. J. Org. Chem. 1988, 53, 3131–3134. [Google Scholar]
- Cunningham M. J.; Nathan D. G. New Developments in Iron Chelators. Curr. Opin. Hematol. 2005, 12, 129–134. [DOI] [PubMed] [Google Scholar]
- Cappellini M. D.; Pattoneri P. Oral Iron Chelators. Annu. Rev. Med. 2009, 60, 25–38. [DOI] [PubMed] [Google Scholar]
- Wanachiwanawin W. Infections in E-beta Thalassemia. J. Pediatr. Hematol./Oncol. 2000, 22, 581–587. [DOI] [PubMed] [Google Scholar]
- Martinez J. S.; Haygood M. G.; Butler A. Identification of a Natural Desferrioxamine Siderophore Produced by a Marine Bacterium. Limnol. Oceanogr. 2001, 46, 420–424. [Google Scholar]
- Yamanaka K.; Oikawa H.; Ogawa H.-O.; Hosona K.; Shinmachi F.; Takano H.; Sakuda S.; Beppu T.; Ueda K. Desferrioxamine E Produced by Streptomyces griseus Stimulates Growth and Development of Streptomyces tanashiensis. Microbiology 2005, 151, 2889–2905. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; Phansteil O. IV The Total Synthesis of Nannochelin: A Novel Cinnamoyl Hydroxamate-Containing Siderophore. J. Org. Chem. 1992, 57, 7140–7143. [Google Scholar]
- Bergeron R. J.; McManis J. S. The Total Synthesis of Bisucaberin. Tetrahedron 1989, 45, 4939–4944. [Google Scholar]
- Bergeron R. J.; McManis J. S.; Perumal P. T.; Algee S. E. The Total Synthesis of Alcaligin. J. Org. Chem. 1991, 56, 5560–5563. [Google Scholar]
- Barbeau K.; Rue E. L.; Bruland K. W.; Butler A. Photochemical Cycling of Iron in the Surface Ocean Mediated by Microbial Iron(III)-Binding Ligands. Nature 2001, 413, 409–413. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; Huang G.; Smith R. E.; Bharti N.; McManis J. S.; Butler A. Total Synthesis and Structure Revision of Petrobactin. Tetrahedron 2003, 59, 2007–2014. [Google Scholar]
- Gardner R. A.; Kinkade R.; Wang C.; Phansteil O. IV Total Synthesis of Petrobactin and Its Homologues as Potential Growth Stimuli for Marinobacter hydrocarbonoclasticus, an Oil-Degrading Bacteria. J. Org. Chem. 2004, 14, 3530–3537. [DOI] [PubMed] [Google Scholar]
- Zawadzka A. M.; Kim Y.; Maltseva N.; Nichiporuk R.; Fan Y.; Joachimiak A.; Raymond K. N. Characterization of Bacillus subtilis Transporter for Petrobactin, an Anthrax Stealth Siderophore. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 21854–21859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persmark M.; Pittman P.; Buyer J. S.; Schwyn B.; Grill P. R. Jr.; Neilands J. B. Isolation and Structure of Rhizobactin 1021, a Siderophore from the Alfalfa Symbiont Rhizobium meliloti 1021. J. Am. Chem. Soc. 1993, 115, 3950–3956. [Google Scholar]
- Drechsel H.; Metzger J.; Freund S.; Jung G.; Boelaert J. R.; Winkelmann G. Rhizoferrin—A Novel Siderophore from the Fungus Rhizopus microsporus var. rhizopodiformis. Biol. Metals 1991, 4, 238–243. [Google Scholar]
- Brandel J.; Humbert N.; Elhabirdi M.; Schalk I. J.; Mislin G. L.; Albrecht-Gary A. M. Pyochelin, a Siderophore of Pseudomonas aeruginosa: Physiochemical Characterizations of the Iron(III), Copper(II) and Zinc(II) Complexes. Dalton Trans. 2012, 41, 2820–2834. [DOI] [PubMed] [Google Scholar]
- Naegeli H.-U.; Zahner H. Metabolites of Microorganisms. Part 193. Ferrithiocin. Helv. Chim. Acta 1980, 63, 1400–1406. [Google Scholar]
- Andrews N. C.; Schmidt P. J. Iron Homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [DOI] [PubMed] [Google Scholar]
- Gunshin H.; Fujiwara Y.; Custodio A. O.; Direnzo C.; Robine S.; Andrews N. C. Slc11a2 is Required for Intestinal Iron Absorption and Erythropoiesis but Dispensable in Placenta and Liver. J. Clin. Invest. 2005, 115, 1258–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandy S.; Williams M.; Leggett A.; Lopez-Jimenez M.; Dedes M.; Ramesh B.; Srai S. K.; Sharp P. Nramp2 Expression is Associated with pH-Dependent Iron Uptake Across the Apical Membrane of Human Intestinal Caco-2 Cells. J. Biol. Chem. 2000, 275, 1023–1029. [DOI] [PubMed] [Google Scholar]
- McKie A. T.; Barrow D.; Latunde-Dada G. O.; Rolfs A.; Sager G.; Mudaly E.; Mudaly M.; Richardson C.; Barlow D.; Bomford A.; Peters T. J.; Raja K. B.; Shirali S.; Hediger M. A.; Farzaneh F.; Simpson R. J. An Iron-Regulated Ferric Reductase Associated with the Absorption of Dietary Iron. Science 2001, 291, 1755–1759. [DOI] [PubMed] [Google Scholar]
- Sharp P.; Surjit K. S. Molecular Mechanisms Involved in Intestinal Iron Absorption. World J. Gastroenterol. 2007, 13, 4716–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter S. W.; Alam J.; Choi A. M. Heme-Oxygenase-1/Carbon Monoxide: From Basic Science to Therapeutic Applications. Physiol. Rev. 2006, 86, 583–650. [DOI] [PubMed] [Google Scholar]
- Oates P. S. The Role of Hepcidin and Ferroportin in Iron Absorption. Histol. Histopathol. 2007, 22, 791–804. [DOI] [PubMed] [Google Scholar]
- Chen H.; Huang G.; Su T.; Gao H.; Attieh Z. K.; McKie A. T.; Anderson G. J.; Vulpe C. D. Decreased Hephaestin Activity in the Intestine of Copper-Deficient Mice Causes Systemic Iron Deficiency. J. Nutr. 2006, 136, 1236–1241. [DOI] [PubMed] [Google Scholar]
- Qian Z. M.; Chang Y. Z.; Leung G.; Du J. R.; Zhu L.; Wang Q.; Niu L.; Xu Y. J.; Yang L.; Ho K. P.; Ke Y. Expression of Ferroportin 1, Hephaestin and Ceruloplasmin in Rat Heart. Biochim. Biophys. Acta 2007, 1772, 527–532. [DOI] [PubMed] [Google Scholar]
- Gaware V.; Kotade K.; Dhamak K.; Somawanshi S. Ceruloplasmin Its Role and Significance: A Review. Int. J. Biomed. Res. 2010, 4, 153–162. [Google Scholar]
- Gouya L.; Muzeau F.; Robreau A.-M.; Letteron P.; Couchi E.; Lyoumi S.; Deybach J.-C.; Puy H.; Fleming R.; Demant P.; Beaumont C.; Grandchamp B. Genetic Study of Variation in Normal Mouse Iron Homeostasis Reveals Ceruloplasmin as an HFE-Hemochromatosis Modifier Gene. Gastroenterology 2007, 132, 679–686. [DOI] [PubMed] [Google Scholar]
- Viatte L.; Nicolas G.; Lou D. Q.; Bennoun M.; Lesbordes-Brion J. C.; Canonne-Hergaux F.; Schonig K.; Bujard H.; Kahn A.; Andrews N. C.; Vaulont S. Chronic Hepcidin Induction Causes Hyposideremia and Alters the Pattern of Cellular Iron Accumulation in Hemochromatic Mice. Blood 2006, 107, 2952–2958. [DOI] [PubMed] [Google Scholar]
- Rivera S.; Liu L.; Gabayan V.; Sorenson O. E.; Ganz T. Hepcidin Excess Induces the Sequestration of Iron and Exacerbates Tumor-Associated Anemia. Blood 2005, 105, 1797–1802. [DOI] [PubMed] [Google Scholar]
- Gkouvatsos K.; Papanikolaou G.; Pantopoulos K. Regulation of Iron Transport and the Role of Transferrin. Biochim. Biophys. Acta 2012, 1820, 188–202. [DOI] [PubMed] [Google Scholar]
- Luck A. N.; Mason A. B. Transferrin-Mediated Cellular Iron Delivery. Curr. Top. Membr. 2012, 69, 3–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knovich M. A.; Storey J. A.; Coffman L. G.; Torti S. V.; Torti F. M. Ferritin for the Clinician. Blood Rev. 2009, 23, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muckenthaler M. U.; Galy B.; Hentze M. W. Systemic Iron Homeostasis and the Iron-Responsive Element/Iron-Regulatory Protein (IRE/IRP) Regulatory Network. Annu. Rev. Nutr. 2008, 28, 197–213. [DOI] [PubMed] [Google Scholar]
- Macedo M. F.; de Sousa M. Transferrin and the Transferrin Receptor: Of Magic Bullets and Other Concerns. Inflammation Allergy: Drug Targets 2008, 7, 41–52. [DOI] [PubMed] [Google Scholar]
- Ganz T. Is TfR2 the Iron Sensor?. Blood 2004, 104, 3839–3840. [Google Scholar]
- Arosio P.; Ingrassia R.; Cavadini P. Ferritins: A Family of Molecules for Iron Storage, Antioxidation, and More. Biochim. Biophys. Acta 2009, 1790, 589–599. [DOI] [PubMed] [Google Scholar]
- Li L.; Fang C. J.; Ryan J. C.; Niemi E. C.; Lebron J. A.; Björkman P. J.; Arase H.; Torti F. M.; Torti S. V.; Nakamura M. C.; Seaman W. E. Binding and Uptake of H-ferritin are Mediated by Human Transferrin Receptor-1. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 3505–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinelli P.; Samuelsson T. Evolution of the Iron-Responsive Element. RNA 2007, 13, 952–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walden W. E.; Selezneva A. I.; Dupuy J.; Volbeda A.; Fontecilla-Camps J. C.; Theil E. C.; Volz K. Structure of Dual Function Iron Regulatory Protein 1 Complexed with Ferritin IRE-RNA. Science 2006, 314, 1903–1908. [DOI] [PubMed] [Google Scholar]
- Gunshin H.; Allerson C. R.; Polycarpou-Schwarz M.; Rofts A.; Rogers J. T.; Kishi F.; Hentze M. W.; Rouault T. A.; Andrews N. C.; Hedinger M. A. Iron-Dependent Regulation of the Divalent Metal Ion Transporter. FEBS Lett. 2001, 509, 309–316. [DOI] [PubMed] [Google Scholar]
- Roseff S. D. Sickle Cell Disease: A Review. Immunohematology 2009, 25, 67–74. [PubMed] [Google Scholar]
- Schrier S. L.Pathophysiology of Alpha Thalassemia. UpToDate, 2014; http://www.uptodate.com/contents/pathophysiology-of-alpha-thalassemia.
- Martell A. E.; Motekaitis R. J.; Sun Y.; Clarke E. T.. Ligand Design of Chelating Agents Effective in the Coordination of Fe(III) and for the Removal of Iron in Cases of Iron Overload. In The Development of Iron Chelators for Clinical Use; Bergeron R. J., Brittenham G. M., Eds.; CRC Press: Boca Raton, FL, 1994; pp 329–351. [Google Scholar]
- Peter H. H.; Bergeron R. J.; Streiff R. R.; Wiegand J.. A Comparative Evaluation of Iron Chelators in a Primate Model. In The Development of Iron Chelators for Clinical Use; Bergeron R. J., Brittenham G. M., Eds.; CRC Press: Boca Raton, FL, 1994; pp 373–394. [Google Scholar]
- Bergeron R. J.; Wiegand J.; Dionis J. B.; Egil-Karmakka M.; Frei J.; Huxley-Tencer A.; Peter H. H. Evaluation of Desferrithiocin and Its Synthetic Analogues as Orally Effective Iron Chelators. J. Med. Chem. 1991, 34, 2072–2078. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; McManis J. S.; Weimar W. R.; Wiegand J.; Eiler-McManis E.. Iron Chelators and Therapeutic Uses. In Burger’s Medicinal Chemistry and Drug Discovery, 6th ed.; Abraham D. J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2003; pp 479–561. [Google Scholar]
- Wolfe L. C.; Nicolosi R. J.; Renaud M. M.; Finger J.; Hegsted M.; Peter H.; Nathan D. G. A Non-Human Primate Model for the Study of Oral Iron Chelators. Br. J. Haematol. 1989, 72, 456–461. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; Streiff R. R.; Creary E. A.; Daniels R. D. Jr.; King W.; Luchetta G.; Wiegand J.; Moerker T.; Peter H. H. A Comparative Study of the Iron-Clearing Properties of Desferrithiocin Analogues with Desferrioxamine B in a Cebus Monkey Model. Blood 1993, 81, 2166–2173. [PubMed] [Google Scholar]
- Bergeron R. J.; Streiff R. R.; Wiegand J.; Vinson J. R. T.; Luchetta G.; Evans K. M.; Peter H.; Jenny H.-B. A Comparative Evaluation of Iron Clearance Models. Ann. N.Y. Acad. Sci. 1990, 612, 378–393. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; McManis J. S.; Wiegand J.; Weimar W. R.; Brittenham G. M.. A Search for Clinically Effective Iron Chelators. In Iron Chelators: New Development Strategies; Badman D. G., Bergeron R. J., Brittenham G. M., Eds.; The Saratoga Group, LLC: Ponte Vedra Beach, FL, 1998; pp 253–292. [Google Scholar]
- Bergeron R. J.; Wiegand J.; Brittenham G. M. HBED: A Potential Alternative to Deferoxamine for Iron-Chelating Therapy. Blood 1998, 91, 1446–1452. [PubMed] [Google Scholar]
- Bergeron R. J.; Wiegand J.; Brittenham G. M. HBED: The Continuing Development of A Potential Alternative to Deferoxamine for Iron-Chelating Therapy. Blood 1999, 93, 370–375. [PubMed] [Google Scholar]
- Bergeron R. J.; Wiegand J.; Brittenham G. M. HBED: Preclinical Studies of a Potential Alternative to Deferoxamine for Treatment of Chronic Iron Overload and Acute Iron Poisoning. Blood 2002, 99, 3019–3026. [DOI] [PubMed] [Google Scholar]
- Yokel R. A.; Datta A. K.; Jackson E. G. Evaluation of Potential Aluminum Chelators in Vitro by Aluminum Solubilization Ability, Aluminum Mobilization from Transferrin and the Octanol/Aqueous Distribution of the Chelators and Their Complexes with Aluminum. J. Pharmacol. Exp. Ther. 1991, 257, 100–106. [PubMed] [Google Scholar]
- Neilands J. B.; Erickson T. J.; Rastetter W. H. Stereospecificity of the Ferric Enterobactin Receptor of Escherichia coli K-12. J. Biol. Chem. 1981, 256, 3831–3832. [PubMed] [Google Scholar]
- Bergeron R. J.; Dionis J. B.; Elliott G. T.; Kline S. J. Mechanism and Stereospecificity of the Parabactin-Mediated Iron-Transport System in Paracoccus denitrificans. J. Biol. Chem. 1985, 260, 7936–7944. [PubMed] [Google Scholar]
- Rastetter W. H.; Erickson T. J.; Venuti M. C. Synthesis of Iron Chelators. Enterobactin, Enantioenterobactin, and a Chiral Analogue. J. Org. Chem. 1981, 46, 3579–3590. [Google Scholar]
- Qiu D.-H.; Huang Z.-L.; Zhou T.; Shen C.; Hider R. C. In Vitro Inhibition of Bacterial Growth by Iron Chelators. FEMS Microbiol. Lett. 2011, 314, 107–111. [DOI] [PubMed] [Google Scholar]
- Sia A. K.; Allred B. E.; Raymond K. N. Siderocalins: Siderophore Binding Proteins Evolved for Primary Pathogen Host Defense. Curr. Opin. Chem. Biol. 2013, 17, 150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. M. T.; Martell A. E. The Kinetics of the Reaction of Iron(III) Chelates of Aminopolycarboxylic Acids with Ascorbic Acid. J. Am. Chem. Soc. 1968, 90, 3386–3389. [DOI] [PubMed] [Google Scholar]
- Martell A. E.; Motekaitis R. J.; Sun Y.; Ma R.; Welch M. J.; Pajeau T. New Chelating Agents Suitable for the Treatment of Iron Overload. Inorg. Chim. Acta 1999, 291, 238–246. [Google Scholar]
- Martell A. E.; Motekaitis R. J.; Clarke E. T.; Sun Y. Comparison of the Drugs Used For the Treatment of Iron Overload with Drugs that Have Potential for the Same Purpose. Drugs Today 1992, 28, 11–18. [Google Scholar]
- Motekaitis R. J.; Martell A. E.; Welch M. J. Stabilities of Trivalent Metal Complexes of Phenolic Ligands Related to N,N′-Bis-(2-hydroxybenzyl)ethylenediamine-N,N′-diacetic Acid (HBED). Inorg. Chem. 1990, 29, 1463–1467. [Google Scholar]
- Powell L. W.; Thomas M. J. Use of Diethylenetriamine Penta-Acetic Acid (D.T.P.A.) in the Clinical Assessment of Total Body Iron Stores. J. Clin. Pathol. 1967, 20, 896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry M.; Cartel G.; Sherlock S. Quantitative Measurement of Iron Stores with Diethylenetriamine Penta-Acetic Acid. Gut 1970, 11, 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma R.; Motekaitis R. J.; Martell A. E. Stability of Metal Ion Complexes of N,N′-Bis-(2-hydroxybenzyl)ethylenediamine-N,N′-diacetic Acid. Inorg. Chim. Acta 1994, 224, 151–155. [Google Scholar]
- Hershko C.; Grady R. W.; Link G. Phenolic Ethylene Derivatives: A Study of Orally Effective Iron Chelators. J. Lab. Clin. Med. 1984, 103, 337–346. [PubMed] [Google Scholar]
- Hershko C.; Grady R. W.; Link G. Development and Evaluation of the Improved Iron Chelating Agents EHPG, HBED and Their Dimethyl Esters. Hematologia 1984, 17, 25–33. [PubMed] [Google Scholar]
- Pandolfo M.; Hausmann L. Deferiprone for the Treatment of Friedreich’s Ataxia. J. Neurochem. 2013, 126, 142–146. [DOI] [PubMed] [Google Scholar]
- Berdoukas V.; Farmaki K.; Carson S.; Wood J.; Coates T. Treating Thalassemia Major-Related Iron Overload: The Role of Deferiprone. J. Blood Med. 2012, 3, 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghaie L.; Sadeghi M. M.; Nikazama A. Synthesis, Analysis, and Determination of Partition Coefficients of N-Arylhydroxypyridinone Derivatives as Iron Chelators. Res. Pharm. Sci. 2006, 1, 40–48. [Google Scholar]
- Epemolu R. O.; Ackerman R.; Porter J. B.; Hider R. C.; Damani L. A.; Singh S. HPLC Determination of 1,2-Diethyl-3-hydroxypyrid-4-one (CP94), Its Iron Complex [Fe(III)(CP94)3] and Glucuronide Conjugate [CP94-GLUC] in Serum and Urine of Thalassemic Patients. J. Pharm. Biomed. Anal. 1994, 12, 923–930. [DOI] [PubMed] [Google Scholar]
- Yokel R. A.; Fredenburg A. M.; Durbin P. W.; Xu J.; Rayens M. K.; Raymond K. N. The Hexadentate Hydroxypyridinonate TREN-(Me-3,2-HOPO) Is a More Orally Active Iron Chelator Than Its Bidentate Analogue. J. Pharm. Sci. 2000, 89, 545–555. [DOI] [PubMed] [Google Scholar]
- Xu J.; O’Sullivan B.; Raymond K. N. Hexadentate Hydroxypyridonate Iron Chelators Based on TREN-Me-3,2-HOPO: Variation of Cap Size. Inorg. Chem. 2002, 41, 6731–6742. [DOI] [PubMed] [Google Scholar]
- Jurchen K. M.; Raymond K. N. Terephthalamide-Containing Analogues of TREN-Me-3,2-HOPO. Inorg. Chem. 2006, 45, 1078–1090. [DOI] [PubMed] [Google Scholar]
- Nick H.; Wong A.; Acklin P.; Faller B.; Jin Y.; Lattmann R.; Sergejew T.; Hauffe S.; Thomas H.; Schnebli H. P. ICL670A: Preclinical Profile. Adv. Exp. Med. Biol. 2002, 509, 185–203. [DOI] [PubMed] [Google Scholar]
- Nisbet-Brown E.; Olivieri N. F.; Giardina P. J.; Grady R. W.; Neufeld E. J.; Sechaud R.; Krebs-Brown A. J.; Anderson J. R.; Alberti D.; Sizer K. C.; Nathan D. G. Effectiveness and Safety of ICL670 in Iron-Loaded Patients with Thalassaemia: A Randomised, Double-Blind, Placebo-Controlled, Dose-Escalation Trial. Lancet 2003, 361, 1597–1602. [DOI] [PubMed] [Google Scholar]
- Yacobovich J.; Stark P.; Barzilai-Birenbaum S.; Krause I.; Yaniv I.; Tamary H. Acquired Proximal Renal Tubular Dysfunction in Beta-Thalassemia Patients Treated with Deferasirox. J. Pediatr. Hematol./Oncol. 2010, 32, 564–567. [DOI] [PubMed] [Google Scholar]
- Hershko C.; Link G.; Pinson A.; Peter H. H.; Dobbin P.; Hider R. C. Iron Mobilization from Myocardial Cells by 3-Hydroxypyridin-4-one Chelators: Studies in Rat Heart Cells in Culture. Blood 1991, 77, 2049–2053. [PubMed] [Google Scholar]
- Porter J. B.; Morgan J.; Hoyes K. P.; Burke L. C.; Huehns E. R.; Hider R. C. Relative Oral Efficiency and Acute Toxicity of Hydroxypyridin-4-one Iron Chelators in Mice. Blood 1990, 76, 2389–2396. [PubMed] [Google Scholar]
- Balfour J. A. B.; Foster R. H. Deferiprone: A Review of its Clinical Potential in Iron Overload in β-Thalassemia Major and Other Transfusion-Dependent Diseases. Drugs 1999, 58, 553–578. [DOI] [PubMed] [Google Scholar]
- Richardson D. R. The Controversial Role of Deferiprone in the Treatment of Thalassemia. J. Lab. Clin. Med. 2001, 137, 324–329. [DOI] [PubMed] [Google Scholar]
- Kalinowski D. S.; Richardson D. R. The Evolution of Iron Chelators for the Treatment of Iron Overload Disease and Cancer. Pharmacol. Rev. 2005, 57, 547–583. [DOI] [PubMed] [Google Scholar]
- Kontoghiorghes G. J.; Aldouri M. A.; Sheppard L.; Hoffbrand A. V. 1,2-Dimethyl-3-hydroxypyrid-4-one, an Orally Active Chelator for Treatment of Iron Overload. Lancet 1987, 1, 1294–1295. [DOI] [PubMed] [Google Scholar]
- Liu Z. D.; Piyamongkol S.; Liu D. Y.; Khodr H. H.; Lu S. L.; Hider R. C. Synthesis of 2-Amido-3-hydroxypyridin-4(1H)-ones: Novel Iron Chelators with Enhanced pFe3+ Values. Bioorg. Med. Chem. 2001, 9, 563–573. [DOI] [PubMed] [Google Scholar]
- Olivieri N. F.; Koren G.; Hermann C.; Bentur Y.; Chung D.; Klein J.; St. Louis P.; Freedman M. H.; McClelland R. A.; Templeton D. M. Comparison of Oral Iron Chelator L1 and Desferrioxamine in Iron-Loaded Patients. Lancet 1990, 336, 1275–1279. [DOI] [PubMed] [Google Scholar]
- Singh S.; Epemolu R. O.; Dobbin P. S.; Tilbrook G. S.; Ellis B. L.; Damani L. A.; Hider R. C. Urinary Metabolic Profiles in Human and Rat of 1,2-Dimethyl- and 1,2-Diethyl-Substituted 3-Hydroxypyridin-4-ones. Drug Metab. Dispos. 1992, 20, 256–261. [PubMed] [Google Scholar]
- Bergeron R. J.; Streiff R. R.; Wiegand J.; Luchetta G.; Creary E. A.; Peter H. H. A Comparison of the Iron-Clearing Properties of 1,2-Dimethyl-3-hydroxypyridin-4-one, 1,2-Diethyl-3-hydroxypyridin-4-one, and Deferoxamine. Blood 1992, 79, 1882–1890. [PubMed] [Google Scholar]
- Porter J. B.; Abeysinghe R. D.; Hoyes K. P.; Barra C.; Huehns E. R.; Brooks P. N. Contrasting Interspecies Efficacy and Toxicology of 1,2-Diethyl-3-hydroxypyridin-4-one, CP94, Relates to Differing Metabolism of the Iron Chelating Site. Br. J. Hamaetol. 1993, 85, 159–168. [DOI] [PubMed] [Google Scholar]
- Porter J. B.; Singh S.; Hoyes K. P.; Epemolu O.; Abeysinghe R. D.; Hider R. C.. Lessons from Preclinical and Clinical Studies with 1,2-Diethyl-3-hydroxypyridin-4-one, CP94 and Related Compounds. In Progress in Iron Research; Hershko C., Konijn A. M., Aisen P., Eds.; Plenum Press: New York, 1994; pp 361–370. [DOI] [PubMed] [Google Scholar]
- Rai B. L.; Dekhordi L. S.; Khodr H.; Jin Y.; Liu Z.; Hider R. C. Synthesis, Physiochemical Properties, and Evaluation of N-Substituted-2-Alkyl-3-hydroxy-4(1H)-pyridinones. J. Med. Chem. 1998, 41, 3347–3359. [DOI] [PubMed] [Google Scholar]
- Liu Z. D.; Khodr H. H.; Liu D. Y.; Lu S. L.; Hider R. C. Synthesis, Physiochemical Characterization, and Biological Evaluation of 2-(1′-Hydroxyalkyl)-3-hydroxypyridin-4-ones: Novel Iron Chelators with Enhanced pFe3+ Values. J. Med. Chem. 1999, 42, 4814–4823. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Roy S.; Kong X.; Chen Y.; Liu D.; Hider R. C. Design and Synthesis of Fluorinated Iron Chelators for Metabolic Study and Brain Uptake. J. Med. Chem. 2012, 55, 2185–2195. [DOI] [PubMed] [Google Scholar]
- Exjade Prescribing Information; Novartis Pharmaceuticals Corporation: East Hanover, NJ, 2014; http://www.pharma.us.novartis.com/product/pi/pdf/exjade.pdf.
- Hay B. P.; Dixon D. A.; Vargas R.; Garza J.; Raymond K. N. Structural Criteria for the Rational Design of Selective Ligands. 3. Quantitative Structure-Stability Relationship for Iron(III) Complexation by Tris-Catecholamide Siderophores. Inorg. Chem. 2001, 40, 3922–3935. [DOI] [PubMed] [Google Scholar]
- Weitl F. L.; Raymond K. N.; Durbin P. W. Synthetic Enterobactin Analogues. Carboxamido-2,3-dihydroxyterephthalate Conjugates of Spermine and Spermidine. Am. Chem. Soc. 1981, 24, 203–206. [DOI] [PubMed] [Google Scholar]
- Weitl F. L.; Raymond K. N. Ferric Ion Sequencing Agents. 1. Hexadentate O-Bonding N,N′,N″-Tris(2,3-dihydroxybenzoyl) Derivatives of 1,5,9-Triazacyclotridecane and 1,3,5-Triaminomethylbenzene. J. Am. Chem. Soc. 1979, 101, 2728–2731. [Google Scholar]
- Hou Z.; Stack T. D. P.; Sunderland C. J.; Raymond K. N. Enhanced Iron(III) Chelation through Ligand Predisposition: Syntheses, Structures, and Stability of Tris-Catecholate Enterobactin Analogs. Inorg. Chim. Acta 1997, 263, 341–355. [Google Scholar]
- Anderegg G.; Räber M. Metal Complex Formation of a New Siderophore Desferrithiocin and of Three Related Ligands. J. Chem. Soc., Chem. Commun. 1990, 1194–1196. [Google Scholar]
- Hahn F. E.; McMurry T. J.; Hugi A.; Raymond K. N. Coordination Chemistry of Microbial Iron Transport. 42. Structural and Spectroscopic Characterization of Diastereomeric Cr(III) and Co(III) Complexes of Desferriferrithiocin. J. Am. Chem. Soc. 1990, 112, 1854–1860. [Google Scholar]
- Bergeron R. J.; Wiegand J.; McManis J. S.; McCosar B. H.; Weimar W. R.; Brittenham G. W.; Smith R. E. Effects of C-4 Stereochemistry and C-4′ Hydroxylation on the Iron Clearing Efficiency and Toxicity of Desferrithiocin Analogues. J. Med. Chem. 1999, 42, 2432–2440. [DOI] [PubMed] [Google Scholar]
- Baker E.; Wong A.; Peter H.; Jacobs A. Desferrithiocin is an Effective Iron Chelator in Vivo and in Vitro but Ferrithiocin Is Toxic. Br. J. Hamaetol. 1992, 81, 424–431. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J. Hydroxylation of Desazadesferrithiocin Analogues: A Structure–Activity Study. Hematology 2001, 1–8. [Google Scholar]
- Bergeron R. J.; Wiegand J.; McManis J. S.; Weimar W. R.; Huang G. Structure–Activity Relationships among Desazadesferrithiocin Analogues. Adv. Exp. Med. Biol. 2002, 509, 167–184. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; Wiegand J.; Weimar W. R.; Vinson J. R.; Bussenius J.; Yao G. W.; McManis J. S. Desazadesmethyldesferrithiocin Analogues as Orally Effective Iron Chelators. J. Med. Chem. 1999, 42, 95–108. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; Liu C. Z.; McManis J. S.; Xia M. X.; Algee S. E.; Wiegand J. The Desferrithiocin Pharmacophore. J. Med. Chem. 1994, 37, 1411–1417. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; Wiegand J.; Wollenweber M.; McManis J. S.; Algee S. E.; Ratliff-Thompson K. Synthesis and Biological Evaluation of Naphthyldesferrithiocin Iron Chelators. J. Med. Chem. 1996, 39, 1575–1581. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; Wiegand J.; McManis J. S.; Weimar W. R.; Park J. H.; Eiler-McManis E.; Bergeron J.; Brittenham G. M. Partition-Variant Desferrithiocin Analogues: Organ Targeting and Increased Iron Clearance. J. Med. Chem. 2005, 48, 821–831. [DOI] [PubMed] [Google Scholar]
- Sangster J.Octanol–Water Partition Coefficients: Fundamentals and Physical Chemistry; John Wiley and Sons: Chichester, England, 1997; Vol. 2. [Google Scholar]
- Bergeron R. J.; Wiegand J.; McManis J. S.; Vinson J. R. T.; Yao H.; Bharti N.; Rocca J. R. (S)-4,5-Dihydro-2-(2-hydroxy-4-hydroxyphenyl)-4-methyl-4-thiazolecarboxylic Acid Polyethers: A Solution to Nephrotoxicity. J. Med. Chem. 2006, 49, 2772–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan J. M.; Plone M.; Dagher R.; Bree M.; Marquis J. Preclinical and Clinical Development of Deferitrin, a Novel, Orally Available Iron Chelator. Ann. N.Y. Acad. Sci. 2005, 1054, 492–494. [DOI] [PubMed] [Google Scholar]
- Brittenham G. M. Pyridoxal Isonicotinoyl Hydrazone (PIH): Effective Iron Chelation after Oral Administration. Ann. N.Y. Acad. Sci. 1990, 612, 315–326. [DOI] [PubMed] [Google Scholar]
- Galanello R.; Forni G.; Jones A.; Kelly A.; Willemsen A.; He X.; Johnston A.; Fuller D.; Donovan J.; Piga A. A Dose Escalation Study of the Pharmacokinetics, Safety, and Efficacy of Deferitrin, an Oral Iron Chelator in Beta Thalassaemia Patients. ASH Annu. Meet. Abstr. 2007, 110, 2669. [Google Scholar]
- Bergeron R. J.; Wiegand J.; McManis J. S.; Bussenius J.; Smith R. E.; Weimar W. R. Methoxylation of Desazadesferrithiocin Analogues: Enhanced Iron Clearing Efficiency. J. Med. Chem. 2003, 46, 1470–1477. [DOI] [PubMed] [Google Scholar]
- Bergeron R. J.; Wiegand J.; Bharti N.; Singh S.; Rocca J. R. Impact of the 3,6,9-Trioxadecyloxy Group on Desazadesferrithiocin Analogue Iron Clearance and Organ Distribution. J. Med. Chem. 2007, 50, 3302–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron R. J.; Wiegand J.; McManis J. S.; Bharti N.; Singh S. Design, Synthesis, and Testing of Non-Nephrotoxic Desazadesferrithiocin Polyether Analogues. J. Med. Chem. 2008, 51, 3913–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hider R.; Kong X.; Luker T.; Conlon K.; Harland R. SPD602 is a Selective Iron Chelator Which Is Able To Mobilise the Non-Transferrin-Bound Iron Pool. Blood 2013, 122, 1673. [Google Scholar]
- Bergeron R. J.; Bharti N.; Wiegand J.; McManis J. S.; Singh S.; Abboud K. A. The Impact of Polyether Chain Length on the Iron Clearing Efficiency and Physiochemical Properties of Desferrithiocin Analogues. J. Med. Chem. 2010, 53, 2843–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron R. J.; Wiegand J.; Bharti N.; McManis J. S.; Singh S. Desferrithiocin Analogue Iron Chelators: Iron Clearing Efficiency, Tissue Distribution, and Renal Toxicity. BioMetals 2011, 24, 239–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rienhoff H. Y. Jr.; Virakasit V.; Tay L.; Harmatz P.; Vichinsky E.; Chirnomas D.; Kwiatkowski J. L.; Tapper A.; Kramer W.; Porter J. B.; Neufeld E. J. A Phase-1 Dose–Escalation Study: Safety, Tolerability, and Pharmacokinetics of FBS0701, a Novel Oral Iron Chelator for the Treatment of Transfusional Iron Overload. Haematologica. 2011, 96, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld E. J.; Galanello R.; Viprakasit V.; Aydinok Y.; Piga A.; Harmatz P.; Forni G. L.; Shah F. T.; Grace R. E. F.; Porter J. B.; Wood J. C.; Peppe J.; Jones A.; Rienhoff H. Y. Jr. A Phase 2 Study of the Safety, Tolerability, and Pharmacodynamics of FBS0701, a Novel Oral Iron Chelator, in Transfusional Iron Overload. Blood 2012, 119, 3263–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suk O. J. Paradoxical Hypomagnesemia Caused by Excessive Ingestion of Magnesium Hydroxide. Am. J. Emerg. Med. 2008, 26, 837.e1–837.e2. [DOI] [PubMed] [Google Scholar]
- Durlach J.; Durlach V.; Bac P.; Bara M.; Guiet-Bara A. Magnesium and Therapeutics. Magnesium Res. 1994, 7, 313–328. [PubMed] [Google Scholar]
- Randall R. E. Jr. Magnesium Toxicity. Ann. Int. Med. 1963, 58, 744. [Google Scholar]
- Bergeron R. J.; Wiegand J.; Bharti N.; McManis J. S. Substituent Effects on Desferrithiocin and Desferrithiocin Analogue Iron-Clearing and Toxicity Profiles. J. Med. Chem. 2012, 55, 7090–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosse G. Metabolites of the Pyrimidine Amine Preladenant as Adenosine A2a Receptor Antagonists. ACS Med. Chem. Lett. 2013, 4, 5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzer R.; Galeazzi R. L.; Karlaganis G.; Bircher J. Rate of Drug Metabolism in Man Measured by 14CO2-Breath Analysis. Eur. J. Clin. Pharmacol. 1978, 14, 293–299. [DOI] [PubMed] [Google Scholar]
- Li X.-Q.; Zhong D.-F.; Huang H.-H.; Wu S.-D. Demethylation Metabolism of Roxithromycin in Humans and Rats. Acta Pharmacol. Sin. 2001, 22, 469–474. [PubMed] [Google Scholar]
- Han W. K.; Bailly V.; Abichandani R.; Thadhani R.; Bonventre J. V. Kidney Injury Molecule-1 (KIM-1): A Novel Biomarker for Human Renal Proximal Tubule Injury. Kidney Int. 2002, 62, 237–244. [DOI] [PubMed] [Google Scholar]
- Bonventre J. V. Kidney Injury Molecule-1 (KIM-1): A Urinary Biomarker and Much More. Nephrol., Dial., Transplant. 2009, 24, 3265–3268. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Vaidya V. S.; Brown R. P.; Zhang J.; Rosenzweig B. A.; Thompson K. L.; Miller T. J.; Bonventre J. V.; Goering P. L. Comparison of Kidney Injury Molecule-1 and Other Nephrotoxicity Biomarkers in Urine and Kidney Following Acute Exposure to Gentamicin, Mercury, and Chromium. Toxicol. Sci. 2008, 101, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya V. S.; Ramirez V.; Ichimura T.; Bobadilla N. A.; Bonventre J. V. Urinary Kidney Injury Molecule-1: A Sensitive Quantitative Biomarker for Early Detection of Kidney Tubular Injury. Am. J. Physiol.: Renal Physiol. 2006, 290, F517–F529. [DOI] [PubMed] [Google Scholar]
- Vaidya V. S.; Ford G. M.; Waikar S. S.; Wang Y.; Clement M. B.; Ramirez V.; Glaab W. E.; Troth S. P.; Sistare F. D.; Prozialeck W. C.; Edwards J. R.; Bobadilla N. A.; Mefferd S. C.; Bonventre J. V. A Rapid Urine Test for Early Detection of Kidney Injury. Kidney Int. 2009, 76, 108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piyamongkol S.; Liu Z. D.; Hider R. C. Novel Synthetic Approach to 2-(1′-Hydroxyalkyl)- and 2-Amido-3-hydroxypyridin-4-ones. Tetrahedron 2001, 57, 3479–3486. [Google Scholar]
- Li M.-J.; Kwok W.-M.; Lam W. H.; Tao C.-H.; Yam V. W.-W.; Phillips D. L. Synthesis of Coumarin-Appended Pyridyl Tricarbonylrhenium (I) 2,2′-Bipyridyl Complexes with Oligoether Spacer and Their Fluorescence Resonance Energy Transfer Studies. Organometallics 2009, 28, 1620–1630. [Google Scholar]
- Hider R. C.; Kong X. Chemistry and Biology of Siderophores. Nat. Prod. Rep. 2010, 27, 637–657. [DOI] [PubMed] [Google Scholar]