

Abstract
Matrix metalloproteinase 13 (MMP-13) has been shown to be the main collagenase responsible for degradation of articular cartilage during osteoarthritis and therefore represents a target for drug development. As a result of high-throughput screening and structure–activity relationship studies, we identified a novel, highly selective class of MMP-13 inhibitors (compounds 1 (Q), 2 (Q1), and 3 (Q2)). Mechanistic characterization revealed a noncompetitive nature of these inhibitors with binding constants in the low micromolar range. Crystallographic analyses revealed two binding modes for compound 2 in the MMP-13 S1′ subsite and in an S1/S2* subsite. Type II collagen- and cartilage-protective effects exhibited by compounds 1, 2, and 3 suggested that these compounds might be efficacious in future in vivo studies. Finally, these compounds were also highly selective when tested against a panel of 30 proteases, which, in combination with a good CYP inhibition profile, suggested low off-target toxicity and drug–drug interactions in humans.
Introduction
According to a National Health Interview Survey1 in 2003–2005, different forms of arthritis affected approximately 46 million adults in the United States. The total cost estimate attributable to arthritis and related conditions is close to $128 billion.2 Osteoarthritis (OA), the most common form of arthritis, is characterized by the destruction of articular cartilage. The main constituents of articular or joint cartilage are type II collagen and various proteoglycans, such as aggrecan, chondroitin sulfate, and hyaluronan.3 The tensile strength of articular cartilage is due to the highly constrained supersecondary triple-helical structure of type II collagen.4 This triple-helical structure also makes collagen resistant to hydrolysis by the majority of human proteinases, with the exception of several matrix metalloproteinases (MMP-1, -8, and -13, known as collagenases, and membrane type 1 MMP).5 In native joint cartilage, type II collagen fibrils are protected from cleavage by tight association with molecules of aggrecan.6 In arthritic cartilage, aggrecan is hydrolyzed by members of another family of metalloproteases, ADAMTS-1, -4, and -5, known as aggrecanases.7 Aggrecanolysis removes aggrecan molecules from type II collagen fibrils, which makes collagenolysis possible.
MMP-13 has been shown to be the main collagenase responsible for degradation of articular cartilage during OA8 and therefore represents a target for drug development. Multiple attempts to develop MMP-13 inhibitor-based drugs have failed mostly due to the dose-limiting side effects collectively known as musculoskeletal syndrome (MSS).9−11 While the exact cause of MSS is not known, it is believed to be due to the lack of selectivity of drug candidates toward other members of the MMP family as well as related metalloenzymes.10,12−14 High structural similarity of the catalytic domains of MMPs and the fact that the majority of chemistry efforts focused on active site Zn-binding groups as a basis for the MMP inhibitors resulted in clinical trial failures mainly due to the off-target inhibition by drug candidates.12,13,15
Despite the prevalence of zinc chelators among MMP inhibitors, there are examples of inhibitors that do not act by binding the active site zinc, but rather bind via so-called exosites or allosteric sites.16−19 Aventis discovered a pyrimidinedicarboxamide that had low micromolar potency for MMP-13 and no activity against other MMPs when tested at 100 μM.16 The potency of this compound was further improved to low nanomolar without loss of selectivity.16 Pfizer reported discovery of highly selective nanomolar range MMP-13 inhibitors based on pyrimidinedione and quinazolinone scaffolds acting via binding to the same S1′ exosite.17,20 Furthermore, pyrimidinedione derivatives were efficacious and safe in rabbit and dog models of OA.20,21 Similarly, Alantos Pharmaceuticals identified a new class of highly selective non-zinc-binding MMP-13 inhibitors.18,19
Although selective MMP-13 inhibitors have been described by Alantos, Aventis, Boehringer, Pfizer, and Wyeth, important pharmacokinetic (PK) and/or other data have not been reported for many of these compounds, and no clinical studies have appeared. For example, no PK or MSS data have been reported for the Aventis and Wyeth compounds.16,22 The first series of Pfizer compounds, while exhibiting good PK and MSS data, were tested against a limited number of MMPs.23−25 In similar fashion, the Boehringer compounds exhibited good PK data but were tested against a limited number of MMPs, and not at all in an MSS model.26,27 The Alantos compounds exhibited excellent MMP selectivity and good PK data, but were not tested in an MSS model.19,28 Only the second series of Pfizer compounds were reported to exhibit excellent MMP selectivity and good PK and MSS data.17,20,29 However, as mentioned above, no clinical studies have been reported for the Pfizer compounds. In our hands, we found the primary Pfizer compound (Table 1) to have low solubility (it could only be tested at a maximal concentration of 2.5 μM), and it inhibited cytochrome P450 1A2. Most recently, the Takeda Pharmaceutical Co. reported yet another non-zinc-binding inhibitor of MMP-13 that acts via binding to the S1′ site.30 The lead of the series, compound 26c, exhibited subnanomolar activity against MMP-13 and good oral availability; however, the type of inhibition was not published. Most of the above inhibitors possess large scaffolds that bind in the MMP-13 S1′ subsite (Chart 1). The structure of a non-zinc-binding mixed inhibitor from Alantos has not been published (compound ALS 1-0635). Neither the type of inhibition nor the binding site has been published for Boehringer compound 3.
Table 1. Mechanistic Characterization of MMP-13 Inhibitors Using fTHP-15 as the Substratec.
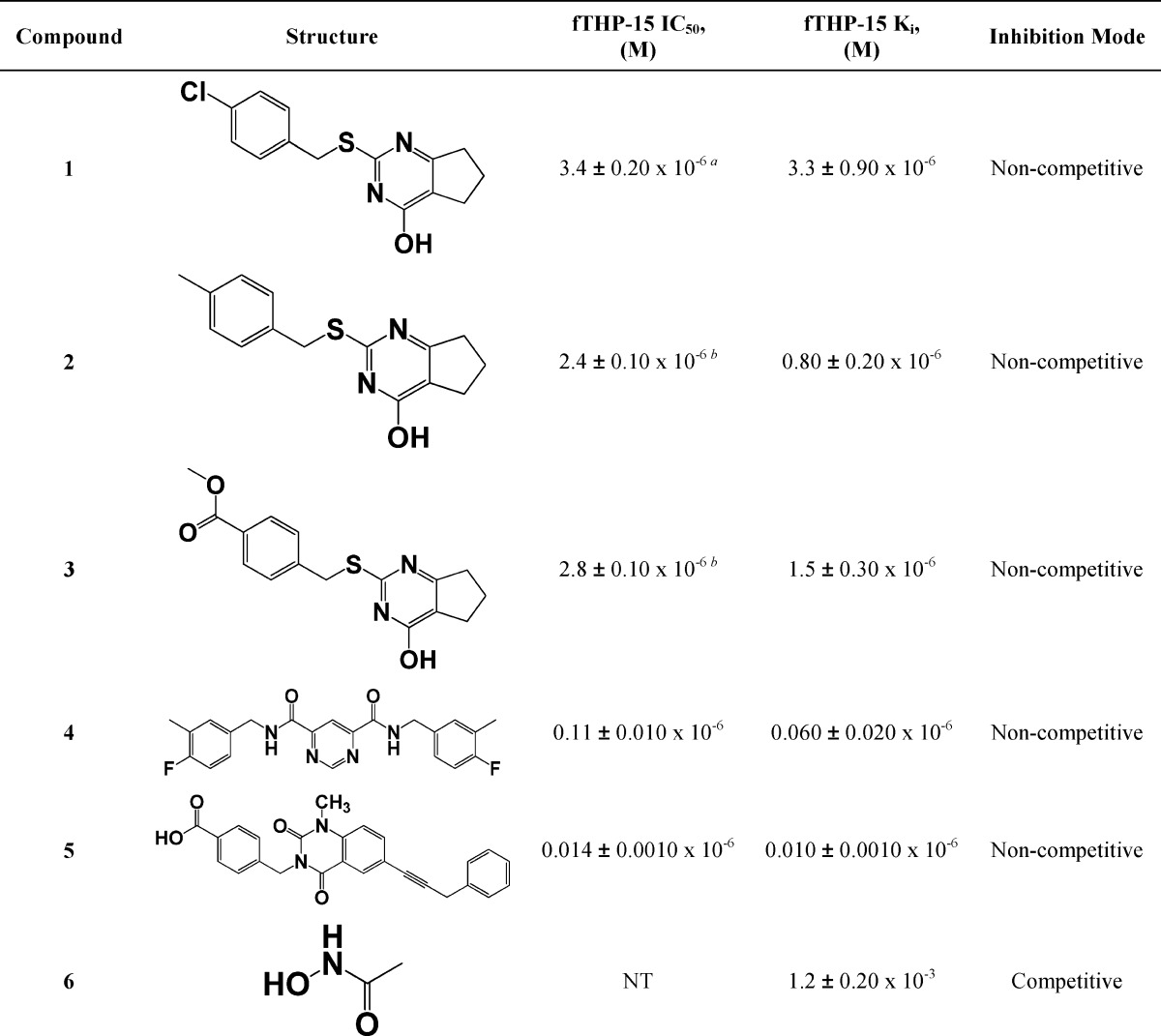
Chart 1. Structures and Inhibition Type Information of Known Selective Inhibitors of MMP-13a.

a Pfizer compound 2 is compound 5 in the present study. Aventis compound 4 is compound 4 in the present study.
Recently, we reported the results of an HTS campaign that resulted in the discovery of a selective MMP-13 inhibitor (1; Table 1),31 which was further optimized as a result of medicinal chemistry efforts (2 and 3; Table 1).32 In the studies reported herein, we mechanistically characterized compounds 1, 2, and 3, assessed their selectivity against an expanded enzymatic panel, and evaluated their ability to prevent type II collagen degradation. The binding of compound 2 to MMP-13 was determined directly by using single-crystal X-ray diffraction.
Results
Single Inhibition Kinetics
Previously, we reported the outcome of an HTS campaign that resulted in a discovery of selective MMP-13 inhibitor 1 (Table 1).31 Interestingly, this was the only compound that was found to be a more effective inhibitor of hydrolysis of a triple-helical, collagen-model substrate compared to a short, linear substrate. This finding suggested that compound 1 was mechanistically different from the rest of the HTS leads. Additionally, as a result of a structure–activity relationship (SAR) study of the 1 chemotype, we identified 2 and 3, which exhibited improved potency against MMP-13 (Table 1) and selectivity against antitargets MMP-1 and MMP-8 (IC50 > 40 μM).32 We have now utilized known selective inhibitors of MMP-13 to benchmark our lead compound. More specifically, we examined (E)-4-((1-methyl-2,4-dioxo-6-(3-phenylprop-1-enyl)-1,2-dihydroquinazolin-3(4H)-yl)methyl)benzoic acid, reported by Pfizer as compound 2,17 and N4,N6-bis(4-fluoro-3-methylbenzyl)pyrimidine-4,6-dicarboxamide, reported by Aventis as compound 4,16 which are referred to herein as 5 and 4, respectively. Compounds 1–3 exhibited low to submicromolar Ki values (see Table 1), whereas 4 and 5 had Ki values of 0.06 ± 0.02 and 0.01 ± 0.0 μM, respectively, when tested with the triple-helical fTHP-15 substrate. Interestingly, all tested compounds, with the exception of 6 (N-hydroxyacetamide (AHA)), inhibited MMP-13 hydrolysis of fTHP-15 via a noncompetitive mechanism (Figure 1). This finding suggests that, similarly to 4 and 5, compounds 1–3 do not bind to the active site Zn and possibly bind outside of an active site (i.e., to an exosite).
Figure 1.

Lineweaver–Burke plot of inhibition of MMP-13 fTHP-15 hydrolysis by (A) 1, (B) 2, (C) 3, (D) 4, (E) 5, and (F) 6. Note the lines of best fit crossing at the y-axis indicative of noncompetitive inhibition for all compounds except AHA. All Ki values were determined by nonlinear regression (hyperbolic equation) analysis using the mixed inhibition model, which allows for simultaneous determination of the mechanism of inhibition. The mechanism of inhibition was qualitatively confirmed using Lineweaver–Burke analysis.
Dual Inhibition Kinetics
To test whether 1 binds to the active site Zn, a known Zn binder, 6 (AHA), a competitive millimolar range MMP-13 inhibitor (see Table 1), was used in combination with 1 following a previously described methodology.336 (AHA) was used in the range of 0–1.25 mM in combination with 1 in the range of 0–2 μM. When initial velocities from this experiment were organized in a Yonetani–Theorell plot, they formed a series of intersecting lines of best fit (Figure 2A). In Yonetani–Theorell plots, the intersecting lines indicate simultaneous (i.e., mutually nonexclusive) binding of both inhibitors to the enzyme.34 For any mutually nonexclusive pair of inhibitors, x = −BKi when [I] is plotted on the x-axis.35 Rearrangement of this equation allows us to calculate the value of B using Ki values obtained from independent experiments with fTHP-15 as the substrate (Table 1). The resulting B value was <1, which indicated that 6 and 1 bind synergistically (i.e., binding of one inhibitor increases the affinity of binding of the second inhibitor).
Figure 2.

Yonetani–Theorell plot of MMP-13 fTHP-15 hydrolysis in the presence of (A) 6 and 1, (B) 4 and 1, and (C) 5 and 1. Note the nonparallel lines of best fit indicating mutually nonexclusive binding by two inhibitors. B < 1, indicating synergistic binding for all three pairs of inhibitors tested.
1 was subsequently tested in combination with 4, which is a noncompetitive, non-Zn-binding inhibitor of MMP-13 with a nanomolar Ki value (Table 1). Compound 4 binds within the S1′ “specificity loop” of the MMP-13 CAT domain, which is a known exosite.16,33 Surprisingly, 1 exhibited mutually nonexclusive binding when cotested with 4 (Figure 2B). The B value was <1, indicating positive cooperativity with 4. Similarly, 1 binding to MMP-13 was synergistic with that of 5 (B < 1, Figure 2C).
Crystal Structure of the MMP-13 CAT–2 Complex
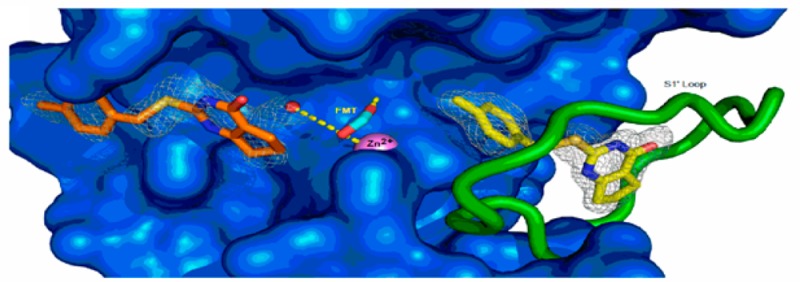
The structure of the MMP-13 CAT–compound 2 inhibitor complex containing two protomers (A and B) in the asymmetric unit was refined to a resolution of 1.66 Å with R and Rfree values of 0.134 and 0.200, respectively (Supplemental Table 3, Supporting Information). As observed in other MMP-13 CAT–inhibitor complexes in this crystal system,16,17,23,36−38 the first eight residues of the two protomers engaged in extensive interprotomer interactions around a ∼153° axis of rotation, an asymmetry that caused these residues to differ somewhat in conformation. Four residues at the C-terminus of protomer B were not observed in protomer A. The two protomers are otherwise remarkably similar, aligning a root-mean-square deviation (rmsd) of 0.18 Å for 391 backbone atom target pairs. Annealed omit maps confirmed that 2 was bound in two distinct binding sites (Figure 3A). Compound 2 is observed in both protomers in the S1′ specificity pocket (Figure 3B), while in protomer A a second 2 binds near the S1 and S2 subsites (designated S1/S2*) within the substrate binding cleft (Figure 3C). Compound 2 does not contact the catalytic zinc ion in either site. The oxygen atoms of formate from the reservoir solution chelated the catalytic zinc ion in a mode similar to that of 6 to prevent protein self-proteolysis.28,33
Figure 3.

Structure of the MMP-13 CAT–compound 2 complex revealing the distinct binding sites in protomer A (see the text). (A) Annealed omit map with coefficients Fo – Fc contoured at 2.5σ superimposed on the refined model of the MMP-13 CAT–2 complex. 2 was left out of the phase calculation. The surface of the protein is blue, and the S1′ loop is green. The catalytic zinc ion is a violet sphere, and formate from the reservoir solution is shown as cyan sticks. The two distinct 2 molecules are represented as yellow and orange sticks. Formate from the reservoir solution chelates the zinc ion and forms hydrogen bonds with a nearby water molecule and the side chain of the catalytic glutamic acid (E233) hidden beneath the surface. (B) S1′ 2 binding site in MMP-13 CAT. The color scheme is the same as that in panel A, and the view is rotated only slightly around the vertical compared to that of panel A. Gray dashed lines represent selected van der Waals contacts (<4.6 Å), and the blue dashed lines represent hydrogen-bonding interactions. (C) σ–A weighted electron density with coefficients 2mFo – dFc superimposed on the S1/S2* 2 binding site in the refined model of the complex. The color scheme is the same as that in panel A except Y176, F189, and P190, which make the majority of contacts with 2, are highlighted as dark green sticks. The green sphere is a calcium ion. Hydrogen bonds are shown as orange dashes, and metal–ligand interactions are shown as yellow dashes. (D) Superposition of compounds 2 (yellow), 4 (white), and 5 (pink) in the S1′ binding site. All three compounds accept a hydrogen bond from the amide nitrogen of Thr247. A second molecule of 2 can be seen on the other side of catalytic zinc in the S1/S2* site. (E) Superposition of compound 2 (yellow) with hydroxamic acid-based inhibitors (PDB code 456C, red; PDB code 830C, cyan36) that do not intrude deeply into the S1′ specificity loop. The difference in positions of residues 248–251 (disordered in structure 830C) in the two classes of inhibitors suggests the S1′ specificity loop is conformationally dynamic in the uninhibited enzyme.
Compound 2 intruded deeply into the hydrophobic S1′ pocket such that its p-methylphenyl moiety pointed toward the substrate binding cleft and contacted the hydrophobic region composed of four residues (Leu185, Val219, Pro242, and Tyr244). There was also a π–π stacking interaction between the imidazole ring of His222 and the compound 2 phenyl ring. The cyclopentene ring moiety pointed toward the S1′ specificity loop and contacted the hydrophobic portions of residues Leu218, Leu239, Thr245, Tyr246, Thr247, Phe252, and Pro255. The central S16 atom of 2 accepted a hydrogen bond from the amide nitrogen of Thr245 (3.4 Å), while the N19 atom of 2 accepted a hydrogen bond from the carbonyl oxygen of Thr245 (2.7 Å). There were two water-mediated hydrogen bond networks that played roles in 2 binding. A cluster of three water molecules (left side of 2 in Figure 3B) connected the carbonyl oxygen atoms of MMP-13 CAT residues Pro236, Ala238, and Ile243 and the side chain hydroxyl moiety of Thr245 to inhibitor 2 atoms S16, N18, and N19. The 2 O17 carbonyl oxygen atom accepted a hydrogen bond from the amide nitrogen of Thr247 (3.3 Å). A second, four-water cluster (right side of 2 in Figure 3B) connected the 2 O17 atom to the MMP-13 S1′ specificity loop (Thr247 side chain hydroxyl oxygen, Met253 amide nitrogen, and Lys249, His251, and Met253 carbonyl oxygens). This hydrogen bond network was essential to stabilize the otherwise flexible S1′ specificity loop to define the S1′ specific pocket size for inhibitor 2.
The second molecule of 2 resided in the substrate binding cleft on the opposite side of the catalytic zinc ion relative to the S1′ specificity pocket. The p-methylphenyl moiety of 2 faced away from the catalytic center and interacted with the hydrophobic region composed of Tyr176, Phe189, and Pro190. The cyclopentene ring pointed toward the catalytic center. An amide nitrogen proton pointed into the aromatic π hole.
Type II Collagen Assay
To assess the potency of lead compounds with a cognate substrate, compounds 1–3 were tested in an assay utilizing type II collagen conjugated to FITC. All compounds exhibited dose-dependent responses in both fTHP-15 (Figure 4A) and type II collagen assays (Figure 4B) with Hill slopes close to 1.0. Compounds 1 and 2 appeared to be more potent in the type II collagen assay than in the fTHP-15 assay (type II collagen IC50 = 0.7 ± 0.2 and 0.4 ± 0.1 μM versus fTHP-15 IC50 = 3.4 ± 0.2 and 2.4 ± 0.1 μM, respectively). Compounds 3 and 4 were equipotent in these two assays (type II collagen IC50 = 2.3 ± 0.2 and 0.07 ± 0.01 μM versus fTHP-15 IC50 = 2.8 ± 0.1 and 0.11 ± 0.01 μM, respectively). Interestingly, while 4 clearly appeared more potent in the fTHP-15 assay than compounds of the 1 chemotype, the difference in apparent potency was much less pronounced in the type II collagen assay. Furthermore, all four compounds exhibited concentration–response curves characteristic of partial inhibitors in the collagen assay (Figure 4B). In the fTHP-15 assay compounds 1–3 appeared to be partial inhibitors, whereas 4 inhibited hydrolysis completely (Figure 4A).
Figure 4.

Results of (A) fTHP-15, (B) type II collagen, and (C) cartilage explant assays. In the fTHP-15 assay (A) compounds of the Q series appear to be partial inhibitors, whereas 4 inhibits hydrolysis completely. In the type II collagen assay (B) all four compounds exhibit concentration–response curves characteristic of partial inhibitors. 4 clearly appeared more potent in the fTHP-15 assay than compounds of the 1 chemotype, while the difference in apparent potency was much less pronounced in the type II collagen assay.
Cartilage Explant Assay
To assess the ability of compounds 1–3 to prevent cartilage degradation, in vitro bovine cartilage digestion was examined. The three compounds inhibited cartilage degradation to different degrees: 69 ± 17%, 59 ± 50%, and 38 ± 20% inhibition at 10 μM 1–3, respectively, and 70 ± 8% inhibition for EDTA at 50 μM (Figure 4C).
Protease Selectivity Panel
One of the primary reasons for failures of MMP inhibitors in clinical trials was the lack of selectivity.13,39,40 To assess the selectivity of compounds 1–3, they were tested against a panel of 30 proteases in 12-point 3-fold dilution dose–response curve format in a single dose. Compounds 1–3 were tested in the 20 μM to 0.1 nM range, while 4 and 5 were tested in the 5 μM to 0.028 nM range. When tested against 15 metalloproteases (Figure 5A), the only enzyme inhibited in excess of 20% by all five tested compounds was MMP-13. Compounds 1–3 appear to have activity in excess of 10% against MMP-2, MMP-3, MMP-9, MMP-12, and ACE; however, examination of the full range of tested concentrations (20 μM to 0.1 nM) revealed the absence of dose-dependent responses (Supplemental Figure 1A, Supporting Information) and therefore must represent an artifact. In addition to the metalloprotease panel, compounds were tested against representatives of the serine, cysteine, and aspartic classes of proteases (Figure 5B). All tested compounds exhibited activity below 10% inhibition for all tested enzymes, except 2 with caspase 5 and cathepsin K and 5 with cathepsin K. Upon further inspection, the activity of 2 against caspase 5 and 5 against cathepsin K proved to be artifactual (Supplemental Figure 1B). In contrast, compound 2 exhibited a semblance of dose-dependent response against cathepsin K, with the highest inhibitory activity reaching 20% at 20 μM.
Figure 5.

Results of protease panel profiling assays. Test compounds were screened in 12-point 3-fold dilution dose–response curve format. The activity of each enzyme was determined as the conversion (%) of the substrate to product after 3 h of incubation. (A) Metalloprotease panel results at the highest compound concentration tested. (B) Serine, cysteine, and aspartic protease panel results at the highest compound concentration tested.
Cytochrome P450 Inhibition Panel
Success or failure of drug candidate molecules in the clinic greatly depends on whether they induce toxicity. To assess possible toxicity, compounds 1–3 were screened against a panel of cytochrome P450 (CYP) enzymes in seven-point dose–response format in duplicate. Compounds 1 and 2 exhibited low micromolar potency against CYP 1A2, 2C9, and 2C19 (Table 3), while 3, 4, and 5 did not inhibit any of the tested CYPs in excess of 50% at inhibitor concentrations of 20, 20, and 2.5 μM, respectively. Examination of the activity of tested compounds at maximal tested concentrations revealed that 3 inhibited CYP 1A2 and 2C9 at 29% and 17%, respectively (Figure 6). Compound 4 inhibited all CYPs in excess of 10%, except 3A4, when tested with midazolam substrate (Figure 6). Compound 5 was tested at a maximal concentration of 2.5 μM due to solubility concerns. At this concentration the only CYP inhibited was 1A2 (17% inhibition).
Table 3. Cytochrome P450 Inhibition Profiling.
| CYP enzyme | substrate | 1 IC50 (μM) | 2 IC50 (μM) | 3 IC50 (μM) | 4 IC50 (μM) | 5 IC50 (μM) |
|---|---|---|---|---|---|---|
| 1A2 | tacrin | 8.6 | 3.7 | >20 | >20 | >2.5 |
| 2B6 | bupropion | >20 | >20 | >20 | >20 | >2.5 |
| 2C8 | amodiaquine | >20 | >20 | >20 | >20 | >2.5 |
| 2C9 | diclofenac | 19 | 8.8 | >20 | >20 | >2.5 |
| 2C19 | (S)-mephentoin | 6.5 | 7. 9 | >20 | >20 | >2.5 |
| 2D6 | dextromethophan | >20 | >20 | >20 | >20 | >2.5 |
| 3A4 | midazolam | >20 | >20 | >20 | >20 | >2.5 |
| 3A4 | testosterone | >20 | >20 | >20 | >20 | >2.5 |
Figure 6.

Results of cytochrome P450 inhibition profiling.
Metabolic Stability
To determine the metabolic stability of 1–3 to predict potential problems, the compounds were incubated with human, rat, and mouse microsomes. Sunitinib was used as a control for this study. Sunitinib is an orally available inhibitor of receptor tyrosine kinases used for the treatment of renal cell carcinoma (RCC) and imatinib-resistant gastrointestinal stromal tumor (GIST) and, therefore, provides a good comparison for metabolic stability studies. Inhibitors 1–3 exhibited low metabolic stability in the microsomes of all three species tested, with the best half-life of 10.4 min exhibited by 1 in human liver microsomes (Table 4). Compounds 1–3 are thioethers and, therefore, could potentially be metabolized in vivo by several members of CYP or flavin-containing monooxygenase (FMO) families to sulfoxides or sulfones,41 which could lead to the limitation of oral bioavailability due to a loss of activity. To test this hypothesis, we attempted to synthesize the sulfone and sulfoxide derivatives of 2; however, they proved to be unstable and decomposed during attempts at chromatographic purification. These results suggest that 1–3 would be unstable in vivo.
Table 4. Summary of Testing of Compounds 1–3 and Sunitinib for Metabolic Stability in Human, Murine, and Rat Microsomesa.
| name | human t1/2 (min) | mouse t1/2 (min) | rat t1/2 (min) |
|---|---|---|---|
| sunitinib | 44.1 | 12.3 | 26.5 |
| 1 | 10.4 | 1.3 | 6.8 |
| 2 | 7.3 | 0 | NT |
| 3 | 0 | 2.0 | 1.8 |
t1/2 = half-life.
Discussion
Kinetic and Structural Studies
As a result of an HTS campaign31 and subsequent SAR studies,32 we identified a series of MMP-13 inhibitors that exhibited a good selectivity profile toward other members of the MMP family. A lack of obvious Zn-binding moieties, preferential inhibition of triple-helical peptide hydrolysis,31 and a partial inhibitor profile42 of a lead of a series, 1, suggested inhibition by a mechanism other than competitive. Indeed, mechanistic characterization of these compounds revealed that they inhibit MMP-13 via a noncompetitive mechanism. The noncompetitive mechanism of inhibition of MMP-13 by our lead compounds in combination with results of the dual inhibition study with a Zn binder (AHA) strongly suggested the possibility of binding outside of an active site at a so-called exosite.43 The importance of exosites for collagenolysis has been shown in multiple studies.44,45 Exploration of these exosites was suggested as a possible way to find selective MMP inhibitors.46 Indeed, Pfizer17 and Aventis16 identified small-molecule inhibitors of MMP-13 that bind within one of these exosites to the so-called “specificity loop” 47 and inhibit catalysis by inducing rigidity. Yonetani–Theorell analysis of dual inhibition kinetic data showed that binding of 1 does not preclude binding of either 4 or 5 to MMP-13, but in fact made it stronger as evidenced by parameter B < 1. This finding suggested either formation of a quaternary complex between two inhibitors, MMP-13, and the fTHP-15 substrate or binding of 1 to an exosite other than the specificity loop. Crystallographic analyses revealed a second binding region for 2 (the S1 and S2 subsites) that is novel compared to those of all known MMP-13 inhibitors. In addition, alignment of all known MMP-13–inhibitor structures to our MMP-13–2 structure showed that the molecular scaffold of 2 is the smallest in the S1′ pocket. Superposition of 2, 4, and 5 in their respective binding poses in the S1′ subsite suggested that 2 and 4 or 5 cannot simultaneously reside in the S1′ subsite (Figure 3D). Since both 4 and 5 have much greater affinity than 2, this suggested that when 4 or 5 is present in the S1′ subsite, 2 can only bind to the second binding site, explaining the lack of mutually exclusive binding (Figure 2B,C) between 2 and 4 or 5.
To understand the effects of zinc-binding and non-zinc-binding inhibitors on MMP-13, we overlaid MMP-13 complexed with 2 with MMP-13 complexed with 4 and 5 (Figure 3D) and two hydroxamate-based inhibitors36 (Figure 3E). The analysis of the superimposed structures revealed that the specificity loop, where the S1 binding site is located, is more disordered in MMP-13 complexed with hydroxamate-based inhibitors, which can be explained by the fact that hydroxamate-based inhibitors do not significantly protrude into the specificity loop.
Future Optimization Studies
Knowledge of the novel binding site of compounds 1–3 will facilitate SAR studies to achieve greater potencies for new analogues. Indeed, despite demonstrating a good selectivity profile comparable to that of compounds developed by Aventis (4) and Pfizer (5), compounds 1–3 have 10–100 times lower affinity than 4 and 5. However, compounds 1–3 are significantly smaller than 4 and 5, suggesting that greater affinity can be achieved by increasing the size of future analogues of 1–3.
Ligand efficiency indices have been utilized as the rational approach to the optimization of compound properties.48 This approach combines affinity toward the target with physicochemical properties. One of the most frequently used methods is based on the combination of the binding efficiency index (BEI, pKi/MW) and surface-binding efficiency index (SEI, pKi/PSA), which allows monitoring of the dependence of affinity on the size and polar surface of the molecule. SEI and BEI values of an idealized compound (Ki = 1 nM, PSA = 50 Å2)49 are used as a way to guide the optimization process. As evidenced by Table 5, the BEI values of 1–3 are similar to those of 4 and 5, whereas the SEI values of 1–3 are greater than those of 4 and 5. Compound 2 exhibits SEI and BEI values that best approximate the values of idealized compound 7. Comparison of SEI versus BEI values (Figure 7) suggested that, to achieve greater potency, both the size and polar surface area of future analogues of 2 should be increased.
Table 5. Summary of Calculations of Ligand Efficiency Indicesa.
| name | fTHP-15 Ki (M) | pKi | PSA (Ǻ2/100) | MW (kDa) | BEI | SEI |
|---|---|---|---|---|---|---|
| 1 | 3.30 × 10–6 | 5.5 | 0.44 | 0.292 | 18.8 | 12.5 |
| 2 | 8.00 × 10–7 | 6.1 | 0.45 | 0.272 | 22.4 | 13.5 |
| 3 | 1.50 × 10–6 | 5.8 | 0.71 | 0.316 | 18.4 | 8.2 |
| 4 | 6.00 × 10–8 | 7.2 | 0.82 | 0.41 | 17.6 | 8.8 |
| 5 | 1.00 × 10–8 | 8.0 | 0.78 | 0.426 | 18.8 | 10.3 |
| 6 | 1.19 × 10–3 | 2.9 | 0.49 | 0.075 | 39.0 | 6.0 |
| 7 (idealized compd) | 1.00 × 10–9 | 9.0 | 0.5 | 0.333 | 27.0 | 18 |
BEI = binding efficiency index. SEI = surface-binding efficiency index.
Figure 7.

Mapping of surface-binding and binding efficiency indices for compounds of the 1 series and study controls. Compound numbering is according to Table 4. Key: red circle, 1; purple circle, 5; blue circle, 4; green circle, 3. The red diagonal line represents an optimization plane as described in ref (49).
Prevention of Type II Collagen and Cartilage Degradation
Degradation of type II collagen is a hallmark of OA. Thus, prevention of collagen degradation in vitro provides an important estimate of the ability of MMP-13 inhibitors to modify the course of the disease. Compounds 1–3 modulated MMP-13 activity with apparent potencies in the submicromolar and low micromolar ranges. To determine whether MMP-13 inhibition translated into prevention of articular cartilage degradation, compounds 1–3 were studied in a cartilage explant assay. The cartilage explant assay is a well-established in vitro system for testing of prevention of cartilage degradation.17,18,50 Compounds 1–3 inhibited cartilage degradation at a level comparable to that of EDTA, a broad-spectrum metalloprotease inhibitor. These data suggested that compounds 1–3 can be potentially protective against cartilage degradation in vivo.
Protease Selectivity Panel
Development of roughly 30 antiarthritic drugs has been discontinued due to the occurrence of MSS as a result of MMP inhibitor dosing in the clinical trials.10,12,13 MSS does not appear to be caused by the off-target inhibition of a single enzyme, but rather a combination of several MMPs and possibly other related enzymes.14 Among enzymes proposed to play role in MSS development are MMP-1,14 MMP-2,51 MMP-9,52 MT1-MMP,53 and adamalysins.14 With that in mind, 1–3 were screened against an expanded panel of proteases, which included proposed arthritis antitargets (MMP-1, MMP-2, MMP-9, and MT1-MMP) and representative adamalysins (ADAM10 and ADAM17 (TACE)). This panel also contained nonmetalloproteinases. Compounds 4 and 5 were also screened. Compound 5 was previously tested in the rat model of MSS and was found to be safe at plasma concentrations well above those needed to protect articular cartilage17 and therefore represents an excellent control for a selectivity study. Importantly, 1–3 exhibited a selectivity profile similar to that of 5; in addition to sparing all arthritis antitargets, they did not inhibit MMP-3 and MMP-8 (important cancer antitargets).12,13 While the usefulness of this protease panel in predicting MSS occurrence has not been established, the similarity of the protease inhibition profile of compounds 1–3 to that of 5 suggested a similar in vivo safety profile. This hypothesis, of course, will have to be tested in one of the existing animal MSS models.17
Cytochrome P450 Inhibition Panel
Adverse effects during animal and human testing account for more than 20% of cases where drug development had to be terminated early.54 Drug–drug or drug–food interactions due to the inhibition or induction of members of the cytochrome P450 enzyme family are believed to be one of the main causes of these effects.55 Approximately 95% of all drug metabolism is attributable to just five representatives of the CYP family, with the 3A4 enzyme being responsible for almost half of these reactions.55 It is therefore encouraging that compounds 1–3 did not inhibit 3A4 when tested with the midazolam substrate. 1 and 2 exhibited some inhibition of 3A4 when testosterone was used as the substrate (14% and 20%, respectively), whereas 3 showed very little activity (3% inhibition). Overall, 3 exhibited significantly lower CYP activity as compared to 1 and 2. Interestingly, the inhibitory activity of compound 3 was either the same as or lower than that of 4 and 5. While the overall good inhibition profile of compounds 1–3 is encouraging, these data suggested the possibility of in vivo drug–drug interactions due to the inhibition of 3A4, 1A2, 2C9, and 2C19.
Metabolic Stability
Compounds 1–3 exhibited low metabolic stability when exposed to microsomes, and their sulfone and sulfoxide metabolites were too unstable to be tested for MMP-13 inhibition; therefore, their activity against MMP-13 could not be ascertained. This suggests that future optimization studies should be focused not just on the improvement of potency, but also on the metabolic stability of future analogues to preempt the lack of in vivo efficacy of future MMP-13 inhibitors deriving from 1–3 due to their metabolic instability. Indeed, compounds 1–3 represent just a starting point in drug development. Future studies will ascertain the need for a thioether in the inhibitors and its effect on metabolic stability. There are examples of thioether-containing drugs and preclinical candidates with good in vivo efficacy,56 suggesting that metabolic stability of the molecule is a function of the structure and properties of the molecule as a whole, and not just one particular atom in the structure.
Conclusion
The lead compound of this novel class of MMP-13 inhibitors, 1, was discovered as a result of an HTS campaign under the auspices of the NIH Roadmap initiative. Additional compounds belonging to the same chemotype were identified as a result of a medicinal chemistry optimization effort as a part of the same program. As a result of the characterization studies presented here, it was determined that compounds 1–3 inhibit MMP-13 via a noncompetitive mechanism with the possibility of binding to two exosites. Type II collagen and cartilage protective effects exhibited by these MMP-13 inhibitors in vitro indicated the possibility of these compounds being efficacious in future in vivo studies. These compounds were also highly selective when tested against a panel of 30 proteases, which, in combination with a good CYP inhibition profile, suggested low off-target toxicity and drug–drug interactions in humans.
Experimental Procedures
MMP-13 inhibitors were synthesized and characterized as described elsewhere.32 [(Pyrimidine-4,6-dicarboxylic acid bis(4-fluoro-3-methylbenzamide)] was obtained from EMD Biosciences/Calbiochem (product no. 444283; San Diego, CA). The synthesis, purification, and characterization of the fluorogenic triple-helical peptide substrate (fTHP-15) have been described.57 Full-length recombinant human pro-MMP-13 (rhMMP-13) was purchased from R&D Systems (catalog no. 511-MM; Minneapolis, MN). The zymogen form of MMP-13 was converted to the active form by incubating pro-MMP-13 with 1 mM (p-aminophenyl)mercuric acid (APMA) for 2 h at 37 °C.58 The stock of active MMP-13 was diluted to 1 μM and stored at −80 °C. Proteases used in the protease profiling panel were from R&D Systems (ACE, ACE2, ADAM10, ADAM17, furin, IDE, neprilysin, thrombin, UPA), Invitrogen (BACE), Biomol (MMP-1–3, -7–9, and -12–14, caspases 1–3 and 5–7), and Calbiochem (Factor Xa, cathepsin D, L, S, and K). CYP450 substrates bupropion, (S)-mephenytoin, and midazolam were obtained from BD Biosciences Discovery Labware (Woburn, MA), while tacrine, amodiaquine, diclofenac, dextromethorphan, and testosterone were purchased from Sigma-Aldrich (St. Louis, MO). CYP450 control inhibitors ketoconazole, sulfaphenazole, and (S)-benzylnirvanol were obtained from BD Biosciences Discovery Labware, while 7,8-benzoflavone and quinidine were purchased from Sigma-Aldrich. Montelukast was purchased from Cayman Chemical (Ann Arbor, MI). Probe substrate metabolites were from BD Biosciences Discovery Labware with the exception of 1-hydroxytacrine, which was purchased from Sigma-Aldrich. Stable labeled isotope internal standards were from BD Biosciences Discovery Labware, and bucetin was from Sigma-Aldrich. All other reagents were purchased from Sigma-Aldrich. Pooled human liver microsomes (HLM, catalog no. 452161; 20 mg/mL protein concentration) were from BD Biosciences Discovery Labware.
Single Inhibitor Kinetics
fTHP-15 and MMP-13 working solutions were prepared in enzyme assay buffer (EAB; 50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 10 mM CaCl2, 0.05% Brij-35 (Sigma-Aldrich)). All reactions were conducted in 384-well white polystyrene plates (Greiner, North Carolina, catalog no. 784076). Determinations of inhibition constants and modalities were conducted by incubating the range of fTHP-15 substrate concentrations (2–25 μM) with 4 nM MMP-13 at room temperature in the presence of varying concentrations of inhibitors. Fluorescence was measured on a Tecan Safire2 monochromator microplate reader using λexcitation = 324 nm and λemission = 393 nm. Rates of hydrolysis were obtained from plots of fluorescence versus time using data points from only the linear portion of the hydrolysis curve.
All kinetic parameters were calculated using GraphPad Prism, version 5.01 (GraphPad Software, Inc., La Jolla, CA). KM values were determined by nonlinear regression analysis using the one-site hyperbolic binding model35 and additionally evaluated by linear analysis. All Ki values were determined by nonlinear regression (hyperbolic equation) analysis using the mixed inhibition model, which allows for simultaneous determination of the mechanism of inhibition.35 The mechanism of inhibition was determined using the “α” parameter derived from a mixed-model inhibition by GraphPad Prism. The mechanism of inhibition was additionally confirmed by Lineweaver–Burke plots.
Dual Inhibition Kinetics
A matrix of two different inhibitor combinations was created in 384-well white polystyrene plates by serially diluting them in EAB. MMP-13 and fTHP-15 were then added, resulting in 4 nM and 8 μM final assay concentrations, respectively. Fluorescence was measured on a Tecan Safire2 monochromator microplate reader. Rates of hydrolysis were obtained from plots of fluorescence versus time using data points from only the linear portion of the hydrolysis curve.
Yonetani–Theorell Analysis of Dual Inhibition Kinetic Data
Data from kinetic experiments utilizing a matrix of two different inhibitor concentrations were fitted to the Yonetani–Theorell equation:
where vij is the initial velocity of fTHP-15 hydrolysis in the presence of both inhibitors, v0 is the initial velocity of fTHP-15 hydrolysis in the uninhibited reaction, Ki and Kj are the dissociation constants for inhibitors I and J, respectively, and B is the parameter that describes the effect of binding of inhibitor I on the affinity of inhibitor J. When two inhibitors bind in a mutually exclusive manner, B = ∞. When two inhibitors bind completely independently of each other, B = 1. Synergistic or antagonistic binding of two inhibitors yields B < 1 or B > 1, respectively. The initial rates of fTHP-15 hydrolysis in the presence of two inhibitors were arranged as Dixon plots with 1/vij plotted as a function of [I] in the presence of varying concentrations of inhibitor J.
Crystallographic Analysis of the MMP-13–2 Complex
The recombinant MMP-13 catalytic domain (MMP-13 CAT, residues 104–274) was expressed, purified, and refolded as described.59 The reconstituted enzyme, which was fully functional in the modified Knight substrate assay,60 was concentrated to 0.5 mg/mL (MMP-13-CAT ε280 = 28 590 mol–1 cm–1) and made 25% (v/v) in glycerol for storage at −80 °C. Immediately prior to crystallization trials, the glycerol was removed from the protein by dialysis against buffer containing 20 mM Tris (pH 8.0), 5 mM CaCl2, and 50 mM NaCl. A 100 mM stock solution of 2 in DMSO was added to obtain a 5:1 inhibitor:protein stoichiometric ratio. After incubation on ice for 1 h, the sample was concentrated to 12 mg/mL and screened in sitting drops using 1344 unique conditions coming from commercially available crystallization screening kits (Qiagen Inc., Valencia, CA). A Phoenix crystallization robot (Art Robbins Instruments, Sunnyvale, CA) was used to dispense the protein sample and crystallization reagents. The screen was performed at room temperature and 4 °C. The initial conditions were systematically optimized, and rod-shaped single crystals suitable for X-ray diffraction work were grown at 4 °C from drops containing protein solution in a 3:1 ratio (v/v) with reservoir solution consisting of 0.1 M Tris (pH 8.5), 1.5 M ammonium formate, and 10–14% PEG4000. Suitable specimens were extracted with nylon loops, swept through reservoir solution made 20% (v/v) in glycerol as a cryoprotectant, and flash-cooled in liquid nitrogen. Diffraction data were measured at beamline 24-ID-C at the Advanced Photon Source, Argonne, IL, equipped with a Pilatus 6M detector and processed with the program XDS.61 Initial phases were obtained via the molecular replacement method as implemented in PHASER62 using the MMP-13 CAT coordinates of Protein Data Bank entry 3ZXH(63) as the search model. Simulated annealing and individual anisotropic temperature factor refinement with the PHENIX suite of programs64 were followed by manual adjustment of the model using the program Coot.65 The positions of 2 molecules bound to MMP-13 CAT were illuminated in difference Fourier electron density maps and were subsequently cross-validated in annealed omit maps. The model was refined using stereochemical restraints generated with the electronic Ligand Builder and Optimization Workbench (eLBOW).66 Structure factors and refined coordinates are deposited in the Protein Data Bank under accession code 4L19.
Ligand Efficiency Calculations
Ligand efficiency indices were calculated according to49
Type II Collagen Assay
Type II collagen conjugated to FITC was purchased from Sigma-Aldrich (catalog no. C4486). All experiments were performed in 384-well white microtiter plates. The assay was initiated by dispensing 10 μL of 222 nM FITC–type II collagen in EAB using an FRD IB workstation (Aurora Discovery, Carlsbad, CA). A 100 nL volume of 75% DMSO/25% water containing the test compounds was dispensed using a 384-head Pintool system (GNF Systems, San Diego, CA). Reactions were initiated by addition of 10 μL of 40 nM MMP-13 in EAB. After 22 h of incubation at 37 °C, the emission fluorescence was read on a PerkinElmer Viewlux (PerkinElmer, Turku, Finland) microplate reader (λexcitation = 480 nm, λemission = 530 nm). Test compounds were assayed in triplicate using 10-point 3-fold serial dilutions. For each compound, fluorescence data were fitted with a four-parameter equation describing a sigmoidal dose–response curve with adjustable baseline using the GraphPad Prism, version 5.01, suite of programs. The IC50 values were generated from fitted curves by solving for the x-intercept at the 50% inhibition level of the y-intercept.
Cartilage Explant Assay
Bovine articular cartilage was procured by Articular Engineering, LLC (Skokie, IL). Cartilage pieces (∼3 mm2) were frozen and thawed three times in LN2 to render them nonviable followed by trypsin digestion to remove aggrecan as described elsewhere.6 After 4 h of digestion, the cartilage was rinsed with 10% FBS and PBS. The cartilage was incubated with 100 nM MMP-13 in a 96-well polystyrene sterile nontreated plate (Falcon no. 1172) in the presence or absence of the compounds for 18 h at 37 °C and 5% CO2. Test compounds were assayed in triplicate at a single concentration, 50 μM for EDTA and 10 μM for compounds 1–3. Collagen degradation products in the buffer were measured using the C1,2C enzyme-linked immunosorbent assay (Ibex, Montreal, Quebec, Canada, catalog no. 60-1002-001). Absorbance data were normalized to wells containing undigested cartilage (100% inhibition) and MMP-13-digested cartilage (0% inhibition) to obtain inhibition values.
General Protease Profiling Protocol
Assays were performed at Nanosyn, http://www.nanosyn.com. The highest concentrations of compounds to be used in the assays were determined. The compounds were solubilized at 50 μM in protease assay buffers, and the absorbance at λ = 540 nm was measured and compared to that of buffer alone. All experiments were performed in a 384-well plate. The assay began by dispensing 5 μL of test compounds in assay buffer followed by 10 μL of the respective protease. Enzymes were allowed to incubate with the test compounds for 30 min at 25 °C. The assays were initiated by addition of 10 μL of various concentrations of the respective peptide substrates. The assays were incubated for 3 h at 25 °C and terminated by addition of 45 μL of quenching solution containing SDS, EDTA, and iodoacetate. Quenched assay solution was used for analysis on the microfluidic-based LabChip 3000 (Caliper Life Sciences, Inc., Hopkinton, MA). Test compounds were screened in 12-point 3-fold dilution dose–response curve format in singlicate. The activity of each enzyme was determined as the conversion (%) of the substrate to product after 3 h of incubation. The activity in the presence of a compound was plotted as a function of the compound concentration, and the curves were fitted using the built-in dose–response model algorithm of XLfit software (IDBS). The IC50 values of the compounds were determined as the concentrations that resulted in 50% enzyme activity when compared to the activity of the control samples (without a compound). Where applicable, the Hill slopes were calculated using the XLFit software.
Cyp450 Panel Inhibition
Experiments were conducted in duplicate at 37 °C using 96-well polypropylene reaction plates incubated on a heating block (Mecour, Groveland, MA). Test compound concentrations were 0, 0.03, 0.1, 0.3, 1, 3, 10, and 20 μM for compounds 1–4 and 0, 0.003, 0.01, 0.03, 0.1, 0.3, 1.0, and 2.5 μM for 5. Incubation mixtures (400 μL) contained probe substrate, inhibitor, HLM, and an NADPH-regenerating system (1.3 mM NADP+, 3.3 mM glucose 6-phosphate, 0.4 U/mL glucose 6-phosphate dehydrogenase, 3.3 mM magnesium chloride) in 0.1 M potassium phosphate buffer (pH 7.4). Reactions were initiated by the addition of diluted HLM and terminated by combining 4 parts sample with 1 part stop solution (stable labeled isotope internal standard containing 0.1% formic acid in acetonitrile) and placing them on ice. Protein concentrations, incubation times, and substrate concentrations are listed in Table 2. Metabolites were quantified by using the BIOCIUS RapidFire ultra-high-throughput mass spectrometer at Biocius Life Sciences Inc. (Woburn, MA) using a proprietary sample purification and injection system coupled to a Sciex API-4000 triple-quadrupole mass spectrometer at the transitions shown in Table 2. Samples were separated on an SPE column (reversed-phase C4 chemistry) with mobile phases of 0.09% formic acid/0.01% trifluoroacetic acid in water and 0.09% formic acid/0.01% trifluoroacetic acid in acetonitrile at a flow rate of 1.5 mL/min. Detection metabolites and internal standard mass-to-charge ratios are listed in Table 2. Test compound activity against CYP450 enzymes was calculated as follows:
where R + i is the area ratio of probe substrate metabolite formed in the presence of the inhibitor and R – i is the area ratio of probe substrate metabolite formed in the absence of the inhibitor.
Table 2. CYP450 Inhibition Assay Conditions.
| CYP450 isoform | substrate | [S] (μM) | [HLM] (mg/mL) | incubation time (min) | metabolite (mass transition, amua) | internal standard (mass transition, amua) |
|---|---|---|---|---|---|---|
| CYP1A2 | tacrine | 1 | 0.2 | 10 | 1-hydroxytacrine (215 → 182) | bucetin (224 → 136) |
| CYP2B6 | bupropion | 80 | 0.2 | 20 | hydroxybupropion (256 → 139) | hydroxybupropion-d6 (262 → 244) |
| CYP2C8 | amodiaquine | 1.5 | 0.02 | 5 | desethylamodiaquine (330 → 285) | desethylamodiaquine-d3 (333 → 285) |
| CYP2C9 | diclofenac | 5 | 0.05 | 5 | 4′-hydroxydiclofenac (312 → 268) | 4′-hydroxydiclofenac-13C6 (316 → 272) |
| CYP2C19 | (S)-mephenytoin | 40 | 0.3 | 10 | 4′-hydroxy-(S)-mephenytoin (235 → 150) | 4′-hydroxy-(S)-mephenytoin-d3 (238 → 150) |
| CYP2D6 | dextromethorphan | 5 | 0.1 | 5 | dextrorphan (258 → 157) | dextrorphan-d3 (261 → 157) |
| CYP3A4 | midazolam | 3 | 0.02 | 5 | 1′-hydroxymidazolam (342 → 203) | 1′-hydroxymidazolam-13C3 (347 → 208) |
| CYP3A4 | testosterone | 50 | 0.05 | 10 | 6β-hydroxytestosterone (305 → 269) | 6β-hydroxytestosterone-d7 (312 → 276) |
Atomic mass units.
IC50 values were calculated by linear interpolation as follows:
where Lconcn is the highest concentration of inhibitor that results in <50% inhibition, Hconcn is the lowest concentration of inhibitor that results in >50% inhibition, L is the inhibition (%) associated with Lconc, and H is the inhibition (%) associated with Hconc.
Hepatic Microsomal Stability
Metabolic stability was evaluated by incubating 1 μM compound with 1 mg/mL hepatic microsomes (human, rat, and mouse) in 100 mM potassium phosphate buffer, pH 7.4. The reactions were held at 37 °C with continuous shaking. The reaction was initiated by adding NADPH, 1 mM final concentration. The final incubation volume was 300 μL, and 40 μL aliquots were removed at 0, 5, 10, 20, 40, and 60 min. The removed aliquot was added to 160 μL of acetonitrile to stop the reaction and precipitate the protein. NADPH dependence of the reaction is evaluated in parallel incubations without NADPH. At the end of the assay, the samples were centrifuged through a 0.45 μm filter plate (Millipore Solventer low binding hydrophilic plates, catalog no. MSRLN0450) and analyzed by LC–MS/MS. The data were log transformed, and the results are reported as half-lives.
Acknowledgments
The National Institutes of Health (Grants MH078948-01, U54MH074404, CA098799, and AR063795) supported this work (Florida). Dr. Louis Scampavia, Pierre Baillargeon, and Lina DeLuca (Lead Identification Division, Translational Research Institute, Scripps Florida) are thanked for their assistance with compound management. This work was also supported in part by a grant from the Robert A. Welch Foundation (AQ-1399) (to P.J.H.) and is based in part upon research conducted at the Northeastern Collaborative Access Team beamlines of the Advanced Photon Source, supported by Award RR-15301 from the National Center for Research Resources at the National Institutes of Health. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. W-31-109-ENG-38. Support for the X-ray Crystallography Core Laboratory by the UTHSCSA Executive Research Committee and by a Cancer Therapy & Research Center (CTRC) Cancer Center Support Grant (NCI P30CA054174) is also acknowledged.
Glossary
Abbreviations Used
- MMP
matrix metalloproteinase
- HTS
high-throughput screening
- AHA
acetohydroxamic acid
- OA
osteoarthritis
- ADAMTS
a disintegrin and metalloprotease with trombospondin motifs
- ADAM
a disintegrin and metalloprotease
- IDE
insulin-degrading enzyme
- ACE
angiotensin-converting enzyme
- UPA
urokinase-type plasminogen activator
- BACE
β-site APP-cleaving enzyme
- fTHP
fluorogenic triple-helical peptide
Supporting Information Available
Enzymes and substrates used in protease profiling of MMP-13 inhibitors, CYP450 inhibition assay conditions, X-ray data collection and protein structure refinement statistics, and MMP-12 and cathepsin K dose–response results for compounds of the 1 series. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hootman J.; B J.; Helmick C.; Langmaid G. Prevalence of doctor-diagnosed arthritis and arthritis-attributable activity limitation—United States, 2003–2005. Morbidity Mortal. Wkly. Rep. 2006, 55, 1089–1092. [PubMed] [Google Scholar]
- CDC. Arthritis. http://www.cdc.gov/arthritis/data_statistics.htm (Accessed February 11, 2014).
- Caterson B.; Flannery C. R.; Hughes C. E.; Little C. B. Mechanisms involved in cartilage proteoglycan catabolism. Matrix Biol. 2000, 19, 333–344. [DOI] [PubMed] [Google Scholar]
- Knudson C. B.; Knudson W. Cartilage proteoglycans. Semin. Cell Dev. Biol. 2001, 12, 69–78. [DOI] [PubMed] [Google Scholar]
- Nagase H.; Woessner J. F. Jr. Matrix metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494. [DOI] [PubMed] [Google Scholar]
- Pratta M. A.; Yao W.; Decicco C.; Tortorella M. D.; Liu R. Q.; Copeland R. A.; Magolda R.; Newton R. C.; Trzaskos J. M.; Arner E. C. Aggrecan protects cartilage collagen from proteolytic cleavage. J. Biol. Chem. 2003, 278, 45539–45545. [DOI] [PubMed] [Google Scholar]
- Tortorella M. D.; Malfait A. M.; Deccico C.; Arner E. The role of ADAM-TS4 (aggrecanase-1) and ADAM-TS5 (aggrecanase-2) in a model of cartilage degradation. Osteoarthritis Cartilage 2001, 9, 539–552. [DOI] [PubMed] [Google Scholar]
- Neuhold L. A.; Killar L.; Zhao W.; Sung M. L.; Warner L.; Kulik J.; Turner J.; Wu W.; Billinghurst C.; Meijers T.; Poole A. R.; Babij P.; DeGennaro L. J. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Invest. 2001, 107, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaishi H.; Kimura T.; Dalal S.; Okada Y.; D’Armiento J. Joint diseases and matrix metalloproteinases: a role for MMP-13. Curr. Pharm. Biotechnol. 2008, 9, 47–54. [DOI] [PubMed] [Google Scholar]
- Fingleton B. MMPs as therapeutic targets—still a viable option?. Semin. Cell Dev. Biol. 2008, 19, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrage P. S.; Mix K. S.; Brinckerhoff C. E. Matrix metalloproteinases: role in arthritis. Front. Biosci. 2006, 11, 529–543. [DOI] [PubMed] [Google Scholar]
- Overall C. M.; Kleifeld O. Tumour microenvironment—opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [DOI] [PubMed] [Google Scholar]
- Overall C. M.; Kleifeld O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br. J. Cancer 2006, 94, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson J. T. The importance of estimating the therapeutic index in the development of matrix metalloproteinase inhibitors. Cardiovasc. Res. 2006, 69, 677–687. [DOI] [PubMed] [Google Scholar]
- Georgiadis D.; Yiotakis A. Specific targeting of metzincin family members with small-molecule inhibitors: progress toward a multifarious challenge. Bioorg. Med. Chem. 2008, 16, 8781–8794. [DOI] [PubMed] [Google Scholar]
- Engel C. K.; Pirard B.; Schimanski S.; Kirsch R.; Habermann J.; Klingler O.; Schlotte V.; Weithmann K. U.; Wendt K. U. Structural basis for the highly selective inhibition of MMP-13. Chem. Biol. 2005, 12, 181–189. [DOI] [PubMed] [Google Scholar]
- Johnson A. R.; Pavlovsky A. G.; Ortwine D. F.; Prior F.; Man C. F.; Bornemeier D. A.; Banotai C. A.; Mueller W. T.; McConnell P.; Yan C.; Baragi V.; Lesch C.; Roark W. H.; Wilson M.; Datta K.; Guzman R.; Han H. K.; Dyer R. D. Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J. Biol. Chem. 2007, 282, 27781–27791. [DOI] [PubMed] [Google Scholar]
- Baragi V. M.; Becher G.; Bendele A. M.; Biesinger R.; Bluhm H.; Boer J.; Deng H.; Dodd R.; Essers M.; Feuerstein T.; Gallagher B. M. Jr.; Gege C.; Hochgurtel M.; Hofmann M.; Jaworski A.; Jin L.; Kiely A.; Korniski B.; Kroth H.; Nix D.; Nolte B.; Piecha D.; Powers T. S.; Richter F.; Schneider M.; Steeneck C.; Sucholeiki I.; Taveras A.; Timmermann A.; Van Veldhuizen J.; Weik J.; Wu X.; Xia B. A new class of potent matrix metalloproteinase 13 inhibitors for potential treatment of osteoarthritis: evidence of histologic and clinical efficacy without musculoskeletal toxicity in rat models. Arthritis Rheum 2009, 60, 2008–2018. [DOI] [PubMed] [Google Scholar]
- Piecha D.; Weik J.; Kheil H.; Becher G.; Timmermann A.; Jaworski A.; Burger M.; Hofmann M. W. Novel selective MMP-13 inhibitors reduce collagen degradation in bovine articular and human osteoarthritis cartilage explants. Inflammation Res. 2010, 59, 379–389. [DOI] [PubMed] [Google Scholar]
- Li J. J.; Nahra J.; Johnson A. R.; Bunker A.; O’Brien P.; Yue W. S.; Ortwine D. F.; Man C. F.; Baragi V.; Kilgore K.; Dyer R. D.; Han H. K. Quinazolinones and pyrido[3,4-d]pyrimidin-4-ones as orally active and specific matrix metalloproteinase-13 inhibitors for the treatment of osteoarthritis. J. Med. Chem. 2008, 51, 835–841. [DOI] [PubMed] [Google Scholar]
- Settle S.; Vickery L.; Nemirovskiy O.; Vidmar T.; Bendele A.; Messing D.; Ruminski P.; Schnute M.; Sunyer T. Cartilage degradation biomarkers predict efficacy of a novel, highly selective matrix metalloproteinase 13 inhibitor in a dog model of osteoarthritis: confirmation by multivariate analysis that modulation of type II collagen and aggrecan degradation peptides parallels pathologic changes. Arthritis Rheum. 2010, 62, 3006–3015. [DOI] [PubMed] [Google Scholar]
- Chen J. M.; Nelson F. C.; Levin J. I.; Mobilio D.; Moy F. J.; Nilakantan R.; Zask A.; Powers R. Structure-based design of a novel, potent, and selective inhibitor for MMP-13 utilizing NMR spectroscopy and computer-aided molecular design. J. Am. Chem. Soc. 2000, 122, 9648–9654. [Google Scholar]
- Blagg J. A.; Noe M. C.; Wolf-Gouveia L. A.; Reiter L. A.; Laird E. R.; Chang S. P.; Danley D. E.; Downs J. T.; Elliott N. C.; Eskra J. D.; Griffiths R. J.; Hardink J. R.; Haugeto A. I.; Jones C. S.; Liras J. L.; Lopresti-Morrow L. L.; Mitchell P. G.; Pandit J.; Robinson R. P.; Subramanyam C.; Vaughn-Bowser M. L.; Yocum S. A. Potent pyrimidinetrione-based inhibitors of MMP-13 with enhanced selectivity over MMP-14. Bioorg. Med. Chem. Lett. 2005, 15, 1807–1810. [DOI] [PubMed] [Google Scholar]
- Reiter L. A.; Freeman-Cook K. D.; Jones C. S.; Martinelli G. J.; Antipas A. S.; Berliner M. A.; Datta K.; Downs J. T.; Eskra J. D.; Forman M. D.; Greer E. M.; Guzman R.; Hardink J. R.; Janat F.; Keene N. F.; Laird E. R.; Liras J. L.; Lopresti-Morrow L. L.; Mitchell P. G.; Pandit J.; Robertson D.; Sperger D.; Vaughn-Bowser M. L.; Waller D. M.; Yocum S. A. Potent, selective pyrimidinetrione-based inhibitors of MMP-13. Bioorg. Med. Chem. Lett. 2006, 16, 5822–5826. [DOI] [PubMed] [Google Scholar]
- Shah M.; Huang D.; Blick T.; Connor A.; Reiter L. A.; Hardink J. R.; Lynch C. C.; Waltham M.; Thompson E. W. An MMP13-selective inhibitor delays primary tumor growth and the onset of tumor-associated osteolytic lesions in experimental models of breast cancer. PLoS One 2012, 7, e29615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim-Riether A.; Taylor S. J.; Liang S.; Gao D. A.; Xiong Z.; Michael August E.; Collins B. K.; Farmer B. T. II; Haverty K.; Hill-Drzewi M.; Junker H. D.; Mariana Margarit S.; Moss N.; Neumann T.; Proudfoot J. R.; Keenan L. S.; Sekul R.; Zhang Q.; Li J.; Farrow N. A. Improving potency and selectivity of a new class of non-Zn-chelating MMP-13 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 5321–5334. [DOI] [PubMed] [Google Scholar]
- Gao D. A.; Xiong Z.; Heim-Riether A.; Amodeo L.; August E. M.; Cao X.; Ciccarelli L.; Collins B. K.; Harrington K.; Haverty K.; Hill-Drzewi M.; Li X.; Liang S.; Margarit S. M.; Moss N.; Nagaraja N.; Proudfoot J.; Roman R.; Schlyer S.; Keenan L. S.; Taylor S.; Wellenzohn B.; Wiedenmayer D.; Li J.; Farrow N. A. SAR studies of non-zinc-chelating MMP-13 inhibitors: improving selectivity and metabolic stability. Bioorg. Med. Chem. Lett. 2010, 20, 5039–5043. [DOI] [PubMed] [Google Scholar]
- Gege C.; Bao B.; Bluhm H.; Boer J.; Gallagher B. M.; Korniski B.; Powers T. S.; Steeneck C.; Taveras A. G.; Baragi V. M. Discovery and evaluation of a non-Zn chelating, selective matrix metalloproteinase 13 (MMP-13) inhibitor for potential intra-articular treatment of osteoarthritis. J. Med. Chem. 2012, 55, 709–716. [DOI] [PubMed] [Google Scholar]
- Jungel A.; Ospelt C.; Lesch M.; Thiel M.; Sunyer T.; Schorr O.; Michel B. A.; Gay R. E.; Kolling C.; Flory C.; Gay S.; Neidhart M. Effect of the oral application of a highly selective MMP-13 inhibitor in three different animal models of rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nara H.; Sato K.; Naito T.; Mototani H.; Oki H.; Yamamoto Y.; Kuno H.; Santou T.; Kanzaki N.; Terauchi J.; Uchikawa O.; Kori M. Thieno[2,3-d]pyrimidine-2-carboxamides bearing a carboxybenzene group at 5-position: highly potent, selective, and orally available MMP-13 inhibitors interacting with the S1″ binding site. Bioorg. Med. Chem. 2014, 22, 5487–5505. [DOI] [PubMed] [Google Scholar]
- Lauer-Fields J. L.; Minond D.; Chase P. S.; Baillargeon P. E.; Saldanha S. A.; Stawikowska R.; Hodder P.; Fields G. B. High throughput screening of potentially selective MMP-13 exosite inhibitors utilizing a triple-helical FRET substrate. Bioorg. Med. Chem. 2009, 17, 990–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J.; Minond D.; Darout E.; Liu Q.; Lauer J.; Hodder P.; Fields G. B.; Roush W. R. Identification of novel, exosite-binding matrix metalloproteinase-13 inhibitor scaffolds. Bioorg. Med. Chem. Lett. 2011, 21, 7180–7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooljarsingh L. T.; Lakdawala A.; Coppo F.; Luo L.; Fields G. B.; Tummino P. J.; Gontarek R. R. Characterization of an exosite binding inhibitor of matrix metalloproteinase 13. Protein Sci. 2008, 17, 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Irujo J. J.; Villahermosa M. L.; Mercapide J.; Cabodevilla J. F.; Santiago E. Analysis of the combined effect of two linear inhibitors on a single enzyme. Biochem. J. 1998, 329Pt 3689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R.Enzymes, 2nd ed.; Wiley-VCH: New York, 2000. [Google Scholar]
- Lovejoy B.; Welch A. R.; Carr S.; Luong C.; Broka C.; Hendricks R. T.; Campbell J. A.; Walker K. A.; Martin R.; Van Wart H.; Browner M. F. Crystal structures of MMP-1 and -13 reveal the structural basis for selectivity of collagenase inhibitors. Nat. Struct. Biol. 1999, 6, 217–221. [DOI] [PubMed] [Google Scholar]
- Wu J.; Rush T. S. III; Hotchandani R.; Du X.; Geck M.; Collins E.; Xu Z. B.; Skotnicki J.; Levin J. I.; Lovering F. E. Identification of potent and selective MMP-13 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 4105–4109. [DOI] [PubMed] [Google Scholar]
- Kohno T.; Hochigai H.; Yamashita E.; Tsukihara T.; Kanaoka M. Crystal structures of the catalytic domain of human stromelysin-1 (MMP-3) and collagenase-3 (MMP-13) with a hydroxamic acid inhibitor SM-25453. Biochem. Biophys. Res. Commun. 2006, 344, 315–322. [DOI] [PubMed] [Google Scholar]
- Fingleton B. Matrix metalloproteinases as valid clinical targets. Curr. Pharm. Des. 2007, 13, 333–346. [DOI] [PubMed] [Google Scholar]
- Yiotakis A.; Dive V. Synthetic active site-directed inhibitors of metzincins: achievement and perspectives. Mol. Aspects Med. 2008, 29, 329–338. [DOI] [PubMed] [Google Scholar]
- Usmani K. A.; Karoly E. D.; Hodgson E.; Rose R. L. In vitro sulfoxidation of thioether compounds by human cytochrome P450 and flavin-containing monooxygenase isoforms with particular reference to the CYP2C subfamily. Drug Metab. Dispos. 2004, 32, 333–339. [DOI] [PubMed] [Google Scholar]
- Nakai R.; Salisbury C. M.; Rosen H.; Cravatt B. F. Ranking the selectivity of PubChem screening hits by activity-based protein profiling: MMP13 as a case study. Bioorg. Med. Chem. 2009, 17, 1101–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall C. M. Molecular determinants of metalloproteinase substrate specificity: matrix metalloproteinase substrate binding domains, modules, and exosites. Mol. Biotechnol. 2002, 22, 51–86. [DOI] [PubMed] [Google Scholar]
- Chung L.; Dinakarpandian D.; Yoshida N.; Lauer-Fields J. L.; Fields G. B.; Visse R.; Nagase H. Collagenase unwinds triple-helical collagen prior to peptide bond hydrolysis. EMBO J. 2004, 23, 3020–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung L.; Shimokawa K.; Dinakarpandian D.; Grams F.; Fields G. B.; Nagase H. Identification of the (183)RWTNNFREY(191) region as a critical segment of matrix metalloproteinase 1 for the expression of collagenolytic activity. J. Biol. Chem. 2000, 275, 29610–29617. [DOI] [PubMed] [Google Scholar]
- Minond D.; Lauer-Fields J. L.; Cudic M.; Overall C. M.; Pei D.; Brew K.; Visse R.; Nagase H.; Fields G. B. The roles of substrate thermal stability and P2 and P1′ subsite identity on matrix metalloproteinase triple-helical peptidase activity and collagen specificity. J. Biol. Chem. 2006, 281, 38302–38313. [DOI] [PubMed] [Google Scholar]
- Bode W.; Fernandez-Catalan C.; Tschesche H.; Grams F.; Nagase H.; Maskos K. Structural properties of matrix metalloproteinases. Cell. Mol. Life Sci. 1999, 55, 639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad-Zapatero C.; Champness E. J.; Segall M. D. Alternative variables in drug discovery: promises and challenges. Future Med. Chem. 2014, 6, 577–593. [DOI] [PubMed] [Google Scholar]
- Abad-Zapatero C.; Metz J. T. Ligand efficiency indices as guideposts for drug discovery. Drug Discovery Today 2005, 10, 464–469. [DOI] [PubMed] [Google Scholar]
- Poole G. D. a. A. Immunohistochemical detection and immunochemical analysis of type II collagen degradation in human normal, rheumatoid, and osteoarthritic articular cartilages and in explants of bovine articular cartilage cultured with interleukin 1. J. Clin. Invest. 1989, 83, 657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martignetti J. A.; Aqeel A. A.; Sewairi W. A.; Boumah C. E.; Kambouris M.; Mayouf S. A.; Sheth K. V.; Eid W. A.; Dowling O.; Harris J.; Glucksman M. J.; Bahabri S.; Meyer B. F.; Desnick R. J. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat. Genet. 2001, 28, 261–265. [DOI] [PubMed] [Google Scholar]
- Vu T. H.; Shipley J. M.; Bergers G.; Berger J. E.; Helms J. A.; Hanahan D.; Shapiro S. D.; Senior R. M.; Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 1998, 93, 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmbeck K.; Bianco P.; Caterina J.; Yamada S.; Kromer M.; Kuznetsov S. A.; Mankani M.; Robey P. G.; Poole A. R.; Pidoux I.; Ward J. M.; Birkedal-Hansen H. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 1999, 99, 81–92. [DOI] [PubMed] [Google Scholar]
- Kola I.; Landis J. Can the pharmaceutical industry reduce attrition rates?. Nat. Rev. Drug Discovery 2004, 3, 711–715. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [DOI] [PubMed] [Google Scholar]
- Ling C.; Fu L.; Gao S.; Chu W.; Wang H.; Huang Y.; Chen X.; Yang Y. Design, synthesis, and structure–activity relationship studies of novel thioether pleuromutilin derivatives as potent antibacterial agents. J. Med. Chem. 2014, 57, 4772–4795. [DOI] [PubMed] [Google Scholar]
- Lauer-Fields J. L.; Fields G. B. Triple-helical peptide analysis of collagenolytic protease activity. Biol. Chem. 2002, 383, 1095–1105. [DOI] [PubMed] [Google Scholar]
- Knauper V.; Lopez-Otin C.; Smith B.; Knight G.; Murphy G. Biochemical characterization of human collagenase-3. J. Biol. Chem. 1996, 271, 1544–15450. [DOI] [PubMed] [Google Scholar]
- Pathak N.; Hu S. I.; Koehn J. A. The expression, refolding, and purification of the catalytic domain of human collagenase-3 (MMP-13). Protein Expression Purif. 1998, 14, 283–288. [DOI] [PubMed] [Google Scholar]
- Neumann U.; Kubota H.; Frei K.; Ganu V.; Leppert D. Characterization of Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2, a fluorogenic substrate with increased specificity constants for collagenases and tumor necrosis factor converting enzyme. Anal. Biochem. 2004, 328, 166–173. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., D: Biol. Crystallogr. 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tommasi R. A.; Weiler S.; McQuire L. W.; Rogel O.; Chambers M.; Clark K.; Doughty J.; Fang J.; Ganu V.; Grob J.; Goldberg R.; Goldstein R.; Lavoie S.; Kulathila R.; Macchia W.; Melton R.; Springer C.; Walker M.; Zhang J.; Zhu L.; Shultz M. Potent and selective 2-naphthylsulfonamide substituted hydroxamic acid inhibitors of matrix metalloproteinase-13. Bioorg. Med. Chem. Lett. 2011, 21, 6440–6445. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., D: Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., D: Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty N. W.; Grosse-Kunstleve R. W.; Adams P. D. Electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr., D: Biol. Crystallogr. 2009, 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.