

Abstract
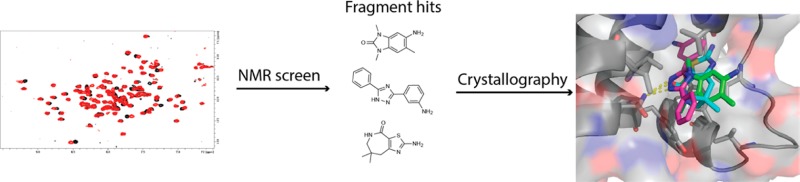
Cellular and genetic evidence suggest that inhibition of ATAD2 could be a useful strategy to treat several types of cancer. To discover small-molecule inhibitors of the bromodomain of ATAD2, we used a fragment-based approach. Fragment hits were identified using NMR spectroscopy, and ATAD2 was crystallized with three of the hits identified in the fragment screen.
Introduction
Bromodomains are epigenetic reader modules of post-translational modifications, responsible for the recognition of ε-N-acetyllysine (Kac) marks on histone proteins. In recent years, bromodomains have been implicated as both attractive and druggable targets for treating cancer, inflammation, and neurological disorders.1 Inhibitors have been discovered against several bromodomains, including the BET (bromodomain and extra-terminal) family members (BRD2, BRD3, BRD4, and BRDT), BAZ2A/B, CREBBP, EP300, and SMARCA2/4.1−3 However, very few reports have appeared on the discovery of inhibitors to other bromodomain targets that are implicated in cancer biology but likely more challenging to drug. One such bromodomain for which an inhibitor could be useful is that of the protein ATAD2 (AAA domain-containing protein 2), also known as ATAD2A or ANCCA (AAA nuclear coregulator cancer-associated protein).
The bromodomain of ATAD2 offers an exciting target for the design of novel cancer therapeutics for several reasons. Overexpression of ATAD2, a protein that possesses both AAA+ ATPase (ATPases associated with various cellular activities) and bromodomain functionalities, has been linked to poor prognosis in prostate, lung, and triple-negative breast cancers, as well as in hepatocellular carcinoma.4−10 ATAD2 serves as a coactivator for transcription factors such as E2F family members, MYC, and estrogen and androgen receptors, whereby driving the expression of a subset of genes that promote cell proliferation and survival, including c-myc, cyclin d1, e2f, and survivin.5,7,8,11 Finally, downregulation of ATAD2 via siRNA shows an increase in apoptotic activity, suggesting that a small-molecule inhibitor of ATAD2 could result in cancer cell death and tumor regression.4
While the cellular and genetic evidence suggests that ATAD2 inhibitors could be useful for cancer therapy, one computational study of the bromodomain family classified ATAD2 as”‘difficult to drug”, as its Kac binding site is quite dissimilar to other druggable bromdomains12 ATAD2 is composed of the typical left-handed four-helix bundle structure, with the ZA and BC loops determining the geometry of the Kac binding pocket (Figure 1A).13 However, when compared to BRD4, only three of seven residues lining the Kac binding pocket that interact with peptide are shared with ATAD2 (Figure 1B, Supporting Information (SI), Table S1, Figure S1).
Figure 1.

Structure of the ATAD2 bromodomain. (A) X-ray structure of ATAD2 bound to histone 4-derived peptide (H4Kac5), highlighting the conserved Kac binding site hydrogen bond donor N1064 in purple spheres (PDB ID 4QUU). (B) Surface view of Kac binding pocket of ATAD2. Residues shown as sticks are those that interact with the Kac residue of H4Kac5, with only three residues sharing identity to BRD4 (colored purple). Polar contacts between the peptide and ATAD2 are shown as yellow dashes, and crystallographic water molecules are shown as blue spheres.
To target challenging proteins, such as those involved in protein–protein interactions, fragment-based drug discovery has been used to effectively identify hits that can be elaborated to high affinity molecules via structure-based design.14,15 While Kac-reading bromodomains possess deeper and more-defined binding pockets than traditional protein–protein interaction surfaces, fragment-based methods could be applied to those bromodomains that are deemed difficult to drug. While a number of techniques are possible for conducting a fragment-based screen, we rely on the use of protein-observed NMR spectroscopy because it offers several advantages over other biochemical or biophysical methods.16 Because the binding observation by NMR is direct, weak binders up to several millimolar in affinity can be identified with very few false positives. In addition, the binding location of the hits can be ascertained if the resonance assignments of the protein are known. NMR can also be used to determine fragment binding affinities, even for very weak-binding hits, allowing for hits to be rank ordered without a secondary assay.
Herein we present the results of a fragment-based screen conducted by NMR against the bromodomain of ATAD2. From this screen, hits were identified representing several chemotypes that have not been previously observed as bromodomain ligands. Although relatively few hits were found, the calculated hit rate suggests that this bromodomain should be druggable. We also present the X-ray crystal structures of ATAD2 bound to the hits identified in the screen. These results serve as useful starting points toward the design of high affinity, selective chemical probes for ATAD2 in order to understand its role in cancer biology.
Results and Discussion
NMR-Based Screen Identified Fragment Hits
Because ATAD2 has been predicted to be challenging to drug,12 it may be difficult to identify hits from traditional high-throughput screening methods. Indeed, it was recently reported that a screen of ATAD2 against a library of Kac mimics revealed no potent hits.17 As an alternative, we chose a fragment-based approach in an effort to identify even weak binding hits that could provide a useful starting point for further optimization. To screen ATAD2 against an unbiased fragment library of approximately 13800 molecules, protein-observed 1H–15N SOFAST-HMQC NMR spectra were recorded on uniformly 15N-labeled ATAD2 in the presence of mixtures containing 12 fragments. A mixture was considered a hit if, relative to the absence of fragments, chemical shift changes were observed for the backbone resonances in the presence of fragments (SI, Figure S2). Hit mixtures were then deconvolved as singletons to isolate the hit fragments, resulting in a total of 65 fragment hits (Figure 2). The binding affinity range for the hits, as determined by SOFAST-HMQC titration experiments, is 350 μM to >2 mM, with 12 hits having a Kd of less than 1 mM. Taken together, very few weak-binding hits have been identified against ATAD2, with a calculated hit rate of 0.1%. However, a previous analysis of fragment-based screens suggests that protein targets with a hit rate of at least 0.1% can result in the design of high affinity, drug-like molecules against the target.18 We therefore conclude that ATAD2 may be a druggable target, but based on the weak affinity of the hits identified, it is likely to still be difficult to drug. This conclusion is in accordance with the computational SiteMap druggability assessment mentioned previously.12
Figure 2.

Representative ATAD2 hits identified by NMR-based fragment screening. Hits are clustered by chemotype and classified as being commonly observed (cluster 1) or novel bromodomain ligands (clusters 2–3). Kd values were determined by SOFAST-HMQC titration experiments.
Although the assignments of ATAD2’s backbone resonances are not known, we observed that one distinct set of resonances shift in the spectra depending on the fragment hit bound to ATAD2, and these residues likely correspond to those within the Kac binding pocket (SI, Figure S2). The 65 hits identified have been clustered into three chemical clusters (Figure 2). Cluster 1 fragment hits (1–4) represent 5,6- and 6,6-fused ring chemotypes that have been previously identified as bromodomain inhibitors, including dihydroquinolinones, dihydroimidazolones, quinazolines, and benzoimidazolylanilines.1 Cluster 2 hits (5–8), on the contrary, are unique to ATAD2 and include tricyclic-containing small molecules such as diphenyltriazoles and diphenyloxadiazoles. A second unique cluster of ATAD2 hits, cluster 3 (9–12), is composed of sulfur-containing fragments such as tetrahydrothiazoloazepinones and benzothiazoles. The listed binding affinities of 1–12 vary only 2-fold despite great differences in chemical structure; however, relative to recently published ATAD2 ligands, the fragments identified herein are significantly greater in potency and thus may represent better starting points for medicinal chemistry optimization.17 On the basis of ligand efficiencies alone (Figure 2), one may choose to optimize the more efficient members of cluster 1. However, for selectivity purposes, medicinal chemistry optimizations of the unique fragments from clusters 2–3 may also be desirable.
X-ray Structures Reveal Critical Intermolecular Contacts
On the basis of high-resolution structures presented in the literature for bromodomains of the BET family and recently for ATAD2,1,17 the binding orientations of cluster 1 molecules to ATAD2 could be modeled to aid in medicinal chemistry optimization of the fragment hits. However, because two clusters of hits identified from the screen have not been previously characterized as bromodomain inhibitors, it was important to understand how these molecules bind within the Kac binding site. To obtain this information, we determined X-ray structures of ATAD2 when complexed with three different fragments (5-amino-1,3,6-trimethyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (1), 3-(5-phenyl-1H-1,2,4-triazol-3-yl)aniline (5), and 2-amino-7,7-dimethyl-5,6,7,8-tetrahydro-4H-thiazolo[5,4-c]azepin-4-one (12)) (Figure 3, SI, Figure S3), which served as representatives of each chemical cluster discussed above (Table 1). All three complexes crystallized in the P6522 space group with one molecule in the asymmetric unit, allowing for direct comparisons between the complexes and also to the ligand-free ATAD2 structure (PDB ID 3DAI). As shown in Figure 3, all three fragments occupy the same general space within the Kac binding pocket and make a critical hydrogen bond to the highly conserved N1064 residue (Figure 1A). When compared to ligand-free ATAD2, minimal structural perturbations were observed upon binding of these three fragments, with the rmsd ranging from 0.117 to 0.176, suggesting that structural changes to the pocket are not inducible.
Figure 3.

X-ray structures of ATAD2 bound to fragment hits. View of the Kac binding site in the presence of fragment 1 (A), 5 (B), and 12 (C) (PDB IDs 4TYL, 4TZ2, and 4TZ8). Residues in contact with each fragment are shown as sticks. Crystallographic water molecules are indicated with blue spheres. Dashed yellow lines depict polar contacts between the bound fragment and ATAD2 residues/crystallographic water molecules.
Table 1. X-ray Data Collection and Refinement Statistics.
| ligand |
|||
|---|---|---|---|
| parameter | 1 | 5 | 12 |
| PDB ID code | 4TYL | 4TZ2 | 4TZ8 |
| no. of chains | 1 | 1 | 1 |
| Data Collection | |||
| space group | P6522 | P6522 | P6522 |
| cell dimensions | |||
| a, b, c (Å) | 79.49, 79,49, 137.51 | 79.20, 79.20, 136.04 | 79.94, 79.94, 138.26 |
| α, β, γ (deg) | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 |
| resolution (Å) | 38.18–1.85 (1.92–1.85) | 38.02–1.70 (1.73–1.70) | 34.62–2.15 (2.19–2.15) |
| no. of unique reflections | 22647 (1100) | 28313 (1400) | 14730 (711) |
| completeness (%) | 99.9 (99.5) | 99.9 (99.8) | 99.27 (97.71) |
| redundancy | 23.47 (23.1) | 23.37 (20.9) | 23.36 (23.5) |
| Rsym | 8.3 (98.2) | 10.6 (96.9) | 14.5 (97.6) |
| I/σ(I) | 20.37 (3.61) | 25.89 (7.41) | 13.19 (4.18) |
| Structure Refinement | |||
| no. of reflections test set | 2000 | 2000 | 1469 |
| Rwork/Rfree | 16.05/19.72 | 16.90/20.11 | 19.83/23.06 |
| Wilson B-factor | 27.76 | 21.03 | 36.46 |
| no. of non-hydrogen atoms/av B-factor | |||
| protein | 1086/31.6 | 1130/29.0 | 1069/41.20 |
| ligand | 23/53.0 | 33/46.2 | 25/68.5 |
| water | 251/47.1 | 200/42.4 | 135/48.4 |
| Ramanchandran | |||
| preferred regions (%) | 100 | 99 | 100 |
| allowed regions (%) | 0 | 1 | 0 |
| generously allowed (%) | 0 | 0 | 0 |
| disallowed regions (%) | 0 | 0 | 0 |
| rms deviations | |||
| bond length (Å) | 0.018 | 0.019 | 0.002 |
| bond angle (deg) | 1.490 | 1.610 | 0.56 |
| coordinate error (mL-based, Å) | 0.18 | 0.15 | 0.23 |
Fragment 1 represents a hit from cluster 1 and has been observed as part of elaborated small-molecule inhibitors for BET family bromodomains.1 On the basis of a map of intermolecular contacts (PDB ID 4TYL), 1 is anchored within the Kac binding site primarily due to the hydrogen bonding interactions between its carbonyl and both N1064 and a water molecule inside the pocket (Figure 3A). Much of the six-membered ring is solvent exposed, with its amine substituent making hydrogen-bonding interactions with two crystallographic waters in the binding site. The remaining interactions are hydrophobic in nature (V1008, V1013, V1018, Y1021, Y1063, and I1074).
From cluster 2, fragment 5 was chosen for crystallization with ATAD2 (PDB ID 4TZ2). This tricyclic fragment takes advantage of the same contacts as 1, being mainly hydrophobic in nature (V1008, V1013, V1018, Y1021, Y1063, and I1074) aside from the hydrogen bonding interactions with N1064 (Figure 3B). Its Kac-mimic triazole warhead does not form a hydrogen bond to any water molecules within the binding site, likely because this larger-sized fragment displaces the water molecule seen both in the complexes with 1 and 12 (SI, Figure S4). Similar to 1, the 5-phenyl portion of 5 is largely solvent exposed and likely contributing very little to binding affinity. However, unlike the binding of 1, the 3-aminophenyl substituent binds deep within the Kac binding pocket, displacing an additional three conserved water molecules (SI, Figure S4A). The displacement of water may account for its higher affinity observed for binding to ATAD2 relative to fragments from clusters 1 and 3. This sum of four displaced water molecules is unprecedented in the bromodomain literature. Only one other example of water displacement is available in the PDB, and in this case, the inhibitor displaces just one water molecule from the Kac binding pocket of BRD2 (PDB ID 2DVV). The 3-aminophenyl portion of 5 that binds deep inside the Kac recognition pocket participates in hydrogen bonds to I1056, M1029, and a crystallographic water molecule. It should be noted that an elaborated analogue of 5 has been described as a dual kinase–bromodomain inhibitor and has been crystallized with BRD4 (PDB ID 4O77).19 However, the analogue and 5 do not share similar binding poses as the analogue does not bind deep within the Kac pocket.
A cluster 3 representative fragment (12) binds in a very similar fashion as to 1, with the carbonyl of its seven-membered ring making hydrogen-bonding interactions to both N1064 and a water molecule (PDB ID 4TZ8). The amine substituent and heteroatoms of the thiazole ring make hydrogen-bonding interactions to V1008, K1011, and two crystallographic waters in the binding site. This amine substituent takes the place of a water molecule deep inside the Kac binding pocket, in agreement with one of the four waters also displaced by 5 (SI, Figure S4B). This fragment also utilizes a number of hydrophobic interactions involving V1008, V1013, V1018, Y1021, Y1063, and I1074 (Figure 3C). The dimethyl substituent on the seven-membered ring is largely solvent exposed and likely not contributing to binding affinity.
Comparing Ligand-Bound Structures for Hit-to-Lead Optimization
Following the submission of this manuscript, a group from the Structural Genomics Consortium (SGC) published structures of ATAD2 bound to a histone-derived peptide (H4Kac5), several solvent molecules, DNA/RNA bases, and their analogues.17 We have prepared structural overlays of our fragment-bound structures with these newly released structures to provide suggestions on how to improve the potency of the fragment hits identified from our NMR screen (Figure 4, SI, Figure S5).
Figure 4.

Kac binding site overlays of ligand-bound ATAD2 structures. (A) Binding pose of 1 (green) (PDB IC 4TYL) overlaid with 1-methylquinolin-2-one (salmon) (PDB ID 4QST). (B) Binding pose of 5 (magenta) (PDB ID 4TZ2) overlaid with thymidine (yellow) (PDB ID 4QSV). (C) Binding pose of 12 (cyan) (PDB ID 4TZ8) overlaid with thymidine (yellow) (PDB ID 4QSV). Residues depicted as sticks and water molecules summarize interactions between ATAD2 and the fragments 1, 5, and 12. (D) Overlay of binding poses for H4Kac5 peptide (orange) (PDB ID 4QUU), thymidine (yellow), 1 (green), 5 (magenta), and 12 (cyan).
On the basis of the chemical similarity of 1 to 1-methylquinolin-2-one (PDB ID 4QST), the binding poses of these two molecules were overlaid for comparison of intermolecular interactions. As seen in Figure 4A, the two small molecules overlay almost exactly in the Kac binding site. Both have a carbonyl group that makes a hydrogen bond to N1064 and a water molecule. Binding affinity data was not provided for 1-methylquinolin-2-one; however, we assume its Kd is greater than 10 mM, as all of the molecules with reported Kd values in the study were at best 10 mM. We rationalize the large difference in Kd (estimated 10 mM relative to 600 μM for 1) is due to the additional two hydrogen bonds formed between 1 and crystallographic water molecules in the Kac binding site. Thus, an electron rich group that can participate in hydrogen bond formation is favored on the side of the pocket formed by the ZA loop.
When the two binding poses of thymidine and 5 are overlaid, they do not share many common binding elements (Figure 4B), as expected. In the case of 5, its Kac-mimic warhead is the triazole ring, allowing the aniline NH2 group to plunge deeper into the Kac binding site. However, the urea portion of thymidine serves as the warhead. Thus, it does not bind deeply inside the Kac pocket. In fact, 5 is positioned more than 3 Å further inside the pocket, and we reason that this unique binding pose drives the substantial difference in affinity for ATAD2 (350 μM vs 10 mM). One interesting insight this provides is that introducing substituents around the aniline moiety may provide unwanted steric bulk and force the aniline to not bind as deep inside the pocket. Although the aniline ring occupies a never before drugged portion of the Kac binding site that could be optimized to increase interactions between the molecule and ATAD2, we suggest only conservative changes to the ring be made to avoid a change in its binding pose.
Fragment 12 identified from our screen does not structurally resemble the solvent molecules or DNA/RNA bases that were crystallized with ATAD2 by the SGC.17 Similar to 5, we chose to overlay 12 with thymidine to highlight how the thiazole of 12 also binds deeper within the Kac binding pocket (Figure 4C). The amino group of the thiazole ring binds nearly 3 Å deeper into the pocket than the methyl substituent of thymidine. In doing so, 12 displaces a water molecule present in both the structures with bound 1 and thymidine (SI, Figure S4B).
From an overlay of binding poses for the H4Kac5 peptide, thymidine, 1, 5, and 12 (Figure 4D), it is apparent that 5 and 12 bind in a portion of the Kac pocket that is unoccupied by the other molecules. These two fragments bind deep into the pocket, which may contribute to their higher affinity for ATAD2. As changes are made to the portions of 5 and 12 that bind deep within the pocket, NMR HMQC spectra could be used to asses whether the binding mode changes significantly, as there appears to be a subset of resonances that report binding deeper inside the pocket (SI, Figure S2). From the structural overlays, it is also evident that the peptide and 5 do not fill the side of the Kac pocket formed by the ZA loop. Modifications to 5 and even 12 to better fill that portion of the Kac pocket could also improve potency. We suggest the addition of a hydrogen-bond donor/acceptor group, such an amine substituent like seen in 1, or a hydroxyl that, similar to thymidine, could bridge through hydrogen bonding to the ZA loop itself.17
Conclusions
Bromodomains represent a highly druggable class of therapeutic targets that have received great interest in recent years. Despite this interest in exploiting bromodomain inhibition for the treatment of several diseases, relatively few chemical probes or inhibitors for bromodomains outside of the BET family have been discovered. On the basis of cellular and genetic studies, inhibition of ATAD2 provides a useful strategy for the treatment of several types of cancer, including prostate, lung, and breast malignancies. We sought to identify small-molecule ligands for the bromodomain of ATAD2 from which chemical probes could be designed to validate ATAD2 as a cancer target. A total of 65 fragment hits for ATAD2 were identified by NMR spectroscopy, and about 1/4 of these fragments bind with affinities less than 1 mM. Co-crystal structures of ATAD2 were determined for three fragment hits, revealing the contacts critical for each cluster of inhibitor. Together with recently published data from the SGC, we propose several strategies to improve ligand binding affinities toward the design of a chemical probe. Chemical probes will be useful to examine the biological impact of ATAD2 bromodomain inhibition and to determine whether this is a viable strategy for treating human cancers.
Experimental Section
All screened compounds were purchased from commercial vendors and the purity was determined to be 95% or higher. HPLC data of all compounds were obtained using an Agilant 1200 series sytem with a UV detector set to 220 nm. Samples were injected onto a Phenomenex 2.0 mm × 50 mm, 2.6 μm, C18 column at 45 °C. A mobile phase of A being H2O + 0.1% trifluoroacetic acid and B being acetonitrile + 0.1% trifluoroacetic acid was used. A linear gradient from 5% to 95% B in 1.95 min was then followed by pumping 95% B for another 0.45 min having a flow rate of 1.0 mL/min. Mass spectral data were obtained in positive ion mode using Agilent 6140 single quadrupole mass spectrometer using an electrospray ionization (ESI) source.
Acknowledgments
This work was supported by the U.S. National Institutes of Health, NIH Director’s Pioneer Award to S.W.F (DP1OD006933/DP1CA174419). M.J.H. gratefully acknowledges postdoctoral support from the Damon Runyon Cancer Research Foundation (DRG 2123-12). The Biomolecular NMR Facility at Vanderbilt University is supported in part by grants for NMR instrumentation from the NSF (0922862), the NIH (S10 RR025677), and matching funds from Vanderbilt University. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under contract no. DE-AC02-06CH11357. The authors thank the Vanderbilt Institute of Chemical Biology (VICB) High-Throughput Screening Core Facility at Vanderbilt University for chemical management of our in-house fragment library.
Glossary
Abbreviations Used
- ATAD2
AAA domain-containing protein 2
- Kac
acetyllysine
- BET
bromodomain and extra-terminal
- SOFAST-HMQC
selective optimized flip angle short transient heteronuclear multiple quantum coherence
- PDB
Protein Data Bank
- SGC
Structural Genomics Consortium
- H4Kac5
histone 4-derived peptide acetylated on lysine 5
Supporting Information Available
Complete experimental details for protein expression and purification, NMR setup and sample spectra, and protein crystallization, data collection, and analyses. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
PDB ID codes: 4TYL, 4TZ2, and 4TZ8.
Author Contributions
M.J.H. collected and analyzed the data. B.A.C., J.P., and S.W.F. assisted in data analysis. M.J.H. and S.W.F. prepared the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Filippakopoulos P.; Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nature Rev. Drug Discovery 2014, 13, 337–356. [DOI] [PubMed] [Google Scholar]
- PFI-3: Selective chemical probe for SMARCA bromodomains; Structural Genomics Consortium: Toronto; http://www.thesgc.org/chemical-probes/PFI-3.
- Ferguson F. M.; Fedorov O.; Chaikuad A.; Philpott M.; Muniz J. R.; Felletar I.; von Delft F.; Heightman T.; Knapp S.; Abell C.; Ciulli A. Targeting low-druggability bromodomains: fragment based screening and inhibitor design against the BAZ2B bromodomain. J. Med. Chem. 2013, 56, 10183–10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron C.; Lestrat C.; Marsal S.; Escoffier E.; Curtet S.; Virolle V.; Barbry P.; Debernardi A.; Brambilla C.; Brambilla E.; Rousseaux S.; Khochbin S. Functional characterization of ATAD2 as a new cancer/testis factor and a predictor of poor prognosis in breast and lung cancers. Oncogene 2010, 29, 5171–5181. [DOI] [PubMed] [Google Scholar]
- Hsia E. Y.; Kalashnikova E. V.; Revenko A. S.; Zou J. X.; Borowsky A. D.; Chen H. W. Deregulated E2F and the AAA+ coregulator ANCCA drive proto-oncogene ACTR/AIB1 overexpression in breast cancer. Mol. Cancer Res. 2010, 8, 183–193. [DOI] [PubMed] [Google Scholar]
- Kalashnikova E. V.; Revenko A. S.; Gemo A. T.; Andrews N. P.; Tepper C. G.; Zou J. X.; Cardiff R. D.; Borowsky A. D.; Chen H. W. ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2. Cancer Res. 2010, 70, 9402–9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revenko A. S.; Kalashnikova E. V.; Gemo A. T.; Zou J. X.; Chen H. W. Chromatin loading of E2F-MLL complex by cancer-associated coregulator ANCCA via reading a specific histone mark. Mol. Cell. Biol. 2010, 30, 5260–5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Huang J.; Luo L.; Chen Z.; Guo Y.; Guo L. Significance of PRO2000/ANCCA expression, a novel proliferation-associated protein in hepatocellular carcinoma. Cancer Cell Int. 2014, 14, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J. X.; Guo L.; Revenko A. S.; Tepper C. G.; Gemo A. T.; Kung H. J.; Chen H. W. Androgen-induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res. 2009, 69, 3339–3346. [DOI] [PubMed] [Google Scholar]
- Zou J. X.; Revenko A. S.; Li L. B.; Gemo A. T.; Chen H. W. ANCCA, an estrogen-regulated AAA+ ATPase coactivator for ERalpha, is required for coregulator occupancy and chromatin modification. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 18067–18072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciro M.; Prosperini E.; Quarto M.; Grazini U.; Walfridsson J.; McBlane F.; Nucifero P.; Pacchiana G.; Capra M.; Christensen J.; Helin K. ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer Res. 2009, 69, 8491–8498. [DOI] [PubMed] [Google Scholar]
- Vidler L. R.; Brown N.; Knapp S.; Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J. Med. Chem. 2012, 55, 7346–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J. P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Muller S.; Pawson T.; Gingras A. C.; Arrowsmith C. H.; Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajduk P. J.; Meadows R. P.; Fesik S. W. Discovering high-affinity ligands for proteins. Science 1997, 278, 497–499. [DOI] [PubMed] [Google Scholar]
- Shuker S. B.; Hajduk P. J.; Meadows R. P.; Fesik S. W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996, 274, 1531–1534. [DOI] [PubMed] [Google Scholar]
- Harner M. J.; Frank A. O.; Fesik S. W. Fragment-based drug discovery using NMR spectroscopy. J. Biomol. NMR 2013, 56, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikuad A.; Petros A.; Fedorov O.; Xu J.; Knapp S.. Structure-based approaches towards indentification of fragments for the low-druggability ATAD2 bromodomain. MedChemComm 2014, DOI 10.1039/C4MD00237G. [DOI] [Google Scholar]
- Hajduk P. J.; Huth J. R.; Fesik S. W. Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 2005, 48, 2518–2525. [DOI] [PubMed] [Google Scholar]
- Ember S. W.; Zhu J. Y.; Olesen S. H.; Martin M. P.; Becker A.; Berndt N.; Georg G. I.; Schonbrunn E. Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.