

Abstract
Background
The limited circulation of many of the agents that are likely to be used in a bioterrorism attack precludes the ready availability of positive controls. This means that only specialized laboratories can screen for the presence of these agents by nucleic amplification assays. Calibrated controls are also necessary for quantitative measurements. Primers and probes to be used in both conventional and real-time PCR assays were designed for the detection of agents likely to be used by a bioterrorist. Three plasmids, each of which contains 4 to 6 specific sequences from agents on the CDC Category A and B list (excluding RNA viruses) were constructed. Two plasmids incorporate the sequences of Category A and B agents, respectively. The third plasmid incorporates sequences from Variola major and organisms that cause rash-like illnesses that may be clinically confused with smallpox. An "exogenic sequence", introducing a NotI restriction site was incorporated in the native sequences of the bioterrorism agents inserted in plasmids. The designed molecular system for detection of bioterrorism agents was tested on each of these agents (except Monkeypox virus, Smallpox virus and 2 Burkholderia species for which no native DNA was available) and a collection of 50 isolates of C. burnetii using constructed plasmids as positive controls.
Results
Designed primers and probes allowed molecular detection, in either single or multiplex assays, of agent-specific targets with analytical sensitivities of between 1 and 100 DNA copies. The plasmids could be used as positive controls. False-positive results due to contamination by the positive control were easily detected by sequencing and eliminated by digestion with NotI.
Conclusion
Plasmid A and B can be used as positive controls in molecular assays for the detection of bioterrorism agents in clinical specimens or environmental samples. Plasmid C can be used as a positive control in differentiation of vesicular rashes. It is also possible to avoid or to ensure immediate detection of false positive results due to contamination by positive controls using these plasmids. These plasmids and the corresponding primers and probes are immediately available for all clinical microbiology laboratories provided they have molecular amplification equipment.
Background
Following press reports on a case of inhalation anthrax in Florida, USA, on October 4, 2001 [1], the bioterrorism threat became more obvious, and health authorities in the different countries have intensified preparedness and response programs. The Centers for Disease Control and Prevention (CDC) in the USA has listed the potential agents of bioterrorism (AB) for public health preparedness efforts. They have been classified by order of priority as categories A and B (Table 1). Detection of these agents will have to be performed from various specimens, including environmental samples (such as powders) and clinical samples of patients with symptoms compatible with infection caused by AB. In fact, 120 reference laboratories in the USA and Canada have the capacity to identify most of AB [2]. Suspect bacterial or viral isolates obtained in culture must also be identified. Molecular assays for the detection and identification of infectious agents can be performed in clinical microbiology laboratories that have specific equipment and facilities.
Table 1.
Genes and specific primers used for amplification of Bioterrorism agents. Bioterrorism agents are classified according to CDC recommendations. Their presence in the 3 constructed plasmids is given.
| Agent | Priority Level | Targeted gene | Plasmid | Genbank access n° | Forward primer | Reverse primer |
| Bacteria | ||||||
| Bacillus anthracis | A | pag | CatA | AF065404 | 5'-AGGCTCGAACTGGAGTGAA-3' | 5'-CCGCCTTTCTACCAGATTT-3' |
| Yersinia pestis | A | pla | CatA | M27820 | 5'-ATGGAGCTTATACCGGAAAC-3' | 5'-GCGATACTGGCCTGCAAG-3' |
| Francisella tularensis | A | fopA | CatA | AF097542 | 5'-TAATAATTTCATTGCTCCTTTTG-3' | 5'-TTCTATCTTGAGGACCCCAA-3' |
| Brucella spp. | B | omp19 | CatB | L27997 | 5'-CCGGTGAACTGGCTAATCT-3' | 5'-TGAAGAATAGAGCGAGGCAA-3' |
| Burkholderia mallei | B | fliC | CatB | AF11790 | 5'-GACGCTGGCGCTGTCGA-3' | 5'-CGGCTTGTTGACCGCGTT-3' |
| B. pseudomallei | B | CatB | U82287 | |||
| Chlamydia psittaci | B | ompA | CatB | AF269265 | 5'-ATTGCTTCGATTCAGATCAAC-3' | 5'-ACCAGTGATTGACCATTTGT-3' |
| Coxiella burnetii | B | sodB | CatB | M74242 | 5'-CTTTTTACCGACTCCGCAAA-3' | 5'-ACGAGCGTTGACAGTGCTT-3' |
| Rickettsia prowazekii | B | ompB | CatB | AF161079 | 5'-GCTCTTATGAATAATAATGCTC-3' | 5'-TGTTGTAACGCAGCATTAACA-3' |
| Rickettsia rickettsii | B | ompA | CatB | U83451 | 5'-TTTGGTGGAGCTCATAAGTTA-3' | 5'-GTACTATATTTGTTGTATTAAAAG-3' |
| Rickettsia akari | - | ompB | CatVEF | AF123707 | 5'-TTGGTGGGAATAATTGTAATGT-3' | 5'-TGCAGCAAAATTGAATTCTG-3' |
| Viruses | ||||||
| Variola virus | A | Hemagglutinin | CatA, CatVEF | AF377893 | 5'-GACKTCSGGACCAATTACTA-3' | 5'-TTGATTTAGTAGTGACAATTTCA-3' |
| Monkeypox virus | - | Hemagglutinin | CatVEF | AF375114 | 5'-AAAGTAGATTATGAAGAATACTC-3' | 5'-CAGAAATAGTTTCGACAATTTCA-3' |
| Orthopoxvirus | - | Hemagglutinin | CatVEF | AF377892 | 5'-ACCAAGGATAAARTATCWTACG-3' | 5'-ATAYATAAGTACCCGGCATCT-3' |
| Varicella- zoster virus | - | IE 62 transactivator | CatVEF | X04370 | 5'-GGTTAAACGTTTGAATCCATC-3' | 5'-CAGCAGACTTTCTCGAACG-3' |
| Herpes simplex virus 1 and 2 | - | DNA polymerase | CatVEF | X14112 | 5'-CCGACCCGGAGAGGGAC-3' | 5'-CCAGGCGCTTGTTGGTGT-3' |
However, several problems limit a large diffusion of molecular techniques for detection of AB: (i) due to safety regulations, it is difficult to obtain nucleic acids of potential AB to calibrate the techniques and serve as positive controls, (ii) several microorganisms would have to be tested simultaneously by using different controls, (iii) in some instance, such as for detection of anthrax spores, quantification would be useful as the size of spore inoculum has been demonstrated to be related with lethal dose in non human primates [3], (iv) false positive results due to contamination from positive controls should be avoided, (v) levels of equipments and methodologies used currently in clinical microbiology laboratories are highly variable. Currently, commercially available rapid tests are limited for the detection of B. anthracis, Brucella sp., Francisella tularensis and Yersinia pestis [4].
In this study, we propose a new strategy for the preparation of positive controls to overcome these problems. We designed 2 plasmids (CatA and CatB) that contain 4 and 6 AB sequences to serve as positive controls. We also designed primers and probes to detect DNA of these agents. We designed a plasmid (CatVEF) to detect variola and other agents that can cause vesicular rash and mimicking it. It contains sequences of Herpes viruses, Varicella-zoster virus, Rickettsia akari (the agent of rickettsialpox) and broad-spectrum orthopox viral sequences. We modified the native sequences by replacement of 8 nucleotides with a 20 nucleotide exogenic sequence that contains a NotI site. This modification allows NotI pre-digestion of the sample to avoid contamination by plasmid DNA. Moreover, amplified DNA from the control can be differentiated from the natural agent by (i) NotI digestion, (ii) sequencing and (iii) analysis of melting curves. NotI pre-digestion of the extracted DNA eliminated plasmid DNA contaminating amplicons. Choice of targeted DNA, validation of constructed plasmids, tests for sensitivity and specificity of the chosen targets, quantification assays and ability to differentiate amplified plasmid DNA from native DNA by using different DNA amplification techniques, including multiplex assay, are herein described.
Results
No viable organisms could be recovered using the Chelex procedure. No colonies were obtained from treated B. anthracis spore suspension.
Targeted genes, sequences, probes and primers are summarized in Table 1, 2 and additional file 1. Targeted sequences of the 3 plasmids demonstrated that they were identical to the in silico predicted sequences. The specific sequences of plasmids CatA, CatB and CatVEF have been deposited in Genbank database under AY333963, AY333965, and AY333966 accession numbers, respectively. The construction of CatA is represented in Figures 1 and 2. E. coli strains harboring these plasmids are deposited at the CNCM collection (CNCM, Paris, France) under numbers I3087, I3088 and I3089, respectively. The sequences of the plasmids were verified after they were sent back by the CNCM for verification in our laboratory.
Table 2.
Sequence of probes used for hybridization in real time PCR assays
| Targeted agent | Probe sequence |
| Variola virus | 5'-AAGATCATACAGTCACAGACACTGT-3' |
| Bacillus anthracis | 5'-TACCGCAAATTCAAGAAACAACTGC-3' |
| Francisella tularensis | 5'-TTGCAAATACTTATAGCGCTTTGACT-3' |
| Yersinia pestis | 5'-AAGGAGTGCGGGTAATAGGTTATAA-3' |
| Burkholderia mallei; B. pseudomallei | 5'-AACCAGCGCGCTGTCCGCGAC-3' |
| Chlamydia psittaci | 5'-ATGAAATCTAGAAAATCTTGTGGTGTA-3' |
| Coxiella burnetii | 5'-ATCATTTCGGCTCGGGATGGGC-3' |
| Brucella spp. | 5'-AATGGCAAGCAACTCGTCCTTTAC-3' |
| Rickettsia prowazekii | 5'-TGGTTCTATTCAGTTAGATGGGAGT-3' |
| Rickettsia rickettsii | 5'-TATATTGTTCAAAGGAGCGGGAGAT-3' |
| Monkeypox virus | 5'-TTGATTGTAAATACAGATAGTGAATCG-3' |
| Orthopoxvirus | 5'-TCCATACGATGATCTAGTTACAACTA-3' |
| Varicella-zoster virus | 5'-ATGCCACCTTTACAGTTGGAGGAA-3' |
| Herpes simplex virus 1 and 2 | 5'-ACCGCCGAACTGAGCAGACACC-3' |
| Rickettsia akari | 5'-ACGTCTTATGGCAGGAACAAATTTAG-3' |
Figure 1.
Schematic representation of plasmid CatA This plasmid contains specific sequences of category A agents of bioterrorism (green/white/pink), exogenic sequences (yellow/blue) with inserted NotI site (blue). Primers (green) have been designed to amplifiate agent specific sequences and probes (pink) to detect specifically each sequences.
Figure 2.
Construction of DNA control plasmid designed for the 4 CDC Category A DNA agents (Smallpox virus [seq1], Bacillus anthracis [seq2], Francisella tularensis [seq3], and Yersinia pestis [seq4]). Assembling of the smallpox virus and B. anthracis sequences is presented as an example. Successive steps are indicated by framed numbers. 1, PCR amplification of the two matrix sequences by primers consisting of the stabilization and the restriction site sequences (italics). PCR reactions were carried out in a volume of 50 μl that included 10 mM Tris-HCl [pH 9.0], 1.5 mM MgCl2, 50 mM KCl, 0.1% Triton X-100, 200 μM each dNTP, 0.4 μM of each oligonucleotide primer, 0.4 μM of the single stranded DNA, and 1.5 U of Taq DNA polymerase (Invitrogen, Cergy-Pontoise, France). The thermocycler (Biometra, Göttingen, Germany) profile was 5 min at 95°C, followed by 35 cycles of 30 sec at 95°C, 30 sec at 55°C, and 1 min at 72°C, and terminated by a final extension for 7 min at 72°C. PCR products were electrophorezed in 3% TAE-agarose gel containing ethidium bromide and visualized under UV transillumination. Column purification of the PCR products. PCR products of the expected size were column-purified by using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions, and eluted in 50 μl of RNase free distillated water. When two bands or more were observed by gel analysis, the band of expected size was excised from the gel and purified by glass milk extraction with the GenClean III Kit (Q-Bio-Gene, Carlsbad CA, USA). 2, assemblage was conducted by pair, seq1 with seq2 (resulting in seq1-2). Equal volumes (10 μl) of purified seq1- and seq2-dsDNA were incubated at 37°C in the presence of Sac I. Sac I site is located at the 3' and 5' ends of seq1 and seq2, respectively. 3, the reaction product was column purified using the protocol aforementioned to discard the 15-nt DNA fragments corresponding to the 5' and 3' ends to avoid their re-ligation to their respective complementary sequences at step 5. 4, Overnight incubation at 4°C in the presence of T4 DNA ligase. Ten μl of the reaction was incubated with T4 DNA ligase (Roche, Basel, Switzerland) according to the manufaturer's instructions. 5, PCR amplification by using the external primers (italics) was performed according to the protocol described at step 1. Then column purification using the protocol detailed at step 2 of the resulting PCR product. At this step the seq1-2 PCR product may be cloned into PGEM-T for storage. The same procedure was performed for seq3 and seq4. Ultimately, seq1-2 and seq3-4 were assembled by using the same protocol (sections 1–9). The final product cloned into PGEM-T plasmid includes seq1-2-3-4 flanked by the two Sseq and restriction sites.
Thirteen viral and bacterial targeted agents were detected by using the primers and probes developed in this study using standard PCR, Taq Man and Light Cycler real-time PCRs (Table 1 and 2). Four agents were not available (Monkeypox, Smallpox virus and 2 Burkholderia species). Only the specific primers with the corresponding DNA gave positive results. Multiplex real-time PCR results were identical. The 15 targeted plasmid sequences were detected using the same primers. Amplicons were differentiated from the native DNA by aspect of melting curves and sequencing of the amplifiates (Figure 3, additional file 2). All 50 C. burnetii strains were detected using the same procedures.
Figure 3.
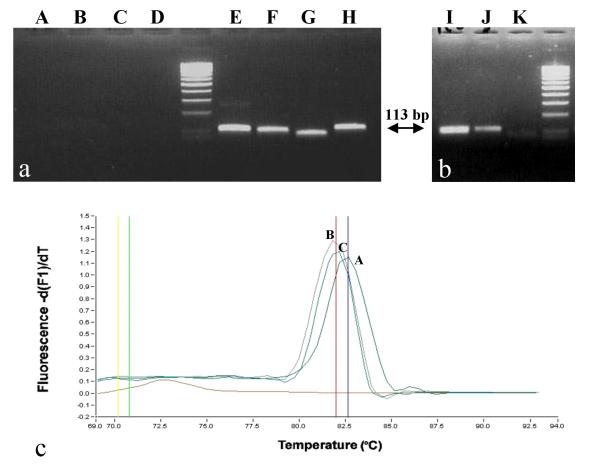
Molecular detection and identification of true and false positives using plasmid CatA as positive control. 3a. NotI predigestion of CatA plasmid DNA. A, B, C, D correspond to Not I digested-CatA plasmid DNA (overnight digestion with 10 U followed by an 1-hour digestion with another 10 U) respectively PCR amplified with Y. pestis, F. tularensis, B. anthracis, and smallpox virus oligonucleotide primers. E, F, G, H correspond to the same DNA samples which have not been digested by NotI. 3b. Discrimination of Y. pestis amplicons by NotI post-PCR digestion in a 3% agarose gel. lane I, NotI post-PCR digestion of amplicon from Y. pestis native DNA (101 bp); lane J, amplicon from CatA plasmid DNA (113 bp); lane K, NotI post-PCR digestion of amplicon from CatA plasmid DNA (digestion result in 2 products of 76 bp and 37 bp, respectively; only the 76 bp product is seen). 3c. Melting curves obtained from SYBR Green Light Cycler reactions. Curve A was obtained with CatA plasmid DNA and Y. pestis primers; curve B was obtained with Y. pestis DNA amplified with Y. pestis specific primers; curve C was obtained with Y. pestis DNA amplified in a multiplex reaction including the 8 primers specific for the category A agents (Y. pestis, F. tularensis, B. anthracis, smallpox virus). The melting temperature observed with A is 82.65 whereas it is 81.97 with native Y. pestis DNA (curves B and C).
Respective sensitivity of the 15 pairs of primers towards the 17 target DNA sequences was evaluated by ten-fold and/or two-fold serial dilutions of a plasmid DNA master solution containing one femtomole/μL of DNA, on a Light Cycler platform with SYBRGreen standard protocol. The results are presented in Table 3. Briefly, sensitivities ranged 5–50 molecules for the 3 plasmids, except for F. tularensis (500 molecules) and R. rickettsii (100 molecules).
Table 3.
Sensitivity of the detection tests performed onto the Light Cycler platform. For Smallpox virus, Monkey pox virus and Burkholderia species, native DNA was not available. For C. psittaci and Varicella-zoster virus titrated micro-organisms were not available.
| Plasmid | Microorganism | Number of molecules detected | |
| Plasmid | Native (strain) | ||
| CatA | Smallpox virus | 5 | NA |
| CatA | Y. pestis | 5 | 5 (Kim5) |
| CatA | B. anthracis | 5 | 5 (Sneath D455) |
| CatA | F. tularensis | 500 | 5 (URFTMUE1) |
| CatB | Brucella sp. | 50 | 50 (B. melitensis B115) |
| CatB | R. prowazeckii | 50 | 100 (Brein l) |
| CatB | R. rickettsii | 100 | 50 (VR 891) |
| CatB | B. mallei/pseudomallei | 50 | NA |
| CatB | C. burnetii | 5 | 100 (Nine Mile) |
| CatB | C. psitacci | 50 | NA |
| CatVEF | Smallpox virus | 5 | NA |
| CatVEF | Monkeypoxvirus | 5 | NA |
| CatVEF | Orthopoxvirus | 5 | 10 (ectromelia TC8) |
| CatVEF | Varicella-zoster virus | 5 | NA |
| CatVEF | Herpesvirus 1/2 | 5 | 10 (MA1) |
| CatVEF | R. akari | 5 | 50 (Kaplan) |
However, sensitivities on native DNA from these species were < 5 and 50 molecules respectively. Combined quantification of the bacterial suspension and its plasmid-derived DNA permitted to estimate that a single E. coli contained 100–200 copies of the plasmid and therefore 100–200 molecules of the target sequence. Quantifying the number of AB by comparison towards an E. coli culture will permit to bypass the plasmid extraction procedure that may be cumbersome to perform in an emergency context. Multiplex and standard PCR protocol displayed similar sensitivities with both native and plasmid DNA.
We tested DNA from BA, their genetically close relatives and common agents in clinical microbiology. None of the PCR tests carried out using the 15 pairs of primers against DNA from these 45 tested agents generated unspecific amplicons. None of the DNA extracted from the non anthracis Bacillus sp. generated amplicons.
No amplicons were obtained from the plasmid DNA suspension digested by NotI, whereas this procedure did not inhibit amplification of the native DNA (Figure 3). Amplicons from plasmid DNA were digested by NotI but not those from native DNA (Figure 3).
Discussion
The CDC has organized detection and diagnosis of biological warfare agents on laboratory testing abilities and facilities by classifying laboratories according to their core-testing capacities [5,6]. Recent developments of commercial kits for detection and quantification of infectious agents, such as Chlamydia trachomatis or HIV [7,8], means that molecular detection techniques are increasingly being used in clinical microbiology laboratories. However, control DNA is necessary for ensuring that there are no inhibitors in the specimens and that the assay is working properly. It is obvious that distribution of AB to all laboratories is not considered. As a matter of fact, this inability to obtain DNA from these agents, make it impossible to test our primers on the native DNA from variola virus, Monkey pox virus, B. mallei and B. pseudomallei.
In this study, we have constructed plasmids containing sequences of different pathogens. This allows limitation of the number of controls to be used. We also introduced a very simple method of decontamination using Chelex [9,10] that kills B. anthracis spores. It can be applied to all tested specimens. We also designed primers to amplify DNA from either plasmids or AB and demonstrated that they ensured AB detection with high sensitivity and specificity. Primarily, the specificity was checked "in silico" and then with a collection of potentially confounding organisms. Multiplex-PCR assay can be performed using these primers and controls. This enables us to detect all AB of a given class (class A and B). The procedure was also demonstrated to be efficient in real-time PCR. Moreover, false positive amplicons resulting from carryover or from preceding amplification of the positive control are easily recognized. As for F. tularensis and R. rickettsii, the number of detected copies from the plasmid DNA was higher than for the other AB. This was not correlated with a lower sensitivity on native DNA.
The strategy we propose may allow detection of AB by all laboratories with molecular biology facilities. The CatA and CatB plasmids could be used for the detection of AB from suspect powders and human samples as in the case of suspected pneumonia (including C. burnetii, C. psittaci, Burkholderia, B. anthracis, Y. pestis, R. prowazekii and F. tularensis). The procedure also allows identification of isolated micro-organism suspected of being an AB.
The CatVEF plasmid is to be used for the detection of AB from humans who are suffering from vesicular eruption resembling that of smallpox. It combines DNA from agents that can be clinically confounding. Few infectious agents cause a vesicular rash. The most common differential diagnosis is the infection by Varicella-zoster virus. Generalized herpes virus infection may also be difficult to differentiate. Moreover, after the anthrax alert in the USA began at the autumn 2001, the number of cases of diagnosed rickettsialpox, a rare disease caused by R. akari, suddenly increased as physicians confronted by patients with vesicular rash or skin eschar wanted to confirm whether it was rickettsialpox or smallpox [11]. Another outbreak of vesicular rashes, resembling smallpox in the USA, was in fact the cases of monkeypox infections [12]. Finally, with increasing diffusion of vaccination campaign, accidental vaccinia will occur [13], challenging the diagnosis of smallpox. Differential laboratory diagnosis of such cases has been tentatively done by using in situ hybridization on cutaneous biopsies [14], but the plasmid CatVEF we propose may also reveal useful.
We believe that our strategy of multiple target plasmids brings two major improvements. It allows to perform quantitative detection that can be of importance, in the case of B. anthracis spores, by helping to evaluate the risk of pulmonary anthrax. It overcomes problems due to PCR contamination by positive control. Discrimination of plasmid DNA from native DNA may be insured a posteriori by observation of different melting curves, sequences or NotI digestion. Such a strategy of target modification for use as a control was previously described for detection of B. anthracis by modification of the size of the amplified fragment [15]. However, our strategy using incorporation of a restriction enzyme site represents an improvement, allowing to perform NotI pre-digestion of the sample. In that case, false positive results due to contamination from previous experiences using the control are avoided. Contaminations by the DNA of native AB may be avoided by incorporation in PCR mix of dUTP rather than that of dTTP and pre-treatment of samples by Uracil N-glycosylase [16]. A combined pre-treatment with NotI would allow to work with an un-contaminable PCR procedure.
Conclusions
To our knowledge, we report the first construction of plasmids to be used as positive controls in amplification reactions for AB. It is also possible to avoid or to ensure immediate detection of false positive results due to contamination by positive controls using these plasmids. These plasmids and the corresponding primers and probes for detection of AB are immediately available for all clinical microbiology laboratories provided they have molecular amplification equipment. However, before routine use, primers and probes will have to be extensively studied on collections of the different targeted pathogens, as we did it on C. burnetii, the sole agent for which we have a significant number of isolates. Due to the increasing availability of complete genome sequences, the primers and the target genes described in this study can be changed to others in order to propose different associations of genes or pathogens.
Methods
For B. anthracis and Y. pestis, previously proposed target genes were used [17,18]. For other agents, we selected genes that were sequenced in several isolates, to ensure that they were conserved in the species. Sequences were aligned using CLUSTALW software at the PBIL http://npsa-pbil.ibcp.fr. Primers, chosen in conserved areas, were designed manually or by using the primer design tool of CyberGene AB http://www.cybergene.se in order to design primers with a Tm of approximately 58° C and a G + C content of less than 50%. Target sequences (Table 1) were compared to the Genbank database using Blastn software http://www.ncbi.nlm.nih.gov/BLAST/ in order to ensure in silico specificity. The fixed cut-off for in silico target sequence similarity was < 25% for any other sequence in any other genera of the GenBank database. Among members of the same genus this cut-off was <75%. Exceptions were R. rickettsii for which similarity with closest relative Rickettsia mongolotimonae was 94% and R. prowazekii for which similarity with closest relative Rickettsia typhi was 90%.
The sequences were separately synthesized by an oligonucleotide manufacturer (Eurogentec, Seraing, Belgium). Each sequence was specific to one agent with exceptions of B. pseudomallei, which was designed to also recognize B. mallei, and Brucella which was designed to recognize all Brucella species. The sequences (129 to 155 nt long) were organized in a similar manner for all constructions (Figures 1 and 2): (i) an ATGCATGCATGC sequence to ensure optimal efficacy of the enzyme during the restriction step (subsequently reported as stabilization sequence [Sseq]), (ii) the 6-nt sequence was specific for the defined restriction enzyme, (iii) the sequence of the agent to be detected which include the sequence of the specific primers at both extremities, (iv) the positive control exogenic sequence ATTATA-GCGGCCGC-TTATTA that includes the Not I site (in bold), (v) a second 6-nt sequence was specific for a restriction enzyme that is distinct from that aforementioned, (vi) the same 12-nt Sseq. Accordingly, all synthetic sequences contain 56 invariable nucleotides.
Three plasmids were constructed such as CatA for category A agents, CatB for category B agents and CatVEF for vesicular eruptive fever (Tables 1 and additional file 1). Construction of DNA control plasmid designed for the 4 CDC Category A DNA agents (Smallpox virus [seq1], Bacillus anthracis [seq2], Francisella tularensis [seq3], and Yersinia pestis [seq4]) is presented as an example (Figure 1).
Once the four single stranded DNA (ssDNA) sequences were synthesized by the manufacturer, they were assembled as a single double-stranded DNA (dsDNA) fragment to be subsequently cloned into the PGEM-T plasmid (PGEM-T Easy Vector System I kit, Promega France SARL, Charbonnieres-les-Bains, France). The sequential steps were as follows (see Figure 2):
1. PCR amplification of each of the 4 single stranded DNA sequences by using primers consisting of the stabilization and the restriction site sequences (italics in additional file 1). PCR reactions were carried out in a volume of 50 μl that included 10 mM Tris-HCl [pH 9.0], 1.5 mM MgCl2, 50 mM KCl, 0.1% Triton X-100, 200 μM each dNTP, 0.4 μM of each oligonucleotide primer, 0.4 μM of the single stranded DNA, and 1.5 U of Taq DNA polymerase (Invitrogen, Cergy-Pontoise, France). The thermocycler (Biometra, Göttingen, Germany) profile was 5 min at 95°C, followed by 35 cycles of 30 sec at 95°C, 30 sec at 55°C, and 1 min at 72°C, and terminated by a final extension for 7 min at 72°C. PCR products were electrophorezed in 3% TAE-agarose gel containing ethidium bromide and visualized under UV transillumination.
2. Column purification of the PCR products. PCR products of the expected size were column-purified by using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions, and eluted in 50 μl of RNase free distillated water. When two bands or more were observed by gel analysis, the band of expected size was excised from the gel and purified by glass milk extraction with the GenClean III Kit (Q-Bio-Gene, Carlsbad CA, USA).
3. Assemblage was conducted by pair, seq1 with seq2 (resulting in seq1-2), seq3 with seq4 (resulting in seq3-4); then seq1-2 with seq-4 (resulting in seq1-2-3-4).
4. Equal volumes (10 μl) of purified seq1- and seq2-dsDNA were incubated at 37°C in the presence of Sac I. Sac I site is located at the 3' and 5' ends of seq1 and seq2, respectively.
5. The reaction product was column purified using the protocol aforementioned to discard the 15-nt DNA fragments corresponding to the 5' and 3' ends to avoid their re-ligation to their respective complementary sequences at step 5.
6. Overnight incubation at 4°C in the presence of T4 DNA ligase. Ten μl of the reaction was incubated with T4 DNA ligase (Roche, Basel, Switzerland) according to the manufaturer's instructions.
7. PCR amplification by using the external primers (italics in additional file 1) was performed according to the protocol described in section 1.
8. Column purification using the protocol detailed in section 2 of the resulting PCR product. At this step the seq1-2 PCR product may be cloned into PGEM-T for storage.
9. The same procedure was performed for seq3 and seq4
Ultimately, seq1-2 and seq-4 were assembled by using the same protocol (sections 1–9). The final product cloned into PGEM-T plasmid includes seq1-2-3-4 flanked by the two Sseq and restriction sites.
Once the first assembled double stranded DNA was obtained, it was cloned into PGEM-T plasmid and sequenced to verify sequence accuracy with primers consisting of the stabilization and the restriction site sequences (italics in additional file 1). Sequencing of each assembled DNA was performed to assess sequence accuracy, and clones that were 100% identical to the reference sequence were used for further assembling. At the end, the complete assembled plasmid was sequenced by using primers located on the PGEM-T plasmid (M13 primers) to verify its 100% identity with the original sequences.
All DNA extractions were performed using Chelex resin under previously described conditions [9,10]. DNA solutions were buffered with 10X PCR buffer without MgCl2 to get a final concentration of 0.5X, and stored at -20°C. The Chelex extraction procedure was additionally tested onto B. anthracis spores in order to verify that it ensure spore killing. Briefly, a heavy suspension of a 96 h agar culture of B. anthracis in sterile PBS was prepared. Ten μl of this suspension was deposited on a slide and stained by Wirtz-Conlin spore staining [19]. Spores, colored in green, were counted. The suspension was then adjusted to a spore concentration of 107/ml. A 100 μl aliquot was then subjected to Chelex extraction and inoculated onto sheep blood agar plate to test growth of B. anthracis.
Amplification conditions were as follows: (I) Standard PCRs were carried out under a classic 35 cycle-protocol with hybridization at 55°C (detailed in Plasmid assembly, section 1); (II) Real-time PCR carried out on the ABI 7000 platform used the protocols recommended by the manufacturer. Briefly, 10 μl of DNA solution was combined with 50 pmol of each primer and 10 pmol of the FAM- and TAMRA-labeled probe (Table 2) in 50-μl total reaction volume by using the TaqMan Universal PCR Master Mix (Applied Biosystems, Roche, USA). The samples were subjected to 40 cycles of amplification in an ABI Prism 7000 or 7700 Sequence Detection System instrument (PE Applied Biosystems) according to the manufacturer's protocol for TaqMan assay cycling conditions. The rate of TaqMan assay positivity was calculated by using the 99.9% confidence level settings in the PE 7000 or 7700 Sequence Detection System software; (III) Real-time PCR carried out on the Light Cycler platform used the SYBRGreen protocols recommended by the manufacturer. Briefly, 1 μl of tenfold serial dilution of DNA solutions was combined with 50 pmol of each primer in a 20-μl total reaction volume by using the LightCycler FastStart DNA Master SYBR Green I Kit (Roche Applied Science). The samples were subjected to 40 cycles of amplification in a LightCycler (Roche) with a 58°C-hybridization step.
Evaluation of the sensitivity of the Light Cycler detection for each of the 15 systems (Tables 1 and 2) were done by testing serial tenfold dilutions of the plasmid and quantified native DNA. NotI digestion of the PCR products was performed for 1 hr at 37°C in a 20 μl volume and results were observed after 3% agarose gel electrophoresis under UV transillumination.
The specificity of the 15 pairs of primers was evaluated by (i) cross-amplification of the 13 cat.A/B and VEF agents of which sequences have been incorporated into the plasmids, (ii) tentative amplification of closely related bacteria or viruses and (iii) tentative amplification of bacteria and viruses frequently isolated/detected in microbiology laboratories. Selected agents were:
(i) the 13 bacteria and viruses to be detected: C. burnetii, F. tularensis, R. rickettsi, R. akari, R. prowazeckii, Y. pestis, B. anthracis, C. psitacci, B. melitensis, vaccinia virus, varicella-zoster virus and herpesvirus 1 and 2.
(ii) a batch of 50 randomly selected C. burnetii isolates from our collection.
(iii) 5 closely related microorganisms: Rickettsia typhi, Rickettsia conorii, Yersinia enterocolitica, Bacillus cereus and Bacillus thurigiensis.
(iv) 28 human pathogens frequently isolated in microbiology laboratories: Corynebacterium ANF group, Nocardia asteroides, Shigella flexneri, Mycobacterium avium, Campylobacter jejuni, Streptococcus agalactiae, streptococcus pyogenes, Pseudomonas paucimobilis, Actinomyces viscosus, Shigella sonnei, Streptococcus bovis, Burkholderia cepacia, Listeria monocytogenes, Clostridium perfringens, Actinomyces meyerii, Fusobacterium nucleatum, Escherichia coli, Staphylococcus aureus, Propionibacterium acnes, Haemophilus influenzae, Bacteroides fragilis, Salmonella enterica Typhi, Kingella kingae, Enterobacter aerogenes, Corynebacterium striatum, Bartonella quintana, Legionella pneumophila and Neisseria meningitidis.
(v) a batch of 12 non anthracis Bacillus sp. isolated in our laboratory from suspect powders [20]
Both sensitivity and quantification tests were performed by using real-time PCR procedure on a Light Cycler platform (Roche) with the SYBRGreen standard protocol recommended by the manufacturer in a 20 μl final volume. Hybridization temperature was set at 58°C for all primers. The three plasmid-harboring E. coli were grown overnight at 37°C under shaking conditions in 10 mL of LB medium containing 100 μg/mL of ampicillin; the cultures were quantified spectrophotometrically and were adjusted to 108 bacteria/mL. For each plasmid, three 3-mL aliquots were independently pelleted by centrifugation. Supernatant was discarded and the pellet was used to extract plasmid DNA by a standard Miniprep procedure (QiaPrep Spin Miniprep Kit, Qiagen, Hilden, Germany) according to the manufacturer's recommendations. This plasmid DNA was spectrophotometrically measured at 260/280 nm, and was adjusted by dilution to a concentration of 1 femtomol/μL constituting the stock solution. The latter was subsequently serially diluted at tenfold, and 1 μL of each dilution was tested by Lightcycler real-time PCR. Final adjustment of the minimal number of molecules detectable by each pair of primers was determined by testing the series of twofold dilutions. Experiments were conducted in triplicate. Combined quantification of the bacterial suspension and its plasmid-derived DNA permitted to estimate the number of plasmid copies harbored by a single bacteria. Sensitivity of the technique to detect AB was determined by testing titrated suspensions of these agents. Titration of agar growing bacteria was performed by macrobroth dilution method [21]. Titration of purified strict intracellular bacteria was performed by flow cytometry counting [22]. Herpes simplex virus 1 was titrated by cell culture infectious dose 90 on Vero cells [23]. Orthopoxvirus was titrated by electronic microscopy negative staining counting.
The suitability of multiplex protocols was evaluated with primers and probes corresponding to each of the three plasmids, respectively. Briefly, serial dilutions of each plasmid and corresponding native DNA were tested in reactions including (i) all primers and probes against plasmid DNA, (ii) all primers and probes against native DNA, (iii) all primers and the specific probe of each agent against plasmid DNA and (iv) the two primers and the probe specific for one agent against native DNA.
To address the possibility of pursuing the testing even whether a laboratory contamination has been detected in previous experiments, we tested the suitability of a decontamination procedure based on NotI treatment of extracted nucleic acids prior to start the PCR protocol. A DNA suspension adjusted to a concentration 106 times higher than the limit detection was prepared for the 4 plasmids. Three test solutions were then either tested directly after nucleic acid extraction, or submitted to a one-hour restriction in the presence of NotI at 37°C: they consisted of (i) plasmid nucleic acid (NA) alone simulating a laboratory-contaminated negative specimen, (ii) plasmid NA mixed with native NA simulating a laboratory-contaminated positive specimen, (iii) native NA simulating a true positive specimen.
Abbreviations
AB: agents of bioterrorism.
Author's contribution
RNC and BL designed primers, probes, and plasmids, conceived experimental design and drafted the manuscript. Both contributed equally to this work. DR conceived and coordinated the study. All authors read and approved the final manuscript.
Supplementary Material
Sequences of DNA fragments used to construct plasmids.Low case sequence are not specific for the select agent, upper case sequence are specific for the select agent. Invariable stabilization sequences are in italics, restriction sites are italics and underlined, specific primers are upper case underlined, exogenic sequence is in bold, the underlined part being the NotI site.
Discrimination between native DNA and control plasmid. Discrimination of amplicons is based on results of electrophoresis in 3% agarose gel and analysis of melting curves obtained on Light Cycler from SYBRGreen assays.
Acknowledgments
Acknowledgments
None.
Contributor Information
Remy N Charrel, Email: rnc-virophdm@pop.gulliver.fr.
Bernard La Scola, Email: bernard.lascola@medecine.univ-mrs.fr.
Didier Raoult, Email: didier.raoult@medecine.univ-mrs.fr.
References
- Centers for Disease Control and Prevention (CDC) Notice to readers : ongoing investigation of anthrax-Florida. MMWR Morbid Mortal Wkly Rep. 2001;50:877. [Google Scholar]
- Morse SA, Kellog RB, Perry S, Meyer RF, Bray D, Nichelson D, Miller JM. Detecting biothreat agents: the laboratory response network. ASM News. 2003;69:433–437. [Google Scholar]
- Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Friedlander AM, Hauer J, McDade J, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Tonat K. Anthrax as a biological weapon. JAMA. 1999;281:1735–1745. doi: 10.1001/jama.281.18.1735. [DOI] [PubMed] [Google Scholar]
- Payne DA, Petersen JW. Rapid molecular testing for bioterrorism agents: targets, tactics and technology. Journal of Clinical Ligand Assay. 2002;25:348–357. [Google Scholar]
- Jortani SA, Snyder JW, Valdes R., Jr The role of the clinical laboratory in managing chemical or biological terrorism. Clin Chem. 2000;46:1883–1893. [PubMed] [Google Scholar]
- Klietman WF, Ruoff KL. Bioterrorism: implications for the clinical microbiologist. Clin Microbiol Rev. 2001;14:363–381. doi: 10.1128/CMR.14.2.364-381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Doornum GJ, Schouls LM, Pijl A, Cairo I, Buimer M, Bruisten S. Comparison between the LCx probe system and the Cobas amplicor system for detection of Chlamydia trachomatis and Neisseria gonorrhoeae infections in patients attending a clinic for treatment of sexually transmitted diseases in Amsterdam the Netherlands. J Clin Microbiol. 2001;39:829–835. doi: 10.1128/JCM.39.3.829-835.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla DJ, Granger S, Jennings C, Bremer JW. Multisite comparison of reproducibility and recovery from the standard and ultrasensitive Roche Amplicor HIV-1 monitor assays. J Clin Microbiol. 2001;39:1121–1123. doi: 10.1128/JCM.39.3.1121-1123.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lamballerie X, Zandotti C, Vignoli C, Bollet C, de Micco P. A one-step microbial DNA extraction method using "Chekex 100" suitable for gene amplification. Res Microbiol. 1992;143:785–790. doi: 10.1016/0923-2508(92)90107-Y. [DOI] [PubMed] [Google Scholar]
- Stein A, Raoult D. A simple method for amplification of DNA from paraffin-embedded tissues. Nucleic Acid Res. 1992;20:5237–5238. doi: 10.1093/nar/20.19.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddock CD, Zaki SR, Koss T, Singleton J, Jr, Sumner JW, Comer JA, Eremeeva ME, Dasch GA, Cherry B, Childs JE. Rickettsialpox in New York city: a persistent zoonosis. Ann N Y Acad Sci. 2003;990:36–44. doi: 10.1111/j.1749-6632.2003.tb07334.x. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC) Update: multistate outbreak of monkeypox-illinois, Indiana, Kansas, Missouri, Ohio and Wisconsin. MMWR Morbid Mortal Wkly Rep. 2003;52:561–564. [PubMed] [Google Scholar]
- Aragon TJ, Ulrich S, Fernyak S, Rutherford GW. Risks of serious complication and death from smallpox vaccination: a systematic review of the United States experience, 1963–1968. BMC Public Health. 2003;3:26. doi: 10.1186/1471-2458-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuovo GJ, Plaza JA, Magro J. Rapid diagnosis of smallpox infection and differentiation from its mimics. Diag Mol Pathol. 2003;12:103–107. doi: 10.1097/00019606-200306000-00006. [DOI] [PubMed] [Google Scholar]
- Brightwell G, Pearce M, Leslie D. Development of internal controls for PCR detection of bacillus anthracis. Mol Cell Probes. 1998;12:367–377. doi: 10.1006/mcpr.1998.0195. [DOI] [PubMed] [Google Scholar]
- Rys PN, Persing DH. Preventing false positives: quantitative evaluation of three protocols for inactivation of polymerase chain reaction amplification products. J Clin Microbiol. 1993;31:2356–2360. doi: 10.1128/jcm.31.9.2356-2360.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellerbrok H, Nattermann H, Ozel M, Beutin L, Appel B, Pauli G. Rapid and sensitive identification of pathogenic and apathogenic Bacillus anthracis by real time PCR. FEMS Microbiol Let. 2002;214:51–59. doi: 10.1016/S0378-1097(02)00837-6. [DOI] [PubMed] [Google Scholar]
- Hinnebusch J, Schwan TG. New method for plague surveillance using polymerase chain reaction to detect Yersinia pestis in fleas. J Clin Microbiol. 1993;31:1511–1514. doi: 10.1128/jcm.31.6.1511-1514.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapin KC, Murray PR. Stains. In: Murray PR, editor. Manual of Clinical Microbiology. Washington, ASM Press; 1999. pp. 1674–1686. [Google Scholar]
- La Scola B, Fournier PE, Raoult D. Searching for Bacillus anthracis in suspect powders: a French experience. J Clin Microbiol. 2003;41:524–525. doi: 10.1128/JCM.41.1.524-525.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Committee for Clinical Laboratory Standards. NCCLS, Wayne Pennsylvania. 2000. pp. M7–A5.
- Davey HM, Kell DB. Flow cytometry and cell sorting of heterogeneous microbial populations: the importance of single-cell analyses. Microbiol Rev. 1996;60:641–696. doi: 10.1128/mr.60.4.641-696.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricout F, Moraillon A, Sonntag P, Hoerner P, Blackwelder W, Plotkin S. Virus-inhibiting surgical glove to reduce the risk of infection by enveloped viruses. J Med Virol. 2003;69:538–545. doi: 10.1002/jmv.10342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequences of DNA fragments used to construct plasmids.Low case sequence are not specific for the select agent, upper case sequence are specific for the select agent. Invariable stabilization sequences are in italics, restriction sites are italics and underlined, specific primers are upper case underlined, exogenic sequence is in bold, the underlined part being the NotI site.
Discrimination between native DNA and control plasmid. Discrimination of amplicons is based on results of electrophoresis in 3% agarose gel and analysis of melting curves obtained on Light Cycler from SYBRGreen assays.