

Abstract
Hepatitis B virus (HBV) is one of the most prevalent pathogens in the world, and infection with this virus is a serious threat for public health. Yunnan is considered as an important endemic center for blood-borne viruses such as human immunodeficiency virus and hepatitis C virus, in China. However, the distribution and diversity of HBV subgenotypes remain unclear in Yunnan province. In the current study, HBV positive samples were collected from different prefectures of Yunnan province and their molecular epidemiological characters were determined. Phylogenetic analysis on the pre-S/S gene (865 bps) showed the prevalence of four HBV genotypes, including genotype B (24 cases, 33.3%), genotype C (45 cases, 62.5%), genotype I (two cases, 2.78%) and C/D recombinants (one case, 1.39%). The most prevalent genotypes B and C could be sub classified into subgenotype B2 and C1, C2, C5, and C7, respectively. Clusters of subgenotype B2 and C2 consisted of strains from China and other East Asian countries, while subgenotype C1, C5, and C7 and genotype I formed a cluster together with strains from Southeast Asia. Using Bayesian inference from phylogenetic, HBV genotypes B and C were estimated to have originated in 1860s and 1910s with an evolutionary rate of 3.26 and 8.01 × 10−4 substitutions/site/year, respectively. These findings indicate that the distribution of HBV genotypes in Yunnan was influenced by strains from the rest of China and the neighboring countries. J. Med. Virol. 86: 1675–1682, 2014. © 2014 The Authors. Journal of Medical Virology Published by Wiley Periodicals, Inc.
Keywords: HBV, genotype distribution, phylogenetic analyses, evolutionary analyses
INTRODUCTION
Hepatitis B virus (HBV) infection is a global health problem; more than two billion people have been infected with HBV in the world. It is estimated that 400 million people have developed into chronic HBV infection, and more than two-thirds live in Asia [Lok, 2000]. HBV infection is highly endemic in China, with approximately 93 million infected individuals [Lu and Zhuang, 2009]. Although the incidence of acute HBV infection has declined due to implementation of vaccination programs in many countries, including China, HBV-related complications are still increasing.
HBV, a prototype member of the family Hepadnaviridae, has a compact structure and a partially double stranded circular DNA that encodes four partially overlapped open reading frames (ORFs): S, C, P, and X. Due to the lack of viral-encoded polymerase proofreading activity, HBV genome has a great diversity [Xu et al., 2013]. Based on intergenotypic divergence of at least 8% in the full-length nucleotide sequence or more than 4% in the S gene, HBV can be classified into at least nine genotypes (A–I), each of them has also been classified into several subgenotypes [Okamoto et al., 1986; Tanaka et al., 2004; Tran et al., 2008]. The various HBV genotypes are associated with differences in pathogenicity [Yuen et al., 2004], disease progression [Mayerat et al., 1999], and responses to antiviral drugs [Halfon et al., 2006].
The distribution of HBV genotypes is different geographically [Norder et al., 2004]. Genotype A is found mainly in Northern and Western Europe, North America, and Africa. Genotypes B and C, with a large number of subgenotypes, are prevalent in Asia [Orito et al., 2001; Sugauchi et al., 2002]. Except for subgenotype B1, B2, C1, and C2, which reported to distribute commonly in the Chinese mainland, the other subgenotypes of genotype B and C are distributed commonly in Southeast Asian countries [Olinger et al., 2008; Shi et al., 2012]. Genotype D and the C/D recombinant are concentrated in Northwest China, where most of the residents are migrated from Central Asia [Zeng et al., 2005]. Genotype E is the most prevalent genotype in Eastern and Central Africa, and sporadically found in Colombia and Northern India. Genotypes F and H are prevalent in the Amerindian population and in Central America, respectively. Recently, the newly designated genotype I has been characterized using phylogenetic analysis, which identified in Vietnam, Laos and Canada [Tran et al., 2008; Osiowy et al., 2010], and seems localized in Southeast Asia.
Southeast and East Asia, with 25% of the world's population, are considered to be most seriously affected regions of infectious diseases. As the most prevalent pathogen, at least four HBV genotypes including genotype B, C, D and I, and multiple patterns of recombinants are circulating in this area. Yunnan province is located in Southwest China and borders with Southeast Asian countries. Because of its special geographical location and common intravenous drug addiction, many blood-borne viruses are endemic in this region. The unique genotypic distribution of HCV, HIV, and GBV-C in Yunnan has been well documented in previous reports [Li et al., 2005; Xiao et al., 2007; Feng et al., 2011]. HBV is considered to be a major causative agent of liver diseases. However, its subgenotypes distribution in Yunnan province and homologous relationship between Yunnan and neighboring regions remain unclear.
MATERIALS AND METHODS
Serum Samples
A total of 80 HBV-positive serum samples were collected from hepatitis patients who sought medical service at the First People's Hospital of Yunnan Province from 2011 to 2012. Among them, 16 cases were from Kunming, the capital city of Yunnan; 12 cases were from Qujing, the central city of east Yunnan; 18 cases were from Yuxi, the central city of middle Yunnan; 10 cases were from Chuxiong, a major industrial city; 12 were from Dehong, the border city of southwest Yunnan; 6 cases were from Honghe, the prefecture in South Yunnan; and 8 cases were from Dali, a renowned tourist city of west Yunnan. Active HBV infection was confirmed by detection of HBsAg, anti-HBsAg, HBeAg, anti-HBeAg and HBcAg, and HBV DNA copies at the clinical laboratory of the hospital. All enrolled patients were confirmed to have chronic hepatitis B by clinical testing, characterized as HBsAg-positive >6 months; serum HBV DNA >105 copies/ml; persistent or intermittent elevation of ALT/AST levels; liver biopsy showing chronic hepatitis, necroinflammatory score >4. Demographic data such as age, ethnicity, and residential address were recorded by interviewing. All participants provided written informed consent for participation in this study, and the research was approved by the Kunming University of Science and Technology Ethics Committee.
PCR Amplification and Sequencing
HBV DNA was extracted from 100 µl sera using an AxyPrep Blood Genomic DNA Miniprep Kit (AXYGEN, Union City, CA). For amplification of the pre-S region and part of the S region (positions: 2,891–540), a nested PCR reaction was performed. First-round PCR was conducted using the outer set of primers SF1 and SR1, followed by a second round of PCR using the inner set of primers SF2 and SR2. The same cycle conditions were used for both rounds of PCR: initial denaturation at 94°C for 1 min; followed by 35 cycles each of denaturation at 94°C for 30 sec, annealing at 50°C for 30 sec, and extension at 72°C for 90 sec. The PCR products were purified and directly submitted for sequencing in the Beijing Genomics Institute (BGI, Beijing, China) with the second-round primers. In addition, to obtain the full-length HBV circular genome, two overlap fragments were amplified by semi-nested PCR. The outer set of primers for the first fragment was F1817F and F374R, and the inner set of primers was F1817F and F192R. The second fragment was amplified by using the outer set of primers SF1 and F1822R and the inner set of primers SF2 and F1822R. The sequences and locations of the primers mentioned above are shown in Table 1. The GenBank/EMBL/DDBJ accession number of the sequence of HBV strains used in this paper are KF917452–KF917522 (pre-S/S) and KF917451 (full genome).
I.
Sequences and Locations of Primers
| Primer name | Sequences (5′–3′) | Location (EU139543) |
|---|---|---|
| SF1 | CTTAATCCTVAACCTVAATGGCAAACTCCTC | 2,513–2,539 |
| SR1 | CCCAAAAGACCCACAATTCKTT | 734–758 |
| SF2 | GCHTCATTTTGCGGGTCACCATATTC | 2,800–2,827 |
| SR2 | GATGGGATGGGAATACARGTGCA | 873–900 |
| F1817F# | CATGCAACTTTTTCACCTCTGCCTARTCA | 1,813–1,841 |
| F1822R# | AAAAGTTGCATGGTGNTGGTGAACA | 1,818–1,843 |
| F192R | AAAAACCCCGCCTGTAACACGA | 188–210 |
| F374R | GATAAAACGCCGCAGACACATCCA | 370–394 |
Primers was first reported by Zhang et al. [2011].
Phylogenetic and Recombination Analyses
Sequences were aligned and edited using the integrated ClustalX 1.83 program [Aiyar, 2000] and BioEdit V7.0.8 [Hall, 1999]. HBV genotypes were determined by phylogenetic analysis of the nucleotide sequence of the pre-S/S fragment. The phylogenetic relationship was characterized using the neighbor-joining method with 1,000 bootstrap replications in the MEGA software (Kimura 2-parameter substitution model) [Tamura et al., 2011]. Recombination signals were detected initially by the Simplot software. The bootscan window sizes were 200 bases, the step size was 20 with 100 replicates. The reference sequences used in phylogenetic and recombination analyses were obtained from the Hepatitis Virus Database (http://s2as02.genes.nig.ac.jp/index.html) and GenBank (S1).
Bayesian Markov Chain Monte Carlo Evolutionary Analyses
The Bayesian Markov chain Monte Carlo (MCMC) method implemented in BEAST package [Drummond and Rambaut, 2007] was used to estimate the evolutionary rate and starting time of diversification of the predominant HBV genotype in Yunnan. The Hasegawa–Kishino–Yano (HKY) nucleotide substitution model with a gamma-distributed model for site rate variation using four rate categories (C4) and a constant population size model [Yang, 1994] were chosen as the best models for the Bayesian coalescence analyses. Each MCMC analysis was run for at least 50 million generations and sampled every 50,000 generations in the BEAST V1.7.5 package. Only ESS values >250 were accepted. For constructing maximum clade credibility (MCC) trees, the first 25% of generated trees were discarded as burn-in and summarized using TreeAnnotator implemented in the BEAST V1.7.5 package. The reconstructed trees were examined and edited by using FigTree V1.3.1 (tree.bio.ed.ac.uk/software/figtree/), which was also used to estimate the evolutionary rates and the dates of various nodes on the MCC tree.
RESULTS
Demographic and Clinical Characteristics
In total, 80 HBV-positive patients were recruited from 7 representative prefectures of Yunnan. The PreS/S fragment of 72 samples, accounting for 90% of the total collected samples, was amplified successfully. Their mean age was 34 years with the range of 19–69 years, and 45 (62.5%) of them were men. The HBV-DNA load was determined in these 72 cases by quantitative PCR with a median value of 7.16 ± 1.05 log copies/ml (range, 5.35–9.29 log copies/ml). 59 cases (75.6%) were identified positive for HBeAg. Regarding the major parameters of liver function, the mean ALT level was 98.8 ± 136 IU/L (range, 10–595 IU/L), and the mean AST level was 81 ± 118 IU/L (range, 12–584 IU/L).
HBV Genotype Distribution
The HBV genotype could be determined by phylogenetic analysis in 72 patients. It accounts for four HBV genotypes (B, C, C/D, and I) identified in this study. Among them, genotype C was the most prevalent with infection of 45 (62.5%) patients, followed by genotype B (24/72, 33.3%) and genotype I (2/72, 2.78%). According to their phylogenetic relationship, HBV genotype B consisted of a single subgenotype B2, while HBV genotype C could be classified into several subgenotypes including 28 cases of C2, 10 cases of C1, 3 cases of C5, and 4 cases of C7 (Fig. 1). Notably, subgenotype C1, C5, and C7 were most prevalent among the ethnic population of Southeast Asia. Sample YN.071, collected at Qujing, the most northeastern prefecture in Yunnan, was clustered as a C/D1 recombinant together with two strains from western China. In comparing with other genotypes, higher level of liver function parameters were observed in genotype C, but not significant (dates not shown).
Fig 1.
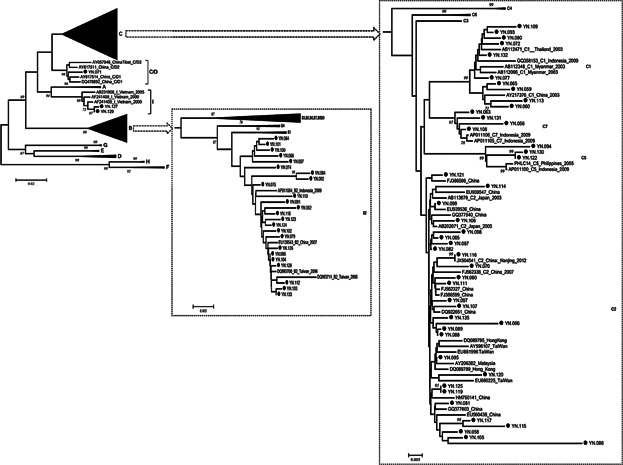
Phylogenetic analysis of pre-S/S sequences of HBV strain in Yunnan compared with reference strains representing genotypes A–I. Accession numbers and countries are shown in each branch, and HBV genotypes or subgenotypes are listed on the right. Bootstrap values were shown along each main branch. The characterized strains in this study are marked with black dots.
The reconstructed neighbor-joining tree also showed that the strains of subgenotype B2 and C2, which were identified as the most prevalent subgenotypes in Yunnan, were mixed together with strains from China and other East Asian countries in their respective clusters. The strains of subgenotype C1, C5, and C7 forming a cluster closely with the strains from Southeast Asia, suggests a close genetic relationship between them. Two strains of genotype I being grouped together with Vietnamese strains in the same branch, indicates that they probably originated from Vietnam. The bootstrap value between sample YN.071 recombinant and C/D1 recombinant from western China was 99%. To confirm further the recombinant nature of YN.071, phylogenetic and bootscan analyses on the sequence of complete genomes were performed. The inter-genotypes recombinant model was verified in the reach of full-length sequences. The breakpoint of C/D1 recombinant (Fig. 2) was similar to that of previous reported recombinants [Wang et al., 2005; Zhou et al., 2011].
Fig 2.
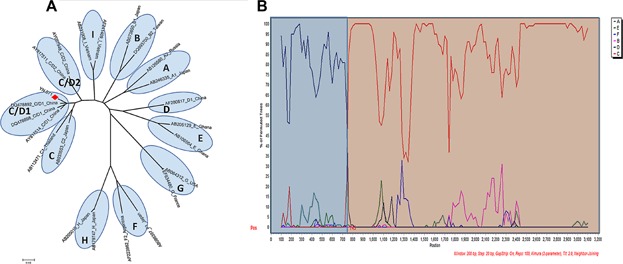
Phylogenetic analysis (A) and SimPlot bootstrap analysis (B) on nucleotide sequence of YN.071 with consensus sequences representing each of the genotypes A–I. YN.071 had high similarity with genotype D from 1 to 745 nt and high similarity with genotype C from 746 to 3,215 nt.
Evolutionary Characters of Genotype B and C
Through Bayesian evolutionary calculation, the mean value of the evolutionary rate of HBV/B pre-S/S fragment was estimated as 3.26 × 10−4 substitutions/site/year (95% HPD, 7.50 × 10−5–5.61 × 10−4). Based on sequences of these strains and other reference strains from other regions, the time-scaled Bayesian tree was reconstructed (Fig. 3A). As the most prevalent subgenotype, most of the strains of subgenotype B2 were intermixed with the other reference strains, but a small significant clad including five strains were distinct. The estimated timing of tree's root suggested that genotype B was originated in the 1860s. Whereas, several significant clades in subgenotype B2 from Yunnan were dated back to 37 years ago, corresponding to the later of 1970s.
Fig 3.
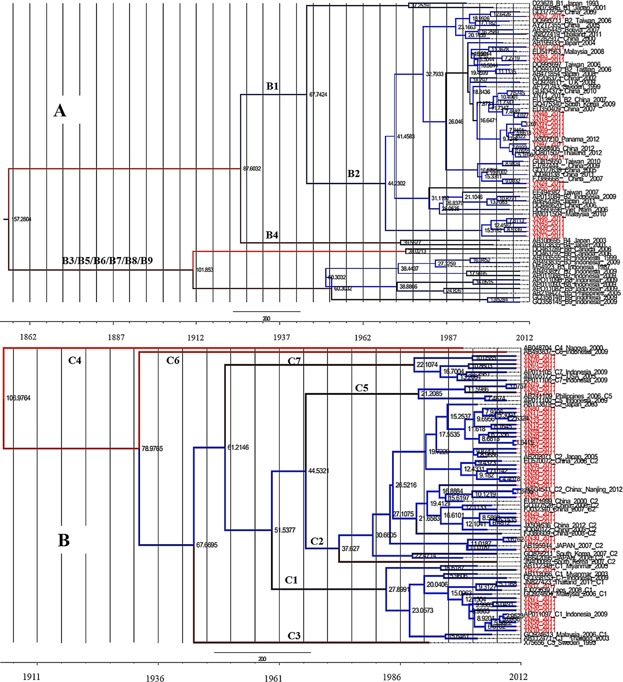
Maximum clade credibility (MCC) trees were estimated by Bayesian analysis on HBV genotype B and C sequences. Yunnan strains are indicated in red. The internal numbers are node ages. The scale at the bottom of the tree represents the time (year).
In the case of genotype C, the estimated mean value of the evolutionary rate was 8.01 × 10−4 substitutions/site/year (95% HPD, 3.82 × 10−4–1.27 × 10−3), almost twice as the evolutionary rate of genotype B. The origin of genotype C was back to 93 years ago in the tMRCA analysis (Fig. 3B). Segregation of these strains according to their geographic origin was not observed. The strains from Yunnan were concentrated mainly in the younger branches of the MCC tree (C1, C2, C5, and C7).
DISCUSSION
Accurate classification of HBV genotypes and subgenotypes is important since HBV genotypes are related to the course of the infection, responses to antiviral treatment regimens, and clinical outcomes [Kramvis and Kew, 2005; Zeng et al., 2005]. Phylogenetic analysis and Bayesian analysis based on the nucleotide sequence of partial genes have been widely accepted for the determination of genotypes and evolutionary characteristics. However, some genotyping methods based on partial genes were not able to accurately distinguish HBV subgenotypes. In the current study, the nucleotide sequence of pre-S gene and part of S gene (865 bps) were used in phylogenetic and evolutionary analyses. HBV genotypes and subgenotypes could be identified accurately using this established method (Phylogenetic trees of the HBV genotypes and subtypes created by comparison of pre-S/S sequences with full-length genome and Pre-S1 sequences in the same reference strains were showed in S2). The classified genotypes and subgenotypes based on this partial sequence were consistent with results of phylogenetic analysis on the full-length genome. The pre-S gene and part of S gene could thus be recommended as target gene for detailed HBV genotyping.
In this study, all participants were recruited from seven different prefectures including the Han Ethnicity majority population in China and five ethnic minorities. Therefore, they were geographically and demographically representative for Yunnan province. Among all enrolled patients, pre-S/S gene amplification was not detected in eight patients because of their low HBV-DNA copy levels determined by quantitative real-time PCR. Due to the limitation of collected samples, a significant difference of HBV genotypes/subgenotypes distribution between the studied prefectures was not found. However, the strains of genotype I and subgenotypes C1, C5, and C7, which are prevalent in Southeast Asia, were more frequently identified in the frontier prefectures.
Genotype B and C are the most prevalent HBV genotypes in China with geographically and demographically different genotypes distributions. Genotype C is predominant in northern China, whereas genotype B is more prevalent in southern China [Zeng et al., 2005]. In addition to the most prevalent HBV subgenotypes B2 and C2, other identified subgenotypes including C1, C5, and C7, were infrequent in China. These subgenotypes were considered to be introduced from the Southeast Asian countries. This could be confirmed further by the close relationship between the strains from Yunnan and reference strains from neighboring Southeast Asia. Notably, two strains of genotype I were identified in this study, and were thought to be originated in Vietnam, where HBV genotype I was reported first in 2008 [Tran et al., 2008]. The HBV C/D1 recombinant, being considered to endemic in the Qinghai-Tibet Plateau in western China [Zhou et al., 2011, 2012], was also identified in Yunnan. Due to the different recruited subjects and limited sample size, this characterized HBV genotypes distribution is a little different with previous report [Kang et al., 2011]. In spite of this, it could be determined that the distribution of hepatitis B virus in Yunnan was influenced by the strains from other provinces of China and strains from Southeast Asian countries. The important role of Yunnan in the epidemic of the blood-borne virus was confirmed further by the molecular epidemiological characters of HBV.
Bayesian analysis is widely used because of their ability to integrate information regarding mean evolutionary rate, dated phylogeny, and coalescent population dynamics. It was deduced the estimated evolutionary rate of the pre-S/S fragment were different between genotypes B and C. Repeated analysis using different model showed that this difference indeed exists. The higher evolutionary rate of genotype C was verified with its documented faster adaptability to the host environment [Shi et al., 2012]. Furthermore, genotype C could be classified into the various subgenotypes, which ranks the most subgenotypes among all genotypes [Chan, 2011]. Infection with genotype C seems related to more aggressive disease course [Lin and Kao, 2011; Chan et al., 2002; Chan et al., 2003; Chan et al., 2009; Chien et al., 2006; Ahn et al., 2010]. In addition, the origin of genotype B was approximately 60 years earlier than genotype C. The MCC tree showed that subgenotype B2 was a young clade, backdate to be introduced into Yunnan approximately 37 years ago. Phylogenetically, the strains of genotype C could not be segregated significantly according to their geographic origin. It may suggest a relatively recent origin and multiple exchanges in these local areas.
The distinct geographical location of Yunnan province results in the diverse epidemiological characters of blood-borne HBV infection in this region. This conclusion was repeated verified by our previously documented multiple genotypes distribution of HIV, HCV and GBV-C [Yang et al., 2002; Zhang et al., 2002; Xia et al., 2008; Feng et al., 2011], and further HBV in this study. As a hub between China and Southeast Asian neighboring countries, Yunnan region requires further epidemiological research.
Acknowledgments
We thank Prof. Tianrui Xu and Dr. Yang Yang in Kunming University of Science and Technology for his helpful manuscript revision, and Prof. Kok Keng Tee in University of Malaya for his kind suggestion.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
REFERENCES
- Ahn SH, Yuen L, Han KH, Littlejohn M, Chang HY, Damerow H, Ayres A, Heo J, Locarnini S, Revill PA. Molecular and clinical characteristics of hepatitis B virus in Korea. J Med Virol. 2010;82:1126–1134. doi: 10.1002/jmv.21844. [DOI] [PubMed] [Google Scholar]
- Aiyar A. The use of CLUSTAL W and CLUSTAL X for multiple sequence alignment. Methods Mol Biol. 2000;132:221–241. doi: 10.1385/1-59259-192-2:221. [DOI] [PubMed] [Google Scholar]
- Chan HL. Significance of hepatitis B virus genotypes and mutations in the development of hepatocellular carcinoma in Asia. J Gastroenterol Hepatol. 2011;26:8–12. doi: 10.1111/j.1440-1746.2010.06514.x. [DOI] [PubMed] [Google Scholar]
- Chan HL, Tsang SW, Liew CT, Tse CH, Wong ML, Ching JY, Leung NW, Tam JS, Sung JJ. Viral genotype and hepatitis B virus DNA levels are correlated with histological liver damage in HBeAg-negative chronic hepatitis B virus infection. Am J Gastroenterol. 2002;97:406–412. doi: 10.1111/j.1572-0241.2002.05478.x. [DOI] [PubMed] [Google Scholar]
- Chan HL, Wong ML, Hui AY, Hung LC, Chan FK, Sung JJ. Hepatitis B virus genotype C takes a more aggressive disease course than hepatitis B virus genotype B in hepatitis B e antigen-positive patients. J Clin Microbiol. 2003;41:1277–1279. doi: 10.1128/JCM.41.3.1277-1279.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan HL, Wong GL, Tse CH, Chim AM, Yiu KK, Chan HY, Sung JJ, Wong VW. HBV genotype C is associated with more severe fibrosis than genotype B. Clin Gastroenterol Hepatol. 2009;7:1361–1366. doi: 10.1016/j.cgh.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Chien RN, Lin CY, Yeh CT, Liaw YF. Hepatitis B virus genotype B is associated with better response to thymosin alpha1 therapy than genotype C. J Viral Hepat. 2006;13:845–850. doi: 10.1111/j.1365-2893.2006.00761.x. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Zhao W, Feng Y, Dai J, Li Z, Zhang X, Liu L, Bai J, Zhang H, Lu L, Xia X. A novel genotype of GB virus C: Its identification and predominance among injecting drug users in Yunnan, China. PLoS ONE. 2011;6:e21151. doi: 10.1371/journal.pone.0021151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfon P, Bourlière M, Pol S, Benhamou Y, Ouzan D, Rotily M, Khiri H, Renou C, Pénaranda G, Saadoun D, Thibault V, Serpaggi J, Varastet M, Tainturier MH, Poynard T, Cacoub P. Multicentre study of hepatitis B virus genotypes in France: Correlation with liver fibrosis and hepatitis B e antigen status. J Viral Hepat. 2006;13:329–335. doi: 10.1111/j.1365-2893.2005.00692.x. [DOI] [PubMed] [Google Scholar]
- Hall T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Kang W, Ding Z, Shen L, Zhao Z, Tian B, Li H, Li Y, Zhang S, BI S. Distribution of hepatitis B virus genotypes and serotypes in people who had a physical examination in Yunnan Province. Chin J Exp Clin Virol. 2011;25:114–116. [PubMed] [Google Scholar]
- Kramvis A, Kew MC. Relationship of genotypes of hepatitis B virus to mutations, disease progression and response to antiviral therapy. J Viral Hepat. 2005;12:456–464. doi: 10.1111/j.1365-2893.2005.00624.x. [DOI] [PubMed] [Google Scholar]
- Li XJ, Kusagawa S, Xia X, Yang C, Wang Q, Yokota Y, Hoshina Y, Onogi T, Nohtomi K, Imamura Y, Shiino T, Yang R, Yamamoto N, Ben K, Takebe Y. Molecular epidemiology of the heterosexual HIV-1 transmission in Kunming, Yunnan Province of China suggests origin from the local IDU epidemic. AIDS Res Hum Retroviruses. 2005;21:977–980. doi: 10.1089/aid.2005.21.977. [DOI] [PubMed] [Google Scholar]
- Lin C, Kao JH. The clinical implications of hepatitis B virus genotype: Recent advances. J Gastroenterol Hepatol. 2011;26:123–130. doi: 10.1111/j.1440-1746.2010.06541.x. [DOI] [PubMed] [Google Scholar]
- Lok AS. Hepatitis B infection: Pathogenesis and management. J Hepatol. 2000;32:89–97. doi: 10.1016/s0168-8278(00)80418-3. [DOI] [PubMed] [Google Scholar]
- Lu FM, Zhuang H. Prevention of hepatitis B in China: Achievements and challenges. Chin Med J. 2009;122:2925–2927. [PubMed] [Google Scholar]
- Mayerat C, Mantegani A, Frei PC. Does hepatitis B virus (HBV) genotype influence the clinical outcome of HBV infection. J Viral Hepat. 1999;6:299–304. doi: 10.1046/j.1365-2893.1999.00174.x. [DOI] [PubMed] [Google Scholar]
- Norder H, Couroucé AM, Coursaget P, Echevarria JM, Lee SD, Mushahwar IK, Robertson BH, Locarnini S, Magnius LO. Genetic diversity of hepatitis B virus strains derived worldwide: Genotypes, subgenotypes and HbsAg subgenotypes. Intervirology. 2004;47:289–309. doi: 10.1159/000080872. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Imai M, Shimozaki M, Hoshi Y, Iizuka H, Gotanda T, Tsuda F, Miyakawa Y, Mayumi M. Nucleotide sequence of a cloned hepatitis B virus genome, subgenotype ayr: Comparison with genomes of the other three subgenotypes. J Gen Virol. 1986;67:2305–2314. doi: 10.1099/0022-1317-67-11-2305. [DOI] [PubMed] [Google Scholar]
- Olinger CM, Jutavijittum P, Hübschen JM, Yousukh A, Samountry B, Thammavong T, Toriyama K, Muller CP. Possible new hepatitis B virus genotype, Southeast Asia. Emerg Infect Dis. 2008;14:1777–1780. doi: 10.3201/eid1411.080437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orito E, Ichida T, Sakugawa H, Sata M, Horiike N, Hino K, Okita K, Okanoue T, Iino S, Tanaka E, Suzuki K, Watanabe H, Hige S, Mizokami M. Geographic distribution of hepatitis B virus (HBV) genotype in patients with chronic HBV infection in Japan. Hepatology. 2001;34:590–594. doi: 10.1053/jhep.2001.27221. [DOI] [PubMed] [Google Scholar]
- Osiowy C, Kaita K, Solar K. Molecular characterization of hepatitis B virus and a 9-year clinical profile in a patient infected with genotype I. J Med Virol. 2010;82:942–948. doi: 10.1002/jmv.21758. [DOI] [PubMed] [Google Scholar]
- Shi W, Zhu C, Zheng W, Carr MJ, Higgins DG, Zhang Z. Subgenotype reclassification of genotype B hepatitis B virus. BMC Gastroenterol. 2012;12:116. doi: 10.1186/1471-230X-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugauchi F, Orito E, Ichida T, Kato H, Sakugawa H, Kakumu S, Ishida T, Chutaputti A, Lai CL, Ueda R, Miyakawa Y, Mizokami M. Hepatitis B virus of genotype B with or without recombination with genotype C over the precore region plus the core gene. J Virol. 2002;76:5985–5992. doi: 10.1128/JVI.76.12.5985-5992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Hasegawa I, Kato T, Orito E, Hirashima N, Acharya SK, Gish RG, Kramvis A, Kew MC, Yoshihara N, Shrestha SM, Khan M, Miyakawa Y, Mizokami M. A case-control study for differences among hepatitis B virus infection of genotypes A (subgenotypes Aa and Ae) and D. Hepatology. 2004;40:747–755. doi: 10.1002/hep.20365. [DOI] [PubMed] [Google Scholar]
- Tran TT, Trinh TN, Abe K. New complex recombinant genotype of hepatitis B virus identified in Vietnam. J Virol. 2008;82:5657–5663. doi: 10.1128/JVI.02556-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liu Z, Zeng G, Wen S, Qi Y, Ma S, Naoumov NV, Hou J. A new intertype recombinant between genotypes C and D of hepatitis B virus identified in China. J Gen Virol. 2005;86:985–990. doi: 10.1099/vir.0.80771-0. [DOI] [PubMed] [Google Scholar]
- Xia X, Lu L, Tee KK, Zhao W, Wu J, Yu J, Li X, Lin Y, Mukhtar MM, Hagedorn CH, Takebe Y. The unique HCV genotype distribution and the discovery of a novel subgenotype 6u among IDUs co-infected with HIV-1 in Yunnan, China. J Med Virol. 2008;80:1142–1152. doi: 10.1002/jmv.21204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Kristensen S, Sun J, Lu L, Vermund SH. Expansion of HIV/AIDS in China: Lessons from Yunnan Province. Soc Sci Med. 2007;64:665–675. doi: 10.1016/j.socscimed.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Wei C, Guo Y, Zhang C, Zhang N, Wang G. An analysis of the molecular evolution of Hepatitis B viral genotypes A/B/D using a Bayesian evolutionary method. Virol J. 2013;10:256. doi: 10.1186/1743-422X-10-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods. J Mol Evol. 1994;39:306–314. doi: 10.1007/BF00160154. [DOI] [PubMed] [Google Scholar]
- Yang R, Xia X, Kusagawa S, Zhang C, Ben K, Takebe Y. On-going generation of multiple forms of HIV-1 intersubgenotype recombinants in the Yunnan Province of China. AIDS. 2002;16:1401–1417. doi: 10.1097/00002030-200207050-00012. [DOI] [PubMed] [Google Scholar]
- Yuen MF, Sablon E, Tanaka Y, Kato T, Mizokami M, Doutreloigne J, Yuan HJ, Wong DK, Sum SM, Lai CL. Epidemiological study of hepatitis B virus genotypes, core promoter and precore mutations of chronic hepatitis B infection in Hong Kong. J Hepatol. 2004;41:119–125. doi: 10.1016/j.jhep.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Zeng G, Wang Z, Wen S, Jiang J, Wang L, Cheng J, Tan D, Xiao F, Ma S, Li W, Luo K, Naoumov NV, Hou J. Geographic distribution, virologic and clinical characteristics of hepatitis B virus genotypes in China. J Viral Hepat. 2005;12:609–617. doi: 10.1111/j.1365-2893.2005.00657.x. [DOI] [PubMed] [Google Scholar]
- Zhang C, Yang R, Xia X, Qin S, Dai J, Zhang Z, Peng Z, Wei T, Liu H, Pu D, Luo J, Takebe Y, Ben K. High prevalence of HIV-1 and hepatitis C virus coinfection among injection drug users in the southeastern region of Yunnan, China. J Acquir Immune Defic Syndr. 2002;29:191–196. doi: 10.1097/00042560-200202010-00014. [DOI] [PubMed] [Google Scholar]
- Zhang T, Chen Q, Yang B, Chen S, Yuan Q, GE S. Amplification of complete hepatitis B virus genome by a sensitive nested PCR. J Xiamen Univ. 2011;50:947–950. [Google Scholar]
- Zhou B, Xiao L, Wang Z, Chang ET, Chen J, Hou J. Geographical and ethnic distribution of the HBV C/D recombinant on the Qinghai-Tibet Plateau. PLoS ONE. 2011;6:e18708. doi: 10.1371/journal.pone.0018708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Wang Z, Yang J, Sun J, Li H, Tanaka Y, Mizokami M, Hou J. Novel evidence of HBV recombination in family cluster infections in western China. PLoS One. 2012;7:e38241. doi: 10.1371/journal.pone.0038241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.