

SUMMARY
A large portion of common variant loci associated with genetic risk for schizophrenia reside within non-coding sequence of unknown function. Here, we demonstrate promoter and enhancer enrichment in schizophrenia variants associated with expression quantitative trait loci (eQTL). The enrichment is greater when functional annotations derived from human brain are used relative to peripheral tissues. Regulatory trait concordance analysis ranked genes within schizophrenia genome-wide significant loci for a potential functional role, based on co-localization of a risk SNP, eQTL and regulatory element sequence. We identified potential physical interactions of non-contiguous proximal and distal regulatory elements. This was verified in prefrontal cortex and induced pluripotent stem cell-derived neurons for the L-type calcium channel (CACNA1C) risk locus. Our findings point to a functional link between schizophrenia-associated non-coding SNPs and 3-dimensional genome architecture associated with chromosomal loopings and transcriptional regulation in the brain.
INTRODUCTION
A recent multi-stage genome-wide association study (GWAS) in schizophrenia (SCZ) identified 22 linkage disequilibrium (LD) independent loci that reached genome-wide significance (Ripke et al., 2013). The majority of identified SNPs reside within non-coding regions of genes or in intergenic regions. Furthermore, the regions were frequently large and often contained multiple implicated SNPs due to local LD patterns. In order to be able to understand these associations mechanistically, it is important to develop strategies for honing in on regions and SNPs more likely to have functional effects. Thus, the elucidation of the function of non-coding disease-associated loci is an important next step towards the development of testable hypotheses regarding biological processes involved in the pathogenesis of SCZ.
Across many phenotypes, non-coding GWAS risk loci are involved in the regulation of transcriptional activity and demonstrate enrichment for expression quantitative trait loci (eQTL) (Nicolae et al., 2010; Richards et al., 2012; Zhong et al., 2010) and cis-regulatory elements (CREs) (Degner et al., 2012; Harismendy et al., 2011; Maurano et al., 2012; Musunuru et al., 2010). A CRE, such as a promoter or an enhancer, is a non-coding DNA sequence in, near or distal to a gene that contains binding sites for regulatory factors and is required for proper spatiotemporal expression of the gene. Long-distance enhancers are thought to interact physically with gene proximal promoters and transcription start sites (TSS) by forming loops through regulatory proteins including cohesins and other repressors and facilitators of transcription (Fanucchi et al., 2013; Kagey et al., 2010). Because these chromosomal loopings can bypass hundreds of kilobases on the linear genome, risk loci positioned within CREs at a distance from the TSS could affect the binding of these regulatory proteins, and ultimately lead to allele-specific differences in target gene expression and subsequent alterations in molecular pathways implicated in disease.
In this study, we use functional annotations to categorize GWAS SNPs and demonstrate that variants increasing risk for SCZ are enriched for alleles that affect gene expression (eSNPs) and lie within promoters or enhancers. The enrichment shows tissue specificity and is greatest when functional annotations derived from human cerebral cortex are used. For ten out of 22 SCZ GWAS hits, we identify functional variants positioned within promoters, enhancers and other regulatory sequences that are associated with expression of 13 genes. For the L-type calcium channel (CACNA1C), a well-established SCZ risk locus, we map non-contiguous physical interactions between enhancer regions and the TSSs using human postmortem brain tissue and human induced pluripotent stem cells (hiPSCs) derived neurons. Overall, this study provides a functional link between SCZ non-coding risk loci and physical interactions of non-contiguous DNA elements associated with transcriptional regulation in the human brain.
RESULTS
Enrichment of functional annotation categories in SCZ
We primarily used publically available data to generate functional annotations. Brain eSNPs were generated by using genotype and gene expression profiles from 8 datasets (Colantuoni et al., 2011; Gibbs et al., 2010; Zhang, 2013) (Table S1). The brain CRE annotation map was generated based on chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) experiments of histone modifications (Cheung et al., 2010; Maurano et al., 2012; Shulha et al., 2013; Shulha et al., 2012; Zhu et al., 2013) (Table S2, S3). We integrated data from specific sets of histone methylation and acetylation markings and DNase I hypersensitive sites (DHS) to define five types of CREs: active promoter, active enhancer, poised promoter, repressed enhancer and open chromatin regions.
Genome-wide SNPs from a published SCZ GWAS data set (Ripke et al., 2013) were classified into four different categories: (i) eSNP: if they affect gene expression of specific transcripts; (ii) CRE: if they lie within a CRE region; (iii) eSNP in a cis regulatory element (creSNP); or, (iv) functionally unannotated variants (FUV) if they did not clustered to any of the above categories (Table S4). Among SCZ nominally associated loci at P ≤ 10−3 (n = 42,253 SNPs,), 37.3% were grouped into the eSNP category (n = 15,762) (Table 1). Among this 37.3%, 4.9% were in active promoters, 9.6% in active enhancers, 3.5% in DHS, 1.0% in poised promoters and 1.5% in repressed enhancers. Relative enrichments for the categories were calculated using an empirical cumulative distribution of the GWAS P values after controlling for genomic inflation as described previously (Schork et al., 2013). Across all P value thresholds tested, the largest enrichment of GWAS SNPs occurs in the following categories: eSNPs and three types of CREs, active promoters, active enhancers and DHS (Table 1; Figure 1). Despite having fewer SNPs, enrichment is greater when the combined creSNP functional category is analyzed for all types of CREs (CRE range: 1.58 – 7.08 fold; creSNP range: 4.03 – 29.51 fold). This indicates that SCZ-associated variants are enriched for SNPs that have stronger support for a functional role (creSNP). Higher enrichment of the creSNP in comparison to CRE categories alone is found for the individual, non-integrated, CRE and creSNP functional annotation categories (Figure S1; Table S5). Among the individual, non-integrated creSNP annotations, H3K4me1, an individual histone mark of enhancers, in fetal and adult brain tissue, is the most enriched category (Table S5).
Table 1.
SNP enrichment for different GWAS P values.
| Functional category | Number of SNPs | Number of SNPs (SNP enrichment) | ||||||
|---|---|---|---|---|---|---|---|---|
| P< 10−3 | P< 10−5 | P< 5×10−8 | ||||||
| CRE | creSNP | CRE | creSNP | CRE | creSNP | CRE | creSNP | |
| SCZ GWAS | 9,815,700 | 42,253 | 4,337 | 692 | ||||
| eSNP | 1,479,508 | 15,762 (3.68) | 2,585 (8.07) | 529 (14.13) | ||||
|
Active promoter [H3K4me3 and (H3K9ac or H3K27ac)] |
209,349 | 51,728 | 1,363 (2.13) |
778 (5.17) |
172 (2.88) |
142 (9.31) |
18 (4.57) |
17 (17.38) |
|
Active enhancer [H3K4me1 and (H3K9ac or H3K27ac)] |
419,981 | 95,701 | 2,858 (2.30) |
1,518 (5.66) |
353 (3.16) |
290 (11.46) |
59 (7.08) |
54 (29.51) |
| DHS | 276,468 | 46,770 | 1,365 (1.58) |
558 (4.06) |
158 (2.39) |
118 (10.94) |
26 (5.13) |
23 (26.92) |
|
Poised promoter [H3K4me3 and H3K27me3] |
81,260 | 13,743 | 357 (1.4) |
155 (4.03) |
27 (1.02) |
25 (5.36) |
3 (2.29) |
3 (13.80) |
|
Repressed enhancer [H3K4me1 and H3K27me3] |
165,117 | 25,547 | 623 (1.13) |
241 (3.12) |
46 (0.82) |
34 (4.67) |
5 (1.51) |
5 (9.55) |
The enrichment of each functional category is given in comparison to FUV.
Figure 1.

Stratified Q-Q plots for eSNP, CRE and creSNP in (a) active promoter, (b) active enhancer, (c) DHS, (d) poised promoter and (e) repressed enhancer functional annotation categories. The numbers for each functional category (blue box: creSNP; green box: CRE) illustrate the estimated increase in terms of the proportion of p-values expected below a genome-wide significant P value (P < 5 × 10−8; red dashed line) compared to the functionally unannotated variant (FUV) category. The estimated enrichment for eSNPs is 14.1. For all functional categories, enrichment is greater when the creSNP functional category is analyzed relative to CREs. All summary statistics were corrected for inflation by using the FUV inflation control. The major histocompatibility complex locus (chr6: 25–35Mb) was excluded from the SCZ dataset.
We provide a single number quantification of enrichment by calculating a categorical enrichment score (CES), which is a conservative estimate of the variance attributable to non-null SNPs (Schork et al., 2013). The CES analysis indicates the following: first, SNPs that cluster within CRE, eSNP and creSNP functional annotation categories show higher CES compared to FUV (Figure 2). Second, the creSNP categories (scaled CES creSNP range: 0.66 – 1) have higher CES than CREs (scaled CES CRE range: 0.11 – 0.29). Third, certain creSNP categories (active promoter, active enhancer and DHS) were the most enriched as measured by the CES. The enrichment was significant for active promoter and enhancer (for both CRE and creSNP), eSNP and DHS creSNP (all P ≤ 0.0001 by permutation). In the individual non-integrated functional categories, the H3K4me1 and H3K4me3 creSNP annotations in fetal brain tissue were the most enriched (26-fold compared to FUV category; P ≤ 0.0001 by permutation), as measured by the CES (Figure S2).
Figure 2.

Categorical enrichment for the combined functional annotations as measured by the CES. On the left side, we show the observed enrichment (red dashed lines) against the null distribution (gray density plots). For each functional category, we performed 10,000 permutations to calculate the null distribution of enrichment for comparison to observed categorical enrichment. Functional categories with empirical P values that survived Bonferroni multiple testing correction are in bold (P corrected: 0.05 / 11 = 4.5 × 10−3). The number is parenthesis indicate the number of SNPs per annotation category. On the right side, the barplot illustrates the scaled CES for each functional category. creSNPs are illustrated in blue color, eSNPs in red, CREs in green and FUV in black. The CES are scaled using the maximum value across functional categories. All summary statistics were corrected for inflation by using the functionally unannotated variant (FUV) inflation control.
Differences in the extent of linkage disequilibrium (LD, estimated based on the sum r2) and minor allele frequency (MAF) were observed between the functional categories (Table S6). Since categories with higher average MAF or larger total amount of LD could spuriously appear enriched for SCZ association, we performed regression analysis which demonstrated that the effect of functional categories remains independently strong with an effect size rank that mirrors enrichment, despite the significant correlation among categories (Table S7). Furthermore, the enrichment was significant for all functional categories compared to FUV after removing SNPs with r2 > 0.1 and those that were <100 kb from a more strongly associated variant in the SCZ study (Table S6; Figure S3).
Tissue specificity of functional annotation categories for GWAS enrichment
We then examined whether functional categories generated in brain tissue can better inform SCZ SNPs (show higher CES) compared to annotations derived from non-brain tissues. We used the following non-brain eSNP -- Lymphoblastoid cell line (LCL) (Xia et al., 2012), liver (Innocenti et al., 2011), peripheral blood mononuclear cells (PBMC) (Westra et al., 2013), skin and adipose tissue (Grundberg et al., 2012) -- and CRE --skin, T-helper, liver, adipose tissue (Maurano et al., 2012; Zhu et al., 2013) -- functional categories to estimate CES for SCZ GWAS as described above. In addition we calculated CES using a previously published GWAS in rheumatoid arthritis (RA) (Stahl et al., 2010). The PBMC eSNP and T-helper CRE functional categories were selected as positive controls for RA since immunological cells are implicated in its etiopathogenesis (Okada et al., 2014; Trynka et al., 2013). The major histocompatibility complex (MHC) locus (chr6: 25–35Mb) was excluded from both GWAS datasets. The brain eSNP and CRE functional category showed the highest enrichment for SCZ (Figure 3A). In contrast, the blood/T-helper functional category showed the highest enrichment for RA. Within the brain and blood/T-helper related functional categories, the active promoter, active enhancer and DHS show the highest enrichment for SCZ and RA SNPs, respectively (Figure 3B).
Figure 3.
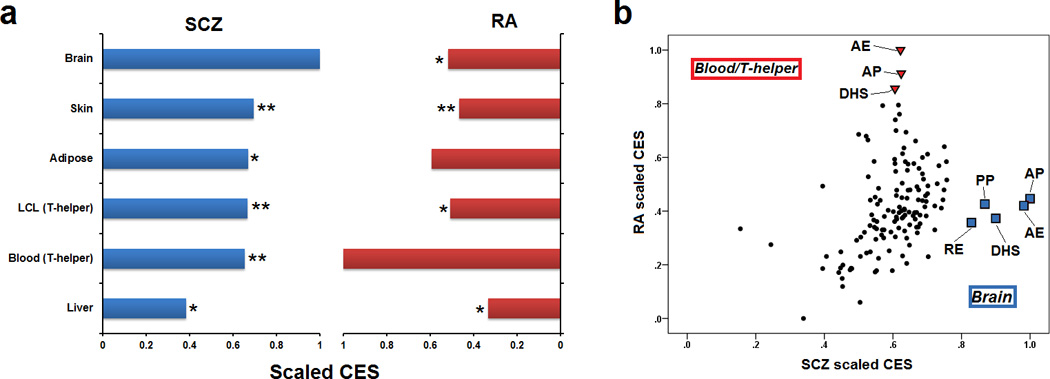
Categorical enrichment in the SCZ and RA GWAS datasets for eSNPs and CREs (brain, LCL/T-helper, liver, PBMC/T-helper, skin and adipose tissue). For each eSNP-CRE combination, the (a) average value of the CES across all functional categories (active promoter, active enhancer, DHS, poised promoter and repressed enhancer) or (b) individual value was calculated. For each GWAS dataset, the CES were scaled using the maximum value across functional categories. The brain and blood/T-helper related eSNP and CRE functional category showed the highest enrichment for SCZ (blue) and RA (red) SNPs, respectively. All summary statistics were corrected for inflation by using the functionally unannotated variant (FUV) inflation control. *P < 0.05, **P < 0.01 by nonparametric Mann-Whitney among the most enriched category and the rest of functional categories for each GWAS dataset. AP: active promoter; AE: active enhancer; PP: poised promoter; RE: repressed enhancer.
Using functional annotations to prioritize risk loci in SCZ
The majority of SCZ genome-wide significant loci (index and SNPs in LD with them) are non-coding (Ripke et al., 2013). Given the enrichment of SCZ loci for eSNPs, we used the regulatory trait concordance (RTC) approach (Grundberg et al., 2012; Nica et al., 2010) to prioritize SNPs and genes within SCZ genome-wide significant loci. RTC scores ≥ 0.9 indicate that overlapping eSNP and GWAS signals likely tag the same variant.
The genome-wide significant SNPs in Ripke et al. were mapped to 22 hotspot intervals (Table 2). We detected overlapping eSNP and GWAS signals at RTC ≥ 0.9 for 10 of 22 intervals, four times the number expected by chance (2.2 expected in the top 10% of scoring intervals under the uniform distribution (Nica et al., 2010); P = 2 × 10−5). The SCZ-related eSNPs are associated with expression of 17 genes (3 intervals had RTC scores with eSNPs of > 0.9 for more than one gene). Given the enrichment of SCZ loci for creSNPs, we examined whether any of the 17 SCZ-associated eSNPs (and tag SNPs within 500kb and r2 > 0.8) lie within the most enriched CRE categories (active promoter, active enhancer and DHS). Thirteen eSNPs out of the 17 are localized within such a CRE (Table 2; Figure 4). The expression of 5 genes is associated with loci within enhancers (C10orf32, CACNA1C, NGEF, RP11–490G2.2 and SDCCAG8). Gene expression of AS3MT is influenced by an eSNP that lies within the promoter region. The expression level of the remaining genes (NT5C2, GATAD2A, HAPLN4, GNL3, NEK4, SPCS1 and TYW5) is associated with multiple eSNPs within both promoter and enhancer sequences.
Table 2.
Annotation of the 22 genome-wide significant loci using the functional eSNP and CRE data.
| SCZ GWAS | Recombination Interval |
RTC analysis | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosomal region | Index SNP | P | Size (kb) |
Genesa | eSNPb | Distance (kb)c |
r2 | Gene | RT C |
beta (SE) d |
creSN Pe |
CRE |
|
Chr. 6: 31,596,138– 32,813,768 |
rs11400214 0 |
9.14 × 10 −14 |
1905.5 | 263 (127) |
NA | - | - | - | - | - | - | |
|
Chr. 10: 104,487,871– 105,245,420 |
rs7085104 |
3.68 × 10 −13 |
4807.9 | 135 (85) |
rs491969 0 |
12.4 | 0.64 | AS3MT | 1 |
−0.72 (0.07) |
1 | AP/DHS |
|
rs491969 0 |
12.4 | 0.64 | C10orf32 | 1 |
−0.51 (0.07) |
1 | AE/DHS | |||||
| rs569468 76 | 24.7 | 0.68 | NT5C2 |
0.9 8 |
−0.90 (0.04) |
14 |
AP/AE/D HS |
|||||
| rs619824 | 47.6 | 0.62 | WBP1L |
0.9 9 |
−0.64 (0.07) |
- | - | |||||
| Chr. 7: 1,827,717–2,346,115 | rs6461049 |
5.93 × 10 −13 |
3196.4 | 66 (39) |
rs121544 73 |
35.3 | 0.85 | SNX8 |
0.9 7 |
−0.34 (0.09) |
- | - |
|
Chr. 1: 98,141,112– 98,664,991 |
rs1198588 |
1.72 × 10 −12 |
775.5 | 24 (2) |
rs119857 2 |
55.7 | 0.87 |
RP11 490G2.2 |
0.9 8 |
−0.40 (0.08) |
1 | AE/DHS |
|
Chr. 12: 2,285,731– 2,440,464 |
rs1006737 |
5.22 × 10 −12 |
1339.0 | 61 (17) | rs758170 | 16.2 | 0.97 | CACNA1C | 1 |
−0.39 (0.09) |
4 | AE/DHS |
|
Chr. 10: 18,601,928– 18,934,390 |
rs17691888 |
1.27 × 10 −10 |
882.1 | 36 (4) |
rs108286 53 |
38.2 | 0.59 | CACNB2 | 1 |
−0.34 (0.08) |
- | - |
|
Chr. 8: 143,297,312– 143,410,423 |
rs4129585 | 2.19 × 10 −10 |
538.9 | 68 (16) | rs1198862 5 |
7.4 | 0.52 | LYPD2 | 0.8 9 |
0.33 (0.09) |
NA | NA |
|
Chr. 1: 73,275,828– 74,099,273 |
rs10789369 | 3.64 × 10 −10 |
1814.3 | 33 (3) | rs1089006 6 |
346.9 | 0.00 2 |
CRYZ | 0.4 8 |
0.30 (0.08) |
NA | NA |
|
Chr. 11: 130,706,918– 130,894,976 |
rs7940866 | 1.83 × 10−9 |
240.4 | 34 (7) | rs1079110 7 |
28.9 | 0.78 | APLP2 | 0.8 8 |
0.36 (0.09) |
NA | NA |
|
Chr. 5: 151,888,959– 152,835,304 |
rs17504622 | 2.65 × 10−9 |
1242.4 | 34 (9) | NA | - | - | - | - | - | - | |
|
Chr. 19: 19,354,937– 19,744,079 |
rs2905424 |
3.44 × 10−9 |
7682.3 |
321 (160) |
rs725423 0 |
39.1 | 0.91 | GATAD2A | 1 |
−0.38 (0.08) |
14 |
AP/AE/D HS |
|
rs207429 5 |
104.0 | 0.45 | GMIP |
0.9 9 |
0.32 (0.08) |
- | - | |||||
|
rs124607 64 |
41.5 | 0.91 | HAPLN4 | 1 |
0.34 (0.08) |
14 |
AP/AE/D HS |
|||||
|
Chr. 2: 37,422,072– 37,592,628 |
rs2373000* | 6.78 × 10−9 |
666.1 | 32 (13) | rs1262127 6 |
293.5 | 0.06 | EIF2AK2 | 0.8 9 |
−0.40 (0.09) |
NA | NA |
|
Chr. 5: 101,581,848– 101,870,822 |
rs6878284* | 9.03 × 10−9 |
3643.4 | 42 (8) | NA | - | - | - | - | - | - | |
|
Chr. 3: 52,215,002– 53,175,017 |
rs4687552 |
1.16× 10 −8 |
7777.1 |
294 (142) |
rs124865 54 |
95.9 | 0.75 | GNL3 |
0.9 9 |
−0.40 (0.08) |
15 |
AP/AE/D HS |
|
rs226802 3 |
19.1 | 0.78 | NEK4 |
0.9 5 |
−0.28 (0.07) |
13 |
AP/AE/D HS |
|||||
|
rs130715 84 |
33.9 | 0.74 | SPCS1 |
0.9 9 |
0.34 (0.08) |
14 |
AP/AE/D HS |
|||||
|
Chr. 2: 145,139,727– 145,214,607 |
rs12991836 | 1.19× 10−8 |
1164.0 | 29 (5) | rs7285849 6 |
68.4 | 0.01 | GTDC1 | 0.8 | 0.38 (0.08) |
NA | NA |
|
Chr. 2: 200,628,118– 201,293,421 |
rs2949006 |
1.21× 10 −8 |
1916.3 | 61 (18) |
rs352204 50 |
65.3 | 0.83 | TYW5 |
0.9 8 |
−0.52 (0.07) |
3 |
AP/AE/D HS |
|
Chr. 18: 52,722,378– 52,827,668 |
rs4801131 | 1.22 × 10−8 |
1047.2 | 33 (10) | rs8084537 | 121.7 | 0.00 2 |
TCF4 | 0.1 5 |
−0.33 (0.08) |
NA | NA |
|
Chr. 2: 233,550,961– 233,808,241 |
rs778371 |
1.51 × 10−8 |
1945.1 | 105 (42) | rs778371 | 0.0 | - | NGEF | 1 |
0.56 (0.07) |
7 | AE |
|
Chr. 1: 243,593,066– 244,025,999 |
rs14403 |
1.80× 10−8 |
1614.2 | 51 (11) |
rs300691 6 |
24.0 | 0.64 | SDCCAG8 |
0.9 8 |
0.42 (0.09) |
3 | AE/DHS |
|
Chr. 12: 123,447,928– 123,913,433 |
rs11532322 | 2.28 × 10−8 |
2236.6 | 101 (50) | NA | - | - | - | - | - | - | |
|
Chr. 1: 243,418,063– 243,627,135 |
rs1538774 | 2.53 × 10−8 |
1614.2 | 51 (11) | rs3006916 | 95.0 | 0.02 | SDCCAG8 | 0.5 7 |
0.42 (0.09) |
NA | NA |
|
Chr. 8: 89,188,454– 89,761,163 |
rs11995572 | 3.33 × 10−8 |
1550.2 | 24 (7) | NA | - | - | - | - | - | - | |
|
Chr. 5: 60,484,179– 60,843,706 |
rs171748 | 3.78 × 10−8 |
2761.2 | 48 (14) | rs295571 | 72.1 | 0.20 | PDE4D | 0.8 7 |
0.31 (0.08) |
NA | NA |
|
Chr. 5: 152,505,453– 152,707,306 |
rs2910032 | 4.12 × 10−8 |
1242.4 | 34 (9) | rs2118792 | 30.2 | 0.07 | FAM114A2 | 0.8 8 |
0.37 (0.08) |
NA | NA |
number of genes (based on the Gencode v16 annotations) that lie within 1Mb upstream or downstream from recombination intervals. In parenthesis are the number of genes that were affected by eSNPs and included in the RTC analysis for each recombination interval.
eSNP with the highest RTC score for each SCZ GWAS chromosomal region. NA indicates that the GWAS SNP was excluded from the eSNP analysis due to MAF < 5% (rs17504622) or increased missingness (>10%) per marker after imputation (rs6878284, rs114002140, rs11532322, rs11995572).
distance among the eSNP and index SCZ GWAS SNP.
standardized beta coefficient and standard error (SE) for the effect of minor allele on gene expression
number of creSNPs that are in strong LD (within 500kb and with r2>0.8) with the eSNP All eSNPs with RTC ≥ 0.9 are illustrated in bold fonts. For GWAS regions with no eSNP at RTC ≥ 0.9, we present the eSNP with the highest RTC. For those SNP, no creSNP or CRE data are presented.
The (*) indicates that this region was not genome-wide significant in the recent PGC2 GWAS analysis (2014).
RTC: regulatory trait concordance; AP: active promoter; AE: active enhancer; DHS: DNase I hypersensitive site
Figure 4.
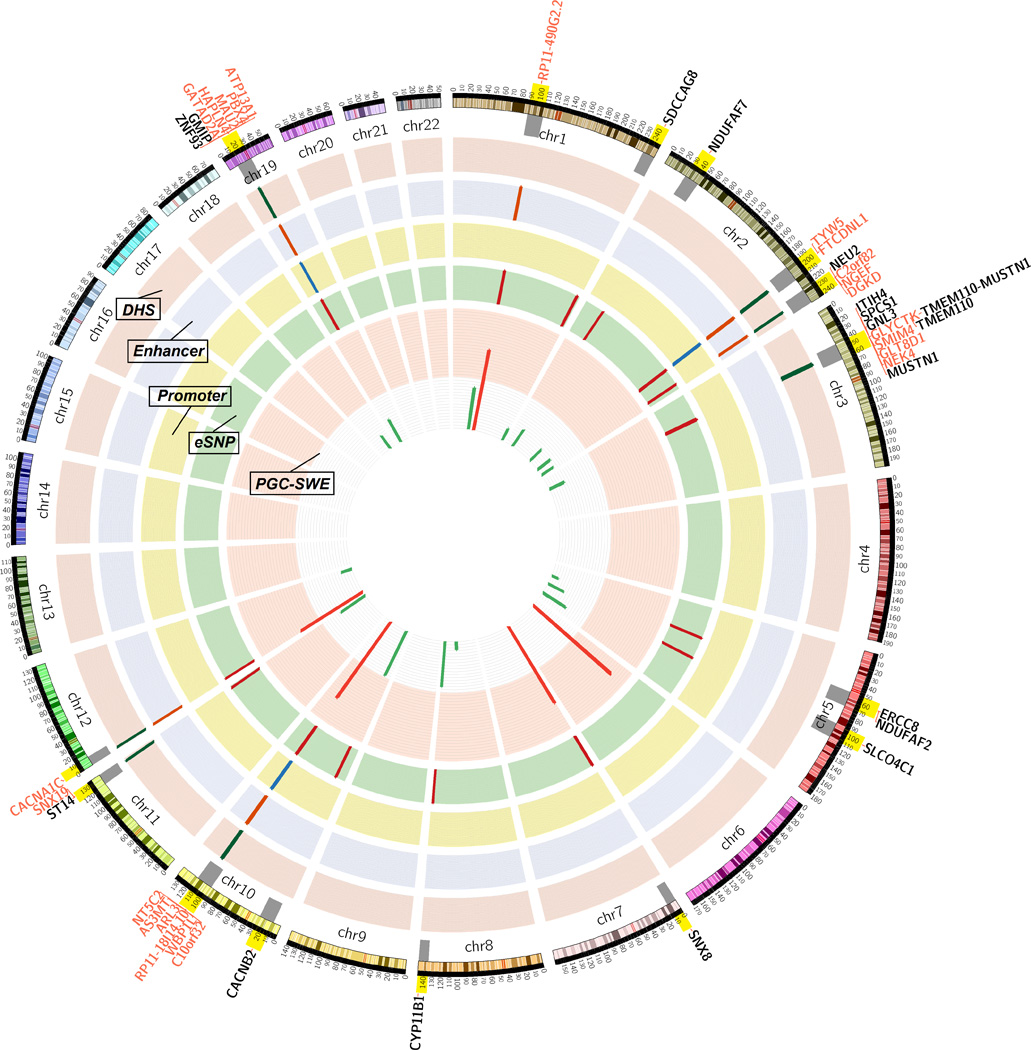
Functional genomic analysis for the SCZ genome-wide significant loci. Layer 1 shows the 24 genome-wide significant loci. Green lines illustrate loci with -log10 P value ≤ 10 and red lines show loci with -log10 P value > 10. Layer 2 (eSNP) illustrates which of the 24 significant loci had eSNP with RTC ≥ 0.9. Layers 3–5 show whether eSNPs (and tag SNPs with r2 > 0.8 within 500kb) lie within active promoter (layer 3; Promoter), active enhancer (layer 4; Enhancer) or DNase I hypersensitive site (layer 5; DHS). Genes affected by functional SNPs are illustrating outside the circo. In black fonts are genes affected by eSNP. In red fonts are genes affected by creSNPs (within active promoter, active enhancer or DHS).
Functional annotations identify risk variants in CACNA1C that lie within putative enhancers and affect gene expression
The step-wise approach described above (Table 2; Figure 4) allows us to formulate a testable hypothesis for the mechanism through which some variants might increase risk for SCZ. For example, the CACNA1C index SNP (rs1006737) and eSNP (rs758170) are in close proximity (16.2 kb) and have an RTC score of 1. Furthermore, the risk allele is associated with decreased CACNA1C gene expression (P = 1.88 × 10−5 at FDR 0.7%).
We identified 4 SNPs (rs2159100, rs12315711, rs11062170, rs4765905) that are in perfect LD with rs758170 and rs1006737, and lie within two predicted enhancers (spanning 1.4Kb and 1.85Kb), ∼185Kb downstream from the gene’s TSS (Figure 5A, Figure 5B). To identify whether the predicted enhancer region might be capable of forming promoter–enhancer loops, we mapped its physical interaction with the CACNA1C TSS in human dorsolateral prefrontal cortex (n = 6) and hiPSC derived-neurons by chromosome conformation capture (3C) assay (Mitchell et al., 2013) (Table S8, S9). In addition, this approach allows us to dissect the LD complexity of the region and identify variants with additional support for a functional role. Of the 17 restriction site regions tested, only one displayed an increased interaction with the TSS in human postmortem tissue (Primer #4 in Figure 5A–D). This enhancer region includes rs2159100 (MAF = 0.35, GWAS P value = 1.1 × 10−10) and rs12315711 (MAF=0.35, GWAS P value = 1.1 × 10−10) and demonstrates increased interaction frequency with the CACNA1C promoter (F (16, 83) = 5.52, P = 8 × 10−8). The rs2159100/rs12315711 3C interaction was confirmed in hiPSC derived-neurons (Figure 5D). In addition to being in an enhancer region, rs2159100 also co-localizes with a 600bp DHS region. Thus, we examined its effect on transcriptional activity in in vitro experiments. Compared to the reference rs2159100 C allele, the risk T variant is associated with decreased transcriptional activity in HEK-293 and Neuro-2a cells (42% (P < 0.0001) and 23% (P < 0.05) reduction in luciferase activity, respectively) (Figure 5E).
Figure 5.
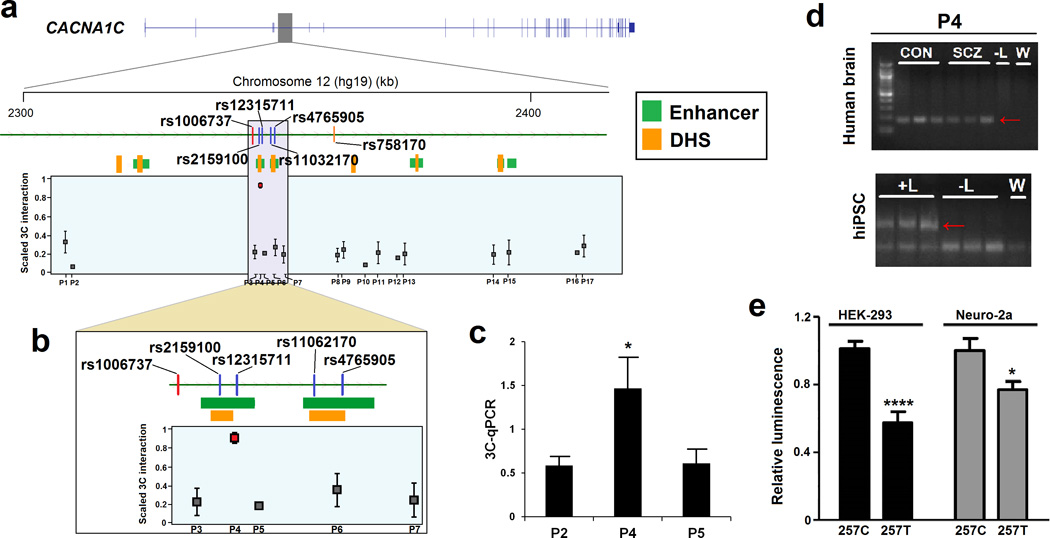
A physical interaction between the CACNA1C promoter and an enhancer region was confirmed by chromosome conformation capture (3C). (a) Part of the genomic region of CACNA1C (chr12:2,303,497–2,419,832) is displayed. The index SNP (red line; rs1006737), eSNP (orange line; rs758170) and tag SNPs (blue line; r2 > 0.8 within 500kb) that lie within CREs (enhancer and DHS) are illustrated. The index SNP and SNPs in LD with them are associated with lower CACNA1C gene expression. 3C–PCR primers were designed at the CACNA1C TSS and multiple regions (primers #1–17). A 3C physical interaction between the CACNA1C TSS and primer #4 was found in 3C libraries made from postmortem brain tissue (n = 6) (F (16, 83) = 5.52, P = 8 × 10-8). (b) More detailed view of the associated region (chr12:2,344,353–2,362,387). The primer #4 includes the rs2159100 creSNP (GWAS P value=1.1 × 10-10), which lies within an active enhancer (green box) and DHS (orange box). (c) 3C–qPCR shows increased interaction for primer #4 in libraries made from postmortem brain tissue (n = 6) (F (2, 15) = 4.54, P = 0.029). (d) Gel images are shown for primer #4 for 3C interactions using prefrontal cortex libraries in controls and SCZ and hiPSC derived-neurons. The red arrow shows the 3C interaction band. All 3C PCR products were sequence verified and the interactions were not present in the no ligase (-L) and water (W) controls. (e) The rs2159100 T risk allele (position 257 in the construct: 257T) affects the relative luciferase activity in HEK-293 and Neuro-2a cells compared to the C allele (257C). Data show the mean and standard error of mean. *P < 0.05, ****P < 0.0001 by unpaired t-test.
DISCUSSION
We have demonstrated that SCZ associated loci are enriched for certain functional annotation categories. As reported previously, we found enrichment with eSNPs (Richards et al., 2012) and CRE annotations highlighting active regulatory regions (Trynka et al., 2013). Here we demonstrate for the first time that integrative analysis of eSNPs with CRE epigenomic annotations identifies greater enrichment of risk loci compared to eSNPs or CREs alone. Given that eSNP (Dimas et al., 2009; Fairfax et al., 2012; Gibbs et al., 2010; Nica et al., 2011) and CRE-mediated epigenetic regulation (Cheung et al., 2010; Heintzman et al., 2009; Maurano et al., 2012) is often specific for tissue and even cell type, our findings add to the growing evidence supporting the importance of studying the human brain tissue for functional genomics analysis in neuropsychiatric illnesses.
The strongest SCZ associated enrichment was observed in fetal and adult brain creSNP annotations for enhancers. Previous studies have established the important regulatory role of the enhancers on transcriptome organization during neurodevelopment and adulthood (Andersson et al., 2014; Visel et al., 2013; Wenger et al., 2013). Overall, these findings suggest that risk allele specific alterations in enhancers could affect the proper spatiotemporal organization of the transcriptome that when combined with other perturbations leads to SCZ.
We have used a step-wise approach for defining functional enrichment and, in so doing, were led to a mechanistic hypothesis (of an interaction between disease signal, expression signal and an enhancer) about the formation of chromosome loops between the enhancer and promoter. We experimentally validated these long-range interactions in human brain tissue and hiPSC derived-neurons for a gene that has been strongly associated with neuropsychiatric disorders. More specifically CACNA1C, a subunit of the L-type calcium channel, is one of the most widely reproduced associations, first identified in bipolar disorder (2011; Ferreira et al., 2008; Sklar et al., 2008), but subsequently also shown to be strongly associated with SCZ (Ripke et al., 2013). The association signal is in the middle of an intron, and does not suggest any immediate functional possibilities. In addition, the associated region is large (154.7 kb) and contains multiple implicated SNPs due to local LD patterns (216 SNPs with P < 10−3). We have now demonstrated in post-mortem human brain, and hiPSC derived-neurons, a specific interaction of a narrow, 1.4 kb region (which is an enhancer) with the proximal gene promoter by chromosome conformation capture. Furthermore, the risk variant is associated with reduced CACNA1C gene expression and transcriptional activity in the eSNP and in vitro experiments, respectively. Another recent study reported that CACNA1C rs1006737 (or rs2159100) is associated with decreased gene expression (Gershon et al., 2013), similar to our findings, although this is not consistently observed (Bigos et al., 2010). Since the initial report of CACNA1C association with bipolar disorder (Ferreira et al., 2008), there has been follow up with studies that report effects on functional connectivity in attention and emotion networks, cognitive performance and personality traits in SCZ and bipolar disorder (Bigos et al., 2010; Erk et al., 2010; Hori et al., 2012; Paulus et al., 2013; Radua et al., 2013; Roussos et al., 2013; Roussos et al., 2011; Tesli et al., 2013).
Two out of the 17 genes identified through the RTC approach are calcium channels subunit genes (CACNA1C and CACNB2). We note that calcium channel subunits have been previously implicated in GWAS of bipolar disorder (Ferreira et al., 2008; Lee et al., 2011; Sklar et al., 2011; Sklar et al., 2008), SCZ (Ripke et al., 2013) and cross-disorder analyses (2013). In addition, an enrichment of rare disruptive variants in calcium channel subunits was reported in a recent exome sequencing study (Purcell et al., 2014). CACNA1C codes for Cav1.2, the most abundant neuronal L-type calcium channel (Obermair et al., 2004). The α1c subunit (CACNA1C) forms the transmembrane pore and directly interacts with the intracellular β2 subunit (CACNB2). The cytosolic β2 subunit has a major role in stabilizing the α1 subunit conformation and delivering it to the cell membrane by its ability to mask an endoplasmic reticulum retention signal in the α1 subunit (Bichet et al., 2000). Calcium-mediated signaling has an important role on neuronal differentiation by regulating axonal growth and guidance and this process is also controlled by glutamatergic signaling (Rosenberg and Spitzer, 2011). It is possible that altered tuning in calcium-mediated signaling triggered by Cav1.2 and glutamatergic neurotransmission (Fromer et al., 2014; Purcell et al., 2014), could lead to alterations in the rate of axon outgrowth and pathfinding, resulting in inefficient neuronal wiring in SCZ.
While our approach can identify functional categories with higher enrichment compared to FUV categories, we still observe a deviation from the null for the FUV category, indicating multiple SCZ-associated loci that were not captured through our current functional annotation categories. Clearly current databases do not yet capture all types of functional variants, including sites of DNA methylation and hydroxymethyaltion (Lister et al., 2013), as well as additional CREs such as insulators (Herold et al., 2012).
Furthermore, there is relatively low genomic resolution of current functional categories and many additional alternative putative mechanisms, such as transcriptional regulation through small and long non-coding RNAs and alternative splicing, likely mediate the effect of some non-coding risk alleles.
In conclusion, these results suggest a tissue-specific regulatory role for many SCZ associated common loci. While this paper was under review, a mega-analyses by the Psychiatric Genomic Consortium (PGC) in SCZ on over 80,000 cases and controls was published (2014). In the new PGC GWAS, 22 out of 24 loci remain genome-wide significant. In addition, 19 out of the 24 SNPs presented in Table 2 were in LD (r2 ≥ 0.3) with a genome-wide significant PGC index SNP. Our analysis provides a framework for future integrative analysis of additional data sets to generate testable hypotheses and derive mechanistic insights for SCZ associated variants.
EXPERIMENTAL PROCEDURES
A brief description of key methods and sample description are provided below, whereas complete details are found in the Extended Experimental Procedures.
GWAS data sets
A large published SCZ GWAS data set (Ripke et al., 2013) in the form of summary statistic P values was obtained from public access website (https://pgc.unc.edu/). A previously published GWAS data set in rheumatoid arthritis (Stahl et al., 2010) was obtained through collaboration with investigators in the form of summary statistic P values. For all the analyses presented here, the major histocompatibility complex (MHC) locus (chr6: 25–35Mb) was excluded from both GWAS datasets.
eSNP data sets
Brain eSNPs were generated using the gene expression and genotyping data of Caucasian samples, included in the Braincloud (Colantuoni et al., 2011) (GEO accession number: GSE30272), NIA/NIH (Gibbs et al., 2010) (GEO accession number: GSE15745) and Harvard Brain Tissue Resource Center (HBTRC) (Zhang, 2013) (GEO accession number: GSE44772) datasets (Table S1). The following non-brain eSNP datasets were downloaded from public access websites: LCL (Xia et al., 2012) (http://www.bios.unc.edu/research/genomic_software/seeQTL/), liver (Innocenti et al., 2011) (http://www.scandb.org/), peripheral blood mononuclear cells (PBMC) (Westra et al., 2013) (http://genenetwork.nl/bloodeqtlbrowser), skin (Grundberg et al., 2012) and adipose tissue (Grundberg et al., 2012) (http://www.muther.ac.uk/Data.html).
cis regulatory element annotations
Multiple CRE annotations were used in the current study (Table S2, S3). ChIP-seq and DHS data generated as part of the ENCODE (Maurano et al., 2012) and REMC (Zhu et al., 2013) projects for human brain/neuron (Table S2), T - helper cells, liver, skin and adipose tissue were downloaded from the NCBI repository (http://www.ncbi.nlm.nih.gov/geo/roadmap/epigenomics/). Additional data for the dorsolateral prefrontal cortex after fluorescence-activated cell sorting (FACS) were generated as described previously (Cheung et al., 2010; Shulha et al., 2013; Shulha et al., 2012) (Table S3).
Definition of functional datasets
All SNP coordinates described here are relative to UCSC hg19. The functional datasets used in the current study were divided into 3 groups: eSNP, CRE and creSNP (eSNP in a cis regulatory element).
eSNP: The brain eSNP dataset was generated by including all significant cis eSNPs derived from each eSNP analyses. For the non-brain eSNPs (LCL, liver, skin, PBMC and adipose tissue), we used the list of cis eSNPs generated as described previously (Grundberg et al., 2012; Innocenti et al., 2011; Westra et al., 2013; Xia et al., 2012) at FDR 10%.
CRE: The ChIP-seq and DHS significant peaks were clustered into subgroups based on assay and origin of tissue (Figure S4). We integrated multiple CRE subgroup annotations for generating functional annotations defining active promoters (overlap of H3K4me3 with H3K9ac or H3K27ac), poised promoters (overlap of H3K4me3 with H3K27me3), active enhancers (overlap of H3K4me3 with H3K9ac or H3K27ac), repressed enhancers (overlap of H3K4me1 with H3K27me3) and open chromatin (DHS).
creSNP: The creSNP functional dataset was defined as the eSNPs that lie within different CRE subcategories.
GWAS Positional Annotation
We used a mixed approach for assigning the GWAS SNPs into functional categories. For the eSNP functional category, we leveraged the eSNP dataset in the densely mapped 1000G to identify the GWAS studied SNP that was tagged, as a result of LD. For generating the CRE or creSNP functional categories we used a positional approach (ignoring the annotation categories of SNPs in LD with the tag SNP). This mixed model allows us to capture all possible SNPs that affect gene expression (tag or SNPs in LD), followed by positional selection of SNPs that lie within putative regulatory DNA regions. GWAS SNP that did not fit in any functional category was assigned as the FUV category. The Table S4 describes the count of SCZ for each functional brain category.
Quantification of Enrichment
Stratified Q-Q plots were generated for assessment of the similarity or differences between the empirical cumulative distribution function of the functional and FUV datasets. In the stratified Q-Q plots, the enrichment of SCZ SNPs for a specific functional category is observed as a horizontal deflection from the FUV category. The quantification of enrichment for each functional category was done using the CES (mean (z2 – 1)), as described previously (Schork et al., 2013). The CES provides a summary score of category-specific enrichment where the mean is taken over all SNP z-scores in the given category. For each functional category, the statistical significance of CES was evaluated by permutation on a combined 10,000 randomized set of CREs and eSNPs.
Regulatory Trait Concordance score
The likelihood of a shared functional effect between a SCZ genome-wide significant SNP and an eSNP was assessed by the RTC approach (Grundberg et al., 2012; Nica et al., 2010). RTC score ranges from 0 to 1, with values ≥ 0.9 indicating the GWAS signal and eSNP tag the same underlying signal, as demonstrated previously (Grundberg et al., 2012; Nica et al., 2010).
Chromosome Conformation Capture
Postmortem prefrontal cortex brain tissue for cases with SCZ and controls was obtained from the University of Maryland and pair-matched for age, sex, PMI, and pH (n = 3/group) (Table S8). Chromosome conformation capture (3C) was performed as described in a recently published protocol applicable to postmortem brain (Mitchell et al., 2014). Physical looping interactions were quantified with PCR. Primers were designed less than 120 bp from the HindIII or NcoI restriction site (Table S9). The PCR products were resolved on a 2% agarose gel and the level of interaction between two primers was measured semiquantitatively using band intensities normalized with the background (raw 3C interaction) with ImageJ (Schneider et al., 2012). For each library (HindIII and NcoI) in the human postmortem brain tissue studies, we transformed the raw 3C interaction to Z scores, followed by scaling (0 to 1) (Scaled 3C interaction). All 3C PCR products were sequence verified and the interactions were not present in the no ligase and water controls. For primers #2, #4 and #5, physical looping interactions were further quantified with qPCR using an ABI Prism 7900 (Applied Biosystems). The reactions were run in triplicate for each sample and DNA PCR product was measured through SYBR Green I (Life Technologies).
Human induced pluripotent stem cells differentiation to neurons
Human induced pluripotent stem cells (hiPSCs) were derived from fibroblasts of a control sample (GM03651) as described previously (Brennand et al., 2011). hiPSC derived-neurons were differentiated for ∼6 weeks in neural differentiation media. The majority of forebrain hiPSC neurons are VGLUT1-positive, and so are presumably glutamatergic, although approximately 30% of neurons are GAD67-positive (GABAergic) (Brennand et al., 2011).
Transient Transfection and Luciferase Assays
We constructed luciferase reporter plasmids by cloning the regulatory sequence containing rs2159100 into the pGL4.24 vector (Promega) upstream of the minP. HEK293 cells or Neuro-2a cells (40%–60% confluent) were transfected with each construct (500 ng) and the Renilla luciferase expression vector pRL-TK (200 ng; Promega). The luciferase activity in the cell lysates was determined using the Dual Luciferase Reporter System (Promega). Firefly luciferase activities were normalized to that of Renilla luciferase and expression relative to the activity of the rs2159100 C allele was noted.
Supplementary Material
Acknowledgments
We thank Iris Cheung and Yin Guo for ChIP-seq assays on sorted brain nuclei. We thank all members of the Rheumatoid Arthritis Consortium for providing access to the GWAS data. Members of Rheumatoid Arthritis Consortium are Steve Eyre, John Bowes, Dorothée Diogo, Annette Lee, Anne Barton, Paul Martin, Alexandra Zhernakova, Eli Stahl, Sebastien Viatte, Kate McAllister, Christopher I Amos, Leonid Padyukov, Rene E M Toes, Tom W J Huizinga, Cisca Wijmenga, Gosia Trynka, Lude Franke, Harm-Jan Westra, Lars Alfredsson, Xinli Hu, Cynthia Sandor, Paul I W de Bakker, Sonia Davila, Chiea Chuen Khor, Khai Koon Heng, Robert Andrews, Sarah Edkins, Sarah E Hunt, Cordelia Langford, Deborah Symmons, Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate, Wellcome Trust Case Control Consortium, Pat Concannon, Suna Onengut-Gumuscu, Stephen S Rich, Panos Deloukas, Miguel A Gonzalez-Gay, Luis Rodriguez-Rodriguez, Lisbeth Ärlsetig, Javier Martin, Solbritt Rantapää-Dahlqvist, Robert M Plenge, Soumya Raychaudhuri, Lars Klareskog, Peter K Gregersen, Jane Worthington, the Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate (BRAGGSS) & the Wellcome Trust Case Control Consortium (WTCCC). We thank Dr. Ron Zielke and staff from the Brain and Tissue Bank for Developmental Disorders, and Dr. Andree Lessard from the Maryland Psychiatric Research Center, University of Maryland, for providing postmortem brain tissue.
We acknowledge funding support from National Institutes of Health (NIH)/National Institute of Mental Health (NIMH) grant R01MH095034 (P.S.), NIMH grant R01MH097276 (P.S. and E.E.S.), NIMH grant U01MH103392 (A.S. and P.S.), NIMH grant P50 MH096890 (S.A.), National Institute of Neurological Disorders and Stroke (NINDS) grant R21 NS076958 (S.A.), NINDS grant R01 NS047229 (N.K.R.), National Institute on Aging (NIA) grant R37 AG017926 (N.K.R.), NIA grant P50 AG005138 (N.K.R. and A.G.), Veterans Affairs Merit grant BX002395 (P.R.), the Brain Behavior Research Foundation (P.R., A.C.M., S.A., E.A.S., K.B.), the American Psychiatric Association-Merck & Co. Early Academic Career Research Award (P.R.), NIMH grant R01 MH101454 (K.B.), the New York Stem Cell Foundation (K.B.), Alzheimer's Association grant IIRG-11–205149 (A.G.), the Friedman Brain Institute at Icahn School of Medicine at Mount Sinai and the Icahn Institute for Genomics and Multiscale Biology at Icahn School of Medicine at Mount Sinai. This work was supported in part through the computational resources and staff expertise provided by the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai. The funders had no role in study design, execution, analysis or manuscript preparation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
P.R., E.E.S., S.A., and P.S. designed and supervised the research. P.R., E.A.S., D.M.R., A.C., M.F., and S.M.P. performed the statistical analysis. A.C.M., J.F.F., V.M.P., J.T. and S.A. performed the chromatin conformation capture experiments. G.V., A.G., and N.K.R. performed the luciferase assay. K.B. generated the hiPSCs. Y.O., K.A.S., J.W., L.P., L.K., P.K.G., R.M.P. and S.R. provided the GWAS in rheumatoid arthritis. P.R., A.S., and P.S. prepared the manuscript. P.R. and A.C.M. contributed equally to this work.
References
- Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichet D, Cornet V, Geib S, Carlier E, Volsen S, Hoshi T, Mori Y, De Waard M. The I-II loop of the Ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron. 2000;25:177–190. doi: 10.1016/s0896-6273(00)80881-8. [DOI] [PubMed] [Google Scholar]
- Bigos KL, Mattay VS, Callicott JH, Straub RE, Vakkalanka R, Kolachana B, Hyde TM, Lipska BK, Kleinman JE, Weinberger DR. Genetic variation in CACNA1C affects brain circuitries related to mental illness. Archives of general psychiatry. 2010;67:939–945. doi: 10.1001/archgenpsychiatry.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung I, Shulha HP, Jiang Y, Matevossian A, Wang J, Weng Z, Akbarian S. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:8824–8829. doi: 10.1073/pnas.1001702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, Colantuoni EA, Elkahloun AG, Herman MM, Weinberger DR, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–523. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482:390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erk S, Meyer-Lindenberg A, Schnell K, Opitz von Boberfeld C, Esslinger C, Kirsch P, Grimm O, Arnold C, Haddad L, Witt SH, et al. Brain function in carriers of a genome-wide supported bipolar disorder variant. Archives of general psychiatry. 2010;67:803–811. doi: 10.1001/archgenpsychiatry.2010.94. [DOI] [PubMed] [Google Scholar]
- Fairfax BP, Makino S, Radhakrishnan J, Plant K, Leslie S, Dilthey A, Ellis P, Langford C, Vannberg FO, Knight JC. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat Genet. 2012;44:502–510. doi: 10.1038/ng.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanucchi S, Shibayama Y, Burd S, Weinberg MS, Mhlanga MM. Chromosomal contact permits transcription between coregulated genes. Cell. 2013;155:606–620. doi: 10.1016/j.cell.2013.09.051. [DOI] [PubMed] [Google Scholar]
- Ferreira MA, O'Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershon ES, Grennan K, Busnello J, Badner JA, Ovsiew F, Memon S, Alliey-Rodriguez N, Cooper J, Romanos B, Liu C. A rare mutation of CACNA1C in a patient with bipolar disorder, and decreased gene expression associated with a bipolar-associated common SNP of CACNA1C in brain. Molecular psychiatry. 2013 doi: 10.1038/mp.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundberg E, Small KS, Hedman AK, Nica AC, Buil A, Keildson S, Bell JT, Yang TP, Meduri E, Barrett A, et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet. 2012;44:1084–1089. doi: 10.1038/ng.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, Ren B, Fu XD, Topol EJ, Rosenfeld MG, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold M, Bartkuhn M, Renkawitz R. CTCF: insights into insulator function during development. Development. 2012;139:1045–1057. doi: 10.1242/dev.065268. [DOI] [PubMed] [Google Scholar]
- Hori H, Yamamoto N, Fujii T, Teraishi T, Sasayama D, Matsuo J, Kawamoto Y, Kinoshita Y, Ota M, Hattori K, et al. Effects of the CACNA1C risk allele on neurocognition in patients with schizophrenia and healthy individuals. Scientific reports. 2012;2:634. doi: 10.1038/srep00634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti F, Cooper GM, Stanaway IB, Gamazon ER, Smith JD, Mirkov S, Ramirez J, Liu W, Lin YS, Moloney C, et al. Identification, replication, and functional fine-mapping of expression quantitative trait loci in primary human liver tissue. PLoS Genet. 2011;7:e1002078. doi: 10.1371/journal.pgen.1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MT, Chen CH, Lee CS, Chen CC, Chong MY, Ouyang WC, Chiu NY, Chuo LJ, Chen CY, Tan HK, et al. Genome-wide association study of bipolar I disorder in the Han Chinese population. Molecular psychiatry. 2011;16:548–556. doi: 10.1038/mp.2010.43. [DOI] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AC, Bharadwaj R, Whittle C, Krueger W, Mirnics K, Hurd Y, Rasmussen T, Akbarian S. The Genome in Three Dimensions: A New Frontier in Human Brain Research. Biological psychiatry. 2013 doi: 10.1016/j.biopsych.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nica AC, Montgomery SB, Dimas AS, Stranger BE, Beazley C, Barroso I, Dermitzakis ET. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet. 2010;6:e1000895. doi: 10.1371/journal.pgen.1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nica AC, Parts L, Glass D, Nisbet J, Barrett A, Sekowska M, Travers M, Potter S, Grundberg E, Small K, et al. The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet. 2011;7:e1002003. doi: 10.1371/journal.pgen.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermair GJ, Szabo Z, Bourinet E, Flucher BE. Differential targeting of the L-type Ca2+ channel alpha 1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. The European journal of neuroscience. 2004;19:2109–2122. doi: 10.1111/j.0953-816X.2004.03272.x. [DOI] [PubMed] [Google Scholar]
- Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus FM, Bedenbender J, Krach S, Pyka M, Krug A, Sommer J, Mette M, Nothen MM, Witt SH, Rietschel M, et al. Association of rs1006737 in CACNA1C with alterations in prefrontal activation and fronto-hippocampal connectivity. Human brain mapping. 2013 doi: 10.1002/hbm.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O'Dushlaine C, Chambert K, Bergen SE, Kahler A, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radua J, Surguladze SA, Marshall N, Walshe M, Bramon E, Collier DA, Prata DP, Murray RM, McDonald C. The impact of CACNA1C allelic variation on effective connectivity during emotional processing in bipolar disorder. Molecular psychiatry. 2013;18:526–527. doi: 10.1038/mp.2012.61. [DOI] [PubMed] [Google Scholar]
- Richards AL, Jones L, Moskvina V, Kirov G, Gejman PV, Levinson DF, Sanders AR, Purcell S, Visscher PM, Craddock N, et al. Schizophrenia susceptibility alleles are enriched for alleles that affect gene expression in adult human brain. Molecular psychiatry. 2012;17:193–201. doi: 10.1038/mp.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, O'Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, Bergen SE, Collins AL, Crowley JJ, Fromer M, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013 doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SS, Spitzer NC. Calcium signaling in neuronal development. Cold Spring Harbor perspectives in biology. 2011;3:a004259. doi: 10.1101/cshperspect.a004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos P, Bitsios P, Giakoumaki SG, McClure MM, Hazlett EA, New AS, Siever LJ. CACNA1C as a risk factor for schizotypal personality disorder and schizotypy in healthy individuals. Psychiatry research. 2013;206:122–123. doi: 10.1016/j.psychres.2012.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos P, Giakoumaki SG, Georgakopoulos A, Robakis NK, Bitsios P. The CACNA1C and ANK3 risk alleles impact on affective personality traits and startle reactivity but not on cognition or gating in healthy males. Bipolar disorders. 2011;13:250–259. doi: 10.1111/j.1399-5618.2011.00924.x. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schork AJ, Thompson WK, Pham P, Torkamani A, Roddey JC, Sullivan PF, Kelsoe JR, O'Donovan MC, Furberg H, Schork NJ, et al. All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet. 2013;9:e1003449. doi: 10.1371/journal.pgen.1003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulha HP, Cheung I, Guo Y, Akbarian S, Weng Z. Coordinated cell type-specific epigenetic remodeling in prefrontal cortex begins before birth and continues into early adulthood. PLoS Genet. 2013;9:e1003433. doi: 10.1371/journal.pgen.1003433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulha HP, Cheung I, Whittle C, Wang J, Virgil D, Lin CL, Guo Y, Lessard A, Akbarian S, Weng Z. Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Archives of general psychiatry. 2012;69:314–324. doi: 10.1001/archgenpsychiatry.2011.151. [DOI] [PubMed] [Google Scholar]
- Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, Edenberg HJ, Nurnberger JI, Jr, Rietschel M, Blackwood D, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklar P, Smoller JW, Fan J, Ferreira MA, Perlis RH, Chambert K, Nimgaonkar VL, McQueen MB, Faraone SV, Kirby A, et al. Whole-genome association study of bipolar disorder. Molecular psychiatry. 2008;13:558–569. doi: 10.1038/sj.mp.4002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FA, Zhernakova A, Hinks A, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesli M, Skatun KC, Ousdal OT, Brown AA, Thoresen C, Agartz I, Melle I, Djurovic S, Jensen J, Andreassen OA. CACNA1C risk variant and amygdala activity in bipolar disorder, schizophrenia and healthy controls. PLoS One. 2013;8:e56970. doi: 10.1371/journal.pone.0056970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trynka G, Sandor C, Han B, Xu H, Stranger BE, Liu XS, Raychaudhuri S. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet. 2013;45:124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Taher L, Girgis H, May D, Golonzhka O, Hoch RV, McKinsey GL, Pattabiraman K, Silberberg SN, Blow MJ, et al. A high-resolution enhancer atlas of the developing telencephalon. Cell. 2013;152:895–908. doi: 10.1016/j.cell.2012.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger AM, Clarke SL, Notwell JH, Chung T, Tuteja G, Guturu H, Schaar BT, Bejerano G. The enhancer landscape during early neocortical development reveals patterns of dense regulation and co-option. PLoS Genet. 2013;9:e1003728. doi: 10.1371/journal.pgen.1003728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia K, Shabalin AA, Huang S, Madar V, Zhou YH, Wang W, Zou F, Sun W, Sullivan PF, Wright FA. seeQTL: a searchable database for human eQTLs. Bioinformatics. 2012;28:451–452. doi: 10.1093/bioinformatics/btr678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang BGG, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Bennett DA, Suver C, Shah H, Mahajan M, Lamb JR, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V. Tracing Multi-System Failure in Alzheimer Disease to Causal Genes. Cell. 2013 doi: 10.1016/j.cell.2013.03.030. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Beaulaurier J, Lum PY, Molony C, Yang X, Macneil DJ, Weingarth DT, Zhang B, Greenawalt D, Dobrin R, et al. Liver and adipose expression associated SNPs are enriched for association to type 2 diabetes. PLoS Genet. 2010;6:e1000932. doi: 10.1371/journal.pgen.1000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, Durham T, Miri M, Deshpande V, De Jager PL, et al. Genome-wide Chromatin State Transitions Associated with Developmental and Environmental Cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.