

Abstract
The ability of science and medicine to control the pathogen Mycobacterium tuberculosis (Mtb) requires an understanding of the complex host environment within which it resides. Pathological and biological evidence overwhelmingly demonstrate how the mammalian steroid cholesterol is present throughout the course of infection. Better understanding Mtb requires a more complete understanding of how it utilizes molecules like cholesterol in this environment to sustain the infection of the host. Cholesterol uptake, catabolism, and broader utilization are important for maintenance of the pathogen in the host and it has been experimentally validated to contribute to virulence and pathogenesis. Cholesterol is catabolized by at least three distinct sub-pathways, two for the ring system and one for the side chain, yielding dozens of steroid intermediates with varying biochemical properties. Our ability to control this worldwide infectious agent requires a greater knowledge of how Mtb uses cholesterol to its advantage throughout the course of infection. Herein, the current state of knowledge of cholesterol metabolism by Mtb is reviewed from a biochemical perspective with a focus on the metabolic genes and pathways responsible for cholesterol steroid catabolism.
Keyterms: catabolism, metabolism, enzyme, pathway, nutrition, persistence
The practical insolubility of cholesterol in water renders the obtaining of an insight into the mechanism of this breakdown process very difficult. In this respect cholesterol cannot be rightly compared with the fats, since a simple hydrolysis is able to convert these latter compounds into more or less water-soluble components. To the contrary the breakdown of cholesterol will ask for a direct action of the desmolytic catalysts of the cells, or in other words this compound must as such be subject to reactions of an oxido-reduction type (Tak, 1942).
J.D. Tak, On Bacteria Decomposing Cholesterol, 1942
One breath is all it takes to inhale and give shelter to the pathogen that has successfully infected more than one third of the global human population—Mycobacterium tuberculosis (Mtb) (2012). Mtb is the causative agent of tuberculosis (TB) disease, historically known as consumption, and is responsible for at least two million deaths each year. Today, anywhere from 5–15% of people infected with Mtb will go on to develop active TB disease, which is highly contagious and often deadly. The average person with active TB disease will spread it to 10–15 people. Half of all people diagnosed with TB in some developing countries will die, usually soon after diagnosis (Dye et al., 1999). Worldwide, TB is the leading killer of people infected with HIV or suffering from AIDS, and this disease disproportionally affects those with compromised immune systems, especially very young children and the elderly.
In the 20th century the hope for a remedy came to fruition for the first time in history when Selman Waksman discovered the aminoglycoside streptomycin and demonstrated its success in treating Mtb infection, and Merck brought it to market in the 1940s (Waksman, 1953). It seemed as if the long-awaited anti-mycobacterial cure for TB had finally arrived. Rates of TB infection, which were already in decline due to improved sanitation, continued to plummet into the 1950s and 1960s, and there was optimism that this disease could finally be cured once and for all. However, Mtb strains resistant to streptomycin were noted very soon after the drug entered the market, in the late1940s. This initial resistance to the first TB drug forebode the current situation more than half a century later.
In order to effectively cure TB today, treatments almost always include the use of multiple antibiotics taken simultaneously. During the initial phase of treatment, lasting two months, patients take a combination of two first-line drugs, rifampicin and isoniazid, and typically additional antibiotics like pyrazinamide and ethambutol. The continuation phase of treatment lasts for an extra four to seven months and includes second-line antibiotics, depending on the severity of disease progression. Despite combination drug therapy for a prolonged period of time, the emergence of drug resistance is increasingly on the rise (Reichman and Tanne, 2002). A major factor contributing to the global problem of drug-resistant TB is the patient's failure to complete a full antibiotic cycle.
First-line drugs isonizazid, rifampicin, and ethambutol are generally only bactericidal in patients with clinically active TB where Mtb is actively dividing. Isoniazid is a prodrug activated through ligation with NADH by the catalase-peroxidase KatG (Rv1908c). The active drug binds enoyl-acyl carrier protein reductase InhA (Rv1484), effectually inhibiting fatty acid biosynthesis. Rifampicin inhibits the function of the mycobacterial RNA polymerase through binding to the β-subunit of the enzyme, RpoB (Rv0667). Pyrazinamide is activated by the pyrazinamidase PncA (Rv2043c) to pyrazinoic acid, whose action is effective for both replicating and non-replicating Mtb. Ethambutol also inhibits cell wall biosynthesis by interfering with the arabinogalactan synthesis enzyme arabinosyltransferase, EmbB (Rv3795). Mutations in prodrug-activating enzymes or the drug target account for much of the resistance to anti-mycobacterial drugs worldwide.
In 2012, almost half a million people developed multi-drug resistant TB (MDR-TB), which is more deadly, more costly, and more difficult to cure than drug susceptible TB. An estimated 9.6% of those with MDR-TB have extensively drug resistant TB (XDR-TB), which responds to even fewer antibiotics than MDR-TB. Recently, clinical strains of TB that are totally drug resistant (TDR-TB) have been isolated in India (Udwadia et al., 2012). Drug resistance has resulted in a modern day epidemic of disease whose intricate and elusive biology is as rich and complex as its extensive history. It is clear that science and medicine need to develop new drugs with novel targets to combat the rise in drug resistant strains of Mtb.
The remarkably slow pace of Mtb replication in the host cells is emblematic of the advantage that Mtb has to adapt to the nutrient-deprived environment of the macrophage. Although the link is not direct, the prolonged treatment with several different antibiotics necessary for effective treatment of Mtb is in part due to this non-replicating/slow growing state in vivo. Current anti-tuberculosis drugs target bacterial machinery that is utilized during cell replication. During the chronic phase of infection, Mtb doubles only once every several days.
Genes that are up-regulated during the chronic phase of infection and their corresponding proteins offer a unique avenue for drug design that would allow treatment of latent TB infections. Recently, mycobacterial-specific inhibitors of the Mtb proteasome, oxathiazol-2-one compounds, have been identified that kill non-replicating Mtb (Lin et al., 2009). These compounds act similarly to human proteasome drugs by acting as suicide-substrate inhibitors via cyclocarbonylation of the proteasome active site threonine. The nitroimidazopyran drug PA-824 currently in Phase II clinical trials shows promising anti-mycobacterial activity against this non-replicating population of bacteria (Stover et al., 2000). Finally, the diarylquinoline Bedaquiline (Sirturo), the first new TB drug approved in 40 years and marketed by Janssen Pharmaceuticals, targets ATP synthase, and is approved specifically for the treatment of MDR-TB (Villemagne et al., 2012). Understanding the environment in which Mtb sustains infection and the biological machinery necessary for the bacterium's survival is requisite for the rational development of new drugs with novel mechanisms of action targeting chronic infection. Mounting evidence suggests that cholesterol metabolism gene products are promising targets for further investigation, and these targets will be discussed in this review.
Pulmonary TB disease is caused by an Mtb infection of the respiratory system, where this pathogen resides in host alveolar macrophages. These tissue bound cells are involved in both the acute and chronic immune response intended to stifle foreign pathogens through various bactericidal mechanisms. Mtb has evolved the ability, through millennia of co-evolution with humans, to not only thwart this powerful immune response, but to also use it to its advantage. The immune system is directed by Mtb to form the granuloma, the clinical hallmark of TB infection, which is a chronic granulomatous inflammatory lesion composed of lymphocytes, macrophages, and multinucleated giant cells (Russell et al., 2009). Here, Mtb can reside for decades in a so-called latent state until the host immune system becomes compromised, often by HIV, old age, or poor nutrition. The infection can then progress to active disease where the caseous necrosis within the granulomatous lesion liquefies, extracellular bacteria are released, and the infection is rapidly spread from person to person (Scanga et al., 1999, Robertson, 1933).
The current paradigm asserts that during the latent phase of infection, Mtb is in a metabolically dormant and non-replicating state within the granuloma, sequestered away by the immune system. This is based on the clinical observation that although a third of the world population is latently infected with TB, just under ten million each year will go on to develop active TB disease (Dye et al., 1999). Recent evidence, however, contradicts the classical school of thought, and this comatose metabolism might not be as real as was once thought. Convergent evidence suggests that an assortment of metabolic genes are in fact significantly up-regulated throughout the course of latent infection and disease. For example, in an in vitro human granuloma model of non-replicating Mtb, the metabolic isocitrate lyase (icl) genes were up-regulated (Peyron et al., 2008). Isocitrate lyase is an essential enzyme in the glyoxylate shunt cycle, which is one of two pathways that can be used for the metabolism of fatty acids, the second pathway being β-oxidation.
The intracellular phagocyotic environment of Mtb is likely limited in energy resources since these immune cells are adept at killing foreign pathogens. However, Mtb infection of macrophages alters the intracellular environment of the macrophage, causing dysregulation of host lipid biosynthesis, uptake, and sequestration. A disproportionately high number of host lipid metabolism genes (compared to other metabolic genes) are up-regulated in caseous TB granulomas from patients with TB disease (Kim et al., 2010). Global expression profiling experiments in Mtb have shown that many hypothetical lipid-metabolizing genes are up-regulated during infection of both macrophages and mice (Camacho et al., 1999, Pandey, 2008, Fontan et al., 2008a).
Infected human pulmonary TB granulomas have an increased abundance of lipids compared to uninfected lung tissue. The abundance of lipid and cholesterol molecules within the granuloma is reflected in the accumulation of foamy macrophages resulting from excess lipid uptake by these cells through an imbalance between the export of low-density lipoprotein (LDL) through macrophage associated efflux pumps like ABCA-1 and the excess uptake of LDL through scavenger receptor A (SRA) and CD36, in addition to other mechanisms like pinocytosis (Russell et al., 2009). Mass spectral analysis of the lipid rich granuloma identified LDL particles composed of triacylglycerides, phospholipids, cholesterol, and cholesterol esters (Kim et al., 2010). This composition is very convenient for Mtb, since Mycobacteria and some of their bacterial relatives like Proteobacteria have an unusual ability to utilize steroids like the mammalian molecule cholesterol as a carbon and energy source.
In a laboratory setting, some Mycobacteria can grow on cholesterol as a sole carbon source. Cholesterol is obtained from the host via an ABC-like ATP-dependent cholesterol import system encoded by the mce4 locus—the bacteria are unable to synthesize this molecule on their own (Mohn et al., 2008). It has been shown in vivo that cholesterol is necessary for persistence of Mtb during the latent stage of infection (Pandey, 2008, Nesbitt et al., 2010). Some evidence even points to the idea that high levels of cholesterol in the host's diet can have an adverse effect on the ability of the host immune system to respond to infection (Han, 2009, Schafer et al., 2009).
Gene expression studies performed in vivo highlight that Mtb relies on lipids, including steroids, as sources of carbon and energy, and that lipid metabolism is crucial for virulence. The mounting lines of evidence suggest that cholesterol metabolism offers this pathogen a unique evolutionary advantage for deriving both energy and other valuable steroid intermediates. Here, we review what is known about the steroid metabolic pathway in Mtb to provide a comprehensive perspective. Sterol metabolism in bacteria has been studied for just over a century, and we highlight major findings in Actinobacteria that have contributed to the elucidation of the pathway in Mtb. Since these topics have been reviewed elsewhere (Donova, 2007, García et al., 2011), we focus primarily on new discoveries from the last decade. A more complete understanding of Mtb cholesterol metabolism is highly relevant for finding new approaches to treating TB. Thus we focus on the biochemistry that has been elucidated to date, and make note of areas in which there are gaps in our current understanding.
Early Actinobacterial Steroid Metabolism Studies
Actinobacteria are generally considered Gram-positive bacteria, and they can inhabit a wide range of environments. A large number of them are soil bacteria, but they can also live in water or as pathogens within a host organism. Most Actinobaceria have high G + C content in their genomes (>65%). However, some fresh water Actinobacteria can have low G + C content as well (<45%) (Ghai et al., 2012). Some Actinobacteria play an important role in the carbon cycle because of their ability to decompose a variety of complex organic compounds including many toxic byproducts of modern industry (Kobayashi and Rittmann, 1982). It is well established that Actinobacteria can metabolize sterols including cholesterol, which contains the familiar tetracycloalkane (gonane) steroid nucleus with an eight-carbon methyl branched side chain (Figure 1). Species of bacteria found throughout nature including Arthrobacter, Mycobacteria, Nocardia, Rhodococcus, Gordonia, and Streptomyces can all utilize cholesterol as a single carbon source for nutrition. Animals, plants, and fungi all biosynthesize and use steroids for various purposes, and although it has been reported that several bacteria might be able to biosynthesize steroids de novo, these results are few and controversial (Bode et al., 2003).
Figure 1.
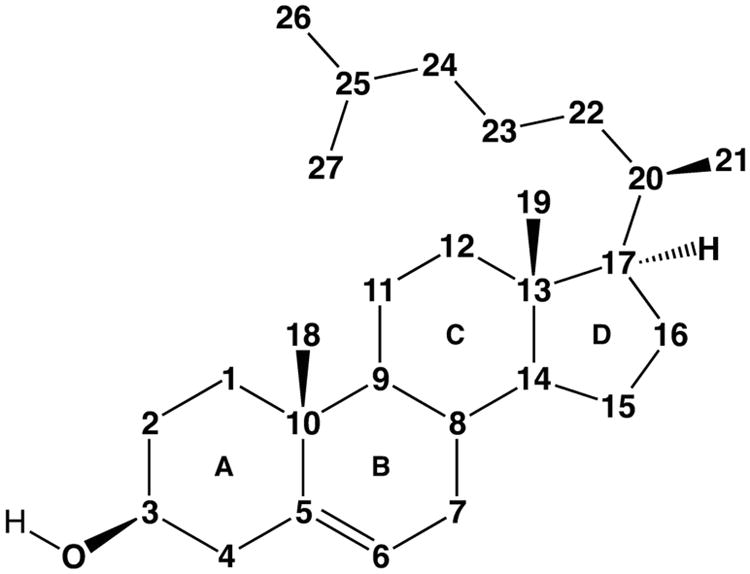
Cholesterol numbering scheme.
Investigation into the degradation of hydrocarbon substrates by microorganisms (mostly fungi, yeast, and bacteria) began in the late 19th century, and has been reviewed (Bushnell and Haas, 1941). The aerobic degradation of alkane substrates specifically by Mycobacteria was described in Germany for the first time just over a century ago (Söhngen, 1913). Both the steroid core and side chain portions of the molecule were found to be catabolized for energy through oxidation of the steroid framework and sidechain. Turfitt demonstrated that carbon C4 of the steroid core (Figure 1) was oxidized and the A-ring of cholesterol was opened in the process, yielding a product deemed Windaus' keto acid with loss of C4 and iso-caproic acid. Even though blocking oxidation of the 3-hydroxyl position with cholesterol acetate prevents oxidative cleavage of the A-ring, iso-caproic acid (C6H12O2) was still observed and presumed to be from side chain catabolism (Turfitt, 1947). Subsequent studies isolated the cholesterol dehydrogenase responsible for oxidation of C4 and determined that C4 and C26 were metabolized to CO2, although the metabolism of C26 was slower than C4 (Stadtman et al., 1954).
Bacterial steroid metabolism research was of particular interest during the late 1940s and 1950s due to the pharmaceutical usefulness of ring-intact sterols with partially metabolized side chains (Malaviya and Gomes, 2008, Van Der Geize and Dijkhuizen, 2004). Androstenedione (AD) and other 17-keto steroids are key starting materials for the preparation of clinically useful steroids such as testosterone, estradiol, progesterone, cortisone, and cortisol (Hogg, 1992). This early work focused on the identification of small molecule metabolic intermediates in order to understand bacterial degradation pathways so that they could be exploited to obtain pharmaceutically valuable compounds. Of particular interest was the controlled enzymatic oxidation of various positions of the steroid framework. Chemical assays were limited to organic extraction of intermediates from bacterial cultures, and it was not until newer methods were developed for identifying intermediates that their structures were confirmed. Efforts focused on microbial fermentation methods to better optimize growth and yield. However, the enzymes that actually catalyzed the chemistry were for the most part not isolated since interest focused on large-scale fermatnation processes for drug development.
Hydroxylated steroids were by far some of the most valuable intermediates isolated during the late 1940s, since these were ultimately used as precursors for valuable steroid molecules and are often unavailable from nature as starting materials. H.C. Murray and D. H. Peterson of The Upjohn Company, who were a bacteriologist and an endocrine biologist, respectively, discovered and patented the microbial conversion of progesterone to C11-hydroxyprogesterone (Peterson, 1952). This coveted process was utilized to synthesize a number of steroids and steroid analogs, most notably the adrenocortical hormone hydrocortisone (Figure 2) (Hogg, 1992). Thus, early studies were focused on isolating ring-intact sterol metabolites with modified side-chains.
Figure 2.
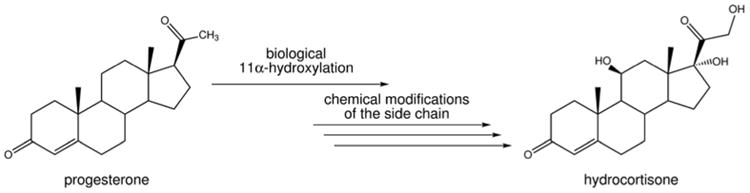
The commerically developed biological/chemical conversion of progesterone to hydrocortisone, pioneered by The Upjohn Company. The biological 11α-hydroxylation of progesterone was carried out by the fungus Rhizopus nigricans, followed by several chemical steps to ultimately give hydrocortisone.
Elucidation of steroid catabolic pathways in bacteria
Nocardia was grown on cholesterol radiolabeled at C4 and C26 to help identify partially metabolized cholesterol intermediates (Sih et al., 1968a, Sih et al., 1968b). These studies demonstrated that cholesterol side chain metabolism to C17 keto-sterols proceeds through intermediates in which the side chain has been cleaved at C24 followed by C22 (Figure 1). Propionyl-CoA is lost from the side chain during the formation of the C24 intermediate, and acetyl-CoA is lost during the formation of the C22 intermediate. Identification of these partially degraded intermediates suggested that the side chain is metabolized via conventional fatty acid β-oxidation, with carbon-carbon cleavage reactions occurring at C24-C25 and C22-C23.
In bacteria, a final loss of C20-C21-C22 as propionyl-CoA results in a C17 keto steroid. Sih et al. proposed dehydrogenation, hydration, and aldolytic fission reactions based on studies performed with Nocardia restrictus (ATCC14887). These early studies proposed an atypical oxidation sequence that ends with the β-hydroxythioester, 3-oxo-17-hydroxy-pregna-4-ene-22-oyl-CoA undergoing a retro-aldol reaction because oxidation of the 17-hydroxyl to a ketone to form a β-ketoester is not possible at C17 (Figure 1).
The net yield of side-chain catabolism is two molecules of propionate (propionyl-CoA) and one molecule of acetate (acetyl-CoA) (Sih et al., 1968a). In contrast, human fatty acyl-CoA thioesters that are branched at the β position, like phytanic acid, undergo α-oxidation in peroxisomes with the loss of a single carbon and the resultant chain is fed into a more typical β-oxidation pathway (Wanders et al., 2011).
A Nature report in the early 1950's described a Gram-negative bacterium able to use testosterone (which lacks a side chain) as a sole source of carbon. The amount of testosterone added to these cultures dictated the total oxygen consumption of the organism, with 40 – 60% of the theoretical consumption going to carbon dioxide or water (Talalay et al., 1952). Paul Talalay isolated Pseudomonas testosteroni (renamed Comamonas testosteroni) and identified its ability to grow on testosterone as a sole carbon source (Talalay et al., 1952). The C. testosteroni Δ5-3-ketosteroid isomerase that converts Δ5-3-ketosteroids to their corresponding Δ4-3-ketosteroid enone products was isolated. By the early 1960s, a complete mechanism for this enzymatic transformation was elucidated using spectroscopy, as well as potential steroid inhibitors of this enzyme identified (Wang et al., 1963).
Likewise tracking the metabolic outcomes for some carbons and trying to harmonize these results with what was known about enzymes involved in degradative pathways was studied in Nocardia. It was found that radiolabeled C4 in the A ring of the steroid framework (cholesterol or cholestenone) was converted to CO2 about four times as rapidly as C26 located on the steroid side chain, indicating different metabolic fates for these two carbons (Stadtman et al., 1954).
A tendency for some bacteria to preferentially utilize one carbon source over another, known as carbon catabolite repression, was observed as early as the 1950s. C. testosteroni grown on testosterone as a sole carbon source was found to be much more readily cultured on acetate than on glucose when the source of carbon was switched, indicating that the enzymes involved in fatty acid metabolism differ from those of sugar metabolism (Santer et al., 1952).
Taken together, the results from the experimental work performed during the first half of the 20th century helped to shape our understanding of how microorganisms like bacteria, and specifically Mycobacteria, are able to adapt to their environments.
Cholesterol metabolism gene annotation in Mtb
Recently, interest in identifying the enzymes involved in cholesterol metabolic pathways has been revived in part due to compounding evidence suggesting cholesterol metabolism by Mtb is important for pathogenesis. Much of what is known about sterol ring metabolism in Mtb was discovered in part based on similarities to better studied steroid-transforming bacteria like C. testosteroni, Rhodococcus sp., and other steroid metabolizing Actinobacteria and Proteobacteria described above. Species of Rhodococcus are of great interest, and therefore, well studied, because of their potential as useful bioremediation agents and the ease of their study. However, many of the strains used are soil bacteria and have prodigious gene redundancy to adapt to many toxic environments. Moreover, most of these strains are not pathogenic. The Mtb genome sequence reported in 1998 opened the door to a better understanding of cholesterol metabolism in the pathogen itself, since preliminary assignments of function to genes could be undertaken (Cole et al., 1998).
Upon sequencing of the Mtb genome, the genes were computationally annotated with proposed biochemical functions (Cole et al., 1998). However, these annotations relied on the limited information in public databases at the time, were missing useful functional assignments, and in addition, contained many incorrect assignments (Schnoes et al., 2009). The Mtb genome contains around four thousand genes and the functions of about 45% were predicted based on similarity to known genes or proteins. Nevertheless, a large portion of the genome was not annotated, and 16% had no similarity to known proteins (Camus et al., 2002, Cole et al., 1998). Furthermore, many of the annotations that were initially ascribed based on sequence similarity were wrong due to an initial incorrect gene assignment that was propagated through early databases, a phenomenon known as genome rot (Schnoes et al., 2009). The genome of Mtb was annotated to contain approximately 250 genes involved in lipid and fatty acid metabolism (Cole et al., 1998). This large number greatly complicated any meaningful assignment of genes that are involved in cholesterol metabolism, and identification of what role they would play in the various cholesterol metabolic pathways in Mtb.
The cholesterol transcriptome
Comparison of transcriptional profiles of Mtb cultured with or without cholesterol identified over 200 genes that are regulated by cholesterol (Nesbitt et al., 2010). Many, but not all of these genes are a subset of the 250 lipid-metabolism genes identified through annotation. At least 52 cholesterol-regulated genes are within an 83-gene region referred to as the “Cho-region” of the Mtb genome (Table) (Nesbitt et al., 2010). The genes in the Cho-region encode primarily homologs of β-oxidation and biphenyl degradation genes from other organisms. A similar set of genes was found to be up-regulated in Rhodococcus RHA1 as well, demonstrating a degree of conservation in this pathway between Actinobacteria (Van Der Geize et al., 2007). Phenotypic profiling has identified 96 genes important for growth on cholesterol (Griffin et al., 2011). This subset includes most of the annotated genes in the Cho-region predicted to degrade the side chain and catabolize the sterol rings, as well as genes in other regions of the genome. Most importantly for our understanding of Mtb pathogenesis, a large portion of these genes overlap with those that are up-regulated in a variety of in vivo models of infection.
Table.
Mtb genes associated with cholesterol metabolism or its regulons.a
| Mtb H37Rv Gene Refb | M. smegmatis ortholog gene number | Gene Name | Enzyme Function | Function Biochemically Validated (☑) Biochemically Predicted (□) Computation Annotated (?) |
Gene Regulation and Essentiality | ||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Mtb Transcript Up-Regulated by Cholc | Gene Required for Growth on Chold | Transcript Up-Regulatede,f or Required for Growthg In Macroϕ | Requiredfor Survival in Miceh | Transcript Repressori | |||||
| Rv0138 | MSMEG_6475 | conserved hypothetical protein | ? | KstR1 | |||||
| Rv0139 | MSMEG_6474 | oxidoreductase | ? | KstR1 | |||||
| Rv0162c | MSMEG_0217 | adhE1 | alcohol dehydrogenase | ? | KstR1 | ||||
| Rv0223c | MSMEG_0309 | Aldehyde dehydrogenase | ? | ✓ | KstR1 | ||||
| Rv0244c | MSMEG_0406 | fadE5 | acyl-CoA dehydrogenase | ? | ✓ | ✓ | Induced | ||
| Rv0468 | MSMEG_0912 | fadB2 | 3-hydroxyacyl-CoA dehydrogenase | ☑ | ✓ | Induced | |||
| Rv0551c | MSMEG_1098 | fadD8 | fatty acyl-CoA synthetase | ? | ✓ | KstR1 | |||
| Rv0687 | MSMEG_1410 | ? | ✓ | Essential | KstR1 | ||||
| Rv0926c | MSMEG_5586 | conserved hypothetical protein | ? | KstR1 | |||||
| Rv0927c | MSMEG_5584 | short-chain type dehydrogenase/reductase | ? | KstR1 | |||||
| Rv0940c | MSMEG_5555 | oxidoreductase | ? | ✓ | KstR1 | ||||
| Rv0953c | MSMEG_5520 | oxidoreductase | ? | ✓ | KstR1 | ||||
| Rv1059 | MSMEG_5286 | dapB | dihydrodipicolinate reductase | ? | KstR1 | ||||
| Rv1071c | MSMEG_5276 | echA9 | enoyl-CoA hydratase | ? | ✓ | Induced | |||
| Rv1106c | MSMEG_5228 | 3β-hsd | 3β-hydroxy steroid dehydrogenase | ☑ | KstR1 (only in M. smeg) | ||||
| Rv1132 | MSMEG_5202 | conserved membrane protein | ? | Induced | KstR1 | ||||
| Rv1193 | MSMEG_5114 | fadD36 | fatty acyl-CoA synthetase | ? | ✓ | ||||
| Rv1323 | MSMEG_4920 | fadA4 | thiolase | ? | Essential | Essential | |||
| Rv1426c | MSMEG_0302 | lipO | esterase | ? | KstR1 | ||||
| Rv1427c | MSMEG_0304 | fadD12 | fatty acyl-CoA synthetase | ? | KstR1 | ||||
| Rv1428c | MSMEG_0305 | conserved hypothetical protein | ? | KstR1 | |||||
| Rv1627c | MSMEG_3844 | non-specific lipid transfer protein | ? | ✓ | KstR1 | ||||
| Rv1628c | MSMEG_3843 | conserved hypothetical protein | ? | ✓ | Induced | KstR1 | |||
| Rv1715 | MSMEG_6791 | fadB3 | 3-hydroxyacyl-CoA dehydrogenase | ? | Induced | ||||
| Rv1894c | MSMEG_3519 | conserved hypothetical protein | ? | ✓ | Induced | KstR1 | |||
| Rv1912c | - | fadB5 | 3-hydroxyacyl-CoA dehydrogenase | ? | Induced | ||||
| Rv1930c | MSMEG_1107 | conserved hypothetical protein | ? | Essential | Mce3R | ||||
| Rv1931c | MSMEG_1106 | transcriptional regulator | ? | Essential | Mce3R | ||||
| Rv1933c | - | fadE18 | steroid acyl-CoA dehydrogenase β subunit | □ | Mce3R | ||||
| Rv1934c | - | fadE17 | steroid acyl-CoA dehydrogenase α subunit | □ | ✓ | Induced | Mce3R | ||
| Rv1935c | - | echA13 | enoyl-CoA hydratase | ? | ✓ | Induced | Mce3R | ||
| Rv1936 | - | monooxygenase | ? | ✓ | Induced | Mce3R | |||
| Rv1937 | - | electron transfer oxygenase | ? | ✓ | Mce3R | ||||
| Rv1938 | - | ephB | epoxide hydrolase | ? | ✓ | Mce3R | |||
| Rv1939 | - | FMN oxidoreductase | ? | ✓ | Induced | Essential | Mce3R | ||
| Rv1940 | - | ribA1 | bifunctional enzyme: GTP cyclohydrolase II and | ? | ✓ | Mce3R | |||
| Rv1941 | - | short-chain dehydrogenase | ? | ✓ | Mce3R | ||||
| Rv1942c | - | conserved hypothetical protein, | ? | Induced | Mce3R | ||||
| Rv1943c | - | conserved hypothetical protein, | ? | Mce3R | |||||
| Rv1963c | - | mce3R | transcriptional repressor (TetR-like) | ☑ | ✓ | ||||
| Rv1964 | - | yrbE3A | Mce-family | ? | ✓ | Mce3R | |||
| Rv1965 | MSMEG_0345 | yrbE3B | Mce-family | ? | ✓ | Mce3R | |||
| Rv1966 | MSMEG_0346 | mce3A | Mce-family | ? | ✓ | Mce3R | |||
| Rv1967 | MSMEG_0347 | mce3B | Mce-family | ? | Mce3R | ||||
| Rv1970 | MSMEG_0350 | lprM | Mce-family lipoprotein LprM (Mce3E) | ? | Induced | Mce3R | |||
| Rv1971 | MSMEG_0351 | mce3F | Mce-family | ? | Mce3R | ||||
| Rv1972 | MSMEG_0352, MSMEG_5208 | Mce-associated | ? | Induced | Mce3R | ||||
| Rv1973 | MSMEG_0353 | Mce-associated | ? | Induced | Mce3R | ||||
| Rv1975 | MSMEG_0355 | protein expressed in hypoxia | ? | Induced | Mce3R | ||||
| Rv2668 | MSMEG_2790 | conserved hypothetical protein | ? | ✓ | ✓ | KstR1 | |||
| Rv2669 | MSMEG_2789 | conserved hypothetical protein | ? | KstR1 | |||||
| Rv2799 | MSMEG_2645 | membrane protein | ? | ✓ | KstR1 | ||||
| Rv2800 | MSMEG_2644 | conserved hypothetical protein | ? | KstR1 | |||||
| Rv3139 | MSMEG_2081 | fadE24 | steroid acyl-CoA dehydrogenase β subunit | □ | ✓ | Induced | sigE | ||
| Rv3140 | MSMEG_2080 | fadE23 | steroid acyl-CoA dehydrogenase α subunit | □ | ✓ | Induced | sigE | ||
| Rv3141 | MSMEG_2033 | fadB4 | 3-hydroxyacyl-CoA dehydrogenase | ? | |||||
| Rv3492c | MSMEG_5893 | conserved hypothetical protein | ? | ✓ | KstR1 | ||||
| Rv3493c | MSMEG_5894 | conserved hypothetical protein | ? | ✓ | KstR1 | ||||
| Rv3494c | MSMEG_5895 | mce4F | mce4 operon: lipid transfer | ☑ | ✓ | KstR1 | |||
| Rv3495c | MSMEG_5896 | mce4E | mce4 operon: lipid transfer | ☑ | ✓ | Essential | KstR1 | ||
| Rv3496c | MSMEG_5897 | mce4D | mce4 operon: lipid transfer | ☑ | ✓ | KstR1 | |||
| Rv3497c | MSMEG_5898 | mce4C | mce4 operon: lipid transfer | ☑ | ✓ | Essential | KstR1 | ||
| Rv3498c | MSMEG_5899 | mce4B | mce4 operon: lipid transfer | ☑ | ✓ | KstR1 | |||
| Rv3499c | MSMEG_5900 | mce4A | mce4 operon: lipid transfer | ☑ | ✓ | Essential | KstR1 | ||
| Rv3500c | MSMEG_5901 | supBh | mce4 operon: lipid transfer | ☑ | ✓ | KstR1 | |||
| Rv3501c | MSMEG_5902 | supAh | mce4 operon: lipid transfer | ☑ | ✓ | Essential | KstR1 | ||
| Rv3502c | MSMEG_5903 | hsd4A | steroid β-hydroxy-acyl-CoA dehydrogenase | □ | ✓ | ✓ | Essential | KstR1 | |
| Rv3503c | MSMEG_5904 | fdxD | ferredoxin | ? | ✓ | Induced | KstR1 | ||
| Rv3504 | MSMEG_5906 | fadE26 | 3-keto-4-cholene-24-oyl-CoA dehydrogenase α subunit | ☑ | ✓ | Essential | KstR1 | ||
| Rv3505 | MSMEG_5907 | fadE27 | 3-keto-4-cholene-24-oyl-CoA dehydrogenase β subunit | ☑ | ✓ | Induced | KstR1 | ||
| Rv3506 | MSMEG_5908 | fadD17 | 3-keto-chol-4-ene-24-oyl-CoA ligase | ☑ | KstR1 | ||||
| Rv3507 | - | PE_PGRS53 | PE family of glycine rich proteins | ? | |||||
| Rv3508 | MSMEG_2146 | PE_PGRS54 | PE family of glycine rich proteins | ? | |||||
| Rv3509c | MSMEG_4348 | ilvX | acetohydroxyacid synthase | ? | |||||
| Rv3510c | - | Rv3510c | conserved hypothetical protein | ? | |||||
| Rv3511 | - | PE_PGRS55 | PE family of glycine rich proteins | ? | |||||
| Rv3512 | - | PE_PGRS56 | PE family of glycine rich proteins | ? | |||||
| Rv3513c | - | fadD18 | acyl-CoA ligase (fragment) | ? | |||||
| Rv3514 | - | PE_PGRS57 | PE family of glycine rich proteins | ? | |||||
| Rv3515c | MSMEG_5914 | fadD19 | 3-keto-4-cholest-4-ene-26-oyl-CoA ligase | ☑ | ✓ | ✓ | KstR1 | ||
| Rv3516 | MSMEG_5915 | echA19 | enoyl-CoA hydratase | ? | ✓ | Induced | KstR1 | ||
| Rv3517 | - | conserved hypothetical protein | ? | ||||||
| Rv3518c | MSMEG_5918 | cyp142 | cholest-4-en-3-one 26-monooxygenase | ☑ | ✓ | Induced | KstR1 | ||
| Rv3519 | MSMEG_5919 | conserved hypothetical protein | ? | Essential | KstR1 | ||||
| Rv3520c | MSMEG_5920 | coenzyme F420-dependent oxidoreductase | □ | ✓ | KstR1 | ||||
| Rv3521 | MSMEG_5921 | conserved hypothetical protein | ? | ✓ | Induced | KstR1 | |||
| Rv3522 | MSMEG_5922 | ltp4 | 3-hydroxy-acyl-CoA aldolase | ? | ✓ | KstR1 | |||
| Rv3523 | MSMEG_5923 | ltp3 | 3-hydroxy-acyl-CoA aldolase | □ | ✓ | Essential | KstR1 | ||
| Rv3524 | - | conserved membrane protein | ? | Induced | |||||
| Rv3525c | - | siderophore binding protein | ? | ||||||
| Rv3526 | MSMEG_5925 | kshA | 3-ketosteroid 9α-hydroxylase oxygenase component | ☑ | ✓ | ✓ | KstR1 | ||
| Rv3527 | MSMEG_5927 | conserved hypothetical protein | ? | ✓ | KstR1 | ||||
| Rv3528c | - | hypothetical protein | ? | ||||||
| Rv3529c | MSMEG_5930 | conserved hypothetical protein | ? | ✓ | KstR1 | ||||
| Rv3530c | MSMEG_5931 | oxidoreductase | ? | ✓ | KstR1 | ||||
| Rv3531c | MSMEG_5932 | conserved hypothetical protein | ? | ✓ | ✓ | KstR1 | |||
| Rv3532 | - | PPE61 | PPE family of glycine rich proteins | ? | |||||
| Rv3533c | - | PPE62 | PPE family of glycine rich proteins | ? | |||||
| Rv3534c | MSMEG_5937 | hsaF | 4-hydroxy-2-oxohexanoate aldolase | □ | ✓ | ✓ | Induced | Essential | KstR1 |
| Rv3535c | MSMEG_5939 | hsaG | propanal dehydrogenase | □ | ✓ | Induced | KstR1 | ||
| Rv3536c | MSMEG_5940 | hsaE | 2-hydroxyhexa-2,4-dienoate hydratase | □ | ✓ | ✓ | Induced | KstR1 | |
| Rv3537 | MSMEG_5941 | kstD | 3-keto-A4-steroid A1-dehydrogenase | ☑ | ✓ | ✓ | Induced, Essential | KstR1 | |
| Rv3538 | MSMEG_5943 | hsd4B | steroid 2-enoyl-CoA hydratase | □ | ✓ | Induced | KstR1 | ||
| Rv3539 | - | PPE63 | PE family of glycine rich proteins | ? | |||||
| Rv3540c | MSMEG_5990 | ltp2 | 3-oxo-17-hydroxy-pregna-4-ene-22-oyl-CoA aldolase | □ | ✓ | ✓ | Induced | Essential | KstR1 |
| Rv3541c | MSMEG_5991 | 3-oxo-pregna-4,17-diene-22-oyl-CoA hydratase α subunit | □ | ✓ | ✓ | Induced, Essential | Essential | KstR1 | |
| Rv3542c | MSMEG_5992 | 3-oxo-pregna-4,17-diene-22-oyl-CoA hydratase β subunit | □ | ✓ | ✓ | Induced, Essential | Essential | KstR1 | |
| Rv3543c | MSMEG_5993 | chsE1 (fadE28) | 3-oxo-pregn-4-ene-22-oyl-CoA dehydrogenase β subunit | ☑ | ✓ | ✓ | Essential | KstR1 | |
| Rv3544c | MSMEG_5994 | chsE2 (fadE29) | 3-oxo-pregn-4-ene-22-oyl-CoA dehydrogenase α subunit | ☑ | ✓ | ✓ | Induced | Essential | KstR1 |
| Rv3545c | MSMEG_5995 | cyp125 | cholest-4-en-3-one 26-monooxygenase | ☑ | ✓ | ✓ | Induced | Essential | KstR1 |
| Rv3546 | MSMEG_5996 | fadA5 | steroid β-keto-acyl-CoA thiolase | ☑ | ✓ | ✓ | Induced | KstR1 | |
| Rv3547 | MSMEG_5998 | conserved hypothetical protein | ? | KstR1 | |||||
| Rv3548c | MSMEG_5999 | short-chain-type dehydrogenase/reductase | ? | ✓ | KstR2 | ||||
| Rv3549c | MSMEG_6000 | short-chain-type dehydrogenase/reductase | ? | ✓ | KstR2 | ||||
| Rv3550 | MSMEG_6001 | enoyl-CoA hydratase | ? | ✓ | KstR2 | ||||
| Rv3551 | MSMEG_6002 | CoA-transferase α subunit | ? | ✓ | ✓ | Essential | Essential | KstR2 | |
| Rv3552 | MSMEG_6003 | CoA-transferase β subunit | ? | ✓ | Induced, Essential | KstR2 | |||
| Rv3553 | MSMEG_6004 | oxidoreductase | ? | ✓ | Induced, Essential | KstR2 | |||
| Rv3554 | - | fdxB | ferridoxin B | ? | Induced | Induced | |||
| Rv3555c | MSMEG_5314 | conserved hypothetical protein | ✓ | Induced | Induced | ||||
| Rv3556c | MSMEG_6008 | fadA6 | thiolase | ? | ✓ | Essential | Induced, Essential | KstR2 | |
| Rv3557 | MSMEG_6009 | kstR2 | transcriptional repressor (TetR-like) | ☑ | ✓ | Induced | KstR2 | ||
| Rv3558 | - | PPE64 | PPE family of glycine rich proteins | ? | |||||
| Rv3559c | MSMEG_6011 | oxidoreductase | ? | ✓ | ✓ | KstR2 | |||
| Rv3560c | MSMEG_6012 | fadE30 | acyl-CoA dehydrogenase | □ | ✓ | ✓ | Induced, Essential | Essential | KstR2 |
| Rv3561 | MSMEG_6013 | fadD3 | 3aα-H-4α(3'-propanoate)-7aβ- methylhexahydro-1,5-indanedione-CoA synthase | ☑ | ✓ | ✓ | Induced | KstR2 | |
| Rv3562 | MSMEG_6014 | fadE31 | steroid acyl-CoA dehydrogenase α subunit | □ | ✓ | Induced | KstR2 | ||
| Rv3563 | MSMEG_6015 | fadE32 | steroid acyl-CoA dehydrogenase β subunit | □ | ✓ | ✓ | Essential | Essential | KstR2 |
| Rv3564 | MSMEG_6016 | fadE33 | steroid acyl-CoA dehydrogenase β subunit | □ | ✓ | ✓ | Induced | KstR2 | |
| Rv3565 | MSMEG_6017 | aspB | aspartate aminotransferase | ? | ✓ | KstR2 | |||
| Rv3566c | MSMEG_0306 | nat | arylamine N-acetyltransferase | □ | ✓ | Induced | Induced | ||
| Rv3567c | MSMEG_6035 | hsaB | 3-hydroxy-9,10-seconandrost-1,3,5(10)-triene-9,17-dione 4-hydroxylase reductase component | ☑ | ✓ | KstR1 | |||
| Rv3568c | MSMEG_6036 | hsaC | 3,4-dihydroxy-9,10-secoandrosta-1,3,5(10)-triene-9,17-dione 4,5-dioxygenase | ☑ | ✓ | ✓ | KstR1 | ||
| Rv3569c | MSMEG_6037 | hsaD | 4,5-9,10-diseco-3-hydroxy-5,9,17-trioxoandrosta-1(10),2-diene-4-oate hydrolase | ☑ | ✓ | ✓ | Essential | KstR1 | |
| Rv3570c | MSMEG_6038 | hsaA | 3-hydroxy-9,10-seconandrost-1,3,5(10)-triene-9,17-dione 4-monooxygenase component | ☑ | ✓ | ✓ | Essential | KstR1 | |
| Rv3571 | MSMEG_6039 | kshB | 3-ketosteroid 9α-hydroxylase reductase component | ☑ | ✓ | ✓ | Induced | KstR1 | |
| Rv3572 | MSMEG_6040 | conserved hypothetical protein | ? | KstR1 | |||||
| Rv3573c | MSMEG_6041 | fadE34 | cholest-4-en-3-one-26-oyl-CoA dehydrogenase | □ | ✓ | ✓ | KstR1 | ||
| Rv3574 | MSMEG_6042 | kstR1 | transcriptional repressor (TetR-like) | ☑ | ✓ | Induced | Essential | KstR1 | |
Mtb genes in the Cho region, KstR1, KstR2, and Mce3R regulons, and selected cholesterol-regulated genes or potential cholesterol metabolizing genes discussed in the text are included.
Genes in the Cho region are shaded with a gray background: (Van Der Geize et al., 2007;Nesbitt et al, 2010).
Transcriptionally up-regulated by cholesterol after 3 or 24 h: (Nesbitt et al, 2010).
Required for growth in cholesterol media: (Griffin et al, 2011).
Upregulated 24 h after THP-1 macrophage infection: (Fontán et al, 2007).
Induced in macrophages: (Schappinger et al, 2003).
Essential in macrophages using TraSH: (Rengaragan et al, 2005).
Essential in mice using TraSH: (Sassetti and Rubin, 2003).
Under control of the transcriptional repressors, KstR1: (Kendall et al., 2007); KstR2: (Kendall et al., 2010); Mce3R: (De La Paz Santangelo et al., 2009).
The transcription of the Cho-region is controlled by at least two transcriptional regulators: KstR1 and KstR2 (Table) (Kendall et al., 2010, Kendall et al., 2007). KstR1 and KstR2 are members of the TetR family of transcriptional repressors. They each contain an N-terminal DNA binding region that recognizes a 14 base pair semi-palindromic sequence, binding of which efficiently blocks RNA polymerase binding (Uhía et al., 2011a, Kendall et al., 2007). The binding of a small molecule inducer, presumably a catabolite, alleviates transcriptional repression of TetR repressors. KstR1 and KstR2 are conserved within Actinobacteria and have been identified in Rhodococcus, Nocardia, and other Mycobacteria species, including M. smegmatis where they were first described (Kendall et al., 2010, Kendall et al., 2007).
The KstR1 regulon in Mtb contains 74 genes, including those involved in A & B ring degradation as well as side chain metabolism (Kendall et al., 2007). In Mtb, a subset of the KstR1 regulon is up-regulated by cholesterol; however, a distinct subset is up-regulated by the fatty acid palmitate (Table). It is likely that KstR1 controls transcription of lipid metabolizing genes in addition to genes for cholesterol metabolism, consistent with the coexistence of lipids and cholesterol in LDL. It is not known if cholesterol or palmitate is the chemical inducer of KstR1. Many of the genes found within these regulons overlap with genes that have been shown to be essential in vivo in mouse or macrophage models of infection, including fadA5, mce4, and the igr operon (Nesbitt et al., 2010, Pandey, 2008, Schnappinger et al., 2003, Rengarajan et al., 2005, Sassetti and Rubin, 2003).
The KstR2 regulon comprises 15 genes in Mtb thought to be responsible for metabolizing the C & D rings of the steroid nucleus (Table). In the case of KstR2, the two-ring cholesterol catabolite 3aα-H-4α(3′-propanoyl-CoA)-7aβ-methylhexa-hydro-1,5-indanedione-CoA (HIP-CoA) has been identified as the inducer molecule and inhibits KstR2 DNA binding, de-repressing the KstR2 regulon, whereas HIP or CoA alone do not (Casabon et al., 2013c).
KstR1 and KstR2 regulate independently indicating separate functions for the genes involved in cholesterol metabolism. Their independence is consistent with the presence of two distinct sub-pathways: one for the degradation of the C & D rings and a second for the degradation of the A & B rings of the steroid nucleus and side chain, sometimes referred to as the higher and lower pathways of cholesterol metabolism. KstR1 and KstR2 have evolved separately, and are only distantly similar to each other (they are in different orthogroups); they have divergent functions (Mcguire et al., 2012).
In some Mycobacteria, Rhodococcus, and Nocardia, there is a single copy of KstR1. However, in more distantly related Actinobacteria often found in the environment like M. ulcerans, M. avium, and M. vanbaalenii there are additional copies of KstR1 that do not exist in Mtb (Mcguire et al., 2012) consistent with their more varied growth environments. TetR-like transcriptional regulators can also be found in Proteobacteria (Ramos et al., 2005). In Mtb, approximately 22 additional TetR-like repressors have been annotated, in addition to a host of other helix-turn-helix transcriptional repressors. At least one of these repressors, Mce3R (Santangelo et al., 2002), regulates genes that are also regulated by cholesterol.
Obtaining functional information for the large number of uncharacterized Mtb genes is a daunting task. Categorization through transcriptomics has helped to delineate genes into sub-pathways of cholesterol or lipid metabolism. Globally profiling genes required for growth under defined conditions can further identify genes required for cholesterol metabolism (Griffin et al., 2011). It should be noted that the latter type of studies do not distinguish between a gene disruption that blocks nutrition-generating catabolism, and a gene disruption that results in accumulation of a toxic metabolite.
To establish fully the biochemical functions of the cholesterol-regulated gene products, direct experimental validation is required. Typically, assignment of function relies on three types of experiments: mutation or deletion of the gene of interest and determination of phenotype, metabolite characterization of gene knockouts, and the expression and purification of recombinant enzyme and synthesis of putative chemical substrates for a biochemical assay.
ATP-dependent cholesterol import
Some of the best evidence substantiating cholesterol metabolism as important for Mtb growth and virulence came from studies performed on the mce4 locus in both R. jostii RHA1 and Mtb. The 11 gene mce4 regulon encodes an ABC-like ATP-dependent transport system that includes two permeases whose function is to actively import cholesterol into the bacterial cell. Deletion of the mce4 locus compromises the uptake of steroids by R. jostii RHA1 (Van Der Geize et al., 2007), and the transporter was demonstrated to bind cholesterol (Mohn et al., 2008). In Mtb, mutation of the ATPase subunit, mceG, or deletion of the mce4 operon fully inhibits in vitro growth on cholesterol, but not on glycerol (Pandey, 2008).
In Mtb, it was demonstrated that an mce4 mutant was unable to grow in IFN-γ activated macrophages (which are a model for the phenotype of a latent or progressive infection), but was able to survive in resting macrophages (Pandey, 2008). An Mtb Δ mce4 mutant was initially able to persist in vivo in a C57BL/6 mouse lung infection, but after four weeks of infection, growth defects became apparent as measured by decreasing cfu counts in the lungs of infected mice. These results clearly indicated a principal role for cholesterol utilization during Mtb infection and disease, especially during the chronic phase of infection.
Characterized enzymes in the Mtb cholesterol catabolic pathway
The bacterial metabolism of cholesterol is long and complicated, The MetaCyc (SRI) curated interactive pathway and database, provides an excellent graphical overview of the metabolite structures and enzymes involved (Caspi et al., 2010) (SRI). Please refer to the superpathway of cholesterol degradation II (cholesterol dehydrogenase) on the MetaCyc website [http://www.biocyc.org/META/NEW-IMAGE?type=PATHWAY&object=PWY-6947&detail-level=4&detail-level=3] to follow the discussion below.
A & B ring metabolism
A & B ring degradation requires ten genes that perform eight enzymatic reactions, kstD, kshAB, and hsaABCDEFG (Figure 3). These enzymes are all EC 1 class oxidoreductases responsible for the sequential oxidation of the A & B carbon framework.
Figure 3. Steroid ring degradation in bacteria.
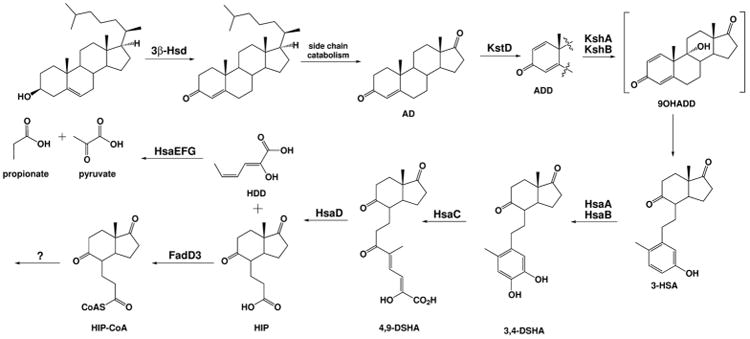
HSD: 3β-hydroxysteroid dehydrogenase; ChoE: cholesterol oxidase; KstD: ketosteroid dehydrogenase; KshA/B: 3-ketosteroid 9α-hydroxylase; HsaA/B: 3-hydroxy-9,10-seconandrost-1,3,5(10)-triene-9,17-dione 4-monooxygenase; HsaC: 3,4-dihydroxy-9,10-secoandrosta-1,3,5(10)-triene-9,17-dione 4,5-dioxygenase; HsaD: 4,5-9,10-diseco-3-hydroxy-5,9,17-trioxoandrosta-1(10),2-diene-4-oate hydrolase; HsaE: 2-hydroxyhexa-2,4-dienoate hydratase; HsaF: 4-hydroxy-2-oxohexanoate aldolase; HsaG: propanal dehydrogenase. Abbreviations of compound names: AD: 4-androstenedione; ADD: 1,4-androstenedione; 9OHADD: 9-hydroxy-androsta-1,4-diene-3,17-dione; 3-HSA: 3-hydroxy-9,10-seconandrost-1,3,5(10)-triene-9,17-dione; 3,4-DSHA: 3,4-dihydroxy-9,10-seconandrost-1,3,5(10)-triene-9,17-dione; 4,9-DSHA: 4,5–9,10-diseco-3-hydroxy-5,9,17-trioxoandrosta-1(10),2-diene-4-oic acid; HIP: 3aα-H-4α(3′-propanoyl-CoA)-7aβ-methylhexa-hydroindane-1,5-dione; HDD: 2-hydroxy-hexa-2,4-dienoic acid.
3β-hydroxysteroid dehydrogenase and cholesterol oxidase
In Actinobacteria the first reaction of ring metabolism is oxidation and isomerization of cholesterol to form cholest-4-ene-3-one. This involves the sequential oxidation of the 3-hydroxy position to a ketone and conversion of the resultant β,γ-unsaturated ketone to the α,β-conjugated enone product. In bacteria, this reaction is catalyzed either by a 3β-hydroxysteroid dehydrogenase (3β-HSD) or cholesterol oxidase (ChOX).
3β-HSD is a member of the short chain dehydrogenase superfamily and uses NAD+ or NADP+ as an electron acceptor. Bacterial cholesterol oxidase (ChOx) is a member of the glucose-methanol-choline (GMC) oxidoreductase family. It is expressed extracellularly as a monomer, binds flavin adenine dinucleotide (FAD) as a cofactor, and uses O2 as an electron acceptor, which gets reduced to hydrogen peroxide in order to regenerate FAD. Although ChOx and 3β-HSD use different reaction mechanisms, both enzymes catalyze the same transformation, and either or both are found within the genomes of all steroid-utilizing bacteria. Nocardia sp. (Horinouchi et al., 1991), C. testosteroni (Horinouchi et al., 2012), and R. jostii (Rosloniec et al., 2009) utilize 3β-HSD while Streptomyces spp. (Ishizaki et al., 1989), Rhodococcus equi (R. equi) (Machang'u and Prescott, 1991), and Gordonia cholesterolivorans (Drzyzga et al., 2011) utilize ChOx.
The genome of Mtb contains an annotated cholesterol oxidase, choD (Rv3409c) and 3β-hydroxysteroid dehydrogenase (Rv1106c). Recombinant 3β-HSD has been enzymatically characterized and is able to convert cholesterol to cholest-4-ene-3-one, and is also able to convert pregnenolone, and dehydroepiandrosterone to their respective 3-keto-4-ene steroid products (Yang et al., 2007). This conversion has not been demonstrated with recombinant ChoD. Knockout experiments indicate 3β-HSD is required for growth of Mtb on cholesterol while growth of a ΔchoD knockout strain was not affected (Yang et al., 2007, Yang et al., 2011). In addition, culture supernatant lost the ability to oxidize cholesterol to cholest-4-ene-3-one in an Rv1106c mutant strain (CDC1551) indicating 3β-HSD is the sole cholesterol-oxidizing enzyme in Mtb (Yang et al., 2007). M. smegmatis culture supernatant from a choD (msmeg_1604) mutant strain was still able to convert cholesterol to cholest-4-ene-3-one (Yang et al., 2011, Uhía et al., 2012). Likewise in Mycobacterium sp. the ΔchoD knockout strain was able to form 3-keto-4-ene steroids (Ivashina et al., 2012). Moreover, in global growth profiling experiments, choD was not required for catabolism of cholesterol, whereas mutation of Rv1106c resulted in reduced fitness (Griffin et al., 2011). These results suggest that 3β-HSD, and not ChoD, is responsible for 3β-hydroxy-5-ene steroid oxidation in Mtb and possibly in other mycobacterial species as well.
A role for ChoD in cholesterol metabolism has not been substantiated and its exact enzymatic function is still unknown. However, recent reports have suggested its importance in Mtb virulence. For example, an Mtb ΔchoD strain showed attenuated virulence in both peritoneal macrophages and mouse models of infection (Klink et al., 2013, Brzostek et al., 2007). The M. smegmatis ΔchoD knockout accumulates a hyperrhamnosylated polar glycopeptidolipid (GPL) called L1334, which lacks acetylation on the 6-deoxytalose moiety (Gao and Sampson, 2014). In M. avium, surface-exposed GPLs stimulate the host Toll-like receptor 2 (TLR-2) mediated inflammatory response, and acetylation of GPLs is obligatory for this activity (Pang et al., 2013, Schorey and Sweet, 2008). These results suggest ChoD is involved in tuning the TLR-2 mediated host inflammatory response and that annotation as a cholesterol oxidase is incorrect (Gao and Sampson, 2014).
Interestingly, 3β-hsd is not regulated by KstR1 in Mtb. In other Mycobacteria including M. smegmatis, 3β-hsd is under regulatory control of KstR1 (Uhía et al., 2011a, Uhía et al., 2011b). In Nocardia, when added to growth media cholesterol induces cholesterol dehydrogenase (ChoA) (Horinouchi et al., 1991, Kishi et al., 2000). This difference in regulation implies that sometime in recent evolutionary history, Mtb lost its ability to specifically up-regulate the gene responsible for the first step in cholesterol metabolism. Up-regulation is certainly not requisite to enzymatic activity since regardless of whether cholesterol is available, 3β-HSD enzymatic activity is always present.
In vitro studies with a CDC1551 mutant suggest that Rv1106c is important for cholesterol metabolism (Yang et al., 2011), but another study suggests that Rv1106c is dispensable for cholesterol metabolism in H37Rv. The reason for this discrepancy may be a result of differences between the strains as has been observed before with cyp125 (Johnston et al., 2010). The possibility that there is an additional dehydrogenase that can compensate for 3β-HSD activity has been investigated. In M. smegmatis, there is at least one additional gene (msmeg_5233) that encodes a dehydrogenase that can convert cholesterol to cholest-4-en-3-one (Uhía et al., 2011b). However, there is not an ortholog of msmeg_5233 in Mtb.
Moreover, a 3β-hsd mutant strain replicated at a similar rate to wild type in macrophages, and infection studies in the guinea pig infection model showed identical cfus in the lungs of wild type, 3β-hsd mutant, and 3β-hsd complement. It was concluded that 3β-hsd is not necessary for nutrition acquisition (Yang et al., 2011), likely because during infection Mtb has access to and utilizes multiple carbon sources (Beste et al., 2013). In most bacteria, carbon sources are catabolized in order of their ability to support growth. However, Mtb can catabolize multiple carbon sources simultaneously (De Carvalho et al., 2010). The essential or primary role of cholesterol metabolism in the pathogenesis of Mtb is unlikely to be solely carbon catabolism.
3-ketosteroid-Δ1-dehydrogenase
The second step of cholesterol A & B ring metabolism is 1,2-desaturation of cholest-4-ene-3-one to a di-α,β enone product, and this reaction is catalyzed by 3-ketosteroid-Δ1-dehydrogenase (KstD). Note, cholesterol side chain β-oxidation and A & B ring oxidation can happen concurrently in vivo, albeit with different kinetics. Thus, we present the order of the steps presented is for clarity of discussion and is not absolute. KstD enzymes involved in steroid metabolism have been identified in Nocardia (Sih, 1962), Rhodococcus (Kaufmann et al., 1992, Knol et al., 2008), and M. smegmatis (Brzostek et al., 2005). Interestingly, some bacteria like R. erythropolis SQ1, have been shown to encode more than one KstD enzyme and deletion of both were required to block 1,2-desaturation (Van Der Geize et al., 2002).
In Mtb, KstD is encoded by Rv3537 (Knol et al., 2008). Its enzyme activity has been demonstrated in vitro with the substrates 5α-androstane-3,17-dione and 17β-hydroxy-5α-androstane-3-one. Genomic analysis has indicated that Rv3537 is the sole KstD encoding gene in Mtb, since an Rv3537 disrupted strain of Mtb accumulates 9-hydroxy-4-androstene-3,17-dione (9-OHAD) (Brzostek et al., 2009). KstD is required for growth of M. tuberuclosis in minimal medium supplemented with cholesterol (Griffin et al., 2011). In resting THP-1 macrophages, growth of a ΔkstD knockout is attenuated compared to H37Rv wild-type strain (Klink et al., 2013).
KshA/B
Next in ring catabolism, a 3-ketosteroid-9α-hydroxylase catalyzes the addition of a hydroxyl group at C9, which leads to subsequent aromatization of the A ring and opening of ring B. This enzyme is a two-component Rieske monooxygenase made up of KshA, the oxygenase component, and KshB, the reductase component. While KshA/B activity has been observed in several Actinobacteria (Andor et al., 2006, Strijewski, 1982, Van Der Geize et al., 2002), it was not until recently that this activity was demonstrated with recombinant enzyme from Rhodococcus for substrates AD and androstadienedione (ADD) (Petrusma et al., 2009). It has also been shown that the substrate specificity and transcriptional regulation of enzymes from Rhodococcus that are involved in steroid degradation differs from Mtb significantly (Petrusma et al., 2011).
The 3-ketosteroid-9α-hydroxylase homologs in Mtb, KshA (Rv3526) and KshB (Rv3571), have been recombinantly expressed in E. coli and purified (Capyk et al., 2009a). Activity was reconstituted in vitro with several substrates including AD, ADD, the CoA thioester of 3-oxo-4-pregnene-20-carboxylic acid, and the CoA thioester of 3-oxo-1,4-pregnadiene-20-carboxylic acid (Capyk et al., 2011).
An Mtb ΔkshA/ΔkshB double mutant was unable to grow on cholesterol, AD, or 5α-androstane-3,17-dione (Hu et al., 2010). It was also shown that the deletion of either kshA or kshB resulted in the rapid clearance of infection in an in vitro macrophage model, or an in vivo mouse infection model. In a mutant where only kshA was deleted there was no observed phenotypic change. Interestingly, the Mtb strain CDC1551 does not contain any kshA orthologs (Fleischmann et al., 2002). However, when kshB was deleted, there was a marked change in cellular wall physiology. This was attributed to an accumulation of tri-acyl-trehaloses (TAT) lipids, and a decrease in the synthesis of di-acyl-trehaloses (DAT), which was measured using radiolabeled 14C-acetic acid and autoradiography. The involvement of the reductase KshB in fatty acid biosynthesis was attributed to its possible interaction with other Rieske-type oxygenase enzymes with structural homology to KshA that are involved in the synthesis of cell wall lipids.
HsaA/HsaB
C. testosteroni is able to utilize testosterone as a sole carbon source and studies elucidating this pathway were some of the first to provide insight into cholesterol ring metabolism in Mtb. For instance, gene disruption of the tesA1 and tesA2 genes of C. testosteroni grown with testosterone was shown to accumulate 3-hydroxy-9,10-secoandrost-1,3,5(10)- triene-9,17-dione (3-HSA) and tesA1 and tesA2 are required for the formation of 3,4-dihydroxy-9,10-seconandrost-1,3,5(10)-triene-9,17-dione (3,4-DSHA) (Horinouchi et al., 2004). TesA1 and TesA2 are assigned as a flavin-dependent oxygenase/reductase pair responsible for converting 3-HSA to 3,4-DSHA.
In Mtb this reaction is assigned to hsaA (Rv3570c) and hsaB (Rv3567c). Conversion of 3-HSA to the catechol-secosteroid derivative 3,4-DSHA has been demonstrated in vitro with recombinant HsaA/B (Dresen et al., 2010). The crystal structure of HsaA demonstrated that there was an elongation of the substrate tunnel at C17, and this information in conjunction with the relatively weak substrate specificity constants of complete side chain degraded substrates suggests that in vivo partially degraded side chain substrates are utilized as well (Hu et al., 2010). Mutagenesis analysis showed that Rv3570c is required for growth of Mtb in macrophages (Rengarajan et al., 2005).
HsaC: 2,3-dehydroxyphenyl dioxygenase
Studies in C. testosteroni identified tesB as the iron-dependent extradiol dioxygenase, responsible for the meta-cleavage of the A ring of 3,4-DHSA to give 4,5-9,10-diseco- α3-hydroxy-5,9,17-trioxoandrosta-1(10),2-diene-4-oic acid (4,9-DSHA) (Horinouchi et al., 2004, Horinouchi et al., 2001). In Rhodococcus sp. HsaC (Rv3568c) is homologous to TesB and a hsaC-disrupted strain grown on cholesterol accumulates 3,4-DHSA (Van Der Geize et al., 2007).
In Mtb, HsaC shares 98% sequence identity to TesB and encodes the dioxygenase responsible for the formation of 4,9-DSHA. Activity has been demonstrated in vitro with recombinant enzyme and substrate 3,4-DHSA (Yam et al., 2009, Van Der Geize et al., 2007). The hsaC gene is required for growth of Mtb on cholesterol, but not on glycerol. Knockout studies reveal that hsaC is important for pathogenesis. Immunocompromised mice and guinea pigs infected with ΔhsaC lived significantly longer than those infected with wild type (Yam et al., 2009). In addition, harvested lungs showed lower bacterial loads after 8 weeks of infection. These results established that hsaC is important for pathogenesis of Mtb.
4,9-DSHA hydrolase
In C. testosteroni, TesD was confirmed as the hydrolase that cleaves 4,9-DSHA to form HIP and 2-hydroxy-hexa-2,4-dienoic acid (HHD). In Mtb, HsaD (Rv3569c) catalyzes the same reaction (Lack et al., 2010, Van Der Geize et al., 2007). The hydrolase activity has been demonstrated with recombinant enzyme and substrate 4,9-DSHA. In addition, hsaD is important for Mtb survival within macrophages (Rengarajan et al., 2005). Like other enzymes that catalyze reactions earlier in the degradation pathway for rings A & B, an hsaD mutant shows significant growth attenuation when grown on minimal medium supplemented with cholesterol (Griffin et al., 2011).
Metabolism of HDD
The genes for the metabolism of HDD by Mtb have been proposed based on homology to enzymes in C. testosteroni. HDD is metabolized to 4-hydroxy-2-oxohexanoic acid in C. testosteroni by 2-hydroxypentadienoate hydratase, TesE (Horinouchi et al., 2005). Next tesG, encoding a 4-hydroxy-2-oxovalerate aldolase, forms pyruvate and propionaldehyde. Pyruvate can be converted to carbohydrates or fatty acids. TesF, an acetaldehyde dehydrogenase, converts propionaldehyde to propionyl-CoA.
In Mtb, HsaEFG (Rv3534c/Rv3535c/Rv3536c) are hypothesized to metabolize HDD (Van Der Geize et al., 2007). HsaE, HsaF, and HsaG share 41%, 47%, and 56% amino acid identity with TesE, TesG, and TesF from C. testosteroni, respectively. Recently HsaF and HsaG were shown to form a heterotetrameric complex, formation of which is required for activity (Carere et al., 2013). Aldolase HsaF catalyzes the cleavage of 4-hydroxy-2-oxohexanoate to propionaldehyde and pyruvate in the presence of NAD+ and CoA. Volatile propionaldehyde is channeled to dehydrogenase HsaG where is it converted to propionyl-CoA. The activity of HsaE has not been verified. Selection studies reveal that hsaE, hsaF and hsaG are not required for growth on cholesterol, although their deletion does result in slower growth on cholesterol as a carbon source (Griffin et al., 2011). Because an hsaE, hsaF, or hsaG mutant still has the requisite genes required for catabolism of the side chain and the C & D rings of cholesterol, growth remains possible in minimal medium supplemented with cholesterol (Griffin et al., 2011). The reason for slower growth could be due to toxicity of accumulated metabolites, or other secondary effects.
Steroid C & D ring degradation
How extensively the C & D rings are degraded by Mtb is not known. After initial catabolism of the A & B rings, it is presumed that the C & D rings are catabolized in an oxidative manner and acetyl-CoA and/or propionyl-CoA are generated. This assumption is supported by the fact that, after a significant lag phase, Mtb is able to grow on HIP, a C & D ring derivative, as a sole carbon source, thus highlighting the fate of at least some of the C & D ring carbons (Casabon et al., 2013a). The precise fate of HIP (Figure 3) in Mtb has not yet been established.
The degradation of the hexahydroindanone C & D steroid ring intermediate begins through the thioesterification of the propionate moiety left over from A & B ring oxidation. FadD3, an acyl-CoA ligase, was recently demonstrated to perform this function on HIP (Casabon et al., 2013a). A Rhodococcus jostii RHA1 ΔfadD3 knockout strain was impaired when grown on cholesterol, but when the Mtb fadD3 (Rv3561) gene was knocked in, the control growth rate was restored. Steady-state kinetic analysis of recombinantly expressed FadD3 from Mtb demonstrated that the specificity constant for HIP, which contains a keto group at the 5 position of the indanone, is 165 times greater than the specificity for 5α-OH HIP, where this is a hydroxyl moiety.
Both cholesterol and cholate degradative genes converge at this point to a single, relatively conserved cluster in Rhodococcus species that are able to degrade both steroids (Mohn et al., 2012). In Rhodococcus spp., it was demonstrated that there is a gene cluster similar to those that exist for other Actinobacteria that is able to degrade bile acids like cholate. The regulation of this process is under the control of the TetR-like transcriptional repressor KstR2 in both Mtb (Rv3557c) and Rhodococcus jostii RHA1 (RHA1_ro04598). This regulon consists of 15 genes in Mtb and M. smegmatis, and 14 genes in RHA1. Contrary to what was proposed in Kendall, et. al, 2010, cholesterol does not relieve binding of the KstR2 repressor to its conserved binding sequence. Rather, it is the hexahydroindanone-CoA metabolite that acts as the chemical inducer to alleviate KstR2 repressor binding (Casabon et al., 2013a). These recent experimental results help to further demonstrate that there is a distinct pathway for the degradation of the C & D rings of the steroid nucleus the regulation of which is dependent on metabolites from A & B ring degradation degradation that occurs first.
Attempts to identify the Proteobacterial genes responsible for the degradation of the C & D rings of steroids have been pursued using gene knockout and metabolic profiling studies. For example, a gene-disrupted mutant of ORF18 in the testosterone degrading bacteria C. testosteroni TA441 was constructed and grown on ADD, chenodeoxycholic acid, and cholic acid in order to identify any buildup of metabolic intermediates (Horinouchi et al., 2006). When incubated with testosterone, no metabolic intermediates accumulated (Horinouchi et al., 2003). However, when the ORF18-disrupted mutant is grown on ADD, it accumulates HIP. This study establishes the importance of this gene as encoding a probable CoA-ligase essential for further degradation of the C & D ring steroid nucleus. Additional substitutions like hydroxyl groups on the steroid ring system, as seen for cholic acid for example, did not preclude metabolism to the step encoded by ORF18.
It is hypothesized that FadE30 of Rhodococcus equi catalyzes the dehydrogenation of HIP because a ΔfadE30 knockout strain of Rhodococcus equi accumulates 7aβ-methyl-hexahydro-5-indanone when cultured with cholesterol. R. equi FadE30 shares 68% amino acid identity with FadE30 (Rv3560c) from Mtb. However, this function has not yet been demonstrated with Mtb FadE30 (Van Der Geize et al., 2011). Expression of the gene triplet fadE31-fadE32-fadE33 is controlled by KstR2 and has been proposed to be involved in C & D ring metabolism (Van Der Geize et al., 2007), although this awaits further biochemical characterization.
Until recently, it was not clear if side chain degradation was prerequisite to C & D ring catabolism. However, current evidence suggests that any partially catabolized bacterial substrate must have the side chain completely removed before any C & D oxidation enzymes are able to function. FadD3 was unable to ligate a CoA onto a side chain-containing metabolite similar to HIP, 1β(2′-propanoate)-3aα-H-4aα(3”-propanoate)-7aβ-methylhexahydro-5-indanone (Casabon et al., 2013a).
Furthermore, an Mtb mutant lacking the igr operon, which removes the last three carbons of the steroid side chain, accumulated a hexahydroindanone metabolite that contained a three-carbon side chain (Thomas et al., 2011). Therefore, the presence of a side chain precludes further catabolism of rings C & D and results in a buildup of metabolites (vide infra). In multiple metabolite profiling studies, AD and ADD were identified when Mtb is cultured with cholesterol (Nesbitt et al., 2010, Thomas et al., 2011). In addition, side chain metabolizing ChsE1-ChsE2 prefers ring intact substrates (Thomas and Sampson, 2013, Thomas et al., 2011), while the ring system metabolizing enzyme KstD prefers fully side chain metabolized 5α-AD to the partially side chain metabolized 5α-pregnane-3,20-dione or progesterone (Knol et al., 2008).
Cholesterol side chain metabolism
Cytochrome P450 125
The side chain of cholesterol was initially proposed to be degraded through a process akin to classical fatty acid β-oxidation (Figure 4). The β-oxidation cycle requires four sequential enzymatic steps catalyzed by an acyl-CoA dehydrogenase (FadE), an enoyl-CoA-hydratase (EchA), a 3-hydroxy-acyl-CoA-dehydrogenase (FadB), and a 3-keto-acyl-CoA thiolase (FadA), respectively (Figure 4). The genome of Mtb encodes a large number of β-oxidation genes, including 35 fadE, 21 echA, 5 fadB, and 6 fadA genes. This redundancy in the genome complicates assignment of function to individual steps in the multiple possible pathways. For example, fadE14 from Mtb is annotated as a fadE, however, it was shown to participate in the synthesis of the iron-scavenging siderophore mycobactin, and is thus not involved in degradation at all (Rodriguez et al., 2002, Lamarca et al., 2004). Further complicating gene assignments, the β-oxidation enzymes responsible for side chain metabolism in other Actinobacteria have not been identified.
Figure 4.
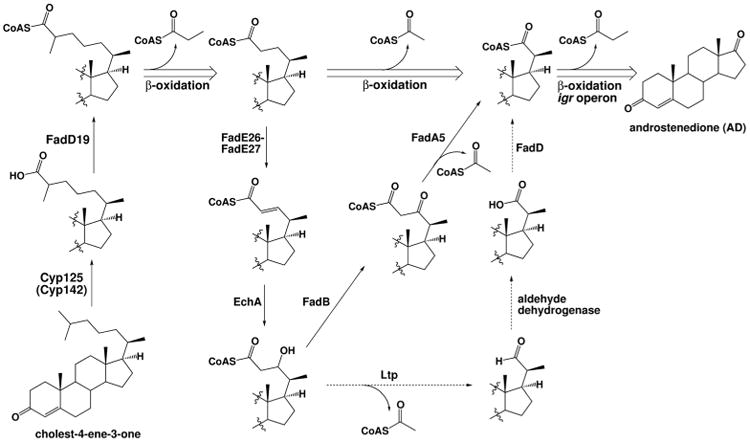
Steroid side chain degradation is initiated by Cyp125, and alternatively Cyp142. A FadD acyl-CoA ligase activates the resultant steroid carboxylic acid through esterification with CoA. The steroid side chain is truncated via three cycles of β-oxidation to yield acetyl-CoA, two units of propionyl-CoA, and a 3,17-dione steroid. The individual steps in the classical β-oxidation pathway are shown for the second cycle with solid arrows. Recently proposed alternate steps via an aldehyde (Holert et al., 2013a, Holert et al., 2013b) are shown as dashed arrows. The individual steps catalyzed in the third cycle are shown in Figure 5. Steps are labeled with specific enzyme names (see Table), if known.
Prior to entering the β-oxidation cycle, the hydrocarbon side chain must first be oxidized to the carboxylic acid and subsequently activated through formation of the CoA thioester (Figure 4). It has long been proposed that a cytochrome P450 is responsible. Actinobacteria have a large number of cytochrome P450s, again making it challenging to assign function—the Mtb genome encodes 20 cytochrome P450 enzymes. The enzyme responsible for catalysis was identified in Rhodococcus jostii (R. jostii) as cyp125 from knockout studies demonstrating cyp125 is required for growth on cholesterol, but not for growth on side chain-activated 5-cholestene-26-oic acid-3β-ol (Rosloniec et al., 2009). In Mtb, cyp125 (Rv3545c), located in the intracellular growth (igr) operon, is the ortholog of cyp125 from R. jostii.
Mtb cyp125 was recombinantly expressed, in separate accounts, in R. jostii RHA1 or in E. coli with coexpression of folding chaperones, GroEL and GroES (Chang et al., 2009, Capyk et al., 2009b). Purified protein was shown to bind cholesterol and cholest-4-ene-3-one with sub-micromolar affinity (Capyk et al., 2009b). Cyp125, with reductase KshB (Rv3571) or spinach ferredoxin reductase, and NADH was able to transform cholesterol and cholest-4-ene-3-one in vitro to the C26 hydroxy and the C26 carboxylic acid products.
The crystal structure of Cyp125 has been reported without ligand as well as with AD and the antitubercular drug econazole (Mclean et al., 2009). These ligands were found to bind within the active-site cavity of the enzyme. Docking was used to incorporate cholesterol into the structure and it was found for the lowest energy structure that the C26 and C27 of cholesterol were 5.3 and 6.3 Å from the heme iron center, respectively. The position of these carbons relative to the active site of the enzyme substantiates that this is where the transformation occurs.
Knockout cyp125 (Δcyp125) Mtb CDC1551 cultures grown with cholesterol accumulate cholest-4-ene-3-one, suggesting that this is the physiological substrate for the enzyme in vivo (Ouellet et al., 2010). The cyp125 gene is required for growth on cholesterol in CDC1551, but interestingly is not required in the H37Rv strain. When expressed recombinantly in vitro, Cyp142 in addition to Cyp125 catalyze the oxidation of C26 in the side chain of cholesterol. Expression of either cyp125 or cyp142 can support the growth of Δcyp125 Mtb on cholesterol (Driscoll et al., 2011, Johnston et al., 2010). Therefore Cyp142 provides compensatory activity in Mtb H37Rv. The cyp142 gene is not expressed in CDC1551 wild type due to a promoter mutation.
Acyl-CoA ligases
Following oxidative activation of the hydrocarbon side chain by Cyp125, an acyl-CoA ligase (FadD) forms an acyl-CoA thioester, which is required for the substrate to enter the β-oxidation pathway since these enzymes require CoA substrates (Figure 4). The Mtb H37Rv genome contains 36 genes annotated as fadDs, a functional redundancy that complicates their respective substrate assignments. E. coli, which is able to metabolize fatty acids as a sole source of carbon, encodes only one FadD that accepts fatty acids of varying length (Kameda and Nunn, 1981). The large difference in fadD numbers between Mtb and E. coli suggests that FadD enzymes in Mtb have potentially many additional functional roles.
In Mtb, several fadD genes located adjacent to polyketide synthase (PKS) genes in the genome, have been shown to encode fatty acyl-AMP ligases with important roles in mycolic acid, DIM, and PGL synthesis (Cox et al., 1999, Khare et al., 2009, Pethe et al., 2010, Simeone et al., 2010, Trivedi et al., 2004). Interestingly, in the opportunistic pathogenic bacterium responsible for many deaths from Cystic fibrosis, Pseudomonas aeruginosa, the presence of a plurality of acyl-CoA ligases was attributed to virulence, since these enzymes are responsible for ‘activating’ all carbon substrates destined to enter a variety of modification pathways (Kang et al., 2010).
Four acyl-CoA synthetase fadD genes are up-regulated during growth of Mtb on cholesterol: fadD3, fadD17, fadD18, and fadD19 (Nesbitt et al., 2010). FadD3 was recently biochemically characterized as being involved in C & D ring metabolism by initiating HIP degradation (Casabon et al., 2013a). FadD19 in Rhodococcus rhodochrous (R. rhodochrous) catalyzes thioesterification of C24-branched steroid-C26-oic acid substrates, and is therefore a sterol-CoA ligase important for degradation of the C24 sterols β-sitosterol and campesterol (Wilbrink et al., 2011). Recombinant R. rhodochrous FadD19 (67% amino acid identity with Mtb FadD19 (Rv3515c) also catalyzes thioesterification of steroid-C26-oic acid substrates. A recent report demonstrated that the ortholog of this protein in Mtb catalyzes thioesterification of 3-oxo-4-cholesten-26-oate with a kcat/Km of 0.33±0.03×105 M-1s-1, thus validating this enzyme as the eight-carbon side chain steroid-CoA ligase (Casabon et al., 2013b).
Casabon et al. also demonstrated that FadD17 (Rv3506) is the steroid-24-oyl-CoA synthetase that ligates CoA onto the 5-carbon side chain of the intermediate metabolite (Casabon et al., 2013b). This activity is required for salvaging cholesterol-24-oic acids that are formed as a result of CoA ester hydrolysis during β-oxidation or that are formed through an alternate pathway (vide infra) (Holert et al., 2013a).
The FadD18 (Rv3513c) sequence is almost identical to the C-terminal half of FadD19 as well as the C-terminal portion of other fatty acid-CoA synthases. No information is available about the function of FadD18 (Lechat et al., 2008). However, it appears that fadD18 could be the result of partial gene duplication (Lew et al., 2011).
Phenotypic profiling experiments using transposon mutant libraries and deep sequencing performed by Griffin et al (2011) suggested a possible role for FadD36 (Rv1193) in Mtb in performing the first step in Mtb steroid side chain thioesterification. However, FadD19 has been shown to perform this function (Casabon et al., 2013b). Moreover, the fadD36 gene is not regulated by KstR1. Therefore, it is unlikely that this functional assignment is correct (Griffin et al., 2011).
Acyl-CoA dehydrogenases
CoA thioesters of lipids including steroids are substrates for β-oxidation, and the first enzyme in this four step catabolic cycle is catalyzed by an acyl-CoA dehydrogenase (ACAD). Degradation of the cholesterol side chain requires three cycles and therefore, three ACADs (Figure 4). Of the 35 acad genes, 13 are up-regulated and 4 are down-regulated by cholesterol (Nesbitt et al., 2010). Eight fadE genes, including chsE1 (fadE28) and chsE2 (fadE29) of the igr operon, and fadE5, fadE25, fadE30, fadE31, fadE32, and fadE33 are required for growth on cholesterol, validating their importance in cholesterol metabolism (Griffin et al., 2011). This functional redundancy of fadE genes is at first perplexing— although Mtb has 35 fadE genes, E. coli only has one (yafH) (Campbell and Cronan, 2002)— and steroid metabolism accounts for about one third of this redundancy.
Recently, our laboratory has identified several heteromeric enzymes encoded by polycistronically expressed fadE genes in Mtb (Wipperman et al., 2013). Interestingly these genes are all regulated by cholesterol, suggesting they are involved in cholesterol transformation and/or metabolism. We have characterized six heteromeric ACADs encoded by cistronic genes in Mtb including chsE1(fadE28)-chsE2(fadE29), fadE17-fadE18, fadE23-fadE24, fadE26-fadE27, fadE31-fadE32, and fadE31-fadE33. All of the identified heteromeric enzymes contain two active sites and bind two FAD cofactors, unlike a traditional homotetrameric ACAD, which has four active sites and binds four FAD cofactors. Each polypeptide of the complex is either insoluble or inactive when recombinantly expressed alone, and both chains are required to form intact FAD cofactor binding sites. The α and β chains are not interchangeable between heterotetramers.
Heterotetrameric ChsE1(FadE28)-ChsE2(FadE29) is encoded by two adjacent genes in the igr operon, Rv3544c and Rv3543c (Figure 5). The igr operon is up-regulated by cholesterol and is located within the cholesterol regulon in the Mtb genome (Nesbitt et al., 2010). The igr operon knockout (Δigr) strain does not grow on cholesterol as a sole carbon source in vitro. However, Δigr can grow just as well as wild-type Mtb on pyruvate, valerate, isovalerate, propionate, palmitate, dodecanoate, glycerol, Tween, and dextrose confirming its role in cholesterol metabolism and not fatty acid metabolism (Chang et al., 2009). The igr operon is conserved in pathogenic and nonpathogenic Mycobacteria, including M. smegmatis, M. avium, and M. bovis. In other Actinobacteria including Rhodococcus, Streptomyces, and Gordonia, the operon is only partially conserved and lacks cyp125.
Figure 5.
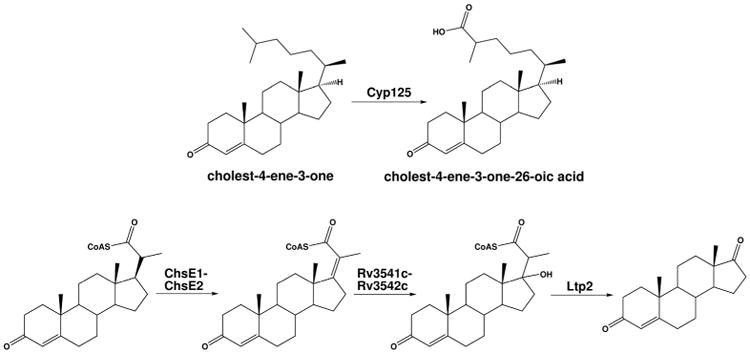
The igr operon in Mtb. The β-oxidation chemistry catalyzed by the igr-operon encoded enzymes.
In a Δigr knockout strain of Mtb, growth was attenuated in resting macrophages and early in the infection process in immunocompetent mice. The cfu levels in the lungs and spleen were greatly reduced compared to wild type. The growth attenuation observed in the Mtb Δigr knockout demonstrates that this operon is important for intracellular growth, which is where igr derives its name (Chang et al., 2007). The in vitro phenotype of Δigr requires cholesterol containing media and the genes encoding Mce4, an ATP-dependent cholesterol transporter (Chang et al., 2009). These studies suggest that the observed attenuation in Δigr is not due to lack of cholesterol, but rather the inability to fully metabolize cholesterol and the accumulation of a toxic intermediate.
Metabolic tracing studies with isotopically labeled [1,7,15,22,26-13C]-LDL cholesterol led to the accumulation of methyl 1β-(2′-propanoate)-3aα-H-4α(3′-propanoic acid)-7aβ-methylhexahydro-5-indanone in the Δigr knockout strain. This metabolite is derived from cholesterol and contains partially metabolized A & B rings, intact C & D rings and a partially metabolized three-carbon side chain (Thomas et al., 2011).
Based on the metabolite studies, the substrate for ChsE1-ChsE2 was hypothesized to be 3-oxo-23,24-bisnorchol-4-en-22-oyl-CoA or 1β-(2′-propanoyl-CoA)-3aα-H-4α(3′-propanoic acid)-7aβ-methylhexahydro-5-indanone. Both substrates were tested as well as short and branched fatty acyl-CoAs. Only the steroid thioesters were oxidized by ChsE1-ChsE2. The four ring 3-oxo-23,24-bisnorchol-4-en-22-oyl-CoA was a significantly better substrate with a kcat/Km that was 255-fold better than for the two-ring indanone. The regiochemistry of dehydrogenation was confirmed by NMR spectroscopy to be at C17-C20 (Thomas and Sampson, 2013) (Figure 5); the double bond that is introduced during the final cycle of cholesterol side chain β-oxidation. This heterotetrameric enzyme was the first characterized ACAD involved in sterol side chain metabolism (Thomas et al., 2011).
We proposed, based on its sequence similarity to ChsE1-ChsE2 and transcriptional regulation by KstR1, that FadE26-FadE27 catalyzes dehydrogenation in the first and/or second cycles of cholesterol side chain β-oxidation. Preliminary activity data demonstrated that FadE26-FadE27 introduced a double bond into the 3-keto-4-cholene-24-oyl-CoA substrate, the cholesterol metabolite with a 5-carbon side chain (Wipperman et al., 2013). With this preliminary activity data for the five-carbon substrate, and the fact that expression of both ChsE1-ChsE2 and FadE26-FadE27 are regulated by KstR1, it is probable that the only other KstR1 regulated ACAD, FadE34, is responsible for the first dehydrogenation step in cholesterol metabolism during the first cycle of β-oxidation. Interestingly, FadE34 has high protein sequence homology with the human very long chain ACAD enzymes, which are rare examples of ACAD dimers rather than tetramers. This protein structure evolved to bind very long chain fatty acids, and it is possible that FadE34 is a dimer with a large enough binding cavity to bind a partially oxidized C27-CoA steroid, although this activity remains to be assessed.
Enoyl-CoA hydratases
The second step of β-oxidation is the hydration of enoyl-CoA thioesters to β-hydroxy-acyl-CoA products by an enoyl-CoA hydratase (Figure 4). In Mtb, 21 genes are computationally annotated as (S)-specific enoyl-CoA hydratases, (Cole et al., 1998). (S)-hydratases, such as the classic example of crotonase, have been well studied, and are typically involved in fatty acid β-oxidation (Stern and Del Campillo, 1956). One or possibly more enoyl-CoA hydratases are required for catabolism of the cholesterol side chain. Of these 21 genes, two are found in the cholesterol degradation cluster, echA19 (Rv3516) and echA20 (Rv3550), though neither is required for Mtb growth on cholesterol (Griffin et al., 2011, Nesbitt et al., 2010). Using a promoter trap screen, echA19 was found to be essential for infection in THP-1 human macrophages (Dubnau et al., 2002).
The echA19 gene is located adjacent to FadD19 in the genome, although it is transcribed in the opposite direction. Its location suggests that EchA19 catalyzes the hydration of the steroid-26-enoyl-CoA, which has an 8-carbon side chain.
The echA20 gene is transcriptionally regulated by KstR2, and was found to be up-regulated significantly in R. jostii RHA1 when the strain is grown on cholesterol or HIP (Casabon et al., 2013c). This regulation suggests that EchA20 plays a role in C & D ring catabolism. It should be noted that R. jostii RHA1 contains an additional hydratase in the KstR2 regulon that is not present in Mycobacteria again highlighting the differences in metabolic capabilities between Mycobacteria and Rhodococcus.
Two additional (S)-enoyl-CoA hydratases have been implicated in cholesterol metabolism. The echA9 gene is required for growth on cholesterol, although its expression is not regulated by cholesterol. The expression of echA9 is under the control of the transcriptional regulator SigE (Rohde et al., 2012). The echA13 gene is in the same operon as fadE17 and fadE18; an operon that is up-regulated by cholesterol and under the control of the transcriptional repressor Mce3R (De La Paz Santangelo et al., 2009). Their role in cholesterol metabolism has yet to be determined.
Two genes of the igr operon, Rv3541c and Rv3542c encode MaoC-like (R)-enoyl-CoA hydratases, which in bacteria are often involved in carbon storage through polyhydroxyalkanoate biosynthesis. The gene products of Rv3541c and Rv3542c form a heteromeric complex, just like ChsE1-ChsE2 (Thomas et al., 2011) also encoded in the igr operon. Based on the metabolite accumulated in the Δigr knockout and the enzymatic activity profile of ChsE1-ChsE2, we proposed that the heteromeric Rv3541c-Rv3542c enzyme catalyzes hydration of 3-oxo-23,24-bisnorchol-4,20-dien-22-oyl-CoA (Thomas et al., 2011). Addition of the (R)-hydratase family to the 21 echA enoyl-coA hydratases encoded in the Mtb genome increases the number of candidate enoyl-CoA hydratases involved in lipid or sterol metabolism.
Characterized (R)-hydratases in bacteria, including those in Aeromonas caviae (A. caviae) (Fukui et al., 1998), Pseudomonas aeruginosa (Tsuge et al., 2000), Rhodospirillum rubrum (Reiser et al., 2000), and E. coli (Park and Lee, 2003) are involved in the biosynthesis of a class of carbon storage polyesters known as polyhydroxyalkanoates (PHA). (3R)-hydroxyacyl-CoA monomers produced by (R)-hydratases are shunted towards PHA synthesis and polymerized by PHA synthase (Fukui et al., 1998, Tsuge et al., 2000). To the best of our knowledge, Mtb does not synthesize PHAs and lacks an obvious PHA synthase, a key enzyme in this pathway.
In mammals, (R)-hydratases are part of multifunctional enzyme 2 (MFE-2), which contains three functional domains with (3R)-hydroxyacyl-CoA dehydrogenase, (R)-hydratase, and sterol carrier protein activities. MFE-2 has been shown to be important for β-oxidation of very-long-chain and α-methyl-branched fatty acids, as well as bile acid synthesis (Koski et al., 2005). For example, MFE-2 in rat liver peroxisomes has been shown to be involved in cholic acid synthesis (Qin et al., 1997). The biosynthesis of cholic acid requires shortening of the side chain of C27 steroids by β-oxidation. Recombinant (R)-hydratase is able to catalyze the hydration of (24E)-3α,7α,12α-trihydroxy-5β-cholest-24-enoyl-CoA, an intermediate in bile acid synthesis. In rats, ΔMFE-2 knockout mice accumulate very long chain fatty acids, branched chain fatty acids, and bile acid intermediates (Baes et al., 2000).
The Mtb gene hsd4B (Rv3538) is annotated as a probable (R)-enoyl-CoA hydratase with a conserved hydratase motif including important catalytic residues. The hsd4B gene is located in the cholesterol-regulated region of the genome and is regulated by KstR1. Furthermore, it is adjacent to kstD, which is known to be involved in the A-ring metabolism of cholesterol by Mtb. However, its substrate has not been identified. Likewise, the enoyl-CoA hydratase(s) responsible for the first two cycles of β-oxidation of the cholesterol side chain have not yet been identified.
β-hydroxy-acyl-CoA dehydrogenases
No β-hydroxy-acyl-CoA dehydrogenase encoding genes are located in the cholesterol regulon, and it is not clear which genes are required for this step in the β-oxidation pathway. Mtb FadB (Rv0860) has been shown to be part of the trifunctional enzyme (TFE) complex required for fatty acid metabolism (Venkatesan and Wierenga, 2013). Recombinantly expressed Mtb FadB2 (Rv0468) can turn over β-hydroxybutyryl-CoA in vitro in the presence of NAD+ (Taylor et al., 2010). The M. smegmatis fadB2 mutant, which shares 84% identity at the amino acid level to Mtb FadB2, was not impaired in growth using various carbon sources including glycerol, glucose, acetate, propionate, caprylic acid, capric acid, lauric acid, myristic acid, palmitic acid, oleic acid, linoleic acid, or cholesterol as the sole carbon source (Taylor et al., 2010). The fadB2 gene is located adjacent to icl1 (Rv0467) in the genome and is conserved among Mycobacteria. FadB3 (Rv1715) is not conserved in Mycobacteria and conflicting reports make its importance for survival questionable (Lamichhane et al., 2003, Williams et al., 2011, Sassetti and Rubin, 2003). Recombinant FadB3 was not active with β-hydroxybutyryl-CoA with NAD+ or NADP+ (Taylor, 2011).
The Mtb genes encoding FadB4 (Rv3141) and FadB5 (Rv1912c) are annotated as probable quinone oxidoreductases and are unlikely to encode the β-hydroxy-acyl-CoA dehydrogenases involved in side chain oxidation. Coincidentally, the fadB4 gene is located directly upstream of fadE23 and fadE24 (which are down-regulated by cholesterol, but up-regulated in a human THP-1 macrophage infection model) and is transcribed in the same direction, although there is no evidence to show that they share an operon (Fontan et al., 2008a).
Mtb Hsd4A (Rv3502c) has been proposed to be the β-hydroxy-acyl-CoA dehydrogenase involved in side chain β-oxidation because it is required for growth on cholesterol and is similar to 17β-hydroxysteroid dehydrogenase (Griffin et al., 2011). The biochemical function of Hsd4A has not been demonstrated, but it is a likely candidate for catalysis of oxidation of β-hydroxy acyl-CoAs in the sterol side chain.
3-ketoacyl-CoA thiolases
Two acyl-CoA thiolases are found in the cholesterol regulon, fadA5 (Rv3546) and fadA6 (Rv3556c). Only fadA5 is required for growth of Mtb on cholesterol, suggesting it is involved in cholesterol metabolism (Nesbitt et al., 2010). Transcription of fadA5 is up-regulated in macrophages and the mouse lung, whereas fadA6 is not (Dubnau et al., 2005, Dubnau et al., 2002). An Mtb ΔfadA5 knockout strain shows growth attenuation in the mouse model of infection; cfu in the lungs showed a marked decrease after 8 weeks of infection compared to wild type and complement strains (Nesbitt et al., 2010).
Nesbitt et al. demonstrated in vitro that this enzyme, which contains an active-site cysteine nucleophile and a general acid/general base histidine and cysteine can catalyze thiolysis of acetoacetyl-CoA and is therefore a thiolase (Nesbitt et al., 2010). Wild-type Mtb cultured with cholesterol accumulates AD and ADD and these oxidized steroids are secreted in small amounts, while these two steroid metabolites are not detected in the ΔfadA5 knockout strain, thus demonstrating that FadA5 is required for their production and likely the thiolase that catalyzes carbon-carbon cleavage in the steroid side chain (Figure 4). This ΔfadA5 knockout strain blocked cholesterol metabolism since it was unable to grow on cholesterol as a sole carbon source, but it did not accumulate any toxic metabolites and was able to grow in the presence of cholesterol. These experiments demonstrate that FadA5 is required for full metabolism of the side chain of cholesterol. However, it is unclear if an additional thiolase like FadA6 is required for β-oxidation of the cholesterol side chain or if two cycles are catalyzed by FadA5.
Lipid transfer proteins and alternative side chain metabolism
What is known about the role of the lipid transfer protein genes (ltp) in Mtb is limited. However, three genes annotated as lipid transfer proteins are regulated by cholesterol: ltp2 (Rv3540c), ltp3 (Rv3523), and ltp4 (Rv3522). We proposed on the basis of the igr operon metabolite profiling and enzymatic activities that Ltp2 is responsible for the thermodynamically downhill retroaldol C17-C20 cleavage in the last step of steroid side chain degradation (Thomas et al., 2011). This catalytic step would provide the C17 ketone, deviating from the classical β-oxidation pathway in this respect (Figure 4). Sih had originally proposed in the 1960s that this step occurred non-enzymatically (Sih et al., 1967, Sih et al., 1968a). However, the genomic and bioinformatic evidence suggest that an enzymatic catalyst that shares some active site similarities with thiolases (FadAs) is responsible. Recombinantly expressed Ltp2 in E. coli was found to be insoluble (Thomas et al., 2011) and biochemical verification of this activity remains to be established.
In Pseudomonas sp. strain Chol1, it has been proposed on the basis of gene knockout and metabolite studies, and assays in cell lysates that cleavage of the 24-oyl-CoA intermediate produces an aldehyde metabolite rather than a thioester via a classical β-oxidation pathway. This cleavage is catalyzed by an aldolase (shy) that is homolgous to Ltp3 from R. rhodochrous DSM 43269 (Holert et al., 2013a, Holert et al., 2013b). The aldehyde is then oxidized to the carboxylic acid by an aldehyde dehydrogenase (sad) and esterified by a CoA ligase. There are Ltp orthologs in Mtb that are both cholesterol and KstR1 regulated (ltp2, ltp3, and ltp4) and acyl-CoA ligases, e.g. FadD17. One apparent aldehyde dehydrogenase ortholog, Rv0223c, is in the KstR1 regulon and regulated by cholesterol. However, it is remote from the Cho region of the genome. The most similar gene to sad in Mtb is Rv0147, but it is also remotely located from the Cho region of the genome, and not regulated by cholesterol or KstR1; thus less likely to be required for side-chain metabolism. The presence in the Mtb genome of both thiolases (FadAs) and aldolases (Ltps) points to the possibility that cholesterol metabolites can be processed through multiple competing pathways in Mtb. However, in Mtb, steroid side chain aldehyde formation and dehydrogenase-catalyzed oxidation activities remain to be established.
Beyond cholesterol degradation
Linkage of Mtb cholesterol degradation to the metabolic pool
Mtb metabolism in the intracellular environment of the host and during conditions of stress, like hypoxia, in vitro, depends upon the up-regulation of oxidative genes that are able to break down cholesterol, fatty acid and complex lipids into C2 (acetate) and C3 (propionate) carbon units (Galagan et al., 2013). Acetate and propionate are then fed into other metabolic pathways to produce ATP and biosynthetic intermediates.
Propionate can be generated as a product of cholesterol, methyl- and odd-branched fatty acid, and amino acid metabolism. Propionate is toxic to bacterial cells and therefore needs to be channeled into appropriate detoxification pathways to prevent accumulation. Mtb incorporates propionate into the biosynthesis of new lipids, some of which contribute to sustaining infection and virulence, like sulfolipid (SL-1) and phthiocerol dimycocerosates (PDIM) (Jain et al., 2007, Yang et al., 2009, Cox et al., 1999, Rousseau et al., 2004). In the absence of exogenous sources of carbon (carbohydrates or sugar), there is an increase in the molecular weight of PDIM for Mtb grown in vitro on cholesterol, and these data in conjunction with our understanding of the lipid diet of Mtb in vivo support the hypothesis that this is an adaptive strategy to deal with a toxic catabolic product in a way that might enhance virulence (Yang et al., 2009). Likewise, phenolic glycolipid (PGL), which is structurally related to PDIM, is hypothesized to cause hypervirulence in certain Mtb isolates, as well as down-regulate the host immune response, specifically the release of important pro-inflammatory cytokines like TNFα, IL-6, and IL-12 (Reed et al., 2004).
Requisite to the utilization of propionate is its conversion from propionyl-CoA to methylmalonyl-CoA through the methylmalonyl pathway or succinate through the methylcitrate cycle (Savvi et al., 2008) (Figure 6). (S)-Methylmalonyl-CoA is generated from propionyl-CoA and bicarbonate in a condensation reaction catalyzed by propionyl-CoA carboxylase (EC 6.4.1.3). In the TCA cycle, condensation of oxaloacetate with acetyl-CoA to generate citrate is catalyzed by citrate (Si)-synthase (EC 2.3.3.1). In the methylcitrate cycle, the enzyme 2-methylcitrate synthase (EC 2.3.3.5) catalyzes the condensation of oxaloacetate with propionyl-CoA resulting in the formation of methylcitrate instead of citrate. This molecule is isomerized to methylisocitrate, which can go on to form succinate and pyruvate that feed into other metabolic pathways.
Figure 6.
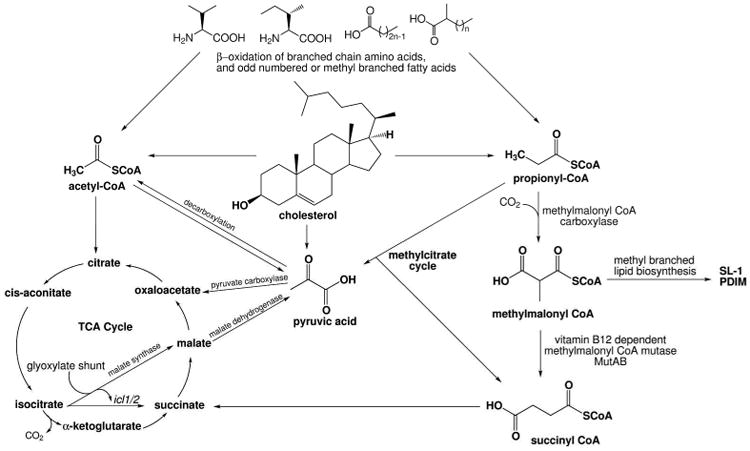
Cholesterol catabolism generates acetyl-CoA, propionyl-CoA, and pyruvate, which are fed into the metabolic pathways shown. PDIM: phthiocerol dimycoserate; SL-1: sulfolipid-1.
Acetate generated during cholesterol, fatty acid, and lipid metabolism can condense with oxaloacetate to form citrate, which is isomerized to isocitrate. Isocitrate is then converted to glyoxylate and succinate by isocitrate lyase (Icl), an essential enzyme in the glyoxylate shunt. This shunt avoids two decarboxylation steps and allows the formation of four carbon subunits from acetate, a necessary process for an organism to obtain carbon and energy from acetate.
The isocitrate lyase genes (icl1 and icl2) are up-regulated when Mtb is grown on fatty acids in vitro, as well as in vivo when harvested from infected mouse lungs (Timm et al., 2003, Munoz-Elias et al., 2006). In knockout models, each icl gene is dispensable and deletion does not affect bacterial viability for infection. However, deletion of both genes (Δicl1/Δicl2) results in fully attenuated virulence in an in vivo mouse infection model (Munoz-Elias et al., 2006).
The Δicl1/Δicl2 Mtb strain is completely unable to grow on propionate or acetate as a carbon source, and this was attributed to the fact that they can act as both isocitrate lyase and methylisocitrate lyase genes. However, because of their dual activity, it was initially unclear whether or not these genes were essential in vivo because they participated in the glyoxylate cycle or the methylcitrate cycle. An Mtb double-mutant deficient in the methylcitrate cycle genes methylcitrate synthase (prpC) and methylcitrate dehydratase (prpD) is completely unable to grow on propionate as a carbon source in vitro or in murine bone marrow-derived macrophages. This phenotype implies that the methyl citrate cycle plays an important role in managing the toxic propionate. It was therefore puzzling that the ΔprpDC knockout was totally viable in a mouse infection model up to 20 weeks post infection, with spleen and lung pathology nearly identical to the wild-type (Munoz-Elias et al., 2006). The researchers concluded that the methylcitrate cycle is dispensable in vivo during infection. The corollary being that propionate flux is managed differently in vivo.
Cholesterol-associated genes remaining to be annotated
In addition to the significant number of oxidation and regulatory genes involved in cholesterol utilization discussed so far, there are still many others that remain to be studied in order to validate their role in the cholesterol utilization pathway. The regulation of cholesterol metabolic genes is complex, and likely reflects an underlying set of sub-pathways, all of which make use of steroid degradation intermediates for various reasons.
In the Cho region, nine genes encode PE (Pro–Glu) or PPE (Pro–Pro–Glu) glycine-rich proteins, part of a family with highly conserved N-terminal proline and glutamine motifs of unclear function (Cole et al., 1998). Of the remaining 74 genes in this region of the Mtb genome, probable functions have been assigned to approximately 60% (Table). These assignments account for the cholesterol metabolism pathway described in Figures 3 and 4. In vitro biochemical assays have been performed to establish function for 27 of the corresponding 43 gene products. Putative enzyme class has been assigned to about half of the 31 (40% of the Cho region) genes for which no preliminary biochemical evidence of function exists.
Outside the Cho region, there are an additional 150 Mtb genes significantly up-regulated in the presence of cholesterol as compared to glycerol (Nesbitt et al., 2010). A large number of genes identified through forward genetics as essential for Mtb pathogenesis and virulence (Sassetti and Rubin, 2003) or essential for growth on cholesterol (Griffin et al., 2011) overlap with these cholesterol-regulated genes that do not have functions within any known cholesterol utilization pathway. Their regulation could be a secondary effect from a metabolite produced during the cholesterol degradation process (propionate, for example), or it could be that these genes are feeding cholesterol metabolites into other yet unknown pathways.
Mce3R
There are three additional mammalian cell entry (mce) regulons in Mtb proposed to be involved in host-TB interactions, which include lipid transport and virulence: mce1, mce2, and mce3 (Santangelo et al., 2002, Mitra et al., 2005, Uchida et al., 2007, Gioffre et al., 2005, Arruda et al., 1993, Mohn et al., 2008). Like Mtb Δmce4, Mtb Δmce3 has an attenuated phenotype in vivo when used to infect C57BL/6 mice (Senaratne et al., 2008). The survival times of the mice, the cfu count in the lungs, and lung post-infection pathology suggest that this repressor is important for virulence. Likewise, BALB/c immunocompetent mice infected with Δmce1, Δmce2, and Δmce3 gene knockouts of Mtb H37Rv all survived, whereas mice infected with the wild-type strain begin to die within five weeks, and after nine weeks were no longer living (Gioffre et al., 2005).
The Mce3 regulon consists of three operons near each other in the genome, including the mce3 operon, whose expression is self-regulated (Table) (Santangelo et al., 2008, Santangelo et al., 2002). The Mce3 regulon is under control of the Mce3R TetR-like transcriptional repressor (Rv1963), as was demonstrated using transcriptional profiling of the Δmce3R knockout strain (De La Paz Santangelo et al., 2009). The bioinformatically-annotated functions of these genes include general oxidoreductases, β-oxidation, and lipid transport genes (De La Paz Santangelo et al., 2009).
Interestingly, one of the three operons that is part of the mce3 regulon is also regulated by cholesterol in Mtb, Rv1933c–Rv1935c (Table) (Nesbitt et al., 2010). These three genes encode an ACAD α subunit and an ACAD β subunit proposed to form a heteterotetrameric ACAD (Wipperman et al., 2013) and an enoyl-CoA hydratase. The substrates for these enzymes are unknown, but a recent finding might offer a clue to their putative function.
Mycobacterium bovis (M. bovis) strain Type 17 is the fastest spreading strain of M. bovis in the United Kingdom, and it costs farmers about 3 billion dollars in lost revenue annually. The only difference between this strain and other types of M. bovis is the absence of seven genes, the orthologs of Mtb Rv1928c-Rv1936, which include the cholesterol-regulated operon in the Mce3 regulon. Lipid radiolabeling studies were performed to compare the lipid profiles of M. bovis wild type to the Type 17. M. bovis Type 17 has a low flux of propionate into acetyl-CoA, which is used for the biosynthesis of mycolic acids (Wheeler et al., 2008). Instead, propionate labeling accumulates in pyruvate, which M. bovis naturally lacks the ability to biosynthesize, since it does not have functional ald or pykA genes. The absence of these β-oxidation genes might therefore offer M. bovis Type 17 a compensatory mechanism to generate an intracellular pool of pyruvate, which could offer an interesting evolutionary advantage for this organism. In the case of Mtb, cholesterol metabolism is known to increase the metabolic pool of propionate (Yang et al., 2009) and the mce3-regulon may be required to distribute the propionate. The fact that the mce3 virulence regulon encodes a heterotetrameric ACAD that appears unique to cholesterol-metabolism pathways (Wipperman et al., 2013) strengthens the linkage between the mce3 regulon and cholesterol metabolism. The direct connection between cholesterol metabolism and mce3-operon encoded enzyme function remains to be elucidated.
SigE
SigE, a stress-induced extracytoplasmic function sigma factor, is a transcriptional activator whose 16-gene regulon includes fadE23 and fadE24 (Fontan et al., 2008b). SigE is regulated by SigH under oxidative stress (Raman et al., 2001) and is predicted to be involved in expression and control of genes involved in cell envelope maintenance and lipid recycling. SigE regulon expression is up-regulated post-phagocytosis, indicating that it may be involved in virulence or evasion of the host's immune system (Schnappinger et al., 2003). Deletion of sigE in Mtb resulted in an attenuated ability for Mtb to kill its murine host (Ando et al., 2003). Interestingly, transcriptional profiling experiments have demonstrated that fadE23-fadE24 is down-regulated by cholesterol (Nesbitt et al., 2010) but up-regulated during THP-1 human macrophage infection (Fontan et al., 2008a). The distinctive heterotetrameric FadE23-FadE24 ACAD structure suggests that the substrate or allosteric regulator contains a steroid framework (Wipperman et al., 2013). The connection between cholesterol metabolism, the predicted structure of FadE23-FadE24, and SigE remains to be established, and is an example of the complexity of cholesterol metabolism in Mtb.
Envoi
Understanding the environment within which an intracellular pathogen resides throughout different stages of its lifecycle is complex, since it is by nature multifactorial and temporal. Evidence continues to accumulate that cholesterol utilization and metabolism contribute to pathogenesis and virulence of Mtb throughout the course of clinical infection and disease. Why Mtb uses this ubiquitous steroid is still a fertile area of enquiry just over a century after the first report of cholesterol metabolism by Mycobacteria (Söhngen, 1913).
Cholesterol is just one molecule present in the intracellular host environment, but it is an important molecule. We have presented what is known of the cholesterol metabolic pathway for which gene and enzyme functions have been elucidated. There are an additional ∼150 genes out of 230-250 genes postulated to have a role involving cholesterol metabolism which have yet to have their function experimentally identified.
The important role that cholesterol plays in the lifecycle of this pathogen is evident, and the need for new antibiotic therapies in the face of bacterial drug resistance is more urgent than ever. A more complete understanding of cholesterol utilizing enzymes coupled with small-molecule inhibitors of their function could be used to either eliminate Mtb outright if these inhibitors were to act as bactericidal antibiotics, or to alter Mtb metabolism to make it more susceptible to current anti-mycobacterial therapies. Future work in this field should continue to focus on elucidating the specific function(s) of recombinantly expressed genes that are found to be important during infection and how these functions contribute to the virulence and pathogenesis of Mtb in order to support strategic development of new treatments for Mtb infection.
Acknowledgments
We thank all the members of the Sampson laboratory as well as our many collaborators who contributed to the work described in this review.
The work in the authors' laboratory was supported by NIH (AI092455, AI085349, AI065251, HL53306, RR021008, N.S.S.), an instrument grant from NSF (BIO1039771) and a DOE-GAANN fellowship (S.T.T.).
Footnotes
Declaration of Interest: The authors declare no conflict of interest.
References
- Global Tuberculosis Report [Online] [Accessed January 4 2014];The World Health Organization. Available: http://apps.who.int/iris/bitstream/10665/75938/1/9789241564502_eng.pdf.
- Ando M, Yoshimatsu T, Ko C, Converse PJ, Bishai WR. Deletion of Mycobacterium tuberculosis sigma factor E results in delayed time to death with bacterial persistence in the lungs of aerosol-infected mice. Infect Immun. 2003;71:7170–2. doi: 10.1128/IAI.71.12.7170-7172.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andor A, Jekkel A, Hopwood DA, Jeanplong F, Ilkoy E, Konya A, Kurucz I, Ambrus G. Generation of useful insertionally blocked sterol degradation pathway mutants of fast-growing mycobacteria and cloning, characterization, and expression of the terminal oxygenase of the 3-ketosteroid 9alpha-hydroxylase in Mycobacterium smegmatis mc(2)155. Appl Environ Microbiol. 2006;72:6554–9. doi: 10.1128/AEM.00941-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda S, Bomfim G, Knights R, Huima-Byron T, Riley LW. Cloning of an M. tuberculosis DNA fragment associated with entry and survival inside cells. Science. 1993;261:1454–7. doi: 10.1126/science.8367727. [DOI] [PubMed] [Google Scholar]
- Baes M, Huyghe S, Carmeliet P, Declercq PE, Collen D, Mannaerts GP, Van Veldhoven PP. Inactivation of the peroxisomal multifunctional protein-2 in mice impedes the degradation of not only 2-methyl-branched fatty acids and bile acid intermediates but also of very long chain fatty acids. J Biol Chem. 2000;275:16329–36. doi: 10.1074/jbc.M001994200. [DOI] [PubMed] [Google Scholar]
- Beste DJ, Noh K, Niedenfuhr S, Mendum TA, Hawkins ND, Ward JL, Beale MH, Wiechert W, Mcfadden J. 13C-flux spectral analysis of host-pathogen metabolism reveals a mixed diet for intracellular Mycobacterium tuberculosis. Chem Biol. 2013;20:1012–21. doi: 10.1016/j.chembiol.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode HB, Zeggel B, Silakowski B, Wenzel SC, Reichenbach H, Muller R. Steroid biosynthesis in prokaryotes: identification of myxobacterial steroids and cloning of the first bacterial 2,3(S)-oxidosqualene cyclase from the myxobacterium Stigmatella aurantiaca. Mol Microbiol. 2003;47:471–81. doi: 10.1046/j.1365-2958.2003.03309.x. [DOI] [PubMed] [Google Scholar]
- Brzostek A, Dziadek B, Rumijowska-Galewicz A, Pawelczyk J, Dziadek J. Cholesterol oxidase is required for virulence of Mycobacterium tuberculosis. FEMS Microbiol Lett. 2007;275:106–112. doi: 10.1111/j.1574-6968.2007.00865.x. [DOI] [PubMed] [Google Scholar]
- Brzostek A, Pawelczyk J, Rumijowska-Galewicz A, Dziadek B, Dziadek J. Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J Bacteriol. 2009;191:6584–91. doi: 10.1128/JB.00488-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzostek A, Sliwinski T, Rumijowska-Galewicz A, Korycka-Machala M, Dziadek J. Identification and targeted disruption of the gene encoding the main 3-ketosteroid dehydrogenase in Mycobacterium smegmatis. Microbiology. 2005;151:2393–402. doi: 10.1099/mic.0.27953-0. [DOI] [PubMed] [Google Scholar]
- Bushnell LD, Haas HF. The Utilization of Certain Hydrocarbons by Microorganisms. J Bacteriol. 1941;41:653–73. doi: 10.1128/jb.41.5.653-673.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho LR, Ensergueix D, Perez E, Gicquel B, Guilhot C. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol Microbiol. 1999;34:257–67. doi: 10.1046/j.1365-2958.1999.01593.x. [DOI] [PubMed] [Google Scholar]
- Campbell JW, Cronan JE., Jr The enigmatic Escherichia coli fadE gene is yafH. J Bacteriol. 2002;184:3759–64. doi: 10.1128/JB.184.13.3759-3764.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camus JC, Pryor MJ, Medigue C, Cole ST. Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology. 2002;148:2967–73. doi: 10.1099/00221287-148-10-2967. [DOI] [PubMed] [Google Scholar]
- Capyk JK, Casabon I, Gruninger R, Strynadka NC, Eltis LD. Activity of 3-ketosteroid 9alpha-hydroxylase (KshAB) indicates cholesterol side chain and ring degradation occur simultaneously in Mycobacterium tuberculosis. J Biol Chem. 2011;286:40717–24. doi: 10.1074/jbc.M111.289975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capyk JK, D'angelo I, Strynadka NC, Eltis LD. Characterization of 3-ketosteroid 9-α-hydroxylase, a Rieske oxygenase in the cholesterol degradation pathway of Mycobacterium tuberculosis. J Biol Chem. 2009a;284:9937–46. doi: 10.1074/jbc.M900719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capyk JK, Kalscheuer R, Stewart GR, Liu J, Kwon H, Zhao R, Okamoto S, Jacobs WR, Jr, Eltis LD, Mohn WW. Mycobacterial cytochrome P450 125 (Cyp125) catalyzes the terminal hydroxylation of C27 steroids. J Biol Chem. 2009b;284:35534–42. doi: 10.1074/jbc.M109.072132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carere J, Mckenna SE, Kimber MS, Seah SY. Characterization of an aldolase-dehydrogenase complex from the cholesterol degradation pathway of Mycobacterium tuberculosis. Biochemistry. 2013;52:3502–11. doi: 10.1021/bi400351h. [DOI] [PubMed] [Google Scholar]
- Casabon I, Crowe AM, Liu J, Eltis LD. FadD3 is an acyl-CoA synthetase that initiates catabolism of cholesterol rings C and D in actinobacteria. Mol Microbiol. 2013a;87:269–83. doi: 10.1111/mmi.12095. [DOI] [PubMed] [Google Scholar]
- Casabon I, Swain K, Crowe AM, Eltis LD, Mohn WW. Actinobacterial acyl-CoA synthetases involved in steroid side chain catabolism. J Bacteriol. 2013b doi: 10.1128/JB.01012-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casabon I, Zhu SH, Otani H, Liu J, Mohn WW, Eltis LD. Regulation of the KstR2 regulon of Mycobacterium tuberculosis by a cholesterol catabolite. Mol Microbiol. 2013c;89:1201–12. doi: 10.1111/mmi.12340. [DOI] [PubMed] [Google Scholar]
- Caspi R, Altman T, Dale JM, Dreher K, Fulcher CA, Gilham F, Kaipa P, Karthikeyan AS, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Paley S, Popescu L, Pujar A, Shearer AG, Zhang P, Karp PD. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2010;38:D473–9. doi: 10.1093/nar/gkp875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC, Harik NS, Liao RP, Sherman DR. Identification of Mycobacterial genes that alter growth and pathology in macrophages and in mice. J Infect Dis. 2007;196:788–95. doi: 10.1086/520089. [DOI] [PubMed] [Google Scholar]
- Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, Sherman DR. igr Genes and Mycobacterium tuberculosis cholesterol metabolism. J Bacteriol. 2009;191:5232–9. doi: 10.1128/JB.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, Mclean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Cox JS, Chen B, Mcneil M, Jacobs WR., Jr Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 1999;402:79–83. doi: 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- De Carvalho LP, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem Biol. 2010;17:1122–31. doi: 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- De La Paz Santangelo M, Klepp L, Nunez-Garcia J, Blanco FC, Soria M, Garcia-Pelayo MC, Bianco MV, Cataldi AA, Golby P, Jackson M, Gordon SV, Bigi F. Mce3R, a TetR-type transcriptional repressor, controls the expression of a regulon involved in lipid metabolism in Mycobacterium tuberculosis. Microbiology. 2009;155:2245–55. doi: 10.1099/mic.0.027086-0. [DOI] [PubMed] [Google Scholar]
- Donova MV. Transformation of steroids by actinobacteria: a review. Prikl Biokhim Mikrobiol. 2007;43:5–18. [PubMed] [Google Scholar]
- Dresen C, Lin LY, D'angelo I, Tocheva EI, Strynadka N, Eltis LD. A flavin-dependent monooxygenase from Mycobacterium tuberculosis involved in cholesterol catabolism. J Biol Chem. 2010;285:22264–75. doi: 10.1074/jbc.M109.099028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll MD, Mclean KJ, Cheesman MR, Jowitt TA, Howard M, Carroll P, Parish T, Munro AW. Expression and characterization of Mycobacterium tuberculosis CYP144: common themes and lessons learned in the M. tuberculosis P450 enzyme family. Biochim Biophys Acta. 2011;1814:76–87. doi: 10.1016/j.bbapap.2010.05.015. [DOI] [PubMed] [Google Scholar]
- Drzyzga O, Fernandez De Las Heras L, Morales V, Navarro Llorens JM, Perera J. Cholesterol degradation by Gordonia cholesterolivorans. Appl Environ Microbiol. 2011;77:4802–10. doi: 10.1128/AEM.05149-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnau E, Chan J, Mohan VP, Smith I. Responses of mycobacterium tuberculosis to growth in the mouse lung. Infect Immun. 2005;73:3754–7. doi: 10.1128/IAI.73.6.3754-3757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnau E, Fontan P, Manganelli R, Soares-Appel S, Smith I. Mycobacterium tuberculosis genes induced during infection of human macrophages. Infect Immun. 2002;70:2787–95. doi: 10.1128/IAI.70.6.2787-2795.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA. 1999;282:677–86. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- Fleischmann RD, Alland D, Eisen JA, Carpenter L, White O, Peterson J, Deboy R, Dodson R, Gwinn M, Haft D, Hickey E, Kolonay JF, Nelson WC, Umayam LA, Ermolaeva M, Salzberg SL, Delcher A, Utterback T, Weidman J, Khouri H, Gill J, Mikula A, Bishai W, Jacobs Jr WR, Jr, Venter JC, Fraser CM. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol. 2002;184:5479–90. doi: 10.1128/JB.184.19.5479-5490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontan P, Aris V, Ghanny S, Soteropoulos P, Smith I. Global transcriptional profile of Mycobacterium tuberculosis during THP-1 human macrophage infection. Infect Immun. 2008a;76:717–25. doi: 10.1128/IAI.00974-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontan PA, Aris V, Alvarez ME, Ghanny S, Cheng J, Soteropoulos P, Trevani A, Pine R, Smith I. Mycobacterium tuberculosis sigma factor E regulon modulates the host inflammatory response. J Infect Dis. 2008b;198:877–85. doi: 10.1086/591098. [DOI] [PubMed] [Google Scholar]
- Fukui T, Shiomi N, Doi Y. Expression and characterization of (R)-specific enoyl coenzyme A hydratase involved in polyhydroxyalkanoate biosynthesis by Aeromonas caviae. J Bacteriol. 1998;180:667–73. doi: 10.1128/jb.180.3.667-673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan JE, Minch K, Peterson M, Lyubetskaya A, Azizi E, Sweet L, Gomes A, Rustad T, Dolganov G, Glotova I, Abeel T, Mahwinney C, Kennedy AD, Allard R, Brabant W, Krueger A, Jaini S, Honda B, Yu WH, Hickey MJ, Zucker J, Garay C, Weiner B, Sisk P, Stolte C, Winkler JK, Van De Peer Y, Iazzetti P, Camacho D, Dreyfuss J, Liu Y, Dorhoi A, Mollenkopf HJ, Drogaris P, Lamontagne J, Zhou Y, Piquenot J, Park ST, Raman S, Kaufmann SH, Mohney RP, Chelsky D, Moody DB, Sherman DR, Schoolnik GK. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature. 2013;499:178–83. doi: 10.1038/nature12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Sampson NS. A GMC Oxidoreductase Homologue Is Required for Acetylation of Glycopeptidolipid in Mycobacterium smegmatis. Biochemistry. 2014;53:611–3. doi: 10.1021/bi4015083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García JL, Uhía I, García E, Galán B. Bacterial degradation of cholesterol and other contaminant sterols. In: Koukkou AI, editor. Microbial bioremediation of non-metals: current research. Norfolk, UK: Caister Academic Press; 2011. [Google Scholar]
- Ghai R, Mcmahon KD, Rodriguez-Valera F. Breaking a paradigm: cosmopolitan and abundant freshwater actinobacteria are low GC. Environ Microbiol Rep. 2012;4:29–35. doi: 10.1111/j.1758-2229.2011.00274.x. [DOI] [PubMed] [Google Scholar]
- Gioffre A, Infante E, Aguilar D, Santangelo MP, Klepp L, Amadio A, Meikle V, Etchechoury I, Romano MI, Cataldi A, Hernandez RP, Bigi F. Mutation in mce operons attenuates Mycobacterium tuberculosis virulence. Microbes Infect. 2005;7:325–34. doi: 10.1016/j.micinf.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han R. Is Hypercholesterolemia a Friend or a Foe of Tuberculosis? Infect Immun. 2009;77:3514–3515. doi: 10.1128/IAI.00469-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg JA. Steroids, the steroid community, and Upjohn in perspective: a profile of innovation. Steroids. 1992;57:593–616. doi: 10.1016/0039-128x(92)90013-y. [DOI] [PubMed] [Google Scholar]
- Holert J, Jagmann N, Philipp B. The essential function of genes for a hydratase and an aldehyde dehydrogenase for growth of Pseudomonas sp. strain Chol1 with the steroid compound cholate indicates an aldolytic reaction step for deacetylation of the side chain. J Bacteriol. 2013a;195:3371–80. doi: 10.1128/JB.00410-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holert J, Kulic Z, Yucel O, Suvekbala V, Suter MJ, Moller HM, Philipp B. Degradation of the acyl side chain of the steroid compound cholate in Pseudomonas sp. strain Chol1 proceeds via an aldehyde intermediate. J Bacteriol. 2013b;195:585–95. doi: 10.1128/JB.01961-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinouchi M, Hayashi T, Koshino H, Kudo T. ORF18-disrupted mutant of Comamonas testosteroni TA441 accumulates significant amounts of 9,17-dioxo-1,2,3,4,10,19-hexanorandrostan-5-oic acid and its derivatives after incubation with steroids. J Steroid Biochem Mol Biol. 2006;101:78–84. doi: 10.1016/j.jsbmb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Horinouchi M, Hayashi T, Koshino H, Kurita T, Kudo T. Identification of 9,17-dioxo-1,2,3,4,10,19-hexanorandrostan-5-oic acid, 4-hydroxy-2-oxohexanoic acid, and 2-hydroxyhexa-2,4-dienoic acid and related enzymes involved in testosterone degradation in Comamonas testosteroni TA441. Appl Environ Microbiol. 2005;71:5275–81. doi: 10.1128/AEM.71.9.5275-5281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinouchi M, Hayashi T, Kudo T. Steroid degradation in Comamonas testosteroni. J Steroid Biochem Mol Biol. 2012;129:4–14. doi: 10.1016/j.jsbmb.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Horinouchi M, Hayashi T, Yamamoto T, Kudo T. A new bacterial steroid degradation gene cluster in Comamonas testosteroni TA441 which consists of aromatic-compound degradation genes for seco-steroids and 3-ketosteroid dehydrogenase genes. Appl Environ Microbiol. 2003;69:4421–30. doi: 10.1128/AEM.69.8.4421-4430.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinouchi M, Kurita T, Yamamoto T, Hatori E, Hayashi T, Kudo T. Steroid degradation gene cluster of Comamonas testosteroni consisting of 18 putative genes from meta-cleavage enzyme gene tesB to regulator gene tesR. Biochem Biophys Res Commun. 2004;324:597–604. doi: 10.1016/j.bbrc.2004.09.096. [DOI] [PubMed] [Google Scholar]
- Horinouchi M, Yamamoto T, Taguchi K, Arai H, Kudo T. Meta-cleavage enzyme gene tesB is necessary for testosterone degradation in Comamonas testosteroni TA441. Microbiology. 2001;147:3367–75. doi: 10.1099/00221287-147-12-3367. [DOI] [PubMed] [Google Scholar]
- Horinouchi S, Ishizuka H, Beppu T. Cloning, nucleotide sequence, and transcriptional analysis of the NAD(P)-dependent cholesterol dehydrogenase gene from a Nocardia sp. and its hyperexpression in Streptomyces spp. Appl Environ Microbiol. 1991;57:1386–93. doi: 10.1128/aem.57.5.1386-1393.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Van Der Geize R, Besra GS, Gurcha SS, Liu A, Rohde M, Singh M, Coates A. 3-Ketosteroid 9alpha-hydroxylase is an essential factor in the pathogenesis of Mycobacterium tuberculosis. Mol Microbiol. 2010;75:107–21. doi: 10.1111/j.1365-2958.2009.06957.x. [DOI] [PubMed] [Google Scholar]
- Ishizaki T, Hirayama N, Shinkawa H, Nimi O, Murooka Y. Nucleotide sequence of the gene for cholesterol oxidase from a Streptomyces sp. J Bacteriol. 1989;171:596–601. doi: 10.1128/jb.171.1.596-601.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashina TV, Nikolayeva VM, Dovbnya DV, Donova MV. Cholesterol oxidase ChoD is not a critical enzyme accounting for oxidation of sterols to 3-keto-4-ene steroids in fast-growing Mycobacterium sp. VKM Ac-1815D. J Steroid Biochem Mol Biol. 2012;129:47–53. doi: 10.1016/j.jsbmb.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Jain M, Petzold CJ, Schelle MW, Leavell MD, Mougous JD, Bertozzi CR, Leary JA, Cox JS. Lipidomics reveals control of Mycobacterium tuberculosis virulence lipids via metabolic coupling. Proc Natl Acad Sci U S A. 2007;104:5133–8. doi: 10.1073/pnas.0610634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JB, Ouellet H, Ortiz De Montellano PR. Functional redundancy of steroid C26-monooxygenase activity in Mycobacterium tuberculosis revealed by biochemical and genetic analyses. J Biol Chem. 2010;285:36352–60. doi: 10.1074/jbc.M110.161117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameda K, Nunn WD. Purification and characterization of acyl coenzyme A synthetase from Escherichia coli. J Biol Chem. 1981;256:5702–7. [PubMed] [Google Scholar]
- Kang Y, Zarzycki-Siek J, Walton CB, Norris MH, Hoang TT. Multiple FadD acyl-CoA synthetases contribute to differential fatty acid degradation and virulence in Pseudomonas aeruginosa. PLoS One. 2010;5:e13557. doi: 10.1371/journal.pone.0013557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann G, Thole H, Kraft R, Atrat P. Steroid-1-dehydrogenase of Rhodococcus erythropolis: purification and N-terminal amino acid sequence. J Steroid Biochem Mol Biol. 1992;43:297–301. doi: 10.1016/0960-0760(92)90164-e. [DOI] [PubMed] [Google Scholar]
- Kendall SL, Burgess P, Balhana R, Withers M, Ten Bokum A, Lott JS, Gao C, Uhía-Castro I, Stoker NG. Cholesterol utilization in mycobacteria is controlled by two TetR-type transcriptional regulators: KstR and KstR2. Microbiology. 2010;156:1362–71. doi: 10.1099/mic.0.034538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall SL, Withers M, Soffair CN, Moreland NJ, Gurcha S, Sidders B, Frita R, Ten Bokum A, Besra GS, Lott JS, Stoker NG. A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis. Mol Microbiol. 2007;65:684–99. doi: 10.1111/j.1365-2958.2007.05827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare G, Gupta V, Gupta RK, Gupta R, Bhat R, Tyagi AK. Dissecting the role of critical residues and substrate preference of a Fatty Acyl-CoA Synthetase (FadD13) of Mycobacterium tuberculosis. PLoS One. 2009;4:e8387. doi: 10.1371/journal.pone.0008387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, Visser A, Wang W, Hsu FF, Wiehart U, Tsenova L, Kaplan G, Russell DG. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med. 2010;2:258–74. doi: 10.1002/emmm.201000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi K, Watazu Y, Katayama Y, Okabe H. The characteristics and applications of recombinant cholesterol dehydrogenase. Biosci Biotechnol Biochem. 2000;64:1352–8. doi: 10.1271/bbb.64.1352. [DOI] [PubMed] [Google Scholar]
- Klink M, Brzezinska M, Szulc I, Brzostek A, Kielbik M, Sulowska Z, Dziadek J. Cholesterol oxidase is indispensable in the pathogenesis of Mycobacterium tuberculosis. PLoS One. 2013;8:e73333. doi: 10.1371/journal.pone.0073333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knol J, Bodewits K, Hessels GI, Dijkhuizen L, Van Der Geize R. 3-Keto-5α-steroid Δ(1)-dehydrogenase from Rhodococcus erythropolis SQ1 and its orthologue in Mycobacterium tuberculosis H37Rv are highly specific enzymes that function in cholesterol catabolism. Biochem J. 2008;410:339–46. doi: 10.1042/BJ20071130. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Rittmann BE. Microbial removal of hazardous organic compounds. Environ Sci Technol. 1982;16:170A–183A. [Google Scholar]
- Koski KM, Haapalainen AM, Hiltunen JK, Glumoff T. Crystal structure of 2-enoyl-CoA hydratase 2 from human peroxisomal multifunctional enzyme type 2. J Mol Biol. 2005;345:1157–69. doi: 10.1016/j.jmb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Lack NA, Yam KC, Lowe ED, Horsman GP, Owen RL, Sim E, Eltis LD. Characterization of a carbon-carbon hydrolase from Mycobacterium tuberculosis involved in cholesterol metabolism. J Biol Chem. 2010;285:434–43. doi: 10.1074/jbc.M109.058081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarca BB, Zhu W, Arceneaux JE, Byers BR, Lundrigan MD. Participation of fad and mbt genes in synthesis of mycobactin in Mycobacterium smegmatis. J Bacteriol. 2004;186:374–82. doi: 10.1128/JB.186.2.374-382.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane G, Zignol M, Blades NJ, Geiman DE, Dougherty A, Grosset J, Broman KW, Bishai WR. A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2003;100:7213–8. doi: 10.1073/pnas.1231432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechat P, Hummel L, Rousseau S, Moszer I. GenoList: an integrated environment for comparative analysis of microbial genomes. Nucleic Acids Res. 2008;36:D469–74. doi: 10.1093/nar/gkm1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew JM, Kapopoulou A, Jones LM, Cole ST. TubercuList--10 years after. Tuberculosis (Edinb) 2011;91:1–7. doi: 10.1016/j.tube.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Lin G, Li D, De Carvalho LP, Deng H, Tao H, Vogt G, Wu K, Schneider J, Chidawanyika T, Warren JD, Li H, Nathan C. Inhibitors selective for mycobacterial versus human proteasomes. Nature. 2009;461:621–6. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machang'u RS, Prescott JF. Purification and properties of cholesterol oxidase and choline phosphohydrolase from Rhodococcus equi. Can J Vet Res. 1991;55:332–40. [PMC free article] [PubMed] [Google Scholar]
- Malaviya A, Gomes J. Androstenedione production by biotransformation of phytosterols. Bioresour Technol. 2008;99:6725–37. doi: 10.1016/j.biortech.2008.01.039. [DOI] [PubMed] [Google Scholar]
- Mcguire AM, Weiner B, Park ST, Wapinski I, Raman S, Dolganov G, Peterson M, Riley R, Zucker J, Abeel T, White J, Sisk P, Stolte C, Koehrsen M, Yamamoto RT, Iacobelli-Martinez M, Kidd MJ, Maer AM, Schoolnik GK, Regev A, Galagan J. Comparative analysis of Mycobacterium and related Actinomycetes yields insight into the evolution of Mycobacterium tuberculosis pathogenesis. BMC Genomics. 2012;13:120. doi: 10.1186/1471-2164-13-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mclean KJ, Lafite P, Levy C, Cheesman MR, Mast N, Pikuleva IA, Leys D, Munro AW. The structure of Mycobacterium tuberculosis CYP125: molecular basis for cholesterol binding in a P450 needed for host infection. J Biol Chem. 2009;284:35524–33. doi: 10.1074/jbc.M109.032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra D, Saha B, Das D, Wiker HG, Das AK. Correlating sequential homology of Mce1A, Mce2A, Mce3A and Mce4A with their possible functions in mammalian cell entry of Mycobacterium tuberculosis performing homology modeling. Tuberculosis (Edinb) 2005;85:337–45. doi: 10.1016/j.tube.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Mohn WW, Van Der Geize R, Stewart GR, Okamoto S, Liu J, Dijkhuizen L, Eltis LD. The actinobacterial mce4 locus encodes a steroid transporter. J Biol Chem. 2008;283:35368–74. doi: 10.1074/jbc.M805496200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn WW, Wilbrink MH, Casabon I, Stewart GR, Liu J, Van Der Geize R, Eltis LD. Gene cluster encoding cholate catabolism in Rhodococcus spp. J Bacteriol. 2012;194:6712–9. doi: 10.1128/JB.01169-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Elias EJ, Upton AM, Cherian J, Mckinney JD. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol. 2006;60:1109–22. doi: 10.1111/j.1365-2958.2006.05155.x. [DOI] [PubMed] [Google Scholar]
- Nesbitt NM, Yang X, Fontan P, Kolesnikova I, Smith I, Sampson NS, Dubnau E. A thiolase of Mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol. Infect Immun. 2010;78:275–82. doi: 10.1128/IAI.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet H, Guan S, Johnston JB, Chow ED, Kells PM, Burlingame AL, Cox JS, Podust LM, De Montellano PR. Mycobacterium tuberculosis CYP125A1, a steroid C27 monooxygenase that detoxifies intracellularly generated cholest-4-en-3-one. Mol Microbiol. 2010;77:730–42. doi: 10.1111/j.1365-2958.2010.07243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci USA. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang L, Tian X, Pan W, Xie J. Structure and function of mycobacterium glycopeptidolipids from comparative genomics perspective. J Cell Biochem. 2013;114:1705–13. doi: 10.1002/jcb.24515. [DOI] [PubMed] [Google Scholar]
- Park SJ, Lee SY. Identification and characterization of a new enoyl coenzyme A hydratase involved in biosynthesis of medium-chain-length polyhydroxyalkanoates in recombinant Escherichia coli. J Bacteriol. 2003;185:5391–7. doi: 10.1128/JB.185.18.5391-5397.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DH, Murray HC. Microbiological oxygenation of steroids at carbon-11. J Am Chem Soc. 1952;74:1871–1872. [Google Scholar]
- Pethe K, Sequeira PC, Agarwalla S, Rhee K, Kuhen K, Phong WY, Patel V, Beer D, Walker JR, Duraiswamy J, Jiricek J, Keller TH, Chatterjee A, Tan MP, Ujjini M, Rao SP, Camacho L, Bifani P, Mak PA, Ma I, Barnes SW, Chen Z, Plouffe D, Thayalan P, Ng SH, Au M, Lee BH, Tan BH, Ravindran S, Nanjundappa M, Lin X, Goh A, Lakshminarayana SB, Shoen C, Cynamon M, Kreiswirth B, Dartois V, Peters EC, Glynne R, Brenner S, Dick T. A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat Commun. 2010;1:57. doi: 10.1038/ncomms1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrusma M, Dijkhuizen L, Van Der Geize R. Rhodococcus rhodochrous DSM 43269 3-ketosteroid 9alpha-hydroxylase, a two-component iron-sulfur-containing monooxygenase with subtle steroid substrate specificity. Appl Environ Microbiol. 2009;75:5300–7. doi: 10.1128/AEM.00066-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrusma M, Hessels G, Dijkhuizen L, Van Der Geize R. Multiplicity of 3-Ketosteroid-9alpha-Hydroxylase enzymes in Rhodococcus rhodochrous DSM43269 for specific degradation of different classes of steroids. J Bacteriol. 2011;193:3931–40. doi: 10.1128/JB.00274-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, Daffe M, Emile JF, Marchou B, Cardona PJ, De Chastellier C, Altare F. Foamy macrophages from tuberculous patients' granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008;4:e1000204. doi: 10.1371/journal.ppat.1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin YM, Haapalainen AM, Conry D, Cuebas DA, Hiltunen JK, Novikov DK. Recombinant 2-enoyl-CoA hydratase derived from rat peroxisomal multifunctional enzyme 2: role of the hydratase reaction in bile acid synthesis. Biochem J. 1997;328(Pt 2):377–82. doi: 10.1042/bj3280377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman S, Song T, Puyang X, Bardarov S, Jacobs WR, Jr, Husson RN. The alternative sigma factor SigH regulates major components of oxidative and heat stress responses in Mycobacterium tuberculosis. J Bacteriol. 2001;183:6119–25. doi: 10.1128/JB.183.20.6119-6125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos JL, Martinez-Bueno M, Molina-Henares AJ, Teran W, Watanabe K, Zhang X, Gallegos MT, Brennan R, Tobes R. The TetR family of transcriptional repressors. Microbiol Mol Biol Rev. 2005;69:326–56. doi: 10.1128/MMBR.69.2.326-356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed MB, Domenech P, Manca C, Su H, Barczak AK, Kreiswirth BN, Kaplan G, Barry CE., 3rd A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 2004;431:84–7. doi: 10.1038/nature02837. [DOI] [PubMed] [Google Scholar]
- Reichman LB, Tanne JH. Timebomb: the global epidemic of multi-drug-resistant tuberculosis. New York: McGraw-Hill; 2002. [Google Scholar]
- Reiser SE, Mitsky TA, Gruys KJ. Characterization and cloning of an (R)-specific trans-2,3-enoylacyl-CoA hydratase from Rhodospirillum rubrum and use of this enzyme for PHA production in Escherichia coli. Appl Microbiol Biotechnol. 2000;53:209–18. doi: 10.1007/s002530050010. [DOI] [PubMed] [Google Scholar]
- Rengarajan J, Bloom BR, Rubin EJ. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci U S A. 2005;102:8327–32. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HE. The persistence of tuberculous infections. Am J Pathol. 1933;9:711–718. 1. [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GM, Voskuil MI, Gold B, Schoolnik GK, Smith I. ideR, An essential gene in Mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect Immun. 2002;70:3371–81. doi: 10.1128/IAI.70.7.3371-3381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde KH, Veiga DF, Caldwell S, Balazsi G, Russell DG. Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Pathog. 2012;8:e1002769. doi: 10.1371/journal.ppat.1002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosloniec KZ, Wilbrink MH, Capyk JK, Mohn WW, Ostendorf M, Van Der Geize R, Dijkhuizen L, Eltis LD. Cytochrome P450 125 (CYP125) catalyses C26-hydroxylation to initiate sterol side-chain degradation in Rhodococcus jostii RHA1. Mol Microbiol. 2009;74:1031–43. doi: 10.1111/j.1365-2958.2009.06915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau C, Winter N, Pivert E, Bordat Y, Neyrolles O, Ave P, Huerre M, Gicquel B, Jackson M. Production of phthiocerol dimycocerosates protects Mycobacterium tuberculosis from the cidal activity of reactive nitrogen intermediates produced by macrophages and modulates the early immune response to infection. Cell Microbiol. 2004;6:277–87. doi: 10.1046/j.1462-5822.2004.00368.x. [DOI] [PubMed] [Google Scholar]
- Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nature Immunol. 2009;10:943–8. doi: 10.1038/ni.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo MP, Blanco FC, Bianco MV, Klepp LI, Zabal O, Cataldi AA, Bigi F. Study of the role of Mce3R on the transcription of mce genes of Mycobacterium tuberculosis. BMC Microbiol. 2008;8:38. doi: 10.1186/1471-2180-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo MP, Goldstein J, Alito A, Gioffre A, Caimi K, Zabal O, Zumarraga M, Romano MI, Cataldi AA, Bigi F. Negative transcriptional regulation of the mce3 operon in Mycobacterium tuberculosis. Microbiology. 2002;148:2997–3006. doi: 10.1099/00221287-148-10-2997. [DOI] [PubMed] [Google Scholar]
- Santer M, Ajl SJ, Turner RA. Steroid metabolism by a species of Pseudomonas. I. J Biol Chem. 1952;198:397–404. [PubMed] [Google Scholar]
- Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A. 2003;100:12989–94. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvi S, Warner DF, Kana BD, Mckinney JD, Mizrahi V, Dawes SS. Functional characterization of a vitamin B12-dependent methylmalonyl pathway in Mycobacterium tuberculosis: implications for propionate metabolism during growth on fatty acids. J Bacteriol. 2008;190:3886–95. doi: 10.1128/JB.01767-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanga CA, Mohan VP, Joseph H, Yu K, Chan J, Flynn JL. Reactivation of latent tuberculosis: variations on the Cornell murine model. Infect Immun. 1999;67:4531–8. doi: 10.1128/iai.67.9.4531-4538.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer G, Guler R, Murray G, Brombacher F, Brown GD. The role of scavenger receptor B1 in infection with Mycobacterium tuberculosis in a murine model. PLoS One. 2009;4:e8448. doi: 10.1371/journal.pone.0008448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol. 2009;5:e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorey JS, Sweet L. The mycobacterial glycopeptidolipids: structure, function, and their role in pathogenesis. Glycobiology. 2008;18:832–41. doi: 10.1093/glycob/cwn076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senaratne RH, Sidders B, Sequeira P, Saunders G, Dunphy K, Marjanovic O, Reader JR, Lima P, Chan S, Kendall S, Mcfadden J, Riley LW. Mycobacterium tuberculosis strains disrupted in mce3 and mce4 operons are attenuated in mice. J Med Microbiol. 2008;57:164–70. doi: 10.1099/jmm.0.47454-0. [DOI] [PubMed] [Google Scholar]
- Sih CJ. Mechanisms of steroid oxidation by microorganisms. Biochim Biophys Acta. 1962;62:541–7. doi: 10.1016/0006-3002(62)90236-6. [DOI] [PubMed] [Google Scholar]
- Sih CJ, Tai HH, Tsong YY. The mechanism of microbial conversion of cholesterol into 17-keto steroids. J Am Chem Soc. 1967;89:1957–1958. doi: 10.1021/ja00984a039. [DOI] [PubMed] [Google Scholar]
- Sih CJ, Tai HH, Tsong YY, Lee SS, Coombe RG. Mechanisms of steroid oxidation by microorganisms. XIV. Pathway of cholesterol side-chain degradation. Biochemistry. 1968a;7:808–18. doi: 10.1021/bi00842a039. [DOI] [PubMed] [Google Scholar]
- Sih CJ, Wang KC, Tai HH. Mechanisms of steroid oxidation by microorganisms. 13. C22 acid intermediates in the degradation of the cholesterol side chain. Biochemistry. 1968b;7:796–807. doi: 10.1021/bi00842a038. [DOI] [PubMed] [Google Scholar]
- Simeone R, Leger M, Constant P, Malaga W, Marrakchi H, Daffe M, Guilhot C, Chalut C. Delineation of the roles of FadD22, FadD26 and FadD29 in the biosynthesis of phthiocerol dimycocerosates and related compounds in Mycobacterium tuberculosis. FEBS J. 2010;277:2715–25. doi: 10.1111/j.1742-464X.2010.07688.x. [DOI] [PubMed] [Google Scholar]
- Söhngen NL. Benzin Petroleum Paraffinöl und Paraffin als Kohlenstoff- und energiequelle für Mikroben. Zentralbl Bakteriol Parasitenkd Infektionskr Hyg Abt. 1913;37:595–609. [Google Scholar]
- Stadtman TC, Cherkes A, Anfinsen CB. Studies on the microbiological degradation of cholesterol. J Biol Chem. 1954;206:511–23. [PubMed] [Google Scholar]
- Stern JR, Del Campillo A. Enzymes of fatty acid metabolism. II. Properties of crystalline crotonase. J Biol Chem. 1956;218:985–1002. [PubMed] [Google Scholar]
- Stover CK, Warrener P, Vandevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, Mcmurray DN, Kreiswirth BN, Barry CE, Baker WR. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405:962–6. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- Strijewski A. The steroid-9 alpha-hydroxylation system from Nocardia species. Eur J Biochem. 1982;128:125–35. doi: 10.1111/j.1432-1033.1982.tb06942.x. [DOI] [PubMed] [Google Scholar]
- Tak JD. On bacteria decomposing cholesterol. Antonie Van Leeuwenhoek. 1942;8:32–40. [Google Scholar]
- Talalay P, Dobson MM, Tapley DF. Oxidative degradation of testosterone by adaptive enzymes. Nature. 1952;170:620–1. doi: 10.1038/170620a0. [DOI] [PubMed] [Google Scholar]
- Taylor RC. Mycobacterial fatty acid metabolism: identification of novel drug targets and chemotherapeutics. University of Birmingham; 2011. Ph.D. [Google Scholar]
- Taylor RC, Brown AK, Singh A, Bhatt A, Besra GS. Characterization of a beta-hydroxybutyryl-CoA dehydrogenase from Mycobacterium tuberculosis. Microbiology. 2010;156:1975–82. doi: 10.1099/mic.0.038802-0. [DOI] [PubMed] [Google Scholar]
- Thomas ST, Sampson NS. Mycobacterium tuberculosis utilizes a unique heterotetrameric structure for dehydrogenation of the cholesterol side chain. Biochemistry. 2013;52:2895–2904. doi: 10.1021/bi4002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas ST, Vanderven BC, Sherman DR, Russell DG, Sampson NS. Pathway profiling in Mycobacterium tuberculosis: elucidation of cholesterol-derived catabolite and enzymes that catalyze its metabolism. J Biol Chem. 2011;286:43668–78. doi: 10.1074/jbc.M111.313643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timm J, Post FA, Bekker LG, Walther GB, Wainwright HC, Manganelli R, Chan WT, Tsenova L, Gold B, Smith I, Kaplan G, Mckinney JD. Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc Natl Acad Sci U S A. 2003;100:14321–6. doi: 10.1073/pnas.2436197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi OA, Arora P, Sridharan V, Tickoo R, Mohanty D, Gokhale RS. Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature. 2004;428:441–5. doi: 10.1038/nature02384. [DOI] [PubMed] [Google Scholar]
- Tsuge T, Fukui T, Matsusaki H, Taguchi S, Kobayashi G, Ishizaki A, Doi Y. Molecular cloning of two (R)-specific enoyl-CoA hydratase genes from Pseudomonas aeruginosa and their use for polyhydroxyalkanoate synthesis. FEMS Microbiol Lett. 2000;184:193–8. doi: 10.1111/j.1574-6968.2000.tb09013.x. [DOI] [PubMed] [Google Scholar]
- Turfitt GE. Microbiological agencies in the degradation of steroids; steroid utilization by the microflora of soils. J Bacteriol. 1947;54:557–62. doi: 10.1128/JB.54.5.557-562.1947. [DOI] [PubMed] [Google Scholar]
- Uchida Y, Casali N, White A, Morici L, Kendall LV, Riley LW. Accelerated immunopathological response of mice infected with Mycobacterium tuberculosis disrupted in the mce1 operon negative transcriptional regulator. Cell Microbiol. 2007;9:1275–83. doi: 10.1111/j.1462-5822.2006.00870.x. [DOI] [PubMed] [Google Scholar]
- Udwadia ZF, Amale RA, Ajbani KK, Rodrigues C. Totally drug-resistant tuberculosis in India. Clin Infect Dis. 2012;54:579–81. doi: 10.1093/cid/cir889. [DOI] [PubMed] [Google Scholar]
- Uhía I, Galán B, Kendall SL, Stoker NG, García JL. Cholesterol metabolism in Mycobacterium smegmatis. Environ Microbiol Rep. 2012;4:168–82. doi: 10.1111/j.1758-2229.2011.00314.x. [DOI] [PubMed] [Google Scholar]
- Uhía I, Galán B, Medrano FJ, García JL. Characterization of the KstR-dependent promoter of the gene for the first step of the cholesterol degradative pathway in Mycobacterium smegmatis. Microbiology. 2011a;157:2670–80. doi: 10.1099/mic.0.049213-0. [DOI] [PubMed] [Google Scholar]
- Uhía I, Galán B, Morales V, García JL. Initial step in the catabolism of cholesterol by Mycobacterium smegmatis mc2155. Environ Microbiol. 2011b;13:943–59. doi: 10.1111/j.1462-2920.2010.02398.x. [DOI] [PubMed] [Google Scholar]
- Van Der Geize R, Dijkhuizen L. Harnessing the catabolic diversity of rhodococci for environmental and biotechnological applications. Curr Opin Microbiol. 2004;7:255–61. doi: 10.1016/j.mib.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Van Der Geize R, Grommen AW, Hessels GI, Jacobs AA, Dijkhuizen L. The steroid catabolic pathway of the intracellular pathogen Rhodococcus equi is important for pathogenesis and a target for vaccine development. PLoS Pathog. 2011;7:e1002181. doi: 10.1371/journal.ppat.1002181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Geize R, Hessels GI, Van Gerwen R, Van Der Meijden P, Dijkhuizen L. Molecular and functional characterization of kshA and kshB, encoding two components of 3-ketosteroid 9alpha-hydroxylase, a class IA monooxygenase, in Rhodococcus erythropolis strain SQ1. Mol Microbiol. 2002;45:1007–18. doi: 10.1046/j.1365-2958.2002.03069.x. [DOI] [PubMed] [Google Scholar]
- Van Der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim E, Dijkhuizen L, Davies JE, Mohn WW, Eltis LD. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A. 2007;104:1947–52. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan R, Wierenga RK. Structure of mycobacterial beta-oxidation trifunctional enzyme reveals its altered assembly and putative substrate channeling pathway. ACS Chem Biol. 2013;8:1063–73. doi: 10.1021/cb400007k. [DOI] [PubMed] [Google Scholar]
- Villemagne B, Crauste C, Flipo M, Baulard AR, Déprez B, Willand N. Tuberculosis: The drug development pipeline at a glance. Eur J Med Chem. 2012;51:1–16. doi: 10.1016/j.ejmech.2012.02.033. [DOI] [PubMed] [Google Scholar]
- Waksman SA. Streptomycin: background, isolation, properties, and utilization. Science. 1953;118:259–66. doi: 10.1126/science.118.3062.259. [DOI] [PubMed] [Google Scholar]
- Wanders RJ, Komen J, Ferdinandusse S. Phytanic acid metabolism in health and disease. Biochim Biophys Acta. 2011;1811:498–507. doi: 10.1016/j.bbalip.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Wang SF, Kawahara FS, Talalay P. The mechanism of the delta5-3-ketosteroid isomerase reaction: absorption and fluorescence spectra of enzyme-steroid complexes. J Biol Chem. 1963;238:576–85. [PubMed] [Google Scholar]
- Wheeler PR, Brosch R, Coldham NG, Inwald JK, Hewinson RG, Gordon SV. Functional analysis of a clonal deletion in an epidemic strain of Mycobacterium bovis reveals a role in lipid metabolism. Microbiology. 2008;154:3731–42. doi: 10.1099/mic.0.2008/022269-0. [DOI] [PubMed] [Google Scholar]
- Wilbrink MH, Petrusma M, Dijkhuizen L, Van Der Geize R. FadD19 of Rhodococcus rhodochrous DSM43269, a steroid-coenzyme A ligase essential for degradation of C-24 branched sterol side chains. Appl Environ Microbiol. 2011;77:4455–64. doi: 10.1128/AEM.00380-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KJ, Boshoff HI, Krishnan N, Gonzales J, Schnappinger D, Robertson BD. The Mycobacterium tuberculosis beta-oxidation genes echA5 and fadB3 are dispensable for growth in vitro and in vivo. Tuberculosis (Edinb) 2011;91:549–55. doi: 10.1016/j.tube.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipperman MF, Yang M, Thomas ST, Sampson NS. Shrinking the FadE proteome of Mycobacterium tuberculosis: insights into cholesterol metabolism through identification of an alpha2beta2 heterotetrameric acyl coenzyme A dehydrogenase family. J Bacteriol. 2013;195:4331–41. doi: 10.1128/JB.00502-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam KC, D'angelo I, Kalscheuer R, Zhu H, Wang JX, Snieckus V, Ly LH, Converse PJ, Jacobs WR, Jr, Strynadka N, Eltis LD. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 2009;5:e1000344. doi: 10.1371/journal.ppat.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Dubnau E, Smith I, Sampson NS. Rv1106c from Mycobacterium tuberculosis is a 3-β-hydroxysteroid dehydrogenase. Biochemistry. 2007;46:9058–67. doi: 10.1021/bi700688x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Gao J, Smith I, Dubnau E, Sampson NS. Cholesterol is not an essential source of nutrition for Mycobacterium tuberculosis during infection. J Bacteriol. 2011;193:1473–6. doi: 10.1128/JB.01210-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Nesbitt NM, Dubnau E, Smith I, Sampson NS. Cholesterol metabolism increases the metabolic pool of propionate in Mycobacterium tuberculosis. Biochemistry. 2009;48:3819–21. doi: 10.1021/bi9005418. [DOI] [PMC free article] [PubMed] [Google Scholar]