

Abstract
Chronic (>24 h) exposure of arsenite, an environmental toxicant, has shown the decreased nitric oxide (NO) production in endothelial cells (EC) by decreasing endothelial NO synthase (eNOS) expression and/or its phosphorylation at serine 1179 (eNOS-Ser1179 in bovine sequence), which is associated with increased risk of vascular diseases. Here, we investigated the acute (<24 h) effect of arsenite on NO production using bovine aortic EC (BAEC). Arsenite acutely increased the phosphorylation of eNOS-Thr497, but not of eNOS-Ser116 or eNOS-Ser1179, which was accompanied by decreased NO production. The level of eNOS expression was unaltered under this condition. Treatment with arsenite also induced reactive oxygen species (ROS) production, and pretreatment with a ROS scavenger N-acetyl-L-cysteine (NAC) completely reversed the observed effect of arsenite on eNOS-Thr497 phosphorylation. Although protein kinase C (PKC) and protein phosphatase 1 (PP1) were reported to be involved in eNOS-Thr497 phosphorylation, treatment with PKC inhibitor, Ro318425, and overexpression of various PKC isoforms did not affect the arsenite-stimulated eNOS-Thr497 phosphorylation. In contrast, treatment with PP1 inhibitor, calyculin A, mimicked the observed effect of arsenite on eNOS-Thr497 phosphorylation. Lastly, we found decreased cellular PP1 activity in arsenite-treated cells, which was reversed by NAC. Overall, our study demonstrates firstly that arsenite acutely decreases NO production at least in part by increasing eNOS-Thr497 phosphorylation via ROS-PP1 signaling pathway, which provide the molecular mechanism underlying arsenite-induced increase in vascular disease.
Keywords: Arsenite, Vascular disease, Nitric oxide, Endothelial nitric oxide synthase, Reactive oxygen species, Protein phosphatase 1
INTRODUCTION
Arsenic, a toxicant in foods and environmental media such as soil and water, is the 20th most abundant element in the earth crust (Mandal and Suzuki, 2002). Because 140 million people worldwide are at risk of exposure to excessive levels of naturally occurring arsenic in well water and groundwater (Hall et al., 2009), exposure of arsenic in drinking water is a serious public health problem. Many epidemiological studies have shown that arsenic exposure is linked to not only cancers but also vascular diseases such as arteriosclerosis, hypertension, and Blackfoot disease (Stea et al., 2014).
Nitric oxide (NO) in endothelial cells (EC) is a key molecule with multiple functions, including vasodilation and many antiatherogenic properties. The production of NO is mainly regulated by endothelial NO synthase (eNOS) and therefore its dysregulation is thought to contribute to the pathogenesis of vasodilation-related diseases such as atherosclerosis and hypertension (Isenovic et al., 2011). It is known that eNOS is mainly controlled at the level of its phosphorylation (Rafikov et al., 2011). Several specific sites of phosphorylation have been identified among which, eNOS at serine 1179 (eNOS-Ser1179; in bovine sequence) has been the most studied. The phosphorylation of eNOS-Ser1179 increases NO production, which is mediated by several specific protein kinases, including Akt, AMP-activated protein kinase (AMPK), calmodulin-dependent kinase II (CaMKII), protein kinase A (PKA), and check point kinase 1 (Park et al., 2011; Rafikov et al., 2011). In addition to kinases, protein phosphatase 2A (PP2A) is also reported to be involved in the level of eNOS-Ser1179 phosphorylation (Park et al., 2013). Conversely, the phosphorylation of eNOS-Thr497 decreases eNOS activity, which is mediated by AMPK (Chen et al., 1999), PKC (Fleming et al., 2001; Matsubara et al., 2003) or ROCK (Watts and Motley, 2009). This site is also dephosphorylated by PP1 and PP2A, which results in an increase in NO production (Michell et al., 2001; Greif et al., 2002). Like eNOS-The497 phosphorylation, the phosphorylation of eNOS-Ser116 decreases eNOS activity and NO production. In basal EC, we reported that the phosphorylation of eNOS-Ser116 is mediated by cyclin-dependent kinase 5 (CDK5) and c-Jun N-terminal kinase 2 (Cho et al., 2010; Park et al., 2012). Very recently, we further reported that the inhibition of CDK5-mediated eNOS-Ser116 phosphorylation is a major mechanism by which valproic acid increases NO production and that this process was mediated by SH2 domain-containing protein tyrosine phosphatase 1 (Cho et al., 2014).
Decreased NO bioavailability in endothelium is implicated in the pathology of arsenic poisoning (Kumagai and Pi, 2004). For example, studies in an endemic area of chronic arsenic poisoning in inner Mongolia (Pi et al., 2000) and in arsenite-administered rats (Lee et al., 2003) revealed that serum concentration of stable NO metabolites was lower in arsenic-exposed subjects than controls. In EC, the treatment with arsenite was also reported to suppress eNOS activity (Pi et al., 2000; Lee et al., 2003) and NO production (Barchowsky et al., 1999), although there are several conflicting reports showing that arsenite increases NO production (Liu and Jan, 2000; Kao et al., 2003). In this study, we reexamined the effect of arsenite on NO production and its underlying molecular mechanism, in particular its acute effect, because so far most studies have evaluated the effect of chronic arsenite exposure on NO production. Our result showed that arsenite acutely decreased NO production at least in part by phosphorylating eNOS-Thr497 via reactive oxygen species (ROS)-stimulated inhibition of PP1 activity.
MATERIALS AND METHODS
Materials
Sodium arsenite (NaAsO2, used as arsenite) was purchased from VWR international (West Chester, PA, USA). Calyculin A, okadaic acid and Ro318425 were obtained from Calbiochem (Nottingham, UK). N-Acetyl-L-cysteine (NAC) and 2′,7′-dichlorofluorescin diacetate (DCFH-DA) were purchased from Sigma (St. Louis, MO, USA). Antibodies against eNOS, p-eNOS-Ser1179, p-eNOS-Thr497, and p-eNOS-Ser116 were obtained from BD Transduction Laboratories (Lexington, KY, USA) and Upstate (Lake Placid, NY, USA), respectively. Antibodies against PP1α, β-actin, and all corresponding secondary antibodies were purchased from Santa Cruz Biotech (Santa Cruz, CA, USA). Lipofectamine 2000, Minimal essential medium (MEM), Dulbecco’s phosphate-buffered saline (DPBS), newborn calf serum (NCS), penicillin-streptomycin antibiotics, L-glutamine, trypsin-EDTA solution, and plasticware for cell culture were obtained from Gibco-BRL (Gaithersberg, MD, USA). All other chemicals were of the purest analytical grade.
Cell culture, drug treatments, and transfection
Bovine aortic EC (BAEC) were isolated and cultured exactly as described previously (Kim et al., 1999) and maintained in MEM supplemented with 5% NCS at 37°C under 5% CO2 in air. EC were confirmed by their typical cobblestone configuration when viewed by light microscopy and by a positive indirect immunofluorescence test for von Willebrand factor VIII. The cells between passages 5 and 10 were used for all experiments. When BAEC were grown to confluence, the cells were further maintained for the indicated times in MEM with 5% NCS containing various concentrations of sodium arsenite. In some experiments, the cells were treated with various chemicals for 0.5 h before arsenite treatment. Transfection was done exactly as described (Cho et al., 2010). Briefly, pcDNA3.1 mammalian expression vector containing cDNA (each 3 μg) encoding haemagglutinin A (HA)-tagged dominant negative (DN)-PKC (a kind gift from Professor Jae-Won Soh, Department of Chemistry, Inha University, Korea) was transfected into the cells grown to 70% confluence in 60 mm dishes using Lipofectamine 2000, according to the manufacturer’s instructions. For control, equal amounts of pcDNA3.1 vector were also transfected.
Western blot analysis
For Western blot analysis, BAEC treated with sodium arsenite in the absence or presence of various chemicals were washed with ice-cold DPBS and lysed in lysis buffer (20 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA) containing 1×Protease Inhibitor Cocktail™ (Roche Molecular Biochemicals, Indianapolis, IN) and 1× Phosphatase Inhibitor Cocktail 2 (Sigma). The protein concentrations were determined using a BCA protein assay kit (Sigma). Equal quantities of protein (20 μg) were separated on sodium dodecyl sulfate polyacrylamide gel under reducing conditions and then electrophoretically transferred onto nitrocellulose membranes. The blots were then probed with the appropriate antibodies, followed by the corresponding secondary antibodies, and finally developed using ECL reagents (Amersham Biosciences, Arlington Heights, IL, USA).
Measurement of NO release
NO released by BAEC was measured as nitrite (a stable metabolite of NO) concentration in cell culture supernatant as described (Cho et al., 2004). The culture medium in 100 mm dish was changed to 1 ml of Kreb’s buffer (pH 7.4; 118 mM NaCl, 4.6 mM KCl, 27.2 mM NaHCO3, 1.2 mM MgSO4, 2.5 mM CaCl2, 1.2 mM KH2PO4, 11.1 mM glucose) and incubate for 1 h at 37°C. Two hundred μl of each supernatant was then carefully transferred into a 96-well plate, with the subsequent addition of 100 μl of Griess reagent (50 μl of 1% sulfanilamide containing 5% phosphoric acid and 50 μl of 0.1% N-(1-naphthyl) ethylenediamine). After color development at room temperature for 15 min, absorbance was measured on a microplate reader at a 530 nm wavelength.
Cell viability assay
Cell viability assay was carried out as described (Park et al., 2011) using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) with minor modifications. BAEC grown in 96-well culture plates were treated with 30 μM sodium arsenite for various times (up to 36 h). After treatments, cells were incubated with 5 mg/ml MTT and further incubated for 2 h at 37°C. Dimethylsulfoxide (DMSO) (200 μL) was added to the cells, incubated for 10 min more and absorbance read at 570 nm using a 96-well microtiter plate reader.
Measurement of intracellular reactive oxygen species (ROS) formation
Production of ROS was measured using an oxidation-sensitive fluorescent probe DCFH-DA, based on the ROS-dependent oxidation of DCFH-DA to 2′,7′-dichlorofluorescein (DCF), as described (Lee et al., 2007). BAEC treated with 30 μM sodium arsenite for various times were detached with trypsin-EDTA, washed with PBS, and then treated with 2 μM DCFH-DA in PBS at 37°C for 15 min. After wash twice with PBS, cells were immediately monitored with FACSCalibur flow cytometry (BD Biosciences, San Jose, CA, USA) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm. ROS production was determined by comparing the changes in fluorescence intensity in drug-treated cells with that in the control.
Measurement of PP1 activity assay
PP1 activity assay was carried out using ProFluor® Ser/Thr PPase Assay kit (Promega, Madison, WI, USA) according to the manufacturer’s instruction. Briefly, cells were treated with 30 μM sodium arsenite or vehicle for 4 h, and lysed with lysis buffer (20 mM Tris-Cl at pH 7.4, 132 mM NaCl, 1% Triton X-100) containing 1×Protease Inhibitor Cocktail™ (Roche Molecular Biochemicals) and 1×PMSF. In some experiments, cells were also preincubated with 10 mM NAC for 0.5 h before sodium arsenite treatment. After treatments, cell lysates were centrifuged at 12,000 g for 10 min, and the supernatant was collected. Protein concentration was determined using the BCA method (Sigma), and equal amount of protein in supernatant (100 μg) was immunoprecipitated using 4 μl of antibody against PP1 or 4 μl of normal rabbit IgG for the control experiment. The immunoprecipitates were washed twice with lysis buffer lacking both protease inhibitor and phosphatase inhibitor, and twice more with 1× the reaction buffer B. Finally, the purified PP1 immunoprecipitates were resuspended in 25 μl of 1× reaction buffer B containing 2 mM MgCl2 and 0.4 mM MnCl2. Reaction was then started by adding 25 μl of the peptide solution containing 10 μMS/T PPase R110 substrate to the samples. The reaction samples were incubated for 10 min at room temperature and followed by further incubation with the protease solution for 90 min. The reaction was then stopped by adding 25 μl of the stabilizer solution containing 3 μM okadaic acid to the reaction mixture. The cellular PP1 activity was quantified with FACSCalibur (BD Biosciences) by measuring the fluorescence intensity at an excitation wavelength of 485 nm and an emission wavelength of 530 nm and normalized to the fluorescence intensity from the control experiment.
Statistical analysis
All results are expressed as means ± standard deviation (S.D.) with n indicating the number of experiments. Statistical significance of difference was determined using Student’s t test for paired data. A value of p<0.05 was considered significant.
RESULTS
Arsenite acutely increases eNOS-Thr497 phosphorylation and decreases NO production
Because chronic (>24 h) exposure of arsenite has shown the increased vascular dysfunction mainly by decreasing eNOS-Ser1179 phosphorylation-mediated NO production, we asked whether acute exposure of arsenite also decreases NO production via decreased eNOS-Ser1179 phosphorylation. As shown in Fig. 1A, however, arsenite (30 μM) acutely (as early as 4 h) increased eNOS-Thr497 phosphorylation, but not of eNOS-Ser1179 phosphorylation, although its longer (at 24 h) exposure considerably decreased eNOS-Ser1179 phosphorylation. No alterations in eNOS-Ser116 phosphorylation and eNOS expression were found under these conditions. However, longer (36 h) exposure of arsenite significantly decreased eNOS expression (Fig. 1B), which was accompanied by significantly decreased cell viability (data not shown). We also found that arsenite acutely increased eNOS-Thr497 phosphorylation in a dose-dependent manner (Fig. 1C). Consistent with phosphorylation status of eNOS-Thr497, arsenite decreased NO production in a time-dependent manner (Fig. 1D). Because a significant increase in eNOS-Thr497 phosphorylation was found in BAEC acutely exposed to arsenite, all subsequent experiments were accomplished with 30 μM arsenite treatment for 4 h, unless otherwise specifically stated.
Fig. 1.

Arsenite increases phosphorylation of eNOS-Thr497 and decreases NO production in BAEC. BAEC were treated with (A) 30 μM sodium arsenite (NaAsO2) for shorter times (4, 8, or 24 h), (B) for longer times (up to 36 h), or (C) with various doses of sodium arsenite (10, 20, or 30 μM) for 4 h (C). Control cells (0 h or 0 μM) were treated with vehicle (H2O) only. The cells were lysed, and the amounts of phosphorylated eNOS (p-eNOS) at multiple sites were measured by Western blot analysis using antibodies specific for eNOS phosphorylated at Ser116 (p-eNOS-Ser116), p-eNOS-Thr497, or p-eNOS-Ser1179. The blots shown are representative of at least three experiments. Densitometry was used to quantify p-eNOS-Ser116, p-eNOS-Thr497, or p-eNOS-Ser1179 relative to the total protein bands, and the graphs show the mean fold alterations above or below control (± S.D.) (n=3). Differences were statistically significant at *p<0.05 and **p<0.01. (D) After BAEC were treated with 30 μM sodium arsenite for the indicated times, NO released by the cells was measured by the Griess method. Each bar represents the mean NO production (after normalization to total cellular protein) as fold decreases below control (vehicle) (± S.D.) (n=3). Differences were statistically significant at *p<0.05 and **p<0.01.
Arsenite increases ROS production and NAC reverses the arsenite-stimulated increase in eNOS-Thr497 phosphorylation in BAEC
Several studies have demonstrated that arsenite generates ROS that leads to alteration in the activity of proteins; for example, arsenite increases ROS, resulting in increased expression or secretion of vascular endothelial growth factor (VEGF) in cancer cells and brain microvascular pericytes, respectively (Gao et al., 2004; Park et al., 2009). Therefore, we first evaluated whether arsenite induced ROS production in BAEC. As shown in Fig 2A, arsenite acutely (as early as 0.5 h) increased ROS production by ∼1.75 fold and this increase was maintained up to 4 h. Next, we tested whether increased ROS affected eNOS-Thr497 phosphorylation. Pretreatment with antioxidant NAC (10 mM) for 0.5 h almost completely reversed the arsenite-induced increase in eNOS-Thr497 phosphorylation (Fig. 2B), suggesting that ROS mediates the observed effect by arsenite.
Fig. 2.

Arsenite increases ROS production and antioxidant NAC reverses arsenite-induced increased eNOS-Thr497 phosphorylation. (A) BAEC were treated with 30 μM sodium arsenite for the indicated times (0, 0.5, 1, 2, or 4 h). ROS was measured by DCFH-DA method using FACSCalibur flow cytometer. Each bar represents mean ROS production as fold increases above control (0 h) (± S.D.) (n=3). Differences were statistically significant at *p<0.05 and **p<0.01. (B) Cells were also preincubated with vehicle or 10 mM NAC for 0.5 h, and then further treated with 30 μM sodium arsenite for 4 h. The level of p-eNOS-Thr497 was measured by Western blot analysis as described in the legend of Fig. 1. The blots are representative and the bar graph shows the mean fold increase above control (± S.D.) (n=3). Differences were statistically significant at *p<0.05 and **p<0.01.
PKC is not involved in the arsenite-stimulated increase in eNOS-Thr497 phosphorylation
PKC has been reported to phosphorylate eNOS-Thr497 in in vitro phosphorylation experiment (Matsubara et al., 2003) and in cultured EC (Fleming et al., 2001; Matsubara et al., 2003). These data, together with previous report that ROS had also been to be capable of activating PKC through oxidation of its N-terminal regulatory domain (Cosentino-Gomes et al., 2012), prompted us to examine whether PKC mediates the arsenite-induced increase in eNOS-Thr497 phosphorylation. Experiment evaluating the effect of PKC-specific inhibitor, Ro318425, however, did not alter the arsenite-stimulated eNOS-Thr497 phosphorylation (Fig. 3A). To further clarify these data, we transfected dominant-negative (DN) PKC isoforms, α, βI, βII, δ, ε and ζ, into BAEC. In accordance with the result from PKC inhibitor experiment, overexpression of DN-PKC genes did not reverse the increased eNOS-Thr497 phosphorylation by arsenite (Fig. 3B), which suggests that PKC is not involved in the arsenite-stimulated increase in eNOS-Thr497 phosphorylation.
Fig. 3.

PKC is not involved in arsenite-induced eNOS-Thr497 phosphorylation, but calyculin A mimics the effect of arsenite on eNOS-Thr497 phosphorylation. BAEC were pretreated with (A) 14 (+) or 28 μM (++) Ro318425 for 0.5 h and then treated with 30 μM sodium arsenite for 4 h. Control cells were treated with vehicle only. The blots shown are representative of at least three experiments. (B) BAEC, transfected with HA-tagged cDNA encoding dominant negative (DN) conventional (α, βI, or βII), novel (δ or ε), or atypical (ζ) PKC gene, were treated with vehicle or 30 μM sodium arsenite for 4 h. Overexpression of the PKC gene after transfection was confirmed by detecting the tagged-HA. The blots shown are representative of at least three experiments. In separate experiments, BAEC were treated with (C) 2.5 or 5 nM okadaic acid for 0.5 h, or (D) 1, 2.5 or 5 nM calyculin A for 0.5 h. Control cells were treated with vehicle (DMSO) alone. (E) In some experiments, cells were pretreated with 5 nM calyculin A, vehicle, or 5 nM okadaic acid for 0.5 h and then treated with 30 μM sodium arsenite for 4 h. The level of p-eNOS-Thr497 was measured by Western blot analysis as described in the legend of Fig. 1. The blots shown are representative of at least three experiments. (A-E) The bar graph shows the mean fold increases above control (± S.D.) (n=3). Differences were statistically significant at *p<0.05 and **p<0.01. ns, not significant.
Calyculin A, but not okadaic acid, mimics the stimulatory effect of arsenite on eNOS-Thr497 phosphorylation
PP1 and PP2A have been also implicated in agonist-induced eNOS-Thr497 dephosphorylation (Michell et al., 2001; Greif et al., 2002). To elucidate whether PP1 or PP2A is associated with the arsenite-induced increase in eNOS-Thr497 phosphorylation, we first treated BAEC with okadaic acid (2.5 or 5 nM) at a concentration known to specifically inhibit PP2A activity. As shown in Fig. 3C, no alteration eNOS-Thr497 phosphorylation was found in okadaic acid-treated cells, indicating no evidence for involvement of PP2A under our condition. In contrast, treatment with calyculin A, a specific PP1 inhibitor, dramatically increased eNOS-Thr497 phosphorylation in a dose-dependent manner (Fig. 3D), mimicking the observed effect of arsenite on eNOS-Thr497 phosphorylation. However, the simultaneous treatment with calyculin A and arsenite led to neither additive nor synergistic effect on the phosphorylation of eNOS-Thr497 when compared with treatment with calyculin A alone (Fig. 3E), implying that arsenite and calyculin A increased eNOS-Thr497 phosphorylation through the same signaling axis for PP1 activity inhibition. All these data suggest that PP1 may be involved in the arsenite-stimulated increase in eNOS-Thr497 phosphorylation.
Arsenite inhibits PP1 that is reversed by NAC
To elucidate the molecular mechanism by which arsenite acutely increases PP1-mediated eNOS-Thr497 phosphorylation, we investigated whether arsenite indeed decreases the protein level and/or the activity of PP1 in BAEC. Although it was reported that arsenite treatment decreased the level of PP1α expression in human lymphoblastoid cells (Tapio et al., 2005), we failed to find decrease in the level of PP1α expression in arsenite-treated BAEC (Fig. 4A). Instead, arsenite significantly decreased PP1 activity by ∼15% (Fig. 4B). Furthermore, the arsenite-induced decrease in PP1 activity was almost completely recovered by the pretreatment of NAC, suggesting that ROS mediates decreased PP1 activity by arsenite.
Fig. 4.
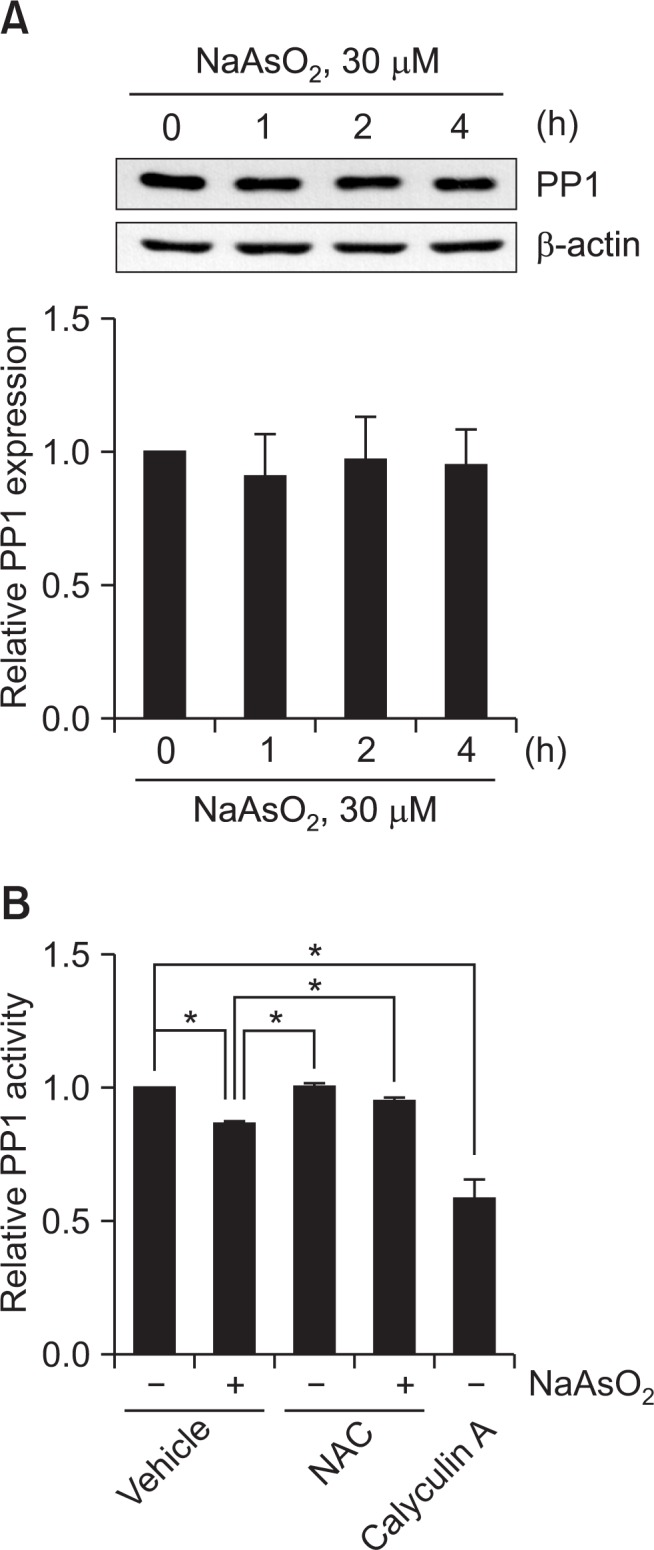
Arsenite decreases PP1 activity and NAC reverses arsenite-induced decrease in PP1 activity. (A) BAEC were treated as described in the legends of Fig. 1. The level of PP1 was measured by Western blot analysis using anti-PP1α antibody as described in the legend of Fig. 1. The blots are representative and the bar graph shows the mean fold alteration above control (± S.D.). (B) BAEC were treated as described in the legend of Fig. 2. PP1 activity was measured by serine/threonine phosphatase assay kit (Promega), quantified by measuring the fluorescence intensity (at 485/530 nm) using FACSCalibur flow cytometer (BD), and normalized to the fluorescence intensity from the control experiment. The bar graph shows the mean fold decreases above control (± S.D.) (n=3). Differences were statistically significant at *p<0.05.
DISCUSSION
Epidemiology studies showed that arsenic exposure is associated with increased risk of cardiovascular diseases including hypertension and peripheral vascular diseases (Stea et al., 2014). Although decreased NO bioavailability may be one important reason for arsenic-derived increase in these diseases, its detailed mechanism has not been defined. In this study, we demonstrate that arsenite attenuates NO production in BAEC via a coordinated interplay of two phosphorylation sites on eNOS; acute (<24 h) exposure of arsenite decreases NO production by increasing eNOS-Thr497 phosphorylation. In contrast, chronic (>24 h) arsenite exposure further decreases NO production via a combination of increased eNOS-Thr497 phosphorylation and decreased eNOS-Ser1179 phosphorylation, together with decreased eNOS expression. Lastly, our data also show that ROS-induced inhibition of PP1 activity provides the mechanism by which arsenite acutely decreases NO production via an increased eNOS-Thr497 phosphorylation.
One of the most important findings in this study is that eNOS-Thr497 phosphorylation mediates the acute effect of NO production. Although previous studies also showed that arsenite decreased NO production and eNOS activity in EC, these studies mostly focused on handling eNOS-Ser1179 phosphorylation and/or eNOS expression to explore its underlying mechanism (Tsou et al., 2005). Our study also showed decreases in eNOS-Ser1179 phosphorylation and eNOS expression particularly in chronic exposure of arsenite, the condition which most of previous studies have adopted. In this regard, our findings bring an important aspect in that dynamic regulation of eNOS activity via its multiple phosphorylation sites determines the net effect of certain agonists on NO production. Several studies have supported this concept showing that there was the coordinated regulation between eNOS-Ser1179 phosphorylation and eNOS-Thr497 dephosphorylation when EC were stimulated by VEGF (Michell et al., 2001), bradykinin (Harris et al., 2001), or H2O2 (Thomas et al., 2002).
It is well accepted that oxidative stress modulating cell signaling pathway has been known as one of the main causes of arsenite-induced toxicity. For example, arsenite increases expression of VEGF and hypoxia-inducible factor 1α via a ROS production in EC (Kao et al., 2003; Kamat et al., 2005) and cancer cells (Gao et al., 2004; Kamat et al., 2005). Concurrently, we reported that arsenite increased VEGF secretion in a ROS-dependent manner in microvascular pericytes. Based on this study, other important findings are shown that ROS induced by arsenite can cause vascular dysfunction via decreased NO production and that eNOS-Thr497 phosphorylation is likely to be one of targets of ROS induced by arsenic toxicity. Whether decreased eNOS-Ser1179 phosphorylation by chronic exposure of arsenite is also ROS-dependent under our condition warrants further investigations.
Previously, PP1, PP2A or PKC was reported to be involved in alteration in eNOS-Thr497 phosphorylation (Fleming et al., 2001; Michell et al., 2001). Based on these data, together with previous finding that NADPH oxidase-derived ROS showed oxidative activation of PKCα in EC, which is essential for vascular cell adhesion molecule-1-dependent transendothelial migration of lymphocytes (Cosentino-Gomes et al., 2012), here, we tested whether arsenite-induced ROS also activated PKC, subsequently increasing eNOS-Thr497 phosphorylation. Several PKC isoforms are known to have N-terminal regulatory domain containing zinc-binding, cysteine-rich motif, which is susceptible to oxidation leading to PKC activation. However, under our experimental condition, a variety of PKC isoforms including PKCα are unlikely to be activated by arsenite-induced ROS, and therefore PKC may not participate in arsenite-induced decrease in NO production.
In contrast with PKC, our data showed clearly that PP1 activity is involved in arsenite-induced increase in eNOS-Thr497 phosphorylation. It was reported that treatment with H2O2 led to inactivation of PP1 in human fibroblasts that was reversed by thiol-specific reagents such as dithiothreitol and NAC (Kim et al., 2003), suggesting a role for cysteine oxidation by ROS in the inactivation of PP1. Like human fibroblasts, we also found in BAEC that NAC almost completely recovered decreased PP1 activity by acute exposure of arsenite, indicating that ROS are upstream molecules of PP1. Disulfide bond formation of PP1 is suggested to be a likely mechanism of PP1 inhibition by ROS because PP1 contains disulfide oxidoreductase active sites (Fetrow et al., 1999). Although ROS-derived decreased PP1 activity mediates arsenite-stimulated increase in eNOS-Thr497 phosphorylation, whether decreased PP1 activity directly plays a role in increasing eNOS-Thr497 phosphorylation needs further study. Previously, it was reported that senescence-associated ERK phosphorylation and prolonged PKA signal induced by mild oxidation are attributable to ROS-dependent oxidative inactivation of PP1 and PP2A (Kim et al., 2003). Therefore, it is also interesting to identify whether ERK or PKA is able to play a role in connecting PP1 to eNOS-Thr497 phosphorylation in pathway responsible for arsenic-stimulated decreases in NO production and vascular function.
In conclusion, arsenite acutely increased eNOS-Thr497 phosphorylation, thereby decreasing NO production; this process is mediated by ROS-dependent reduction of PP1 activity. Our results shed light on the molecular mechanisms underlying arsenite-induced eNOS dysregulation associated with disruption of vascular integrity and subsequent development of vascular diseases. Furthermore, based on our results, care should be also exercised even when acutely exposed to arsenite.
Acknowledgments
This work was carried out with the support of “Cooperative Research Program (Risk assessment research in livestock on exposure to biological, chemical and environmental hazard substance. PJ01052302) for Agriculture Science & Technology Development, Rural Development Administration, Republic of Korea
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no competing interests
REFERENCES
- Barchowsky A, Klei LR, Dudek EJ, Swartz HM, James PE. Stimulation of reactive oxygen, but not reactive nitrogen species, in vascular endothelial cells exposed to low levels of arsenite. Free Radic Biol Med. 1999;27:1405–1412. doi: 10.1016/s0891-5849(99)00186-0. [DOI] [PubMed] [Google Scholar]
- Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443:285–289. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- Cho DH, Choi YJ, Jo SA, Jo I. Nitric oxide production and regulation of endothelial nitric-oxide synthase phosphorylation by prolonged treatment with troglitazone: evidence for involvement of peroxisome proliferator-activated receptor (PPAR) gamma-dependent and PPARgamma-independent signaling pathways. J Biol Chem. 2004;279:2499–2506. doi: 10.1074/jbc.M309451200. [DOI] [PubMed] [Google Scholar]
- Cho DH, Park JH, Joo Lee E, Jong Won K, Lee SH, Kim YH, Hwang S, Ja Kwon K, Young Shin C, Song KH, Jo I, Han SH. Valproic acid increases NO production via the SH-PTP1-CDK5-eNOS-Ser signaling cascade in endothelial cells and mice. Free Radic Biol Med. 2014;76:96–106. doi: 10.1016/j.freeradbiomed.2014.07.043. [DOI] [PubMed] [Google Scholar]
- Cho DH, Seo J, Park JH, Jo C, Choi YJ, Soh JW, Jo I. Cyclin-dependent kinase 5 phosphorylates endothelial nitric oxide synthase at serine 116. Hypertension. 2010;55:345–352. doi: 10.1161/HYPERTENSIONAHA.109.140210. [DOI] [PubMed] [Google Scholar]
- Cosentino-Gomes D, Rocco-Machado N, Meyer-Fernandes JR. Cell signaling through protein kinase C oxidation and activation. Int J Mol Sci. 2012;13:10697–10721. doi: 10.3390/ijms130910697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetrow JS, Siew N, Skolnick J. Structure-based functional motif identifies a potential disulfide oxidoreductase active site in the serine/threonine protein phosphatase-1 subfamily. FASEB J. 1999;13:1866–1874. doi: 10.1096/fasebj.13.13.1866. [DOI] [PubMed] [Google Scholar]
- Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68–75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- Gao N, Shen L, Zhang Z, Leonard SS, He H, Zhang XG, Shi X, Jiang BH. Arsenite induces HIF-1alpha and VEGF through PI3K, Akt and reactive oxygen species in DU145 human prostate carcinoma cells. Mol Cell Biochem. 2004;255:33–45. doi: 10.1023/b:mcbi.0000007259.65742.16. [DOI] [PubMed] [Google Scholar]
- Greif DM, Kou R, Michel T. Site-specific dephosphorylation of endothelial nitric oxide synthase by protein phosphatase 2A: evidence for crosstalk between phosphorylation sites. Biochemistry. 2002;41:15845–15853. doi: 10.1021/bi026732g. [DOI] [PubMed] [Google Scholar]
- Hall MN, Liu X, Slavkovich V, Ilievski V, Pilsner JR, Alam S, Factor-Litvak P, Graziano JH, Gamble MV. Folate, cobalamin, cysteine, homocysteine, and arsenic metabolism among children in bangladesh. Environ Health Perspect. 2009;117:825–831. doi: 10.1289/ehp.0800164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MB, Ju H, Venema VJ, Liang H, Zou R, Michell BJ, Chen ZP, Kemp BE, Venema RC. Reciprocal phosphorylation and regulation of endothelial nitric-oxide synthase in response to bradykinin stimulation. J Biol Chem. 2001;276:16587–16591. doi: 10.1074/jbc.M100229200. [DOI] [PubMed] [Google Scholar]
- Isenovic E, Soskic S, Dungen HD, Dobutovic B, Elvis T, Simone I, Marche P. Regulation of endothelial nitric oxide synthase in pathophysiological conditions. Cardiovasc Hematol Disord. 2011;11:109–118. doi: 10.2174/187152911798346972. [DOI] [PubMed] [Google Scholar]
- Kamat CD, Green DE, Curilla S, Warnke L, Hamilton JW, Sturup S, Clark C, Ihnat MA. Role of HIF signaling on tumorigenesis in response to chronic low-dose arsenic administration. Toxicol Sci. 2005;86:248–257. doi: 10.1093/toxsci/kfi190. [DOI] [PubMed] [Google Scholar]
- Kao YH, Yu CL, Chang LW, Yu HS. Low concentrations of arsenic induce vascular endothelial growth factor and nitric oxide release and stimulate angiogenesis in vitro. Chem Res Toxicol. 2003;16:460–468. doi: 10.1021/tx025652a. [DOI] [PubMed] [Google Scholar]
- Kim HP, Lee JY, Jeong JK, Bae SW, Lee HK, Jo I. Nongenomic stimulation of nitric oxide release by estrogen is mediated by estrogen receptor alpha localized in caveolae. Biochem Biophys Res Commun. 1999;263:257–262. doi: 10.1006/bbrc.1999.1348. [DOI] [PubMed] [Google Scholar]
- Kim HS, Song MC, Kwak IH, Park TJ, Lim IK. Constitutive induction of p-Erk1/2 accompanied by reduced activities of protein phosphatases 1 and 2A and MKP3 due to reactive oxygen species during cellular senescence. J Biol Chem. 2003;278:37497–37510. doi: 10.1074/jbc.M211739200. [DOI] [PubMed] [Google Scholar]
- Kumagai Y, Pi J. Molecular basis for arsenic-induced alteration in nitric oxide production and oxidative stress: implication of endothelial dysfunction. Toxicol Appl Pharmacol. 2004;198:450–457. doi: 10.1016/j.taap.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Oh YK, Rhee M, Lim JY, Hwang JY, Park YS, Kwon Y, Choi KH, Jo I, Park SI, Gao B, Kim WH. The role of STAT1/IRF-1 on synergistic ROS production and loss of mitochondrial transmembrane potential during hepatic cell death induced by LPS/d-GalN. J Mol Biol. 2007;369:967–984. doi: 10.1016/j.jmb.2007.03.072. [DOI] [PubMed] [Google Scholar]
- Lee MY, Jung BI, Chung SM, Bae ON, Lee JY, Park JD, Yang JS, Lee H, Chung JH. Arsenic-induced dysfunction in relaxation of blood vessels. Environ Health Perspect. 2003;111:513–517. doi: 10.1289/ehp.5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Jan KY. DNA damage in arsenite- and cadmium-treated bovine aortic endothelial cells. Free Radic Biol Med. 2000;28:55–63. doi: 10.1016/s0891-5849(99)00196-3. [DOI] [PubMed] [Google Scholar]
- Mandal BK, Suzuki KT. Arsenic round the world: a review. Talanta. 2002;58:201–235. [PubMed] [Google Scholar]
- Matsubara M, Hayashi N, Jing T, Titani K. Regulation of endothelial nitric oxide synthase by protein kinase C. J. Biochem. (Tokyo) 2003;133:773–781. doi: 10.1093/jb/mvg099. [DOI] [PubMed] [Google Scholar]
- Michell BJ, Chen Z, Tiganis T, Stapleton D, Katsis F, Power DA, Sim AT, Kemp BE. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J Biol Chem. 2001;276:17625–17628. doi: 10.1074/jbc.C100122200. [DOI] [PubMed] [Google Scholar]
- Park JH, Kim WS, Kim JY, Park MH, Nam JH, Yun CW, Kwon YG, Jo I. Chk1 and Hsp90 cooperatively regulate phosphorylation of endothelial nitric oxide synthase at serine 1179. Free Radic Biol Med. 2011;51:2217–2226. doi: 10.1016/j.freeradbiomed.2011.09.021. [DOI] [PubMed] [Google Scholar]
- Park JH, Park M, Byun CJ, Jo I. c-Jun N-terminal kinase 2 phosphorylates endothelial nitric oxide synthase at serine 116 and regulates nitric oxide production. Biochem Biophys Res Commun. 2012;417:340–345. doi: 10.1016/j.bbrc.2011.11.112. [DOI] [PubMed] [Google Scholar]
- Park JH, Sung HY, Lee JY, Kim HJ, Ahn JH, Jo I. B56alpha subunit of protein phosphatase 2A mediates retinoic acid-induced decreases in phosphorylation of endothelial nitric oxide synthase at serine 1179 and nitric oxide production in bovine aortic endothelial cells. Biochem Biophys Res Commun. 2013;430:476–481. doi: 10.1016/j.bbrc.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Park JS, Seo J, Kim YO, Lee HS, Jo I. Coordinated regulation of angiopoietin-1 and vascular endothelial growth factor by arsenite in human brain microvascular pericytes: implications of arsenite-induced vascular dysfunction. Toxicology. 2009;264:26–31. doi: 10.1016/j.tox.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Pi J, Kumagai Y, Sun G, Yamauchi H, Yoshida T, Iso H, Endo A, Yu L, Yuki K, Miyauchi T, Shimojo N. Decreased serum concentrations of nitric oxide metabolites among Chinese in an endemic area of chronic arsenic poisoning in inner Mongolia. Free Radic Biol Med. 2000;28:1137–1142. doi: 10.1016/s0891-5849(00)00209-4. [DOI] [PubMed] [Google Scholar]
- Rafikov R, Fonseca FV, Kumar S, Pardo D, Darragh C, Elms S, Fulton D, Black SM. eNOS activation and NO function: structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J Endocrinol. 2011;210:271–284. doi: 10.1530/JOE-11-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stea F, Bianchi F, Cori L, Sicari R. Cardiovascular effects of arsenic: clinical and epidemiological findings. Environ Sci Pollut Res Int. 2014;21:244–251. doi: 10.1007/s11356-013-2113-z. [DOI] [PubMed] [Google Scholar]
- Tapio S, Danescu-Mayer J, Asmuss M, Posch A, Gomolka M, Hornhardt S. Combined effects of gamma radiation and arsenite on the proteome of human TK6 lymphoblastoid cells. Mutat Res. 2005;581:141–152. doi: 10.1016/j.mrgentox.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Thomas SR, Chen K, Keaney JF., Jr Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J Biol Chem. 2002;277:6017–6024. doi: 10.1074/jbc.M109107200. [DOI] [PubMed] [Google Scholar]
- Tsou TC, Tsai FY, Hsieh YW, Li LA, Yeh SC, Chang LW. Arsenite induces endothelial cytotoxicity by down-regulation of vascular endothelial nitric oxide synthase. Toxicol Appl Pharmacol. 2005;208:277–284. doi: 10.1016/j.taap.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Watts VL, Motley ED. Role of protease-activated receptor-1 in endothelial nitric oxide synthase-Thr495 phosphorylation. Exp. Biol. Med. (Maywood) 2009;234:132–139. doi: 10.3181/0807-RM-233. [DOI] [PubMed] [Google Scholar]