

Abstract
As the mechanisms underlying neuronal development and degeneration become clarified, a number of common effectors and signaling pathways are becoming apparent. Here we describe the identification of Aβ, long considered a pathologic mediator of Alzheimers Disease and Down Syndrome, as similarly over-expressed in the neurodevelopmental disease, Fragile X Syndrome. We also show that mGluR5 inhibitors, currently employed for the treatment of Fragile X, reduce Aβ production in rodent models of Fragile X and AD as well as reduce disease phenotypes including seizures. Thus seemingly disparate neurologic diseases may share a common pathologic instigator and be treatable with a common, currently available class of therapeutics.
Introduction
Over the past fifteen years, increased attention has focused on the role of soluble Aβ, composed predominantly of small to medium sized oligomers, to influence neuronal function. Soluble oligomers have been shown to better correlate with neuronal dysfunction than plaques [1], be increased in the brain and CSF of AD patients as well as animal models [2], and have potent in vitro affects on LTP and LTD elicited from hippocampal and cortical slice preparations [3,4]. These studies have implicated soluble Aβ in the disruption of synaptic function and suggested that these changes preceed tau disease or neuronal degeneration. As such, there is growing interest in identifying how Aβ is produced in the microenvironment of the synapse and which signaling cascades it affects. Such studies will likely generate insights into the intitial phases of Aβ-mediated, cognitive impairment and hopefully generate novel therapuetic approaches capable of reversing these events. In this review we discuss new data showing that APP and Aβ are produced in dendritic spines under the regulatory control of the mGluR5-fragile X mental retardation protein (FMRP) signaling pathway. We also discuss in vivo data showing reductions in CNS Aβ by chronic treatment with mGluR5 antagonists.
mGluRs
Activation of metabotropic glutamate receptors (mGluRs) modulates neuroplasticity and neuronal excitability, suggesting involvement of these receptors in a diverse set of acute and chronic neurologic diseases including ischemia, schizophrenia, pain, neurodegeneration and Fragile X Syndrome (FXS)[For review see 5]. mGluRs are members of the type C superfamily of G-protein-coupled receptors. They are subdivided into one of three groups (I-III) according to peptide sequence, type of signal transduction and agonist selectivity [6, 7]. Group I receptors include mGluR1 and mGluR5 and are mainly excitatory. After binding glutamate, they preferentially activate phosphoinositide-specific phospholipase C, culminating in the generation of IP3 and calcium release from intracellular stores. Increased free calcium activates multiple PKC isoforms, Erk, CREB and mTOR culminating in local changes in the synaptic distribution of glutamate receptors and dendritic protein synthesis and more distant effects on nuclear gene transcription [8,9]. Type II and III mGluR’s (mGluRs 2, 3, and 4, 6–8, respectively), are negatively coupled to adenylate cyclase, leading to signaling through alterations in cAMP. mGluR signaling can be further modulated by adaptor or scaffolding proteins. For example, Homer proteins organize postsynaptic proteins by binding group I mGluRs, inositol triphosphate receptors (IP3Rs), Shank, and the TRPC1 cation channel [10].
mGluR1 and 5 are differentially expressed within the CNS with the former predominantly in the thalamus, hippocampus and cerebellum and the latter diffusely throughout the forebrain and hippocampus but absent from the cerebellum. At the ultrastructural level, mGluR1 and mGluR5 show the highest receptor density in an annular pattern on the post-synaptic side [11,12]. Thus the distribution and biology of group I mGluRs makes them attractive therapeutic targets to modify synaptic signaling and function. It is worth noting however, that mGluRs are expressed outside of the CNS by hepatocytes [13], immune cells [14] and endothelium [15]. While the functionality of these receptors is poorly understood in non-neuronal cell types, their existance may enhance off-target effects or unexpected pharmacokinetics.
mGluR agonists and antagonists
A variety of chemically and pharmacologically distinct mGluR5 agonists and antagonists have been identified or developed. The latter include 2-methyl-6-(phenylethynyl)-pyridine (MPEP), E-2-methyl-6-(2-phenylethenyl)pyridine (SIB-1893) or 1-(3-chlorophenyl)-3-(3-methyl-5-oxo-4H-imidazol-2-yl)urea (fenobam) while the former include 2-chloro-5-hydroxyphenylglycine (CHPG). Both MPEP and fenobam act as allosteric modulators and thus are noncompetitive antagonists of mGluR5 [16]. The functional or physiologic consequences of mGluR5 signaling are complex. mGluR5 agonists block neuronal apoptosis in vitro [17] and have potent immuno-suppressive effects on microglia [18]. CHPG significantly reduced NMDA-mediated currents after a stretch-injury in co-cultures of neurons and astrocytes [19]. Paradoxically, antagonism of mGluR5 by MPEP may also provide neuroprotection after glutamate or NMDA excitotoxicty [20]. Similarly, both mGluR5 agonists or antagonists reduced stroke size in rodents after middle cerebral artery occlusion [21]. MGluR5 knockout mice show similar effects consistent with the notion that at least some of the protective effects of MPEP may reflect noncompetitive inhibition of NMDA receptor function, rather than from mGluR5 blockade [22].
In the context of neurodegenerative diseases generally, and AD in particular, there have been increasing attempts to assess the therapeutic utility of mGluR5 modulation. APP processing towards non-amyloidogenic products can be enhanced by mGluR5 agonists [23], demonstrating an interconnection between metabotropic signaling and Aβ production. Pretreatment of cultured neurons with CHPG markedly reduced Aβ induced apoptosis. In this system, MPEP attenuated the effects of CHPG, demonstrating a dependence on mGluR5 rather than NMDA-R [24]. Patients with clinical AD have shown both reduced [25] as well as enhanced mGluR5 mRNA and protein expression [26]. Patients with Down Syndrome have increased cortical mGluR5 expression [27]. Thus it is likely there is significant and physiologically relevant cross-talk between APP and Aβ production and mGluR5 induced signaling. As mGluR5 is predominantly expressed post-synaptically on dendritic spines, these results also suggest that APP and Aβ are produced locally in this critical anatomical location.
FXS and FMRP
Fragile X Syndrome (FXS) is the most prevalent form of inherited mental retardation, affecting one in 4,000 men and one in 8,000 women. This X chromosome–linked disorder is characterized by moderate to severe mental retardation (overall IQ <70), autism, seizures, facial abnormalities (large, prominent ears and long, narrow face) and macroorchidisim [28]. Microscopically, neurons from patients with FXS show increased numbers of long, thin, tortuous dendritic spines with prominent heads and irregular dilations [29,30]. The increased length, density, and immature morphology of dendritic spines in FXS suggest an impairment of synaptic pruning and maturation and likely underlies abnormalities in learning and neuronal function seen in both patients and rodent models [31]. Indeed, hippocampal slices from fmr-1 KO mice show abnormal DHPG induced, long term depression (LTD) [32,33] as well as long term potentiation (LTP) [34] Interestingly, neurons in AD model mice also show dendritic spine dysmorphogenesis [for review see 35]. Changes in extracellular Aβ concentrations and oligomeric status can negatively affect learning, LTP, LTD and synaptic function [1–4]. Thus AD and FXS, two seemingly distinct entities show considerable phenotypic overlap, suggesting some commonality in their respective pathogenesis.
In the majority of cases, FXS results from a trinucleotide (CGG) repeat expansion in the 5′-UTR of the fmr-1 gene (located at Xq27.3) [36]. The CGG expansion to >200 copies is pathogenic and induces hypermethylation and subsequent transcriptional silencing of the fmr-1 gene, resulting in the loss of expression of fragile X mental retardation protein (FMRP) [37]. FMRP is ubiquitously expressed throughout the brain with highest levels in dendritic spines and shafts. Structurally, FMRP contains multiple mRNA binding domains including two heterogeneous nuclear ribonucleoprotein (hnRNP) K homology domains and one RGG box. FMRP interacts with a number of RNA-binding proteins, including nucleolin, YB1 and the FMRP homologs FXR1 and FXR2 [38] as well as components of the miRNA processing system [39].
FMRP has been implicated in the translational repression of dendritic mRNAss [40, 41]. Using in vitro RNA selection and enrichment (SELEX) with recombinant RGG boxes, Darnell and associates identified FMRP ligands as G-rich mRNAs [42]. Structural modeling suggested G rich binding domains were arranged in a stacked planar array or so-called G-quartet. Ligands identified by these approaches included mRNAs coding for MAP1B, munc13 and semaphorin 3F. Others have shown FMRP also binds to uridine-rich mRNAs [43] as well.
While the above experiments helped define possible mRNA ligands, FMRP immunoprecipitates from cell lysates have been analyzed by CLIP assays [44]. These are extremely rigorous and all-but certainly exclude non-specific interactors. Using these techniques about 2 dozen bonefide FMRP mRNA ligands have been identified including Arc, PSD-95, CamKII and FMRP itself [for review see 45]. Consistent with their normal regulation by FMRP, proteins from these mRNAs are upregulated in neurons from fmr-1 KO mice.
Under normal conditions, FMRP binds to and suppress the translation of its mRNA ligands. Repression is temporarily lost after the activation of mGluR5 and FMRP mRNA ligands are rapidly translated. Within 30–60 minutes, repression is re-established, possibly through the local translation of FMRP [46]. Thus, in FXS, the loss of FMRP leads to both constitutive, elevated translation and the loss of pulsatile production after mGluR5 signaling [47]. These abnormalities are selective as dendritic mRNAs not regulated by FMRP are normally expressed.
Given these features of FXS and FMRP regulation, Bear et al [48] proposed that FXS was characterized by excessive mGluR5 signaling. Reductions in tonic signaling could reduce the aberrant translation of FMRP mRNA ligands and positively impact the clinical features of the disease. Both treatment of fmr-1 KO mice as well as a small group of full mutation FXS adults positively affected dendritic spine dysmorphogeneis, seizure propensity and behavior without significant side-effects [49–51]. These results are consistent with the mGluR5 hypothesis but do not clarify how or why this pathway is tonically hyperactive. One attractive hypothesis is that the overexpression of an FMRP mRNA ligand triggers mGluR5 signaling. Thus, mGluR5 blockade could have a positive effect on neuronal and nervous system function despite the absence of FMRP.
Identification of APP mRNA as a ligand for FMRP
Clearly, identifying the gamut of FMRP mRNA ligands remains one of the highest priorities for unraveling the cause of FXS. As discussed above, several mRNA cis motifs have been identified as FMRP targets. Thus, we performed informatics searches designed to identify G-quartet domains in neuronal expressed mRNAs. Both PSD-95 and FMRP mRNAs contain putative G-quartets in their 3′-UTR and coding sequence, respectively [46,52] and thus served as positive controls for the search. Somewhat suprisingly, rat, human and mouse APP mRNA all possessed a highly conserved, G-quartet–like motif in their coding region (position 825–846 of the mouse sequence) embedded within a larger guanine-rich domain (694–846) as well as a second G-rich domain within the 3′UTR. APP mRNAs (70% of APP695 and 50% of APP751/770) are associated with polyribosomes in rat brain [53], suggesting that translational regulation could play an important role in their expression. Therefore, we prepared cortical lysates as well as synaptosomes (SN) from WT mice and immunoprecipitated FMRP. Contrary to a previous report utilizing a different protocol for the preparation of SNs [54], APP mRNA was present in SNs. Reverse transcription (RT)–PCR revealed that APP mRNA was brought down with anti-FMRP antisera but not by control, preimmune IgG from cortical lysates as well as SNs. Thus, APP mRNA interacts with FMRP [55].
To determine the likely interaction site, in vitro, RNase protection assays were performed on FMRP immunoprecipitates from whole cortex lysates. Residual, protected APP mRNA was mapped by RTqPCR with primers immediately surrounding the predicted G-quartet and adjacent sequences. Surprisingly, the G-rich area immediately preceding the G-quartet (nt 699–796) was 2 to 4-fold more protected from nuclease digestion than fragments containing the predicted G-quartet (nt 825–846). Although this protected area does not contain a canonical G-quartet motif, the sequence is very G-rich and contains several closely spaced DWGG repeats. As all these regions contained equivalent and substantial GC content, the observed differences could not be a result of variable cleavage. In addition, a second, RNAse protected domain (nt 2318–2416) was also identified in the 3′UTR. Thus, APP mRNA possess two highly conserved, G-rich sequences that likely bind to FMRP.
It remained possible that the interaction between APP mRNA and FMRP was indirect. Thus, we performed a modified CLIP assay [44]. Intact SN were cross-linked with ultraviolet light (UVL), lysed and immunoprecipitated with anti-FMRP prior to ribonuclease digestion and SDS-PAGE. FMRP immunoreactive material (80 kDa) was excised from the gel and analyzed by RTqPCR. The amplicon encompassing the G-rich sequence (nucleotides 699–796) of APP mRNA gave a positive signal that was approximately five-fold greater than that of the adjacent G-quartet motif containing sequence (nt 774–871). Thus, our data defines the G-rich region immediately preceding the predicted G-quartet as a bone fide binding site between FMRP and APP mRNA. While the 3′UTR site was not analyzed by CLIP, it is also a likely interaction point.
FMRP Regulates APP Translation
A regulatory interaction between FMRP and APP mRNA would be predicted to suppress dendritic APP translation under basal conditions but allow a burst of new synthesis after mGluR5 activation. If so, treatment of cells or animals with mGluR5 antagonists could also suppress dendritic APP synthesis. Therefore, we asked if APP translation was altered from WT in fmr-1 KO mice, a rodent model for FXS. Cortical SN were prepared from both WT and fmr-1 KO mice and overall protein synthesis analyzed in response to DHPG (100 μM)-induced mGluR5 activation. Aggregate translation was identical demonstrating that FMRP was not required for overall protein synthesis, which is in agreement with prior reports [56].
To assess de novo APP synthesis, 35S-labeled WT or KO SN were immunoprecipitated with anti-APP. APP translation significantly increased (2.7 fold) after DHPG treatment in WT preparations while less synthesis and a minimal response to DHPG was seen in KO preparations. Steady state APP levels after DHPG were also assessed by western blot analysis. In WT SN, there was a rapid increase in total APP levels within 5 min of DHPG treatment (1.6-fold, n=3), which was completely absent in KO SN. In the absence of DHPG, steady state levels of APP were substantially higher in KO SN compared to WT. APP mRNA levels and decay rate were equal in WT and KO preparations under basal conditions or after DHPG, excluding differences in gene expression as a cause. Within 20 min of DHPG, APP levels in WT SN approached those seen in unstimulated KO SN. These data suggest that APP mRNA is translationally repressed by FMRP in unstimulated WT dendritic compartments. mGluR activation rapidly de-represses APP synthesis as shown for other FMRP mRNA ligands including FMRP and PSD95 [42,46]. APP levels during maximal de-repression (maximal mGluR5 signaling) approach those seen constitutively in fmr1 KO.
FMRP Regulates Dendritic APP Levels in Cultured Neurons
SN are a relatively crude preparation of pre- and post-synaptic densities that are contaminated with other cell types, such as astrocytes, which form synapses with neurons. Thus, we prepared primary E18 cortical neuron cultures from WT and fmr-1 KO brains and assessed dendritic APP levels by immunofluorescence. APP was found in the cell body as well as dendritic puncta of both WT and fmr-1 KO neurons. There was a highly significant 21% increase in the basal level of APP in untreated fmr-1 KO neurons compared to WT. Neurons stimulated with DHPG for 10 and 20 min prior to cell fixation showed a 18–25% increase in dendritic APP levels in WT but no increase in fmr-1 KO cultures. This data confirms our findings in SN that (1) fmr-1 KO mice have higher basal synaptic levels of APP and (2) DHPG increases APP levels selectively in WT samples.
Overall, our data demonstrates that FMRP and APP mRNA interact, likely through several G-rich domains located in the coding region and 3′ UTR. While it has not been formally demonstrated, presumably the RGG box is the RNA binding region for FMRP. Under resting conditions, FMRP binds to and represses the translation of APP within dendrites. mGluR5 signaling positively and negatively controls the translation of APP. These results suggest the novel possibility that mGluR5 antagonists could be used to reduce APP at synaptic sites.
We tested this possibility in vitro by treating SN or intact neurons with mGluR5 antagonists (MPEP or fenobam). After as little as 15 minutes, APP was reduced by ~40% in SN or dendritic spines of intact neurons. Interestingly, APP remained decreased for as much as 1 day after the administration of antagonist. These results suggest that mGluR antagonists could have significant effects on steady state APP levels.
Soluble Aβ1–40 and Aβ1–42 are increased in Fmr-1 KO
Increased translation of APP provides more target for cleavage by β- and γ-secretases. Therefore, we would expect fmr-1 KO mice to have exacerbated β-amyloid production with aging. Right brain hemispheres from middle-aged FVB mice (11–13 months old) were homogenized in protein extraction buffer containing 1% Triton X-100 and protease inhibitors and the soluble material was analyzed by ELISA for Aβ1–40 and Aβ1–42. The fmr-1 KO mouse brain contained 1.6X more Aβ40 and 2.5X more Aβ42 than WT controls. Similar results were seen with fmr-1 KO mice in a C57BL/6 background. Thus excess APP generated by the loss of FMRP was converted to Aβ levels.
mGluR5 antagonists reduce Aβ in WT and AD model mice
Based on the above results, we asked if acute or chronic in vivo administration of mGluR5 antagonists could reduce some or all of Aβ. As discussed above, a variety of orally available mGluR5 antagonists have been developed with fenobam being the prototype [57]. This agent was never marketed due to side effects which included amnesia and psychotomimetic symptoms [58]. However, due to the loss of patent protection for the drug, and its potential for the treatment of FXS, the FRAXA Research Foundation commissioned the synthesis and oversaw the distribution of fenobam to interested investigators including ourselves. Our strategy was to incorporate fenobam into a refined chow diet which the experimental mice would ingest (Fig 1). Models would include both WT as well as AD model animals that overexpress human APP and Aβ. The endpoints would be decreased pathologic phenotypes as well as reductions in CNS Aβ. Drug delivery via the chow has the major advantage of eliminating the need to perform oral gavage on a large number of animals as well as normalizing the caloric, fat and protein inputs to all mice in the study. A potential concern was that different mice would ingest different amounts of food and fenobam and have somewhat variable drug levels.
Figure 1.

Description of the strategy to generate fenobam containing food and anticipated target of inhibition on the post-synaptic dendritic spines within the brain.
Before testing the effects of chronic fenobam administration, we performed a pilot 4 week trial in 1 month old Tg2576 mice. This is a well established AD model expressing a single copy of the mutant human APP cDNA coding region (with double mutations at K670M/N671L) [59]. Tg2576 shows ~5 fold greater APP and Aβ40 than WT and ~14 fold increased Aβ42. As the endogenous mouse gene is also intact, we would also be able to determine its level of expression as a function of fenobam therapy. Food was generated that contained 0.2 g-fenobam/kg. We assumed that mice ingest approximately 3 g/day of food. Thus the mice should be receiving approximately 600 μg fenobam/day. This dose was calculated from prior rodent and human pharmacokinetic data and is slightly higher than used in a recent human trial of FXS patients [49].
Over the trial period, there were no obvious differences between the control and experimental groups with similar cage behaviours and weight gain. After 4 weeks, the mice were harvested and the levels of human and mouse brain Aβ determined by ELISA [55]. Somewhat disappointedly, the levels of human APP and Aβ were similar in both groups. In retrospect, this result is not suprising. As discussed above, an interaction between 2 cis-acting regulatory elements in APP mRNA and the G-quartet of FMRP is required for normal regulation. The APP cDNA used in Tg2576 only contains one (coding region) of these G-rich domains, not two. Thus we rationalized the failure of fenobam to reduce transgenic APP via this mechanism. If true, we would need an AD model mouse where the entire 3′ UTR as well as the coding region was present. In order to substantiate this hypothesis, we evaluated levels of endogenous mouse APP and Aβ in the fenobam treated mice. As the mouse mRNA is full-length, it would contain all essential cis-acting regulatory elements. Strikingly, mouse Aβ40 was reduced by nearly 7 fold after 1 month of fenobam (Fig 2). Due to its very low initial level, we were unable to determine if Aβ42 was similarly reduced. However, these results established proof of principle that mGluR5 antagonists can be safely delivered for extended periods to experimental mice without side-effects and achieve a reduction in endogenous Aβ40 as predicted from the prior work in fmr-1 KO mice.
Figure 2.

Murine Aβ is dramatically reduced in fenobam treated Tg2576. Tg mice were maintained on fenobam food for 2 weeks prior to harvest and ELISA determination of total, CNS Aβ.
Given the success and caveats of the pilot study, we elected to initiate a trial of 0.2 g-fenobam/kg food for a longer (3 month) period. As we had established that AD mice transgenic with incomplete APP cDNA would probably not recapitulate the biology we observed with the endogenous mRNA, we sought a genomic model. Lamb and colleagues [60] have introduced FAD mutations into YAC’s containing the entire human APP gene. The resultant, Tg mice overexpress human Aβ by ~ 7 fold compared to endogenous WT mice and thus are comparable to that seen with Tg2576. Male mice were obtained from Jackson Labs and started on fenobam containing chow shortly after weaning. After 2 months, a small group were sacrificed and sera evaluated for fenobam. As shown in Fig 3, fenobam was readily detectable with females showing >3 fold higher levels than males. Thus, persistant and approximaately therapeutic levels of fenobam were present in the experimental mice. As before, we did not observe any differences between control and experimental animals in terms of cage behavior or weight gain.
Figure 3.
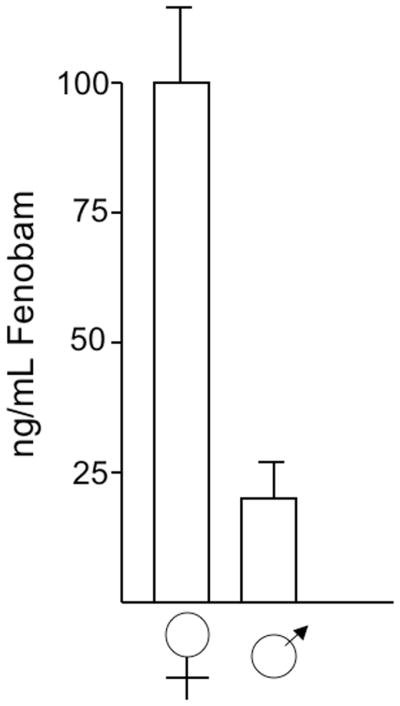
Serum fenobam levels in chronically treated mice. Line R1.40 transgenic mice [ref 60] were fed chow containing 1 g fenobam/kg for 2 months prior to euthanasia. Serum levels were measured by ELISA.
At 4 months of age (3 months of fenobam), mice were sacrificed and assessed for brain Aβ. As shown (Fig 4), there was a statistically significant 20% decrease in Aβ40 but no change in Aβ42. While these results were not as striking as seen for endogenous Aβ (see above), they confirm that mGluR5 antagonism can attenuate Aβ accumulation. We suspect that with more potent inhibitors with more consistent CNS penetration, the results could well have been more impressive. Finally, it is worth noting that the Tg model employed vastly overproduces Aβ to levels never achieved by humans with AD or Down syndrome. Thus any positive effects in this scenario provides strong evidence that the approach is sound and should be considered for patients with cognitive decline and MCI.
Figure 4.

Line R1.40 transgenic mice show significant reductions in whole brain Aβ40 but not Aβ42. Mice were fed control (C) or fenobam (F) containing food for 3 months prior to sacrifice and measurement of CNS Aβ as shown.
Summary and conclusions
The recent discovery that FMRP regulates APP translation in dendritic spines through an mGluR5 mediated process opens new avenues for the possible treatment of dementias related to or caused by the dysregulated production of Aβ. As mGluR5 is located predominantly at synapses, inhibitors directed at this pathway could reduce the toxic accumulation of Aβ in this vulnerable region increasingly identified in the initiation of memory and cognitive impairment seen in AD. In addition, mGluR5 antagonists appear to suppress seizure incidence in animal models overproducing Aβ. Thus fenobam and related agents appear to both block the producttion of and toxic signaling by Aβ. The side-effects of mGluR5 antagonists appear modest with reasonable therapeutic windows and a number of newly developed agents are about to or have recently entered clinical trials. The major unanswered questions are the mechanisms by which Aβ affects synaptic and dendritic function and when mGluR5 inhibitors should be given to patients during the development of AD.
Acknowledgments
The authors would like to thank members of the laboratory for their thoughtful comments. Supported by NIH (HD03352 and DA026067), FRAXA and ADDF to J.S.M. and FRAXA to C.J.W.
Bibliography
- 1.Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008 Sep 1;192(1):106–13. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cerpa W, Dinamarca MC, Inestrosa NC. Structure-function implications in Alzheimer’s disease: effect of Abeta oligomers at central synapses. Curr Alzheimer Res. 2008 Jun;5(3):233–43. doi: 10.2174/156720508784533321. [DOI] [PubMed] [Google Scholar]
- 3.Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spooren WP, Gasparini F, Salt TE, Kuhn R. Novel allosteric antagonists shed light on mGluR5 and CNS disorders. Trends Pharmacol Sci. 2001;22:331–337. doi: 10.1016/s0165-6147(00)01694-1. [DOI] [PubMed] [Google Scholar]
- 6.Bräuner-Osborne H, Egebjerg J, Nielsen EO, Madsen U, Krogsgaard-Larsen P. J Med Chem. 2000;43 doi: 10.1021/jm000007r. [DOI] [PubMed] [Google Scholar]
- 7.Conn PJ, Pin JP. Annu Rev Pharmacol Toxicol. 1997;37 doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 8.Karim F, Wang CC, Gereau RW., 4th Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.HK, Warwick S, Nahorski R, Challiss RA. Group I metabotropic glutamate receptors, mGlu1a and mGlu5a, couple to cyclic AMP response element binding protein (CREB) through a common Ca2+- and protein kinase C-dependent pathway. J Neurochem. 2005;93:232–245. doi: 10.1111/j.1471-4159.2005.03012.x. [DOI] [PubMed] [Google Scholar]
- 10.Yuan JP, Kiselyov K, Shin DM, et al. Cell. 2003;114:777–789. doi: 10.1016/s0092-8674(03)00716-5. [DOI] [PubMed] [Google Scholar]
- 11.Lujan R1, Roberts JDB, Shigemoto R, Ohishi H, Somogyi P. Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1alpha, mGluR2 and mGluR5, relative to neurotransmitter release sites. Journal of Chemical Neuroanatomy. 1997;13(4):219–241. doi: 10.1016/s0891-0618(97)00051-3. [DOI] [PubMed] [Google Scholar]
- 12.Bortolotto ZA, Fitzjohn SM, Collingridge GL. Curr Opin Neurobiol. 1999;9:299–304. doi: 10.1016/s0959-4388(99)80044-0. [DOI] [PubMed] [Google Scholar]
- 13.Storto M, de Grazia U, Knöpfel T, et al. Selective blockade of mGlu5 metabotropic glutamate receptors protects rat hepatocytes against hypoxic damage. Hepatology. 2000;31:649–655. doi: 10.1002/hep.510310315. [DOI] [PubMed] [Google Scholar]
- 14.Pacheco R, Ciruela F, Casadó V, et al. Group I metabotropic glutamate receptors mediate a dual role of glutamate in T cell activation. J Biol Chem. 2004;279:33352–33358. doi: 10.1074/jbc.M401761200. [DOI] [PubMed] [Google Scholar]
- 15.Collard CD, Park KA, Montalto MC, et al. Neutrophil-derived glutamate regulates vascular endothelial barrier function. J Biol Chem. 2002;277:14801–14811. doi: 10.1074/jbc.M110557200. [DOI] [PubMed] [Google Scholar]
- 16.Porter RH, Jaeschke G, Spooren W, et al. Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharmacol Exp Ther. 2005;315 (2):711–21. doi: 10.1124/jpet.105.089839. [DOI] [PubMed] [Google Scholar]
- 17.Vincent AM, TenBroeke M, Maiese K. Metabotropic glutamate receptors prevent programmed cell death through the modulation of neuronal endonuclease activity and intracellular pH. Exp Neurol. 1999;155:79–94. doi: 10.1006/exnr.1998.6966. [DOI] [PubMed] [Google Scholar]
- 18.Biber K, Laurie DJ, Berthele A, et al. Expression and signaling of group I metabotropic glutamate receptors in astrocytes and microglia. J Neurochem. 1999;72:1671–1680. doi: 10.1046/j.1471-4159.1999.721671.x. [DOI] [PubMed] [Google Scholar]
- 19.Lea PM, Custer SJ, Vicini S, Faden AI. Neuronal and glial mGluR5 modulation prevents stretch-induced enhancement of NMDA receptor current. Pharmacol Biochem Behav. 2002;73:287–298. doi: 10.1016/s0091-3057(02)00825-0. [DOI] [PubMed] [Google Scholar]
- 20.O’Leary DM, Movsesyan V, Vicini S, Faden AI. Selective mGluR5 antagonists MPEP and SIB-1893 decrease NMDA or glutamate-mediated neuronal toxicity through actions that reflect NMDA receptor antagonism. Br J Pharmacol. 2000;131:1429–1437. doi: 10.1038/sj.bjp.0703715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bao WL, Williams AJ, Faden AI, Tortella FC. Selective mGluR5 receptor antagonist or agonist provides neuroprotection in a rat model of focal cerebral ischemia. Brain Res. 2001;922:173–179. doi: 10.1016/s0006-8993(01)03062-1. [DOI] [PubMed] [Google Scholar]
- 22.Lea PM, 4th, Movsesyan VA, Faden AI. Neuroprotective activity of the mGluR5 antagonists MPEP and MTEP against acute excitotoxicity differs and does not reflect actions at mGluR5 receptors. Br J Pharmacol. 2005;145:527–534. doi: 10.1038/sj.bjp.0706219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee RK, Wurtman RJ, Cox AJ, Nitsch RM. Proc Natl Acad Sci U S A. 1995;15;92(17):8083–7. doi: 10.1073/pnas.92.17.8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Movsesyan Vilen A, Stoica Bogdan A, Faden Alan I. mGLuR5 activation reduces [beta]-amyloid-induced cell death in primary neuronal cultures and attenuates translocation of cytochrome c and apoptosis-inducing factor. Journal of Neurochemistry. 2004;89(6):1528–1536. doi: 10.1111/j.1471-4159.2004.02451.x. [DOI] [PubMed] [Google Scholar]
- 25.Minuzzia L, Quiriona R, Gauthiera S, Rosa-Netoa P. Declines of hippocampal metabotropic glutamate receptor in Alzheimer’s disease. Alzheimer’s and Dementia. 2009 Jul;5(4, Supplement 1):267. [Google Scholar]
- 26.Casley CS, Lakics V, Lee HG, Broad LM, Day TA, Cluett T, Smith MA, O’Neill MJ, Kingston AE. Up-regulation of astrocyte metabotropic glutamate receptor 5 by amyloid-beta peptide. Brain Res. 2009 Jan 17; doi: 10.1016/j.brainres.2008.12.082. [DOI] [PubMed] [Google Scholar]
- 27.Oka A, Takashima S. The up-regulation of metabotropic glutamate receptor 5 (mGluR5) in Down’s syndrome brains. Acta Neuropathol. 1999 Mar;97(3):275–8. doi: 10.1007/s004010050985. [DOI] [PubMed] [Google Scholar]
- 28.Hagerman RJ, Hagerman PJ. In: Physical and behavioral phenotype. Hagerman RJ, Cronister A, editors. Baltimore: John Hopkins University Press; 2002. pp. 3–109. [Google Scholar]
- 29.Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, et al. Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol (Berl) 1985;67:289–295. doi: 10.1007/BF00687814. [DOI] [PubMed] [Google Scholar]
- 30.Wisniewski KE, Segan SM, Miezejeski CM, Sersen EA, Rudelli RD. The Fra(X) syndrome: Neurological, electrophysiological, and neuropathological abnormalities. Am J Med Genet. 1991;38:476–480. doi: 10.1002/ajmg.1320380267. [DOI] [PubMed] [Google Scholar]
- 31.Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- 32.Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Hou L, Klann E, Nelson DL. Altered hippocampal synaptic plasticity in the FMR1 gene family knockout mouse models. J Neurophysiol. 2009;101(5):2572–80. doi: 10.1152/jn.90558.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shang Y, Wang H, Mercaldo V, Li X, Chen T, Zhuo M. Fragile X mental retardation protein is required for chemically-induced long-term potentiation of the hippocampus in adult mice. J Neurochem. 2009 Jul 30; doi: 10.1111/j.1471-4159.2009.06314.x. [DOI] [PubMed] [Google Scholar]
- 35.Tackenberg C, Ghori A, Brandt R. Thin, stubby or mushroom: spine pathology in Alzheimer’s disease. Curr Alzheimer Res. 2009;6(3):261–8. doi: 10.2174/156720509788486554. [DOI] [PubMed] [Google Scholar]
- 36.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 37.Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 38.Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: The roots of fragile X syndrome. Nat Rev Neurosci. 2005;6:376–387. doi: 10.1038/nrn1667. [DOI] [PubMed] [Google Scholar]
- 39.Cheever A, Ceman S. Phosphorylation of FMRP inhibits association with Dicer. RNA. 2009;15(3):362–6. doi: 10.1261/rna.1500809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, et al. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003;112:317–327. doi: 10.1016/s0092-8674(03)00079-5. [DOI] [PubMed] [Google Scholar]
- 41.Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10:329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- 42.Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, et al. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–499. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- 43.Chen L, Yun SW, Seto J, Liu W, Toth M. The fragile X mental retardation protein binds and regulates a novel class of mRNAs containing U rich target sequences. Neuroscience. 2003;120:1005–1017. doi: 10.1016/s0306-4522(03)00406-8. [DOI] [PubMed] [Google Scholar]
- 44.Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, et al. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 45.Bassell Gary J, Warren Stephen T. Fragile X Syndrome: Loss of Local mRNA Regulation Alters Synaptic Development and Function. Neuron. 2008;60(2):201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Todd P, Mack K, Malter JS. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci U S A. 2003;100:14374–14378. doi: 10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Greenough WT, Klintsova AY, Irwin SA, Galvez R, Bates KE, et al. Synaptic regulation of protein synthesis and the fragile X protein. Proc Natl Acad Sci U S A. 2001;98:7101–7106. doi: 10.1073/pnas.141145998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 49.Berry-Kravis E, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, Hutchison J, Snape M, Tranfaglia M, Nguyen DV, Hagerman R. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46(4):266–71. doi: 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, Willemsen R. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis. 2008;31(1):127–32. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Westmark CJ, Westmark PR, Beard AM, Hildebrandt SM, Malter JS. Seizure Susceptibility and Mortality in Mice that Over-Express Amyloid Precursor Protein. Int J Clin Exp Pathol. 2008;1;1(2):157–68. [PMC free article] [PubMed] [Google Scholar]
- 52.Schaeffer C, Bardoni B, Mandel JL, Ehresmann B, Ehresmann C, et al. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. EMBO J. 2001;20:4803–4813. doi: 10.1093/emboj/20.17.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Denman R, Potempska A, Wolfe G, Ramakrishna N, Miller DL. Distribution and activity of alternatively spliced Alzheimer amyloid peptide precursor and scrapie PrP mRNAs on rat brain polysomes. Arch Biochem Biophys. 1991;288:29–38. doi: 10.1016/0003-9861(91)90161-b. [DOI] [PubMed] [Google Scholar]
- 54.Sung YJ, Weiler IJ, Greenough WT, Denman RB. Selectively enriched mRNAs in rat synaptoneurosomes. Brain Res Mol Brain Res. 2004;126:81–87. doi: 10.1016/j.molbrainres.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 55.Westmark CJ, Malter JS. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007 Mar;5(3):e52. doi: 10.1371/journal.pbio.0050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.D’Agata V, Warren ST, Zhao W, Torre ER, Alkon DL, et al. Gene expression profiles in a transgenic animal model of fragile X syndrome. Neurobiol Dis. 2002;10:211–218. doi: 10.1006/nbdi.2002.0506. [DOI] [PubMed] [Google Scholar]
- 57.Wållberg A, Nilsson K, Osterlund K, Peterson A, Elg S, Raboisson P, Bauer U, Hammerland LG, Mattsson JP. Phenyl ureas of creatinine as mGluR5 antagonists. A structure-activity relationship study of fenobam analogues. Bioorganic and Medicinal Chemistry Letters. 2006;1;16(5):1142–5. doi: 10.1016/j.bmcl.2005.11.092. [DOI] [PubMed] [Google Scholar]
- 58.Jacob W, Gravius A, Pietraszek M, Nagel J, Belozertseva I, Shekunova E, Malyshkin A, Greco S, Barberi C, Danysz W. The anxiolytic and analgesic properties of fenobam, a potent mGlu5 receptor antagonist, in relation to the impairment of learning. Neuropharmacology. 2009 doi: 10.1016/j.neuropharm.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 59.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 60.Lamb BT, et al. Altered metabolism of familial Alzheimer’s disease-linked amyloid precursor protein variants in yeast artificial chromosome transgenic mice. Hum Mol Genet. 1997;6:1535–1541. doi: 10.1093/hmg/6.9.1535. [DOI] [PubMed] [Google Scholar]