

Abstract
Subnetwork detection is often used with differential expression analysis to identify modules or pathways associated with a disease or condition. Many computational methods are available for subnetwork analysis. Here, we compare the results of eight methods: simulated annealing–based jActiveModules, greedy search–based jActiveModules, DEGAS, BioNet, NetBox, ClustEx, OptDis, and NetWalker. These methods represent distinctly different computational strategies and are among the most widely used. Each of these methods was used to analyze gene expression data consisting of paired tumor and normal samples from 50 breast cancer patients. While the number of genes/proteins and protein interactions detected by the eight methods vary widely, a core set of 60 genes and 50 interactions was found to be shared by the subnetworks identified by five or more of the methods. Within the core set, 12 genes were found to be known breast cancer genes.
Keywords: subnetwork detection, pathway analysis, breast cancer, TCGA, network biology
Introduction
With the advent of high-throughput measurements in biotechnology, cancer biologists are able to dissect the complicated pathology of cancers from multiple directions. These measured molecular profiles include genetic mutations, copy number variance, messenger RNA (mRNA) expression, microRNA expression, DNA methylation, protein abundance, etc.1 However, multidimensional data also bring a tremendous challenge to the computational biology community. What can these data tell us about cancer? Differential analysis is a straightforward method in which differences in the molecular profiles of tumor and normal cells are identified. These analyses rely on a large number of samples and result in the identification of thousands of differences in molecular profiles. How to interpret these molecular variations as a whole is still under investigation.
Alternatively, molecular interaction data have shown powerful potential for connecting isolated molecular variations into a meaningful framework. These analyses usually start with differential analysis of molecular profiles, eg, differential gene expression, and score the extent of the difference for each gene. Next, biological network data that indicate the association of genes are collected, and then the scores are overlaid on the network. Now the task is to extract a subset of the network, ie, a subnetwork of the global network, such that the subnetwork is as small as possible while connecting as many highly scored genes as possible. This subnetwork enriched in differentially expressed genes can be used to discover, for example, that the upregulation of one gene is caused by the overexpression of its upstream regulator or dysfunction of its suppressor.
Subnetwork detection is a crucial analysis since it is capable of linking multiple individual molecular variations into an insightful wiring diagram showing how one individual variation is related to the others. Many methods for subnetwork detection have been developed. In 2002, Ideker et al first proposed a computational model for subnetwork detection based on simulated annealing.2 They also proved that subnetwork detection is an NP-Hard problem.2 As reviewed by Mitra et al.3, many attempts have been made during recent the decade to solve this problem efficiently using approximation algorithms. Due to the diversity of subnetwork scoring functions used by the different approximation algorithms, it is unlikely that different programs will obtain identical or even very similar subnetworks given the same expression and network data.
In this study, we propose a pipeline to comprehensively evaluate the performance of subnetwork detection methods from multiple aspects. We first select eight methods and assess them equally using an authoritative data set of breast cancer from The Cancer Genome Atlas (TCGA).1 Then we perform a differential expression analysis using DESeq4 and score the significance of expression change for each gene. Next, we extract subnetworks using the eight methods and compare their outputs based on their coverage of significant genes, network modularity, mutual similarities, and functional enrichment. Finally, we compare their computational costs, user friendliness, and discuss their strengths and weaknesses, respectively.
Results
Overview of subnetwork detection methods
Over 40 computational models have been developed during the past decade based on various algorithms, as reviewed by Mitra et al.3, and Berger et al.5 We selected eight of them (Table 1) for further comprehensive assessment based on the following three rules. First, the input of the models must be a network, and an expression set or a list of gene weights based on the expression. The models were ruled out if they required genetic mutation data or integration of co-expression data. Second, the selected models must be accessible either with open source code or a well-maintained online Graphical User Interface (GUI). Third, the selected models must represent diversity of methodology, and similar or integrative models are excluded. We summarize the eight selected methods and discuss their advantages and limitations in Table 2.
Table 1.
Overview of eight methods.
| METHOD | ALGORITHM | TOOL TYPE | REF. | INPUT NETWORK | INPUT EXPRESSION | RUNNING TIME (MIN) |
|---|---|---|---|---|---|---|
| jAM.SA | Simulated annealing | Cytoscape | 2 | HPRD | Adjusted P-values | ∼40 |
| jAM.GR | Greedy search | Cytoscape | 28 | HPRD | Adjusted P-values | ∼4 |
| DEGAS | Greedy heuristic | GUI | 29 | HPRD | Normalized counts | ∼3 |
| BioNet | Integer-Linear Programming | R package | 30 | HPRD | P-values | ∼7 |
| NetBox | Shortest path | Python, Java | 31 | Preload | Seed genes | ∼100 |
| ClustEx | Clustering, shortest path | C & GUI | 32 | HPRD | Seed genes | ∼150 |
| OptDis | Color coding | C | 33 | HPRD | Normalized counts | ∼1560 |
| NetWalker | Random walks | GUI | 34 | Preload | Adjusted P-values | ∼0.1 |
Notes: jAM.SA denotes jActiveModules using Simulated Annealing; jAM.GR denotes jActiveModules using Greedy Search.
Table 2.
Performance summary of the eight methods.
| METHOD | DESCRIPTION | ADVANTAGE | LIMITATION |
|---|---|---|---|
| jAM.SA | Uses simulated annealing to search for the most highly scored subnetwork | Accepts low-scored genes with a certain probability | Produces large subnetwork; Slow |
| jAM.GR | Extends a subnetwork by adding one of its neighboring genes that maximizes a mutual information–based objective function | Fast; uses mutual information to evaluate subnetwork quality | Does not accept low-scored genes, high tendency to be trapped into a suboptimal solution |
| DEGAS | Models subnetwork detection as a Connected Set Cover problem and solves it using a greedy heuristic | Fast; able to detect differentially expressed genes; does not require weights of genes as inputs | Many parameters that need to be tuned |
| BioNet | The first exact approach. Models subnetwork detection as a Prize-Collecting Steiner Tree problem and solves it using Integer Linear Programming | Fast; produces a single small subnetwork with high coverage of significant genes | Produces single small output subnetwork with a high false-negative rate (low recall) |
| NetBox | Computes the shortest paths between genes in a given seed set and optimizes the size of subnetwork by adding the smallest number of linker genes on those paths | High coverage of significant genes (true positive rate) with the smallest number of insignificant genes (false positive rate) | Produces multiple small and isolated subnetworks |
| ClustEx | First, performs a hierarchical clustering to split the whole network into co-expressed modules, and second, extract subnetworks from the modules using shortest paths to connect significant genes | Combines clustering and shortest paths to detect highly co-expressed subnetworks | Produces multiple isolated subnetworks involving many genes |
| OptDis | Uses color coding technique to search for optimally discriminative subnetworks | Good coverage over significant genes, with small subnetworks | Cannot detect large subnetworks (over 20 genes); very slow |
| NetWalker | Diffuses information flows by random walks to prioritize important genes and interactions in the stationary state | Very fast, friendly GUI | Only produces scores for interactions, no subnetwork search, per se, without additional functional annotations |
In order to perform a fair assessment, we kept the input data of the eight models as similar as possible (see Table 1). On one hand, we used the protein–protein interaction network from Human Protein Reference Database (HPRD)6 as model input if there is no preloaded network data in the models. On the other hand, if the models used their preloaded networks and output a subnetwork including genes not in the HPRD network, we pruned them from the subnetwork. In terms of expression data, we first utilized DESeq to normalize the raw counts of mRNA sequencing from TCGA breast carcinoma data set. Then we performed differential expression analysis across the 50 case and 50 control samples and assigned each gene an adjusted P-value for its significance of differential expression. Those P-values can be directly used as the input for subnetwork detection, be ranked to select a seed gene set, or be converted into a set of particular weights tailored to the requirement of the model (see Table 1 and Methods). Next, we ran each program to detect subnetworks and tuned the parameters to control the size of subnetworks to be approximately 1,000 genes. Finally, we obtained eight subnetworks from the models and performed an assessment of their coverage of significant genes, network modularity, hits of true breast cancer genes, and functional enrichment in Kyoto Encyclopedia of Genes and Genomes (KEGG)7 pathways and Gene Ontology8 terms.
Assessment of subnetwork quality
We assess the quality of subnetworks output by the eight methods from two aspects: coverage of significant genes and network modularity. First, we prepared volcano plots with log2(fold change) versus −log10(P-values) for each method and highlighted the found genes in the eight subnetworks in red, as shown in Figure 1. We find that jActiveModules using Greedy Search (jAM. GR), BioNet, and NetBox cover most of the significant genes in their subnetworks, while excluding insignificant genes. In contrast, jActiveModules using Simulated Annealing (jAM. SA), ClustEx, and NetWalker cover a large number of genes regardless of their significance. DEGAS covers more upregulated genes, whereas OptDis covers more downregulated genes.
Figure 1.

Volcano plots of differential gene expression showing −log10 of the P-values evaluated by DESeq as a function of the log2 fold change (shown in the [−6, 6] only, 99th percentile). The dots highlighted in red are the genes involving in each subnetwork produced by the eight methods.
To further examine the specificity and sensitivity of significant gene coverage of each method, we label each detected gene as a positive sample for each method and examined whether the expression P-values predict the eight subnetworks. We plot eight Receiver Operating Characteristic (ROC) curves in Figure 2 to show the predictability of the P-values for the eight subnetworks. From Figure 2, we find that the top method is BioNet since it achieves an area under the curve (AUC) of 0.93, the highest AUC for any method. This is particularly interesting since BioNet does not depend on a seed gene set. NetBox achieves comparably high AUC (0.89), but there is an obvious kink point on the curve due to the selection of input seed genes based on P-values. The AUC of OptDis ranks the third, probably due to the small size of the subnetwork. jAM.SA detects the largest subnetwork but does not perform well in covering high P-value genes since it accepts a low P-value gene with a specific probability at each iteration in order to avoid suboptimality. ClustEx does not perform as well as NetBox, even though they use the same seed gene set and network data. This is because we only consider the largest subnetwork (210 seeds out of 801 genes) found by ClustEx as the output and discard the smaller subnetworks, which include 455 seeds.
Figure 2.
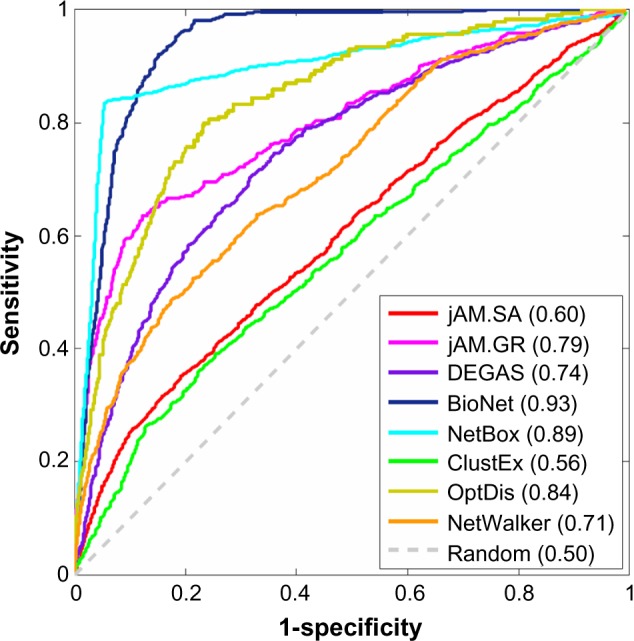
ROC curves of −log10 of the P-values predicting the eight subnetworks. The numbers in the brackets are the AUC.
To examine modularity of the eight subnetworks, we used two different measures: Global Clustering Coefficient (GCC)9 and Cut-Based Ratio (CBR).10 GCC measures how close a subnetwork is to a completely connected graph. And CBR measures the degree to which a subnetwork consists of more edges between nodes within the subnetwork and fewer edges between nodes inside and outside the subnetwork. Both modularity scores were scaled to the interval [0, 1] by dividing by the maximum quantities (Fig. 3). We can see that the OptDis subnetwork has the highest GCC, probably because there are many small (3 to 5 genes) fully connected modules in the subnetwork. In contrast, the ClustEx subnetwork has the highest CBR, probably due to the hierarchical clustering step used before growing the subnetwork within the clusters. The subnetworks of jAM.GR and DEGAS have moderately high modularity scores; both methods search for subnetworks using greedy strategies.
Figure 3.
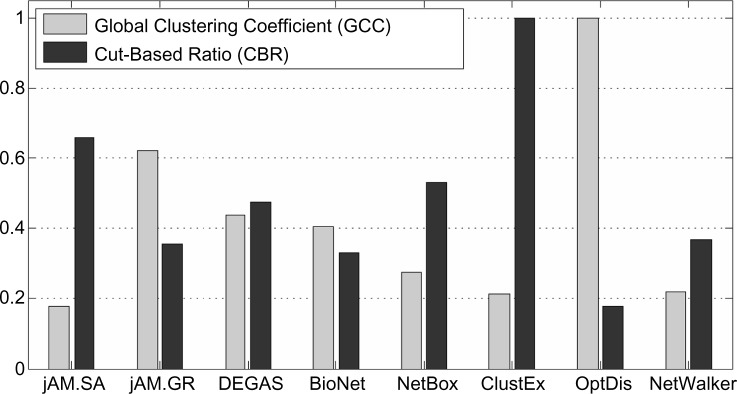
Modularity of the eight subnetworks.
Cross-model comparison and functional analysis of subnetworks
To investigate the similarity of the eight output subnetworks detected by the different methods, we first performed a pairwise comparison of the subnetworks using Jaccard similarity, in terms of nodes (Table 3) and interactions (Table 4). Surprisingly, it was found that the subnetworks of BioNet and NetBox were the most similar even though they used different subnetwork detection strategies. Methods using similar subnetwork detection algorithms have moderate similarities in their output subnetworks, such as jAM.GR and DEGAS. In contrast, methods with the same input expression and network data often detect very dissimilar subnetworks, for instance DEGAS and OptDis, and NetBox and ClustEx. The pairwise similarities of the subnetworks suggest that the use of similar algorithms and/or similar input data do not guarantee a similar output. This is because the different methods use different objective functions to evaluate a subnetwork in optimization.
Table 3.
Common genes identified by the eight methods.
| jAM.SA | jAM.GR | DEGAS | BioNet | NetBox | ClustEx | OptDis | NetWalker | |
|---|---|---|---|---|---|---|---|---|
| jAM.SA | 1290 | 144 | 182 | 164 | 285 | 158 | 52 | 160 |
| jAM.GR | 0.0936 | 393 | 137 | 168 | 213 | 63 | 40 | 146 |
| DEGAS | 0.1025 | 0.1484 | 667 | 143 | 247 | 85 | 57 | 136 |
| BioNet | 0.1038 | 0.2474 | 0.1462 | 454 | 356 | 78 | 64 | 190 |
| NetBox | 0.1526 | 0.2042 | 0.1925 | 0.3704 | 863 | 162 | 107 | 246 |
| ClustEx | 0.0817 | 0.0557 | 0.0615 | 0.0663 | 0.1079 | 801 | 34 | 11 5 |
| OptDis | 0.0365 | 0.0743 | 0.0717 | 0.1113 | 0.1137 | 0.0357 | 185 | 50 |
| NetWalker | 0.0872 | 0.1534 | 0.1100 | 0.1961 | 0.1861 | 0.0827 | 0.0595 | 705 |
Notes: The numbers on the diagonal indicate the numbers of genes identified by the corresponding method alone. The numbers above the diagonal are the numbers of genes identified by both the indicated methods. And the numbers below the diagonal are Jaccard similarities between the gene sets in the subnetworks of the indicated methods (similarities >0.2 are shown in bold).
Table 4.
Common interactions identified by the eight methods.
| jAM.SA | JAM.GR | DEGAS | BioNet | NetBox | ClustEx | OptDis | NetWalker | |
|---|---|---|---|---|---|---|---|---|
| jAM.SA | 2141 | 118 | 152 | 105 | 234 | 97 | 26 | 90 |
| jAM.GR | 0.0433 | 702 | 133 | 123 | 178 | 18 | 15 | 82 |
| DEGAS | 0.0446 | 0.0668 | 1421 | 105 | 256 | 34 | 27 | 84 |
| BioNet | 0.0397 | 0.1035 | 0.0545 | 609 | 429 | 39 | 46 | 173 |
| NetBox | 0.0686 | 0.0878 | 0.0960 | 0.2549 | 1503 | 100 | 94 | 215 |
| ClustEx | 0.0318 | 0.0107 | 0.0142 | 0.0248 | 0.0415 | 1004 | 12 | 51 |
| OptDis | 0.0110 | 0.0160 | 0.0164 | 0.0567 | 0.0567 | 0.0097 | 249 | 34 |
| NetWalker | 0.0316 | 0.0580 | 0.0394 | 0.1405 | 0.1032 | 0.0292 | 0.0337 | 795 |
Notes: The numbers on the diagonal indicate the numbers of interactions identified by the corresponding method alone. The numbers above the diagonal are the numbers of interactions found by both the indicated methods. And the numbers below the diagonal are the Jaccard similarities between the interaction sets selected by the indicated methods (similarities >0.1 are in bold).
We tested whether the detected subnetworks contain putative breast cancer genes. First, we collected 462 breast cancer genes from the KEGG Orthology Based Annotation System (KOBAS) version 2.011 functional enrichment list, which integrates Online Mendelian Inheritance in Man (OMIM),12 KEGG DISEASE,7 Functional Disease Ontology (FunDO),13 Genetic Association Database (GAD),14 and the National Human Genome Research Institute (NHGRI) Genome-Wide Association Studies (GWAS) Catalog15 disease databases. With those 462 genes as ground truth, we calculated the precision and recall of each of the eight subnetworks (Fig. 4) and found that the top subnetworks in identifying the true breast cancer genes are those produced by BioNet, NetWalker, NetBox, and jAM.GR. Surprisingly, these four methods use totally different algorithms for subnetwork detection (see Table 1). And NetWalker displayed its potential for predicting true disease genes, even though its coverage of significantly differentially expressed genes was relatively poor; this may be due to its use of random walks to diffuse information through the whole network without any restriction to shortest paths and greedy search.
Figure 4.
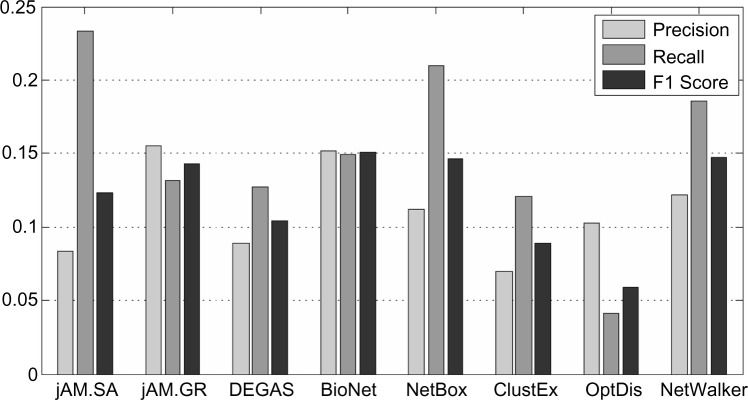
Prediction of the 462 breast cancer genes by the eight subnetworks. F1 score is defined as 2 × precision × recall/(precision + recall).
Then we used the list of true breast cancer genes to investigate if cancer-related genes are more likely to be detected by multiple methods. The distribution of all genes and the breast cancer genes is shown in Figure 5A in terms of how many different methods detect genes in these classes. We can see in Figure 5A that many genes are detected by only a few methods, whereas a small number of genes are detected by almost every method. Surprisingly, the percentage of breast cancer genes in the reported subnetworks increases with the number of methods detecting those genes, suggesting that the genes detected by more methods are more likely to be a true breast cancer genes. And also it suggests that an ensemble method that integrates multiple methods may be a better way of detecting subnetworks covering more disease genes. Similarly, we collected 2,058 interactions enriched in breast cancer pathways using KOBAS 2.0 from the KEGG pathway,7 Pathway Interaction Database (PID),16 BioCarta,17 Reactome,18 BioCyc,19 and Protein ANalysis THrough Evolutionary Relationships (PANTHER)20 databases. The distribution of interactions in terms of the number of methods detecting those interactions is shown in Figure 5B. We found that no interactions were commonly detected by more than six methods. The interactions commonly detected by more methods are slightly more likely to be enriched in pathways related to breast cancer.
Figure 5.
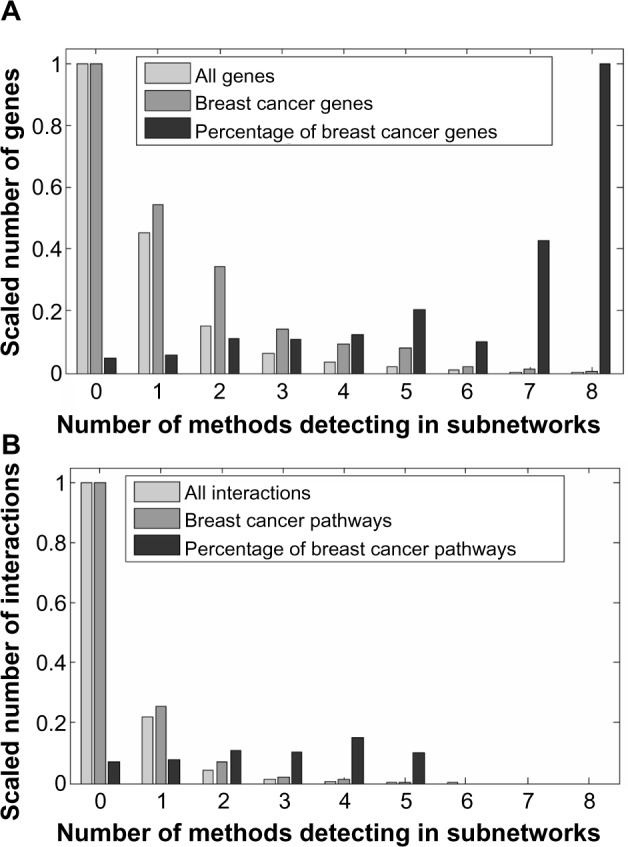
Number of methods detecting genes and interactions in subnetworks. Histograms of the number of genes (A) and interaction counts (B) versus the number of methods that detect them. (A) All genes denote the 7,369 genes in the HPRD network. Breast cancer genes are the 462 genes found by KOBAS in multiple disease databases. Both the gene counts are scaled to [0, 1] by dividing by the maximum count. The percentage of breast cancer genes is the breast cancer gene count divided by the count of all the genes in each category (genes found by a certain number of methods). (B) All interactions denote the 28,571 interactions in the HPRD network. Breast cancer pathways are the 2,058 interactions found by KOBAS in multiple pathways databases. Both the interaction counts are scaled to [0, 1] by dividing by the maximum count. The percentage of breast cancer pathways is the interaction count in breast cancer pathways divided by the total interaction count in each category.
To examine functional enrichment of commonly detected genes, we used KOBAS to annotate the 553 genes detected by at least three methods (Supplementary Table 1). The top enriched KEGG pathways of these genes are cell cycle (hsa04110), MicroRNAs in cancer (hsa05206), and Pathways in cancer (hsa05200), all with the corrected P-values less than 0.05. Cancers are enriched as the topmost disease in KEGG DISEASE database with corrected P-values less than 0.1. And the top GO terms enriched in this gene set are extracellular matrix (GO:0031012), cell division (GO:0051301), and their relevant terms. Note that there is no breast cancer–specific term significantly enriched in terms of pathways, diseases, and functions.
Finally, we used Cytoscape 3.021 to visualize a prominent subnetwork in which each interaction is detected by at least five methods. This subnetwork consists of 60 genes and 50 interactions (Fig. 6). Within those 60 genes, there are 12 true breast cancer genes (red border) detected by KOBAS 2.0 in the multiple databases. Notably, the breast cancer gene Nuclear Receptor Subfamily 3, Group C, Member 2 (NR3C2), a gene encoding the mineralocorticoid receptor, was the only gene detected by all the eight methods. An RNA interference (RNAi) experiment has verified that the depletion of NR3C2 increases cell death in breast.22 This evidence is consistent with Figure 6 in which NR3C2 is downregulated in breast cancer cells (log2(fold change) = −2.2). We also found that actin alpha 1 (ACTA1), one of the interactors of NR3C2, was detected by five methods and was downregulated as well. ACTA1 is a highly conserved protein responsible for cell motility and a major constituent of the contractile apparatus.23 This suggests that downregulation of ACTA1 causes increased cell motility and cancer metastasis. Similarly, inhibin, beta A (INHBA), pleiotrophin (PTN), and seven in absentia homolog family E3 (siah E3) ubiquitin protein ligase 2 (SIAH2), which were detected by seven methods, have been experimentally verified to be associated with breast cancer development. Overexpression of INHBA in mesenchymal cells increases colony formation potential of breast epithelial cells.24 PTN, a secretory cytokine, has been found to stimulate breast cancer progression through remodeling of the tumor microenvironment.25 Downregulation of SIAH2 has been found to be associated with resistance to endocrine therapy in breast cancer.26
Figure 6.

Prominent subnetwork whose interactions are detected by at least five methods. Node color indicates log2 fold change of differential expression (yellow: upregulated in tumor samples; blue: downregulated in tumor samples). The 12 genes in red border are in the list of 462 known breast cancer genes. Visualized by Cytoscape 3.0 version.6
Conclusion
We have performed a comprehensive assessment of a broad spectrum of state-of-the-art methods for subnetwork detection using up-to-date gene expression data specific for breast cancer. The key findings in this study can be summarized in the following three main points.
First, based on the functional enrichment analysis, the subnetworks detected by the individual methods offer only limited information on breast cancer pathology. However, the prominent subnetwork detected by the majority of the methods offers a very specific and relevant result that is clearly related to breast cancer pathology. The data used here are probably as good as or better than what is currently available for most kinds of tumors and are therefore representative of typical situations. Even though each of the eight methods were claimed to be effective in their original publications, based on the data sets they used, the subnetwork detection problem still cannot be considered to be solved and needs further investigation.
Second, the enrichment in known breast cancer–related genes in the set of genes identified by many independent methods suggests that investigators should use several different methods based on different principles. For the data set used here, we suggest that a combination of BioNet, jAM.GR, NetBox, and NetWalker could be used, although it is not clear that this would be true for all data sets or types.
Third, in terms of ease of use, some of the methods are available only as source code, which must be compiled and installed, typically on a UNIX-based system; this may be an obstacle for some experimental biologists. A GUI is highly recommended for the purpose of wide use, or perhaps implementation within a widely used system such as R.
We suggest that the definition of subnetwork needs to be refined to be something more than a simple subset of a global network. Interactome data need to be dissected and reorganized using high-level structures, such as pathways and protein complexes. Those interactome structures ensure that the output subnetworks are biologically meaningful and guide subnetwork detection methods to prune a global network without losing the important biological structures.
Methods
Data preprocessing
Subnetwork detection usually requires two input data sets, a gene expression data set and a network data set. In this study, gene expression was measured by mRNA sequencing (RNA-Seq), and were obtained from TCGA breast invasive carcinoma category.1 The expression data consist of raw counts, normalized median transcript lengths, and Reads Per Kilobase of transcript per Million mapped reads for 20,532 genes in 50 tumor samples, paired with 50 normal samples from the same patients. The network data set was downloaded from HPRD.6 After gene ID matching using BioNet, 7,369 nonredundant genes remained (Supplementary Table 2) and 28,571 interactions were recorded among the encoded proteins after removal of self-loops and isolated interactions (Supplementary Table 3). DESeq4 was used to normalize the raw counts and to detect differentially expressed genes between the tumor and normal samples based on a negative binomial model. The P-values were then adjusted for multiple testing with Benjamini–Hochberg procedure27 (Supplementary Table 1)
Subnetwork detection methods
Unless further specified, we used default setting of parameters for all eight models. The input expression and network data are summarized in Table 1, and the gene and interaction lists of the eight subnetworks are in shown Supplementary Tables 2 and 3, respectively.
jActiveModules2,28 requires a weighted gene list with the weights ranging from 0 to 1. Hence, we directly used the adjusted P-values from DESeq as the weights. Within jActiveModules, there are two different search strategies for subnetworks: simulated annealing and greedy search. For simulated annealing, we increased the default number of iterations from 2,500 to 10,000. Default parameter settings were used for greedy search. For both kinds of searches, we set the maximum number of modules as 1.
DEGAS29 has multiple optional algorithms, and we used the CUSP (Covering Using Shortest Paths) heuristic algorithm to detect subnetworks. Dysregulation direction was selected to be DIFF, and maximum number of modules was set to 1. The number of covered genes k was set to increase from 100 to 1,000 with a step size of 100. The other parameters were kept at their default values.
BioNet30 requires the raw P-values (not adjusted for multiple testing) as the input from differential expression analysis by DESeq. Intrinsically, BioNet first aggregates two lists of P-values from two pairs of comparisons (case 1 vs. control and case 2 vs. control) into one list. Since we only had one comparison between tumor and normal samples, we input one more replicate list of P-values to meet the requirement. We set the False Discovery Rate (FDR) cutoff as 0.00001 other than the default value 0.001. A low FDR cutoff has effects on reducing the size of an output subnetwork.
NetBox31 is provided with a preloaded Human Interaction Network, and therefore, the only input data needed are a list of seed genes. We used only the genes with the adjusted P-value less than 0.0001 in the differential expression analysis as the seed gene set, which selected 1,063 (14.4%) out of 7,369 genes. The shortest path threshold was set to 2 rather than the default value 1.
ClustEx32 provides preloaded network data and also supports customized network uploading. For comparative purposes, we used the trimmed HPRD network described above. It also requires a seed gene set; we used the same set used with NetBox. We considered only the largest output cluster (801 genes) as the final output subnetwork, since all the other 354 clusters contained less than 40 genes.
OptDis33 needs three input data sets: a network, a gene expression profile, and a gene ID conversion list linking the network and expression sets. As shown in Table 1, OptDis ran slowly. To keep the computational cost tractable, we set the maximum size of modules to 10. OptDis returned 50 modules, all with sizes less than 10 genes. We consider the union of these modules to be a single subnetwork in our analysis.
NetWalker34 has a preloaded network database called the NetWalker Interactome Knowledgebase. After matching our 7,369 genes with the 13,328 genes in the preloaded network, we obtained 7,354 matched genes. NetWalker requires an expression ratio for each gene centered around 1. We defined the ratio as r = 2*logit(log2(FC)), where FC denoted the fold change of gene expression in tumor over that in normal cells, and the logit() function was defined as logit(x) = 1/(1 + exp(−x)). The unmatched genes were assigned expression ratios of 1, denoting no significant expression change. After running, NetWalker returned an Edge Flux value ranging from −10.04 to 2.41 for each of the 327,599 interactions in the preloaded network. We selected 2,210 (0.67%) interactions with the values lower than −5.5 or higher than 1.5 as the output subnetwork. Then the interactions not present in the HPRD network were removed, and there remained 795 interactions as the final subnetwork produced by NetWalker.
Subnetwork quality assessment and functional enrichment analysis
Majority of network analysis and graphing were done using MATLAB. And the functional enrichment analysis of subnetworks was performed by KOBAS version 2.0.11 We identified 462 breast cancer genes out of the 7,369 genes (Supplementary Table 2) in multiple disease databases using KOBAS, and used them as the ground truth to evaluate the predictability of the eight subnetworks (see Figs. 4, 5A, and 6). Similarly, we combined the 462 breast cancer genes with 227 genes enriched in cancer pathways to query the HPRD network and found 2,058 interactions (Supplementary Table 3) that connect the 689 genes in the querying list as a positive set of breast cancer pathways (see Fig. 5B). For the functional analysis of commonly detected genes by at least three methods, we input those genes in KOBAS and set the 7,369 genes to the background gene set (Supplementary Table 1).
Supplementary Data
Supplementary Table 1. Functional enrichment analysis of the genes detected by at least three methods.
Supplementary Table 2. Gene list of the eight subnetworks and their P-values in differential expression analysis.
Supplementary Table 3. Interaction list of the eight subnetworks.
Acknowledgments
The authors thank Doug Yatcilla for his advice and assistance installing and running software. The authors also gratefully acknowledge use of the facilities of the Purdue Rosen Center for Advanced Computing.
Footnotes
Author Contributions
Conceived, designed, and conducted the experiments: BJ. Contributed to the writing and revision of the manuscript: BJ, MG. Both authors reviewed and approved of the final manuscript.
ACADEMIC EDITOR: JT Efird, Editor in Chief
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
REFERENCES
- 1.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18(suppl 1):S233–40. doi: 10.1093/bioinformatics/18.suppl_1.s233. [DOI] [PubMed] [Google Scholar]
- 3.Mitra K, Carvunis AR, Ramesh SK, Ideker T. Integrative approaches for finding modular structure in biological networks. Nat Rev Genet. 2013;14(10):719–32. doi: 10.1038/nrg3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger B, Peng J, Singh M. Computational solutions for omics data. Nat Rev Genet. 2013;14(5):333–46. doi: 10.1038/nrg3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keshava Prasad TS, Goel R, Kandasamy K, et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009;37(Database issue):D767–72. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris MA, Clark J, Ireland A, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32(Database issue):D258–61. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luce RD, Perry AD. A method of matrix analysis of group structure. Psychometrika. 1949;14(2):95–116. doi: 10.1007/BF02289146. [DOI] [PubMed] [Google Scholar]
- 10.Whang JJ, Gleich DF, Dhillon IS. Overlapping community detection using seed set expansion; Proceedings of the 22nd Association for Computing Machinery (ACM) International Conference on Information & Knowledge Management; Burlingame, CA, USA. 2013. pp. 2099–108. http://www.cikm2013.org/ [Google Scholar]
- 11.Xie C, Mao X, Huang J, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39(Web Server issue):W316–22. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33(Database issue):D514–7. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du P, Feng G, Flatow J, et al. From disease ontology to disease-ontology lite: statistical methods to adapt a general-purpose ontology for the test of gene-ontology associations. Bioinformatics. 2009;25(12):i63–8. doi: 10.1093/bioinformatics/btp193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becker KG, Barnes KC, Bright TJ, Wang SA. The genetic association database. Nat Genet. 2004;36(5):431–2. doi: 10.1038/ng0504-431. [DOI] [PubMed] [Google Scholar]
- 15.Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(Database issue):D1001–6. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaefer CF, Anthony K, Krupa S, et al. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009;37(Database issue):D674–9. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishimura D. Biotech Software and Internet Report. Vol. 2. New Rochelle, NY: Mary Ann Liebert, Inc; 2001. BioCarta; pp. 117–20. [Google Scholar]
- 18.Joshi-Tope G, Gillespie M, Vastrik I, et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33(Database issue):D428–32. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caspi R, Altman T, Billington R, et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014;42(Database issue):D459–71. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mi H, Lazareva-Ulitsky B, Loo R, et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005;33(Database issue):D284–8. doi: 10.1093/nar/gki078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saito R, Smoot ME, Ono K, et al. A travel guide to Cytoscape plugins. Nat Methods. 2012;9(11):1069–76. doi: 10.1038/nmeth.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silva JM, Marran K, Parker JS, et al. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science. 2008;319(5863):617–20. doi: 10.1126/science.1149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sajnani MR, Patel AK, Bhatt VD, et al. Identification of novel transcripts deregulated in buccal cancer by RNA-seq. Gene. 2012;507(2):152–8. doi: 10.1016/j.gene.2012.07.036. [DOI] [PubMed] [Google Scholar]
- 24.Duss S, Brinkhaus H, Britschgi A, et al. Mesenchymal precursor cells maintain the differentiation and proliferation potentials of breast epithelial cells. Breast Cancer Res. 2014;16(3):R60. doi: 10.1186/bcr3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang Y, Zuka M, Perez-Pinera P, et al. Secretion of pleiotrophin stimulates breast cancer progression through remodeling of the tumor microenvironment. Proc Natl Acad Sci USA. 2007;104(26):10888–93. doi: 10.1073/pnas.0704366104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jansen MP, Ruigrok-Ritstier K, Dorssers LC, et al. Downregulation of SIAH2, an ubiquitin E3 ligase, is associated with resistance to endocrine therapy in breast cancer. Breast Cancer Res Treat. 2009;116(2):263–71. doi: 10.1007/s10549-008-0125-z. [DOI] [PubMed] [Google Scholar]
- 27.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 28.Chuang HY, Lee E, Liu YT, Lee D, Ideker T. Network-based classification of breast cancer metastasis. Mol Syst Biol. 2007;3:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ulitsky I, Krishnamurthy A, Karp RM, Shamir R. DEGAS: de novo discovery of dysregulated pathways in human diseases. PLoS One. 2010;5(10):e13367. doi: 10.1371/journal.pone.0013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beisser D, Klau GW, Dandekar T, Müller T, Dittrich MT. BioNet: an R-Package for the functional analysis of biological networks. Bioinformatics. 2010;26(8):1129–30. doi: 10.1093/bioinformatics/btq089. [DOI] [PubMed] [Google Scholar]
- 31.Cerami E, Demir E, Schultz N, Taylor BS, Sander C. Automated network analysis identifies core pathways in glioblastoma. PLoS One. 2010;5(2):e8918. doi: 10.1371/journal.pone.0008918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu J, Chen Y, Li S, Li Y. Identification of responsive gene modules by network-based gene clustering and extending: application to inflammation and angiogenesis. BMC Syst Biol. 2010;4:47. doi: 10.1186/1752-0509-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dao P, Wang K, Collins C, Ester M, Lapuk A, Sahinalp SC. Optimally discriminative subnetwork markers predict response to chemotherapy. Bioinformatics. 2011;27(13):i205–13. doi: 10.1093/bioinformatics/btr245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komurov K, Dursun S, Erdin S, Ram PT. NetWalker: a contextual network analysis tool for functional genomics. BMC Genomics. 2012;13:282. doi: 10.1186/1471-2164-13-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Functional enrichment analysis of the genes detected by at least three methods.
Supplementary Table 2. Gene list of the eight subnetworks and their P-values in differential expression analysis.
Supplementary Table 3. Interaction list of the eight subnetworks.