

Abstract
Cytochrome c oxidases (Ccoxs) are the terminal enzymes of the respiratory chain in mitochondria and most bacteria. These enzymes couple dioxygen (O2) reduction to the generation of a transmembrane electrochemical proton gradient. Despite decades of research and the availability of a large amount of structural and biochemical data available for the A-type Ccox family, little is known about the channel(s) used by O2 to travel from the solvent/membrane to the heme a3-CuB binuclear center (BNC). Moreover, the identification of all possible O2 channels as well as the atomic details of O2 diffusion is essential for the understanding of the working mechanisms of the A-type Ccox. In this work, we determined the O2 distribution within Ccox from Rhodobacter sphaeroides, in the fully reduced state, in order to identify and characterize all the putative O2 channels leading towards the BNC. For that, we use an integrated strategy combining atomistic molecular dynamics (MD) simulations (with and without explicit O2 molecules) and implicit ligand sampling (ILS) calculations. Based on the 3D free energy map for O2 inside Ccox, three channels were identified, all starting in the membrane hydrophobic region and connecting the surface of the protein to the BNC. One of these channels corresponds to the pathway inferred from the X-ray data available, whereas the other two are alternative routes for O2 to reach the BNC. Both alternative O2 channels start in the membrane spanning region and terminate close to Y288I. These channels are a combination of multiple transiently interconnected hydrophobic cavities, whose opening and closure is regulated by the thermal fluctuations of the lining residues. Furthermore, our results show that, in this Ccox, the most likely (energetically preferred) routes for O2 to reach the BNC are the alternative channels, rather than the X-ray inferred pathway.
Author Summary
Cytochrome c oxidases (Ccoxs), the terminal enzymes of the respiratory electron transport chain in eukaryotes and many prokaryotes, are key enzymes in aerobic respiration. These proteins couple the reduction of molecular dioxygen to water with the creation of a transmembrane electrochemical proton gradient. Over the last decades, most of the Ccoxs research focused on the mechanisms and energetics of reduction and/or proton pumping, and little emphasis has been given to the pathways used by dioxygen to reach the binuclear center, where dioxygen reduction takes place. In particular, the existence and the characteristics of the channel(s) used by O2 to travel from the solvent/membrane to the binuclear site are still unclear. In this work, we combine all-atom molecular dynamics simulations and implicit ligand sampling calculations in order to identify and characterize the O2 delivery channels in the Ccox from Rhodobacter sphaeroides. Altogether, our results suggest that, in this Ccox, O2 can diffuse via three well-defined channels that start in membrane region (where O2 solubility is higher than in the water). One of these channels corresponds to the pathway inferred from the X-ray data available, whereas the other two are alternative routes for O2 to reach the binuclear center.
Introduction
Cytochrome c oxidases (Ccoxs) are the terminal enzymes of the respiratory chain in eukaryotes and in aerobic prokaryotes (reviewed in [1]). These integral membrane proteins belong to the heme-copper oxidases superfamily and couple dioxygen (O2) reduction to the translocation of protons across the membrane. Ccox takes up four electrons from cytochrome c (cyt c) in the positively charged side of the membrane (the inter-membrane space in mitochondria or the periplasm in bacteria) and eight protons from the negatively charged side (eq. 1) [2], [3]:
| (1) |
where the subscripts P and N refer to the positive and negative sides of the membrane, respectively.
Four of the eight protons reported in equation 1 are used to reduce one O2 molecule and form two water molecules [2], [3], whereas the remaining protons are pumped from the negative to the positive side of the membrane. This overall process contributes to the generation and maintenance of a transmembrane electrochemical proton gradient, which can be further utilized for several energy-requiring processes, such as ATP synthesis [4].
Based on structural and phylogenetic analysis, the heme-copper oxidases superfamily is currently divided into three major subfamilies [5]: A, B and C. The main differences between the three families are the pathways and mechanisms of proton transfer/pumping. The A-type Ccoxs, which are the subject of this work, are widespread through all kingdoms of life [5] and among them are the most thoroughly explored Ccoxs [3], [6], such as the bovine heart mitochondria, the Paracoccus (P.) denitrificans and the Rhodobacter (R.) sphaeroides enzymes. These Ccoxs contain, in the catalytic subunit (subunit I), a low spin heme a and a heterodinuclear center named binuclear center, BNC (Fig. 1A). The BNC is deeply buried in the core of the protein and it is formed by a high-spin heme a3 and a copper ion (CuB). In subunit II, these Ccoxs contain only one redox center, a binuclear copper center named CuA, which accepts electrons from the soluble cyt c and then transfers them to the BNC via heme a.
Figure 1. Structure of the membrane bound Ccox from R. sphaeroides [15] .

. For simplicity, only subunits I and II are represented. A) Architecture of the Ccox core functional unit formed by subunits I (colored in green) and II (colored in cyan). The small blue spheres represent the water molecules resolved in the D- and K-pathways in the X-ray structure [15]. During an O2 reduction cycle, the electrons are sequentially donated from cyt c and delivered to the BNC, via CuA and heme a, like in the scheme: cyt c → CuA → heme a → BNC (heme a3/CuB) → O2. B) Putative O2 channel inferred from the X-ray data available. The grey mesh represents the putative O2 channel and it was calculated with the program HOLLOW [92].
It is believed that protons (both chemical and pumped) are transported from the N-side of the membrane to the BNC via two special proton conducting pathways [3]: the D- and K-pathways (Fig. 1A). A third putative proton-conducting pathway, the H-pathway, was proposed for the mammalian Ccox only [6], [7], and it was suggested to be exclusively used for the transfer of the pumped protons [8].
Several high-resolution crystallographic structures of the A-type family are nowadays available in the literature (e.g. mammalian [7], [9]–[11] and bacterial Ccoxs [12]–[15]) and, based on these structures, it is known that all A-type members share a remarkable structural similarity of the core functional unit formed by subunits I and II (Fig. 1A). Subunit I consists of twelve transmembrane α-helices and contains the BNC and the heme a center. Subunit II is formed by a solvent exposed globular β-sheet domain (which functions as a docking surface for cyt c) and two transmembrane α-helices. It contains only one redox center, the binuclear copper center (CuA). Moreover, at the interface between subunits I and II, Ccox has one Mg+2 ion whose function is still not well understood, but it was suggested to be part of the exit pathway for the pumped protons and for water formed in the BNC [16], [17]. Subunit III, although not considered to be part of the core functional unit, is also highly conserved among the A-type subfamily. Nevertheless, its absence significantly increases the probability of suicide inactivation [18], [19] and thereby reduces the catalytic lifespan of Ccox (in 600-fold or more) [19].
Based in the X-ray data available (eg. [7], [12], [13]), a putative O2 channel for the A-type family was proposed (Fig. 1B). Iwata and co-workers, after pressurizing R. sphaeroides Ccox crystals with xenon, were able to identify a continuous hydrophobic channel that starts in the membrane region of subunit I [13]. This putative O2 channel has two possible entrances that merge together in a region close to the proton-gating residue, E286I (the residues are numbered according to the R. sphaeroides Ccox sequence and the subscript indicates the subunit number). This pathway presents a constriction point which does not allow the access of O2 to the BNC, at least without the occurrence of some conformational change in the protein. Unfortunately, until now, none of the mutagenesis and biochemical studies performed in this channel [20]–[22] was able to clearly demonstrate that it serves as an O2 route into the BNC. All the tested mutations were located too close to the BNC [20], [21], which made the interpretation of the results difficult and did not allow to unambiguously distinguish between the structural obstruction of the O2 channel and the perturbation of the BNC binding kinetics. However, and contrary to the A-type family, in the B-type family the channel used by O2 to reach the BNC is nowadays considered to be well established. The crystallographic studies (with xenon pressurization) performed in the Thermus (T.) thermophilus ba3 enzyme [23]–[25], lead to the identification of a “Y-shaped” hydrophobic channel that runs from the membrane region towards the BNC. This channel, although located roughly at the same position of the putative O2 channel in the A-type Ccox, does not possess a constriction point close to the BNC. In the A-type Ccoxs, the narrowing of the O2 channel is mainly caused by two conserved bulky residues (W172I and F282I in R. sphaeroides [13]), whereas in the B-type Ccox, smaller residues occupy these positions (Y133I and T231I in T. thermophilus [23], [24]). The differences between the A- and B-type regarding the O2 channel are thought to reflect the different functional environments of each type of Ccox.
Although the static crystal structures have been a valuable tool for providing insights into the O2 diffusion and for identifying potential O2 channels in Ccox, the elucidation of the molecular basis of O2 diffusion requires the knowledge of the Ccox conformational dynamics. Transiently formed cavities and openings inside the protein (frequently regulated by side chain rotation or by water movements) are not visible in the static X-ray structures, but have already been shown to be very relevant for ligand diffusion (see for example [26]). In this context, molecular dynamics (MD) simulation techniques (with sufficient simulation time and conformational sampling) appear as an alternative for studying the dynamic behavior of proteins and to determine their ligand occupation probabilities inside the protein. In the last decade, computational methods have been widely used to study gas migration in a number of proteins and MD simulations have successfully allowed the identification of several alternative routes for ligand diffusion (e.g. hydrogenase [26]–[28], myoglobins [29], [30], oxidases [31], [32] and laccases [33]). Moreover, the combination of MD simulations with Implicit Ligand Sampling (ILS) [29] calculations allows the calculation of the energy cost of transferring any small, apolar molecule (like O2 or H2) from the solvent to the protein and consequently to compute a 3D free-energy landscape for that specific ligand molecule (e.g [29], [33], [34]).
Over the last decades, most of the Ccox research using computational methods focused on the mechanisms and energetics of reduction and/or proton pumping (e.g [17], [35]–[56]). In the A-type Ccox, little emphasis has been given to the identification of the routes used by O2 to move from the solvent towards the BNC, a question only addressed, to our knowledge, by Hofacker and Schulten [31] and by Farantos and co-workers [31]. In the first work, Hofacker and Schulten [31] used MD simulations to study O2 diffusion in the vicinity of the BNC in a bacterial Ccox from P. denitrificans and in the bovine CcOx enzyme. Their simulations revealed a unique, well-defined O2 diffusion channel starting in the membrane-spanning surface of subunit I, close to the interface with subunit III. More recently, Farantos and co-workers [32] have applied the ILS method in order to study the binding of several small gas molecules around the BNC region in the A-type Ccox from P. denitrificans and in the B-type Ccox enzyme from T thermophilus. From these calculations, the authors were able to identify several cavities around the heme a3 region that are conserved in both the A-type and B-type enzymes. This study is however limited to the BNC region, not including other parts of the protein and, consequently, not allowing the analysis of the whole O2 permeation process.
The main objective of this work is to identify the O2 channels in the fully reduced Ccox from R. sphaeroides [15] using a combination of MD simulations (with and without explicit O2) and ILS calculations. Our results revealed the existence of three putative O2 diffusion channels. One of channels correlates very well with the channel inferred from the X-ray data available, whereas the other two are alternative routes for O2 to reach the BNC, and were not observed in the X-ray structures pressurized with xenon. Both alternative channels start in the membrane phase and terminate close to Y288I.
Results/Discussion
In this work, we investigate the diffusion of O2 molecules from the solvent to Ccox using MD simulations. We started our study by performing simulations with the enzyme (in the fully reduced state) with explicit O2 molecules, as described in detail in the Materials and Methods. Although all O2 molecules were initially placed randomly in the solvent, as time progresses, the gas enters the membrane and concentrates in the lipid tails region (see S8 Figure in Text S1), similarly to what has been described experimentally [57], [58]. After 100 ns of simulation, 46 O2 molecules were internalized in the membrane, which corresponds to more than 50% of the O2 placed originally inside the simulation box.
Furthermore, during the simulation time, some O2 molecules move from the membrane into the protein. The number of O2 inside Ccox increased slowly during the first 30 ns of simulation until it stabilizes at ∼8 molecules on average (see S9 Figure in S1 Text). In general, before entering Ccox, the O2 molecules explore the protein's surface and bind briefly to the cavities and niches formed mainly by hydrophobic residues. However, after 100 ns, none of the internalized O2 molecules was able to reach the BNC in any replicate. Nonetheless, and in order to determine which regions of the protein are more populated by the O2 molecules during our simulations, we calculated the O2 probability density maps [59] over the 100 ns (for all five replicates) and the results are depicted in Fig. 2.
Figure 2. O2 probability density maps obtained from all-atom MD simulations.

A) The probability density contours at 0.00015 Å−3 are depicted as a grey surface. The X-ray structure is represented as a ribbon with subunit I colored in green and subunit II in cyan. The hemes are depicted as green sticks. The yellow, light blue and green spheres represent the Cu, Fe (from the heme groups) and the Mg atoms, respectively. The residues forming the putative O2 channel inferred from the X-ray structure (according to [13]) are colored in orange and the residues forming the constriction point in this channel are highlighted in magenta. B) Zoom image of the putative O2 channel inferred from the X-ray structure pressurized with xenon [13]. The red arrow identifies the two possible entrance points for this O2 channel.
As it can be seen, most of the high-affinity regions are located in the membrane phase (Fig. 2A) and some of them show a good correlation with the putative O2 channel inferred from the X-ray data pressurized with xenon [13] (Fig. 2B). This channel starts at the membrane spanning region in direct contact with the hydrophobic tails of the lipids and it is formed predominately by uncharged and aromatic residues. It has a Y shape with the two entrances located between helices 5 and 8 and helices 11 and 13 in subunit I. The side chains of I104I, L105I, A153I, L157I, F108I, L174I, V194I, L246I, and I250I contribute to form this channel. The two possible entrances merge together into a constriction point located close to E286I. This reduction in the diameter of the channel is caused by the phenyl ring of F282I and the indole ring of W172I (see Fig. 2B). It has been suggested that during the O2 reduction cycle, some protein rearrangement or side chain rotation (of F282I and/or W172I) is required in order to open the constriction point and allow O2 access to the BNC [7], [23]. In our O2 simulations, the side chains of F282I and W172I, although showing some flexibility and being able to slightly change their conformation, did not open an entrance point large enough to allow the passage of O2 into the BNC. For this reason, it was impossible to directly observe O2 diffusion towards the catalytic site.
Recent computational studies have suggested that the hydration level of the internal hydrophobic cavity located after the constriction point (formed by F282I, W172I and E286I) could be important for the regulation of proton transfer in Ccoxs (e.g [55], [56]). It was also suggested that the water distribution inside this cavity is modulated by the protonation of the heme a3 D-propionate and that these hydration changes strongly affect the E286I proton affinity [56]. This hydrophobic cavity bridges between the end of the D-pathway and the BNC region and it has been predicted to contain several water molecules, at least transiently (e.g. [38], [60]–[62]). However, in our simulations, no water molecules were observed in this region and no.direct and visible connection between E286I and the BNC was identified.
Since the simulations with explicit O2 were not able to properly sample the diffusion events (probably due to the simulation timescales), and in order to identify possible alternative O2 diffusion channels in fully reduced Ccox, the ILS method [29] was used. This method uses the protein conformations obtained from a ligand-free MD trajectory and calculates, for all the positions inside the protein, the potential of mean force for placing a small, apolar, low-interacting molecule at that point. The 3D energy map generated in this case represents the Gibbs free energy of moving a O2 molecule from water into any place inside the protein (ΔG wat→prot(O2)) and it can be used to infer about the energetically favourable diffusion paths inside a given protein. In these maps, the regions where ΔG wat→prot(O2) is low represent the positions where O2 has a high probability of residing, meaning that the O2 affinity in that position is high. The ILS method reduces significantly the sampling problems and it takes into account the dynamic conformational changes of the protein and all the transiently formed cavities, which can be combined to form transitory diffusion pathways.
From the 3D affinity map for Ccox obtained from the ILS calculations (Fig. 3A), we can see that Ccox possesses a complex free energy landscape with several possible O2 binding cavities, most of them located at the protein surface and that do not directly connect to the BNC. Moreover, the calculated affinity map from ILS not only correlates well with some of the high probability regions found in the explicit O2 simulations (see Fig. 3B) but also provides a more complete description of the free energy landscape for O2 inside Ccox due to the sampling of lower affinity zones, not sufficiently sampled by the normal MD simulations. Indeed, the ILS approach allowed us to identified three major routes (named channel 1, channel 2 and channel 3) with high probability of O2 occupancy, interconnecting the protein surface to the BNC. Two of the channels (channel 1 and channel 2) approach the BNC from the subunit I side whereas the third one (channel 3) approaches the BNC from the subunit II side (Fig. 3A). Interestingly all three channels start in the membrane spanning region where the O2 concentration is higher than in aqueous phase, which makes physical sense.
Figure 3. O2 free energy landscape obtained from the ILS calculations and lowest free energy pathways obtained from the ILS energy landscape.

A-O2 free energy landscape obtained from the ILS calculations. Two isosurface contours are shown, with free energy levels at -5 (blue inner contour) and 5 (cyan contour) kJ·mol−1. B- Comparison of the O2 free energy landscape obtained from the ILS calculations (blue maps) and high-affinity regions from the O2 explicit MD simulations (red maps). The probability density contours at 0.00015 Å−3 are represented as a red isosurface. C- O2 lowest free energy pathways obtained from the ILS energy landscape. The spheres represent the local free energy minima while the tubes connecting the minima represent the pathways. The size of the spheres representing the local energy minima scales linearly along the displayed free energy range (small spheres indicates high free energy while large spheres indicate low free energy). The diameter of the pathways scales linearly with the free energy and thus with the oxygen affinity (thinner radius represent high free energies and low oxygen affinity). The free energy at the minima and along the pathways follows the same color scale. The channel 1 coincides with the channel inferred from the X-ray data [7], [13] whereas the channel 2 and channel 3 are alternative routes to reach the BNC.
Furthermore and in order to identify the energetically preferred routes for O2 to access the BNC, we extracted, from the ILS affinity map, the lowest free energy pathways connecting the exterior with the O2 high affinity sites (basins in the O2 free energy landscape) inside Ccox, using the same methodology as Damas et al [33] (for details, see the Data Analysis section of Materials and Methods). From the analysis of the O2 free energy landscape (Fig. 3C), we observe that there are many energy minima and low free energy pathways connecting the solvent/membrane region to the interior of the protein. Nonetheless, all these low free energy entrance channels converge into three distinct pathways as we approach the BNC.
The O2 channel 1 approaches the BNC from the subunit I side and corresponds to the channel inferred from the X-ray data (for R. sphaeroides [13] or for bovine [7]). This pathway has two entry points that are fused together in a free energy minimum located in the constriction point just before the BNC (M10 in Fig. 4A). The free energy profile for this pathway (Fig. 4A) is characterized by a very high permeation free energy barrier in the constriction point (associated to the bulky side chain of W172I) and three deep local minima (M8, M9 and M10 in Fig. 4A) located between W172I, F282I and E286I for M10, L243I, F282I and L283I for M8, and I250I, V194I and F108I for M9. The local minima observed just below the Ccox surface (M1 and M6) can probably act as scavengers for the O2 freely diffusing in the membrane and help to create an O2 reservoir inside the protein. In this pathway, W172I and E286I seem to act as the gateway residues that control O2 access to the catalytic site. Nevertheless, the passage from the constriction point to the BNC (moving from M10 to M11 in Fig. 4A) implies the overcoming of a very high free energy barrier of 39.5 kJ·mol−1 (see Fig. 4A and Fig. 5A), which makes O2 diffusion via this pathway very slow and difficult.
Figure 4. Overview of the O2 lowest free energy pathways obtained from the ILS free energy landscape.

For legend details related with the representation of the free energy minima and the pathways connecting them, see Fig. 3C. Heme a3 is represented in grey sticks whereas the grey spheres correspond to the Cu atom from the CuB center and to the Fe atom from heme a3. The residues forming the O2 channels are shown in sticks with a sequence label. A- Lowest free energy pathway and free energy minima for the O2 channel 1. The free energy values of the represented minima (in kJ·mol−1) are: −21.07 (M1), −11.79 (M2), −10.58 (M3), −17.97 (M4), −17.53 (M5), −16.25 (M6), −13.87 (M7), −20.07 (M8), −20.47 (M9), −20.47 (M10) and −7.12 (M11). B- Lowest free energy pathways and minima for the O2 channel 2 and channel 3. For the O2 channel 2, the values (in kJ·mol−1) for the minima are: −13.87 (M12), 0.14 (M13), −22.77 (M19) and −11.03 (M20). The free energy values (in kJ·mol−1) of the minima forming the O2 channel 3 are: −17.24 (M14), −10.46 (M15), −15.81 (M16), −10.40 (M17), −10.18 (M18), −22.77 (M19) and −11.03 (M20).
Figure 5. Free energy barriers experienced by O2.

Free energy barriers experienced by O2 when moving from the membrane region to the BNC, along Channel 1 (A), Channel 2 (B) and Channel 3 (C). For details related to the errors calculation see the data analysis section of the Materials and Methods. The “*” in the fig. indicates the transitions for which the errors could not be calculated. In Fig. A, the black lines correspond to Channel 1, which starts between helices 5 and 8 of subunit I, whereas the orange lines correspond to the second entrance point located between helices 11 and 13 of subunit I. In A and C, the dashed lines correspond to alternative routes for O2 inside the same channel (for example in Fig. C, O2 can move directly from M14 to M16, or it can go from M14 to M15 and only then to M16). The numbers inside the plots on top of the transition states indicate the free energy barriers experienced by O2 when moving from the membrane in the direction of the BNC (i.e. between the different minima on their immediate left and the transition states in question).
Over the last 20 years, several biochemical studies [20]–[22] have tried to demonstrate that this pathway is the one used for O2 diffusion in the A-type Ccoxs, but, until now, no direct measurement proved this unequivocally. All the tested mutations (e.g V287II [20], [21] and G283IV [22]) were located too close to the two metals (Fe in heme a3 and Cu in CuB) in the BNC, which made the interpretation of the results very difficult. It was impossible to unambiguously distinguish between the steric hindrance of the O2 channel (introduced by the mutations) and the perturbation in the structure and local binding kinetics at the BNC. Moreover, until now, only one X-ray structure of the A-type Ccoxs has been pressurized with xenon (considered to be an O2 analog) [13]. In this structure [13], two hydrophobic xenon binding sites were identified, none of them located beyond the constriction point of channel 1. One xenon was observed at the entrance of the X-ray inferred channel while the other was close to E286I. Since no xenon was observed after the constriction region, there is still no consensus whether this pathway is indeed a functional channel for O2 diffusion in these Ccoxs. Additionally, Hofacker and Schulten [31], with the objective of studying O2 diffusion towards the BNC, performed several MD simulations of Ccoxs with several explicit O2 molecules. The authors used the locally enhanced sampling (LES) technique [63], [64] to identify the pathway used by O2 both in a bacterial (P. denitrificans) and in the bovine Ccox enzyme. In their simulations, some of the O2 molecules reached the BNC via a unique and well-defined channel located in the same region as the X-ray inferred channel from [13]. Given the very short duration of their simulations (picoseconds), the observation of such events was only possible due to the lowered free energy barriers experienced by the O2 molecules when using the LES technique. Nevertheless, a single protein trajectory is still effectively employed in the LES approach, making the protein conformational sampling of that study more limited than the one in the present study. Finally, Farantos et al. [32] by performing ILS calculations in the A-type Ccox from P. denitrificans and in the B-type Ccox enzyme from T thermophilus, were able to calculate the free energy surfaces for the interaction of several small polar (CO, NO) and apolar (O2 and Xe) around the BNC region. Regarding the X-ray inferred channel, their results correlate very well with the ones reported in the present study and the high affinity cavity Xe1 observed by the authors in [32] coincides with minima M11 in the O2 channel 1.
The channel 2 (Fig. 3A, Fig. 4C and 4D) also starts at the membrane region and has only one possible entrance point located between the transmembrane helices 13 and 16 of subunit I, around residues V234I, L238I and V325I. Moreover, this channel terminates close to Y288I, a tyrosine residue covalently bonded to H284I, one of the CuB-coordinating histidines. Y288I is located in the end of the K proton-conducting pathway and it has been suggested to be an essential residue for the catalytic mechanism, supplying the proton used in the cleavage of the O-O bond (see [65] for a review). The energy profile for this alternative channel (Fig. 4C and 4D) is characterized by one very low local free energy minimum (M19 in Fig. 4C) located between Y288I and T359I and by a high free energy minimum (M13 in Fig. 4C) around L292I and I323I. The highest energy barriers for this small pathway are 20.4 kJ·mol−1 (Fig. 5B), which are substantially smaller than the energy barrier in the constriction point of the O2 channel 1 (39.5 kJ·mol−1). However, the high energy minimum M13 located in the middle of the channel (see Fig. 4C) may hinder O2 diffusion via this pathway.
Lastly, the O2 channel 3 approaches the BNC from the subunit II side (Fig. 3A, Fig. 4C and 4D) and its entrance is located between the transmembrane helices 28 and 30 of subunit II, around residues F71II and V167II. This channel runs parallel to the heme a3 hydroxylethylfarnesyl tail and also terminates just bellow Y288I. It is generally formed by hydrophobic and aromatic residues, such as I363I, F391I and V272II (see Fig. 4D). The energy profile for this alternative channel (Fig. 4C) is characterized by only one deep local energy minimum (M19 in Fig. 4B) located between Y288I and T359I and by several smaller (in comparison with the O2 channel 1) energy barriers, which suggest a higher probability of O2 passage. In this pathway, the largest energy barriers (19.9–22.3 kJ·mol−1 in Fig. 5C) are related with the zone of the hydroxylethylfarnesyl tail of heme a3 and the side chains of I363I and F391I. This alternative O2 channel was firstly suggested by Tsukihara et al. [7], based on the X-ray structure of the bovine heart Ccox, as one of the three possible O2 entrance points in the A-type oxidases. More recently, Farantos et al. [32] were also able to observe the final part of this channel (located close to heme a3) in the A-type Ccox from P. denitrificans.
Even though only subunits I and II were considered in this work, the entry points for all the channels described above are not obstructed by the missing subunits.
Moreover, it is also interesting to notice that all O2 channels overlap in space with canonical proton pathways. The O2 channel 1, which corresponds to the X-ray inferred channel, overlies with the end of the D-pathway, whereas the two alternative channels (O2 channel 2 and channel 3), which end close to Y288I, overlap with the final part of the K-pathway.
For the two alternative channels identified here, the O2 pathway is not permanently open and by this reason escaped detection when the static X-ray structures were inspected (e.g [7], [12], [13]). These channels resemble a chain of separate, multiple and transiently formed hydrophobic cavities that are interconnected with each other via the fluctuations of the residues lining the pathway. During the diffusion process, O2 enters the protein and probably due to the thermal fluctuations of the heme a3 tail and residues lining the pathways, it is allowed to diffuse further into the protein core until it reaches the BNC.
Deciphering the actual role of these O2 channels will require further mutational and biochemical analysis. Several mutations sites can be suggested (see Table 1), on the three O2 channels, in order to clarify the role of each pathway in O2 diffusion towards the BNC in the A-type Ccoxs.
Table 1. Point mutations suggested for the three putative O2 channels.
| Residue | Location | Suggested Mutations |
| W172I | Channel 1 | less bulkier side-chain residues (e.g. Y or F) |
| F282I | Channel 1 | less bulkier side-chain residues (e.g. T, V, A, G) |
| L292I | Channel 2 | longer and bulkier side-chain residues (e.g. M or F) |
| I323I | Channel 2 | longer and bulkier side-chain residues (e.g. M or F) |
| I363I | Channel 3 | longer side-chain residues (e.g. L or M) |
| V72II | Channel 3 | longer side-chain residues (e.g. L or M) |
| V107II | Channel 3 | bulkier side-chain residue (e.g. F) |
For the X-ray inferred channel, it is clear that alternative mutation sites, located farther away from the metal ions in the BNC, need to be constructed and studied in order to clarify the actual role of this pathway in O2 diffusion. W156I and F282I are excellent candidates for mutation experiments and their mutation for less bulkier residues, such as tyrosine and threonine (similar to what is observed in the T. thermophilus ba3 enzyme [23]–[25]) would give important insights into how O2 diffusion occurs via this channel. Furthermore, for the alternative channels, the residues lining both pathways are also good candidates for mutational studies, because longer and/or bulkier side-chains could, in principle, obstruct the alternative channels and thus clarify the question of whether these channel are, indeed, used to supply O2 to the BNC in the A-type Ccoxs. Nevertheless, until new experimental data is available, we cannot rule out the hypothesis that all three channels may be working under physiological conditions.
Concluding remarks
Although A-type Ccoxs have been widely studied during the last four decades, the details of the O2 diffusion mechanism are still very incomplete. In particular, the existence and the characteristics of the channel(s) used by O2 to travel from the solvent/membrane to the BNC are still unclear. In this study, we have used an integrated strategy of all-atom MD simulations (with and without explicit O2 molecules) and ILS calculations, designed to examine and characterize the O2 delivery channels in fully reduced Ccox from R. sphaeroides. Altogether, our results suggest that O2 does not diffuse unspecifically inside this protein and instead, uses three well-defined channels running from the interior of the membrane (where O2 solubility is higher than in the aqueous phase) towards the Ccox core. The first pathway has two entrance points, located between helices 5 and 8 and helices 11 and 13 of subunit I, which converges into the constriction point just before the BNC. This channel correlates very well with the channel inferred from the available X-ray structures. The second pathway has only one entry located between the transmembrane helices 13 and 16 of subunit I and it terminates close to Y288I. The third identified pathway approaches the BNC from the subunit II side. This channel runs parallel to the heme a3 hydroxylethylfarnesyl tail and also terminates just below Y288I. According to our observations, the hydrophobic channel detected in the X-ray structures does not constitute the most likely (energetically preferred) entrance point for the O2 molecules in this Ccox. From the O2 affinity map, O2 accesses the BNC via the alternative dynamic channels formed by transient hydrophobic cavities, whose opening and closure is regulated by the thermal fluctuations of the protein. This may be the reason why these channels were not visible in the static X-ray structures.
In summary, our results suggest that the original hypothesis (based on static X-ray structures and mutational studies on A-type Ccox) that proposed, that O2 permeation occurs via a unique, continuous and permanently open channel, is indeed a simplification. Our current work does not rule out the role of the X-ray inferred channel, but suggests other alternative routes to the BNC. Furthermore, it emphasizes the need to take into account the dynamic behavior of the protein in order to obtain a more complete description of the O2 putative channels and a more detailed picture of the mechanisms underlying O2 diffusion in these Ccoxs.
Materials and Methods
Starting structure
The 2.15 Å resolution crystal structure of the fully reduced Ccox from R. sphaeroides (pdb code: 3FYE) [15] was used as the starting point for this work. This X-ray structure only contains the minimum functional unit (subunits I and II) for Ccox. Only the water molecules with a relative accessibility to the solvent lower than 50% were kept. The relative accessibility of water was computed using the program ASC [66], [67], resulting in the selection of 240 water molecules.
Since the GROMOS 54A7 force-field [68] lacks the proper parameterization for the Ccox redox centers, the atomic partial charges for reduced CuA, heme a and BNC centers were calculated using quantum mechanical calculations with the software Gaussian09 [69] and RESP fitting [70], as described in detail in S1 Text in section 1. The van der Waals parameters for the iron atom (located in the two heme groups) were taken from the universal force field [71] whereas the remaining bonded and van der Waals parameters for the metal centers were adapted from the GROMOS 54A7 force field [68].
The protonation state of each individual protonable group at pH 7.0 was determined using a combination of Poisson-Boltzmann calculations, performed with the package MEAD (version 2.2.5) [72]–[74], and Metropolis Monte Carlo simulations, using the program PETIT (version 1.3) [75]. These calculations were performed using the methodologies described in [75], [76]. For details related with the determination of the protonation state of the protonatable residues, see section 2 in S1 Text.
Ccox insertion into a lipid membrane
Subunits I and II of Ccox were inserted in a pre-equilibrated dimysristoylphosphatidylcholine (DMPC) lipid membrane (for details related with the membrane construction, equilibration and characterization see [77]). The optimal position of the protein relative to the membrane was determined using the location of the charged residues in the transmembrane helices as a reference. After Ccox insertion into the membrane, all the DMPC molecules located within a cut-off distance of 1.2 Å from the protein atoms were removed, as described in detail elsewhere [77], [78]. Subsequently, the system (protein, membrane and crystallographic waters) was hydrated in a orthorhombic box using a pre-equilibrated box of SPC water molecules [79]. The water molecules misplaced in the center of the membrane (formed by the highly hydrophobic lipid tails), were removed upon visual inspection. The final system contained the reduced Ccox embedded in a 175 DMPC lipid membrane surrounded by 19,645 water molecules, in a total of 75,178 atoms.
MD simulations of Ccox inserted in a lipid membrane
All MD simulations were performed using the software package GROMACS 4.0.4 [80] together with the united atom GROMOS 54A7 force-field [68] for the protein and lipids and the previously described atomic partial charges and parameters for the redox centers. The simple point charge (SPC) water model was used [79]. Periodic boundary conditions were applied to all simulations. Non-bonded interactions were calculated using a twin range method [81] with short and long-range cut-offs of 8 and 14 Å, respectively. A reaction field correction [82], [83] was applied for the truncated electrostatic interactions, considering a dielectric constant of 62 [84]. The SETTLE algorithm [85] was used to constraint the bond lengths and angle in water molecules, while the LINCS algorithm [86] was used to keep all remaining bonds constrained. The time step for integrating the equations of motion was 0.002 ps and the neighbor list was updated every 5 steps. The simulations were performed at the constant temperature of 310 K, which is above the phase transition temperature for the DMPC lipids (Tm = 296–297 K) in order to ensure that the membrane is in the liquid crystalline state [87]. A Berendsen heat bath [88] was used, with separate couplings for the protein, membrane and solvent, using a relaxation time constant of 0.1 ps. The pressure was coupled semi-isotropically (coupling constant of 5.0 ps and isothermal compressibility of 4.6×10−5 bar−1 [84]), resulting in an independent coupling of the lateral (Px+y) and perpendicular (Pz) pressures. For all simulations, the x+y and z pressure components were kept at 1 atm and no surface tension was applied [84]. These simulation conditions were shown by Poger et al. [84], [89] to correctly reproduce several experimental measurements for this type of membranes.
The system was energy minimized with the steepest-descent method in order to remove excessive strain by performing 5000 steps of steepest-descent minimization with harmonic restraints applied to all non-hydrogen atoms (protein and lipids), followed by further 5000 steps restraining the non-hydrogen atoms of the protein, ending with 5000 steps with restraints applied to the Cα atoms only. After the minimization procedure, and in order to allow proper repacking of the lipids around the protein, a 20 ns MD relaxation was executed in three steps. First, a 0.5 ns simulation was performed with position restraints to all non-hydrogen atoms of the protein and solvent, at constant temperature and pressure. Afterwards, an additional 0.5 ns simulation was performed, with position restraints applied to the non-hydrogen atoms of the protein only. Finally, only the Cα atoms were restrained for a period of 19 ns. A force constant of 1000 kJ mol−1nm−2 was used for all the steps that included harmonic position restraints. The unrestrained simulations started after these 20 ns of restrained simulation.
In order to reduce the well known sampling problems in membrane-protein simulations, five MD simulations, 100 ns each, were performed, resulting in 0.5 µs of total simulation time. All replicates were initiated with different sets of random velocities. These simulations will be hereafter designated as O2-free simulations.
MD simulations of Ccox inserted in a lipid membrane with explicit O2
After 20 ns of restrained simulations, we randomly added 84 molecules of dioxygen (O2) in the solvent zone of each system. No O2 was placed inside the protein nor inside the hydrophobic core of the membrane (see S4 Figure in S1 Text). This new set of simulations will be, hereafter, designated as O2 simulations. The water molecules within a 2 Å distance from the O2 molecules were deleted, similarly to the procedure described in [33].
In order to allow the solvent to adapt to the newly added O2 molecules, a 0.5 ns MD simulation with position restraints on all non-hydrogen atoms (force constant of 1000 kJ mol−1nm−2) was performed. After this initialization procedure, unrestrained MD simulations were carried out and the simulation conditions and parameters were similar to the ones described previously for the MD simulations without O2, except for the temperature coupling groups used. In this set of simulations, the O2 molecules were included in the same group as the protein. 5 MD simulations, 100 ns each, were performed. The parameters for the O2 molecules were taken from the previously published work of Victor et al [90].
The 84 O2 molecules added to the system corresponds to an O2 concentration of ∼0.235 M, which is higher than the experimental solubility of this gas in water. However, this high O2 concentration does not affect the structural properties of the protein as shown in S5 Figure in S1 Text.
Moreover, the use of this high number of O2 molecules is necessary to obtain reliable statistics within a reasonable simulation time.
Implicit ligand sampling (ILS) calculations in Ccox
Sites with high O2 affinity were determined using the ILS method, as previously described in [29]. In this method, the potential of mean force for placing an O2 molecule in any position inside the protein is calculated according to:
| (2) |
In equation 2, the implicit ligand potential of mean force, 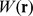 , is an average over a finite number of protein and solvent configurations (
, is an average over a finite number of protein and solvent configurations (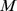 ) and over a number of different equally probable orientations of the ligand (
) and over a number of different equally probable orientations of the ligand ( ). Moreover,
). Moreover, 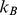 is the Boltzmann constant,
is the Boltzmann constant, 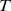 is the absolute temperature,
is the absolute temperature, 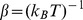 and
and 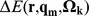 is the interaction energy between the protein and solvent configuration (
is the interaction energy between the protein and solvent configuration (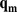 ) with the ligand located at position
) with the ligand located at position  with the orientation
with the orientation 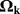 .
.
In our case, the O2 free energy map was constructed using the last 50 ns (for each replicate) of the O2-free simulations. For the calculations, all 50005 conformations (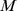 = 10001 conformations x 5 replicates in equation 2) were fitted to the X-ray structure using the Cα atoms. A grid of 51×55×87 dimensions was used with a grid spacing of 1 Å and 400 O2 insertions were performed per grid point (
= 10001 conformations x 5 replicates in equation 2) were fitted to the X-ray structure using the Cα atoms. A grid of 51×55×87 dimensions was used with a grid spacing of 1 Å and 400 O2 insertions were performed per grid point ( = 400 in equation 2). All calculations were carried out using a version of GROMACS 4.0.4 Widom TPI algorithm, modified almost in the same way as described in [33]. The only difference is that the ligand insertions here were made within the whole space of the grid cube (the grid cube is centered at the insertion point and with edge length equal to the grid spacing), while in the previous work (described in [33]) the insertions were only possible within the inscribed sphere on the grid cube.
= 400 in equation 2). All calculations were carried out using a version of GROMACS 4.0.4 Widom TPI algorithm, modified almost in the same way as described in [33]. The only difference is that the ligand insertions here were made within the whole space of the grid cube (the grid cube is centered at the insertion point and with edge length equal to the grid spacing), while in the previous work (described in [33]) the insertions were only possible within the inscribed sphere on the grid cube.
The 3D free energy map obtained describes the Gibbs free energy of moving an O2 molecule from vacuum to a given position in the system, ΔG vac→prot(O2). This map was then converted into the ΔG wat→prot(O2) map of interest using a ΔG wat→prot(O2) calculated as described in [33].
Data analysis
The secondary structure assignment was performed with the program DSSP [91]. To determine the percentage of secondary structure loss relative to the X-ray structure, the secondary structure classes considered were: α-helix, 310-helix, 5-helix, β-sheet and β-bridge.
For the energy landscape analysis, we used the method described in [33]. In short, this method classifies the energy landscape into energy basins through a steepest-descent tessellation and, afterwards, identifies the lowest-energy point within the boundaries between each pair of neighboring basins, i.e. the saddle point between those basins. After this procedure, a network of paths between all energy minima of the landscape can be constructed using the steepest-descent paths from the saddle points to the minima. A cutoff of 20 kJ·mol−1 was used for the network construction.
The errors of the free energy profiles were calculated using two blocks: the first block corresponds to the frames ranging from 50 ns to 75 ns (for all five replicates) whereas the second block contains all the frames ranging from 75 ns to 100 ns. The errors were determined as half the difference of the energies observed between the two blocks for each minina and for each transition. The method used for error calculation assumes that similar minima and pathways can be identified in the two blocks. However, for the O2 channel 1, the pathways connecting M6 to M8 and M10 to M11 were not visible in one of the blocks and by this reason their associated errors could not be calculated.
Supporting Information
Supporting information. This file includes 6 distinct sections: 1-Parameterization of the redox centers. 2- Protonation state of protonable residues at pH = 7.0. 3- Ccox inserted in a lipid membrane with explicit O2. 4- Ccox conformational drift during the MD simulations. 5- O2 diffusion in a DMPC membrane. 6- O2 molecules inside Ccox.
(PDF)
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by Fundação para a Ciência e a Tecnologia (Project PTDC/QUI-BIQ/113446/2009 and Pest-OE/EQB/LA0004/2011). ASFO was supported by a FCT post-doc fellowship (SFRH/BPD/76621/2011) whereas JMD was supported by a FCT PhD fellowship (SFRH/BD/41316/2007). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Siletsky SA, Konstantinov AA (2012) Cytochrome c oxidase: Charge translocation coupled to single-electron partial steps of the catalytic cycle. Biochim Biophys Acta-Bioenergetics 1817: 476–488. [DOI] [PubMed] [Google Scholar]
- 2. Wikstrom MKF (1977) Proton Pump Coupled to Cytochrome-C Oxidase in Mitochondria. Nature 266: 271–273. [DOI] [PubMed] [Google Scholar]
- 3. Brzezinski P, Gennis RB (2008) Cytochrome c oxidase: exciting progress and remaining mysteries. J Bioenerg Biomembr 40: 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. FergusonMiller S, Babcock GT (1996) Heme/copper terminal oxidases. Chem Rev 96: 2889–2907. [DOI] [PubMed] [Google Scholar]
- 5. Sousa FL, Alves RJ, Ribeiro MA, Pereira-Leal JB, Teixeira M, et al. (2012) The superfamily of heme-copper oxygen reductases: Types and evolutionary considerations. Biochim Biophys Acta-Bioenergetics 1817: 629–637. [DOI] [PubMed] [Google Scholar]
- 6. Yoshikawa S, Muramoto K, Shinzawa-Itoh K (2011) Proton-Pumping Mechanism of Cytochrome c Oxidase. Annu Rev Biophys 40: 205–223. [DOI] [PubMed] [Google Scholar]
- 7. Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, et al. (1996) The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 angstrom. Science 272: 1136–1144. [DOI] [PubMed] [Google Scholar]
- 8. Shimokata K, Katayama Y, Murayama H, Suematsu M, Tsukihara T, et al. (2007) The proton pumping pathway of bovine heart cytochrome c oxidase. Proc Natl Acad Sci U S A 104: 4200–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, et al. (1998) Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science 280: 1723–1729. [DOI] [PubMed] [Google Scholar]
- 10. Tsukihara T, Shimokata K, Katayama Y, Shimada H, Muramoto K, et al. (2003) The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc Natl Acad Sci U S A 100: 15304–15309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Muramoto K, Hirata K, Shinzawa-Itoh K, Yoko-O S, Yamashita E, et al. (2007) A histidine residue acting as a controlling site for dioxygen reduction and proton pumping by cytochrome c oxidase. Proc Natl Acad Sci U S A 104: 7881–7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iwata S, Ostermeier C, Ludwig B, Michel H (1995) Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature 376: 660–669. [DOI] [PubMed] [Google Scholar]
- 13. Svensson-Ek M, Abramson J, Larsson G, Tornroth S, Brzezinski P, et al. (2002) The X-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J Mol Biol 321: 329–339. [DOI] [PubMed] [Google Scholar]
- 14. Koepke J, Olkhova E, Angerer H, Muller H, Peng GH, et al. (2009) High resolution crystal structure of Paracoccus denitrificans cytochrome c oxidase: New insights into the active site and the proton transfer pathways. Biochim Biophys Acta-Bioenergetics 1787: 635–645. [DOI] [PubMed] [Google Scholar]
- 15. Qin L, Liu J, Mills DA, Proshlyakov DA, Hiser C, et al. (2009) Redox-dependent conformational changes in cytochrome C oxidase suggest a gating mechanism for proton uptake. Biochemistry 48: 5121–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sharpe MA, Krzyaniak MD, Xu S, McCracken J, Ferguson-Miller S (2009) EPR evidence of cyanide binding to the Mn(Mg) center of cytochrome c oxidase: support for Cu(A)-Mg involvement in proton pumping. Biochemistry 48: 328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sugitani R, Stuchebrukhov AA (2009) Molecular dynamics simulation of water in cytochrome c oxidase reveals two water exit pathways and the mechanism of transport. Biochim Biophys Acta-Bioenergetics 1787: 1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bratton MR, Pressler MA, Hosler JP (1999) Suicide inactivation of cytochrome c oxidase: Catalytic turnover in the absence of subunit III alters the active site. Biochemistry 38: 16236–16245. [DOI] [PubMed] [Google Scholar]
- 19. Mills DA, Hosler JP (2005) Slow proton transfer through the pathways for pumped protons in cytochrome c oxidase induces suicide inactivation of the enzyme. Biochemistry 44: 4656–4666. [DOI] [PubMed] [Google Scholar]
- 20. Riistama S, Puustinen A, GarciaHorsman A, Iwata S, Michel H, et al. (1996) Channelling of dioxygen into the respiratory enzyme. Biochim Biophys Acta-Bioenergetics 1275: 1–4. [DOI] [PubMed] [Google Scholar]
- 21. Riistama S, Puustinen A, Verkhovsky MI, Morgan JE, Wikstrom M (2000) Binding of O-2 and its reduction are both retarded by replacement of valine 279 by isoleucine in cytochrome c oxidase from Paracoccus denitrificans. Biochemistry 39: 6365–6372. [DOI] [PubMed] [Google Scholar]
- 22. Salomonsson L, Lee A, Gennis RB, Brzezinski P (2004) A single-amino-acid lid renders a gas-tight compartment within a membrane-bound transporter. Proc Natl Acad Sci U S A 101: 11617–11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luna VM, Chen Y, Fee JA, Stout CD (2008) Crystallographic studies of Xe and Kr binding within the large internal cavity of cytochrome ba3 from Thermus thermophilus: structural analysis and role of oxygen transport channels in the heme-Cu oxidases. Biochemistry 47: 4657–4665. [DOI] [PubMed] [Google Scholar]
- 24. Luna VM, Fee JA, Deniz AA, Stout CD (2012) Mobility of Xe atoms within the oxygen diffusion channel of cytochrome ba(3) oxidase. Biochemistry 51: 4669–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McDonald W, Funatogawa C, Li Y, Szundi I, Chen Y, et al. (2013) Ligand access to the active site in Thermus thermophilus ba(3) and bovine heart aa(3) cytochrome oxidases. Biochemistry 52: 640–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cohen J, Kim K, King P, Seibert M, Schulten K (2005) Finding gas diffusion pathways in proteins: Application to O-2 and H-2 transport in Cpl [FeFe]-hydrogenase and the role of packing defects. Structure 13: 1321–1329. [DOI] [PubMed] [Google Scholar]
- 27. Teixeira VH, Baptista AM, Soares CM (2006) Pathways of H2 toward the active site of [NiFe]-hydrogenase. Biophys J 91: 2035–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baltazar CSA, Teixeira VH, Soares CM (2012) Structural features of [NiFeSe] and [NiFe] hydrogenases determining their different properties: a computational approach. J Biol Inorg Chem 17: 543–555. [DOI] [PubMed] [Google Scholar]
- 29. Cohen J, Arkhipov A, Braun R, Schulten K (2006) Imaging the migration pathways for O-2, CO, NO, and Xe inside myoglobin. Biophys J 91: 1844–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cohen J, Schulten K (2007) O-2 migration pathways are not conserved across proteins of a similar fold. Biophys J 93: 3591–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hofacker I, Schulten K (1998) Oxygen and proton pathways in cytochrome c oxidase. Proteins 30: 100–107. [PubMed] [Google Scholar]
- 32. Porrini M, Daskalakis V, Farantos SC (2012) Exploring the topography of free energy surfaces and kinetics of cytochrome c oxidases interacting with small ligands. Rsc Adv 2: 5828–5836. [Google Scholar]
- 33. Damas JM, Baptista AM, Soares CM (2014) A New Pathway for O2 diffusion inside CotA Laccase and Implications on the Multicopper Oxidases Family. J Chem Theory Comput 10: 3525–3531. [DOI] [PubMed] [Google Scholar]
- 34. Saam J, Rosini E, Molla G, Schulten K, Pollegioni L, et al. (2010) O-2 Reactivity of Flavoproteins: Dymanic acess of dioxygen to the active site and role of a H+ relay system in D-amino acid oxidase. J Biol Chem 285: 24439–24446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pomes R, Roux B (1998) Free energy profiles for H+ conduction along hydrogen-bonded chains of water molecules. Biophys J 75: 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Backgren C, Hummer G, Wikstrom M, Puustinen A (2000) Proton translocation by cytochrome c oxidase can take place without the conserved glutamic acid in subunit I. Biochemistry 39: 7863–7867. [DOI] [PubMed] [Google Scholar]
- 37. Wikstrom M, Verkhovsky MI, Hummer G (2003) Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim Biophys Acta 1604: 61–65. [DOI] [PubMed] [Google Scholar]
- 38. Zheng X, Medvedev DM, Swanson J, Stuchebrukhov AA (2003) Computer simulation of water in cytochrome c oxidase. Biochim Biophys Acta 1557: 99–107. [DOI] [PubMed] [Google Scholar]
- 39. Olkhova E, Hutter MC, Lill MA, Helms V, Michel H (2004) Dynamic water networks in cytochrome C oxidase from Paracoccus denitrificans investigated by molecular dynamics simulations. Biophys J 86: 1873–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Popovic DM, Stuchebrukhov AA (2004) Electrostatic study of the proton pumping mechanism in bovine heart cytochrome C oxidase. J Am Chem Soc 126: 1858–1871. [DOI] [PubMed] [Google Scholar]
- 41. Cukier RI (2005) A molecular dynamics study of water chain formation in the proton-conducting K channel of cytochrome c oxidase. Biochim Biophys Acta 1706: 134–146. [DOI] [PubMed] [Google Scholar]
- 42. Seibold SA, Mills DA, Ferguson-Miller S, Cukier RI (2005) Water chain formation and possible proton pumping routes in Rhodobacter sphaeroides cytochrome c oxidase: a molecular dynamics comparison of the wild type and R481K mutant. Biochemistry 44: 10475–10485. [DOI] [PubMed] [Google Scholar]
- 43. Xu J, Voth GA (2005) Computer simulation of explicit proton translocation in cytochrome c oxidase: the D-pathway. Proc Natl Acad Sci U S A 102: 6795–6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wikstrom M, Ribacka C, Molin M, Laakkonen L, Verkhovsky M, et al. (2005) Gating of proton and water transfer in the respiratory enzyme cytochrome c oxidase. Proc Natl Acad Sci U S A 102: 10478–10481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Popovic DM, Stuchebrukhov AA (2005) Proton exit channels in bovine cytochrome c oxidase. J Phys Chem B 109: 1999–2006. [DOI] [PubMed] [Google Scholar]
- 46. Song Y, Michonova-Alexova E, Gunner MR (2006) Calculated proton uptake on anaerobic reduction of cytochrome C oxidase: is the reaction electroneutral? Biochemistry 45: 7959–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu J, Voth GA (2006) Free energy profiles for H+ conduction in the D-pathway of Cytochrome c Oxidase: a study of the wild type and N98D mutant enzymes. Biochim Biophys Acta 1757: 852–859. [DOI] [PubMed] [Google Scholar]
- 48. Xu J, Sharpe MA, Qin L, Ferguson-Miller S, Voth GA (2007) Storage of an excess proton in the hydrogen-bonded network of the d-pathway of cytochrome C oxidase: identification of a protonated water cluster. J Am Chem Soc 129: 2910–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu J, Voth GA (2008) Redox-coupled proton pumping in cytochrome c oxidase: further insights from computer simulation. Biochim Biophys Acta 1777: 196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Henry RM, Yu CH, Rodinger T, Pomes R (2009) Functional hydration and conformational gating of proton uptake in cytochrome c oxidase. J Mol Biol 387: 1165–1185. [DOI] [PubMed] [Google Scholar]
- 51. Kaila VR, Verkhovsky MI, Hummer G, Wikstrom M (2009) Mechanism and energetics by which glutamic acid 242 prevents leaks in cytochrome c oxidase. Biochim Biophys Acta 1787: 1205–1214. [DOI] [PubMed] [Google Scholar]
- 52. Lee HJ, Svahn E, Swanson JM, Lepp H, Voth GA, et al. (2010) Intricate role of water in proton transport through cytochrome c oxidase. J Am Chem Soc 132: 16225–16239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang J, Gunner MR (2010) Multiconformation continuum electrostatics analysis of the effects of a buried Asp introduced near heme a in Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry 49: 8043–8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Buhrow L, Ferguson-Miller S, Kuhn LA (2012) From static structure to living protein: computational analysis of cytochrome c oxidase main-chain flexibility. Biophys J 102: 2158–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Woelke AL, Galstyan G, Galstyan A, Meyer T, Heberle J, et al. (2013) Exploring the possible role of Glu286 in CcO by electrostatic energy computations combined with molecular dynamics. J Phys Chem B 117: 12432–12441. [DOI] [PubMed] [Google Scholar]
- 56. Goyal P, Lu J, Yang S, Gunner MR, Cui Q (2013) Changing hydration level in an internal cavity modulates the proton affinity of a key glutamate in cytochrome c oxidase. Proc Natl Acad Sci U S A 110: 18886–18891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lissi EA, Caceres T (1989) Oxygen Diffusion-Concentration in Erythrocyte Plasma-Membranes Studied by the Fluorescence Quenching of Anionic and Cationic Pyrene Derivatives. J Bioenerg Biomembr 21: 375–385. [DOI] [PubMed] [Google Scholar]
- 58. Dzikovski BG, Livshits VA, Marsh D (2003) Oxygen permeation profile in lipid membranes: Comparison with transmembrane polarity profile. Biophys J 85: 1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Victor BL, Baptista AM, Soares CM (2004) Theoretical identification of proton channels in the quinol oxidase aa 3 from Acidianus ambivalens . BiophysJ 87: 4316–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Riistama S, Hummer G, Puustinen A, Dyer RB, Woodruff WH, et al. (1997) Bound water in the proton translocation mechanism of the haem-copper oxidases. Febs Lett 414: 275–280. [DOI] [PubMed] [Google Scholar]
- 61. Tuukkanen A, Kaila VR, Laakkonen L, Hummer G, Wikstrom M (2007) Dynamics of the glutamic acid 242 side chain in cytochrome c oxidase. Biochim Biophys Acta 1767: 1102–1106. [DOI] [PubMed] [Google Scholar]
- 62. Kaila VRI, Verkhovsky MI, Wikstrom M (2010) Proton-Coupled Electron Transfer in Cytochrome Oxidase. Chem Rev 110: 7062–7081. [DOI] [PubMed] [Google Scholar]
- 63. Elber R, Karplus M (1990) Enhanced sampling in molecular dynamics: Use of the time-dependent Hartree approximation for a simulation of carbon monoxide diffusion through myoglobin. J Am Chem Soc 112: 9161–9175. [Google Scholar]
- 64. Roitberg A, Elber R (1991) Modelling side chains in peptides and proteins: Application of the locally enhanced sampling technique and the simulated annealing methods to find minimum energy conformations. J Chem Phys 95: 9277–9287. [Google Scholar]
- 65. Siletsky SA (2013) Steps of the coupled charge translocation in the catalytic cycle of cytochrome c oxidase. Front Biosci (Landmark Ed) 18: 36–57. [DOI] [PubMed] [Google Scholar]
- 66. Eisenhaber F, Argos P (1993) Improved Strategy in Analytic Surface Calculation for Molecular-Systems - Handling of Singularities and Computational Efficiency. J Comput Chem 14: 1272–1280. [Google Scholar]
- 67. Eisenhaber F, Lijnzaad P, Argos P, Sander C, Scharf M (1995) The Double Cubic Lattice Method - Efficient Approaches to Numerical-Integration of Surface-Area and Volume and to Dot Surface Contouring of Molecular Assemblies. J Comput Chem 16: 273–284. [Google Scholar]
- 68. Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, et al. (2011) Definition and testing of the GROMOS force-field versions 54A7 and 54B7. European biophysics journal: EBJ 40: 843–856. [DOI] [PubMed] [Google Scholar]
- 69.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al. (2009) Gaussian 09, Revision A.02. Wallingford, CT: Gaussian, Inc. [Google Scholar]
- 70. Bayly CI, Cieplak P, Cornell WD, Kollman PA (1993) A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges - the Resp Model. J Phys Chem 97: 10269–10280. [Google Scholar]
- 71. Rappe AK, Casewit CJ, Colwell KS, Goddard WA, Skiff WM (1992) Uff, a Full Periodic-Table Force-Field for Molecular Mechanics and Molecular-Dynamics Simulations. J Am Chem Soc 114: 10024–10035. [Google Scholar]
- 72.Bashford D (1997) An Object-Oriented Programming Suite for Electrostatic Effects in Biological Molecules. In: Ishikawa Y, Oldehoeft RR, Reynders JVW, Tholburn M, editors. Scientific Computing in Object-Oriented Parallel Environments. Berlin: ISCOPE97, Springer. pp.233–240. [Google Scholar]
- 73. Bashford D, Gerwert K (1992) Electrostatic Calculations of the pKa Values of Ionizable Groups in Bacteriorhodopsin. J Mol Biol 224: 473–486. [DOI] [PubMed] [Google Scholar]
- 74. Bashford D, Karplus M (1990) pKa's of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry 29: 10219–10225. [DOI] [PubMed] [Google Scholar]
- 75. Baptista AM, Soares CM (2001) Some theoretical and computational aspects of the inclusion of proton isomerism in the protonation equilibrium of proteins. J Phys Chem 105: 293–309. [Google Scholar]
- 76. Teixeira VH, Soares CM, Baptista AM (2002) Studies of the reduction and protonation behavior of tetraheme cytochromes using atomic detail. J Biol Inorg Chem 7: 200–216. [DOI] [PubMed] [Google Scholar]
- 77. Oliveira AS, Baptista AM, Soares CM (2011) Inter-domain Communication Mechanisms in an ABC Importer: A Molecular Dynamics Study of the MalFGK2E Complex. PLoS computational biology 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kandt C, Ash WL, Tieleman DP (2007) Setting up and running molecular dynamics simulations of membrane proteins. Methods 41: 475–488. [DOI] [PubMed] [Google Scholar]
- 79. Hermans J, Berendsen HJC, Vangunsteren WF, Postma JPM (1984) A Consistent Empirical Potential for Water-Protein Interactions. Biopolymers 23: 1513–1518. [Google Scholar]
- 80. Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4: 435–447. [DOI] [PubMed] [Google Scholar]
- 81. van Gunsteren WF, Berendsen HJC (1990) Computer simulation of molecular dynamics: methodology, applications, and perspectives in chemistry. Angew Chem Int Ed Engl 29: 992–1023. [Google Scholar]
- 82. Barker JA, Watts RO (1973) Monte-Carlo Studies of Dielectric Properties of Water-Like Models. Mol Phys 26: 789–792. [Google Scholar]
- 83. Tironi IG, Sperb R, Smith PE, van Gunsteren WF (1995) A generalized reaction field method for molecular-dynamics simulations. J Phys Chem 102: 15451–15459. [Google Scholar]
- 84. Poger D, Van Gunsteren WF, Mark AE (2010) A New Force Field for Simulating Phosphatidylcholine Bilayers. J Comput Chem 31: 1117–1125. [DOI] [PubMed] [Google Scholar]
- 85. Miyamoto S, Kollman PA (1992) SETTLE: An Analytical Version of the SHAKE and RATTLE Algorithms for Rigid Water Models. J Comput Chem 13: 952–962. [Google Scholar]
- 86. Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: A Linear Constraint Solver for molecular simulations. J Comput Chem 18: 1463–1472. [Google Scholar]
- 87. Koyama TM, Stevens CR, Borda EJ, Grobe KJ, Cleary DA (1999) Characterizing the Gel to Liquid Crystal Transition in Lipid-Bilayer Model Systems. Chem Educ 4: 12–15. [Google Scholar]
- 88. Berendsen H, Postma J, van Gunsteren W, Dinola A, Haak J (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81: 3684–3690. [Google Scholar]
- 89. Poger D, Mark AE (2012) Lipid Bilayers: The Effect of Force Field on Ordering and Dynamics. J Chem Theory Comput 8: 4807–4817. [DOI] [PubMed] [Google Scholar]
- 90. Victor BL, Baptista AM, Soares CM (2009) Dioxygen and nitric oxide pathways and affinity to the catalytic site of rubredoxin: oxygen oxidoreductase from Desulfovibrio gigas. J Biol Inorg Chem 14: 853–862. [DOI] [PubMed] [Google Scholar]
- 91. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22: 2577–2637. [DOI] [PubMed] [Google Scholar]
- 92. Ho BK, Gruswitz F (2008) HOLLOW: Generating Accurate Representations of Channel and Interior Surfaces in Molecular Structures. Bmc Struct Biol 8: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information. This file includes 6 distinct sections: 1-Parameterization of the redox centers. 2- Protonation state of protonable residues at pH = 7.0. 3- Ccox inserted in a lipid membrane with explicit O2. 4- Ccox conformational drift during the MD simulations. 5- O2 diffusion in a DMPC membrane. 6- O2 molecules inside Ccox.
(PDF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.