

Abstract
Fluorescence imaging methods that achieve spatial resolution beyond the diffraction limit (super-resolution) are of great interest in biology. We describe a super-resolution method that combines two-photon excitation with structured illumination microscopy (SIM), enabling three-dimensional interrogation of live organisms with ~150 nm lateral and ~400 nm axial resolution, at frame rates of ~1 Hz. By performing optical rather than digital processing operations to improve resolution, our microscope permits super-resolution imaging with no additional cost in acquisition time or phototoxicity relative to the point-scanning two-photon microscope upon which it is based. Our method provides better depth penetration and inherent optical sectioning than all previously reported super-resolution SIM implementations, enabling super-resolution imaging at depths exceeding 100 μm from the coverslip surface. The capability of our system for interrogating thick live specimens at high resolution is demonstrated by imaging whole nematode embryos and larvae, and tissues and organs inside zebrafish embryos.
1. INTRODUCTION
The speed, gentleness, molecular specificity, and contrast of fluorescence microscopy make it a powerful and versatile research tool, enabling diverse applications from cell biology to neuroscience. Although fluorescence imaging continues to improve [1,2], technical barriers still prevent these methods from reaching their full potential. Blurring introduced by diffraction limits the spatial resolution of widefield microscopy to ~250 nm laterally and ~500–750 nm axially, obscuring much detail at the subcellular scale. The effects of diffraction may be overcome with super-resolution techniques [3], but imaging three-dimensional (3D) samples presents additional challenges. At sufficient distances from the coverslip, the increasingly severe effects of optical aberrations [4] and scattering [5] degrade resolution and the signal-to-noise ratio (SNR) so that obtaining even diffraction-limited performance is difficult. Emerging techniques that provide super-resolution at depth and in 3D are thus of great practical interest in biology.
Stimulated emission depletion (STED) microscopy can provide sub-100-nm lateral resolution in vivo [6], but its resolution is easily degraded by depth-dependent aberrations or scattering that distort the shape of excitation and depletion beams. Combining STED microscopy with adaptive optics can compensate for or reduce aberrations [7], and using two-photon (2P) excitation reduces scattering [8]; either approach improves STED microscopy at depth. Single-molecule imaging techniques enable sub-100-nm resolution in live cells [9], but out-of-focus background in 3D samples confounds the ability to localize individual molecules precisely (or at all), ultimately reducing imaging resolution and SNR. Using 2P [10] or selective plane [11] illumination can help to reduce background, thus extending the useful imaging range of these techniques. Despite these advances, neither STED nor single-molecule imaging has matured to the point that 3D acquisition in thick samples (i.e., imaging stacks >10 μm) is routine: the slow acquisition speed and high photobleaching/toxicity entailed by each of these super-resolution methods have limited the vast majority of live imaging applications to single imaging planes at or near the coverslip surface.
In contrast, linear structured illumination microscopy (SIM) offers a more modest ~2× improvement in resolution beyond the diffraction limit than other super-resolution methods, but is far gentler and faster, thus facilitating the collection of tens to hundreds of live cellular volumes [12]. Linear SIM has historically been implemented by exciting the sample with a series of sinusoidal illumination patterns, imaging the fluorescence onto a multipixel detector, and computationally postprocessing and combining the resulting raw images to produce a super-resolution image. Sinusoidal illumination has the advantage of requiring relatively few illumination pattern positions, but suffers from an inability to physically reject out-of-focus light (i.e., optical sectioning is achieved entirely with computation). By using a single excitation focus in combination with a pinhole, as in image scanning microscopy (ISM) [13] (or multiple point-like excitation foci [14,15] in combination with pinholes) instead of sharp sinusoidal excitation patterns [16], out-of-focus background is inherently rejected by the microscope (i.e., without computation) [see Note S1 in Supplement 1], and depth penetration can be improved to enable 3D super-resolution imaging ~50 μm from the coverslip surface [14]. Despite the apparent differences between sinusoidal SIM and point-based SIM implementations derived from ISM [13], the mechanism of resolution enhancement is the same [17] and, in fact, the same software can be used to process data generated by the two types of SIM [18]. Using multifocal 2P illumination has been shown to further improve SIM performance in thick samples, due to the inherently lower background afforded by 2P excitation [19]. Very recently, methods that perform the majority of postprocessing optically [20–22], instead of computationally, have been developed. The power of these methods is their speed: by eliminating the need for excess raw images, live, optically-sectioned super-resolution imaging at video rate or faster is possible (e.g., “instant SIM,” ISIM [20]).
Here we present the first implementation of instant SIM that uses 2P excitation (2P ISIM), enabling imaging with ~150 nm lateral and ~400 nm axial resolution, at depths exceeding ~100 μm. Relative to point-scanning 2P excitation microscopy (on which our microscope is based), our method offers improved resolution with no drawbacks in imaging speed or phototoxicity. Relative to all previous super-resolution SIM implementations, 2P ISIM offers better inherent optical sectioning, background rejection, and depth penetration, which we demonstrate on thick samples including bead phantoms, live nematode embryos and larvae, and zebrafish embryos.
2. RESULTS
Resolution enhancement in point-based SIM techniques can be understood first by considering resolution enhancement in a standard point-scanning confocal microscope. By closing the pinhole almost completely down in a confocal microscope, the point spread function (PSF) is reduced laterally by ~1.4×, as the microscope’s PSF becomes the product of approximately equal excitation and emission PSFs [22,23], and the frequency support of the microscope is doubled. However, this resolution is never achieved in practice in confocal microscopy as the almost-closed pinhole rejects far too much emission light, and any improvement in resolution is abrogated by the severely diminished SNR.
Point-based SIM [13–15,19–22] methods deliver ~1.4× improvement in resolution over widefield microscopy without sacrificing signal by collecting emission with a multipixel detector, treating each pixel as a small pinhole, reassigning the light from each pixel onto a common origin, and summing the result. One method of reassignment is to shrink each emission focus locally before [20] or after [14,19] image acquisition, without changing the distance between foci. Alternatively, reassignment may be implemented by increasing the distance between adjacent emission foci while leaving their size unchanged (Fig. S1), which provides a convenient method for instant, point-based SIM, as has been demonstrated with single-photon (1P) excitation (“rescan microscopy” [21]). We implemented this same method of reassignment to enhance the resolution of 2P excitation microscopy, using an emission-side galvanometric mirror (galvo) to double the distance between adjacent scan points before image acquisition with a camera (Fig. 1 and Figs. S1 and S6 and Note S2 in Supplement 1, and the Methods section below. Use of the 2× expansion factor was determined empirically from measurements of the system excitation and emission PSFs, and from the performance of the system at various expansion factors (Note S2 in Supplement 1).
Fig. 1.
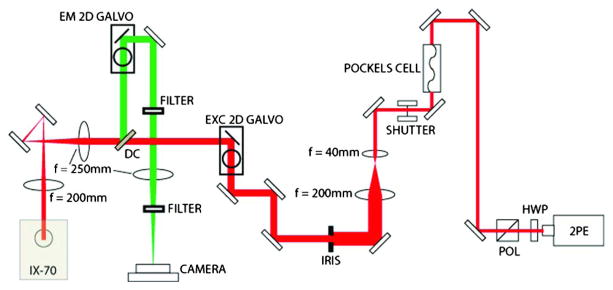
Pulsed femtosecond laser (2PE) provides two-photon excitation to the sample (red), and fluorescence (green) is collected and imaged onto a camera. The remaining elements are used to shape, modulate, shutter, or scan the excitation, or scan and filter the emission (see text for more detail). Symbol key: HWP, half wave plate; POL, polarizer; EXC 2D GALVO, galvanometric mirror used to scan the excitation through the sample; DC, dichroic mirror; IX-70, microscope frame used to house the objective and sample (not shown); EM 2D GALVO, galvanometric mirror used to rescan the emission. Reflective mirrors are shown as rectangles, and other lenses referred to in the text are shown as ellipsoids with focal lengths as indicated. Note that the drawing is not to scale.
To study the resolution improvement enabled by 2P ISIM, we fixed U2OS human osteosarcoma cells and immunolabeled the microtubule cytoskeleton with Alexa Fluor 488 [Fig. 2(a)]. Compared to imaging in conventional, diffraction-limited 2P mode (i.e., scanning only the excitation-side galvo while holding the emission-side galvo fixed), the additional scanning introduced in 2P ISIM sharpened the image and resulted in microtubules with finer apparent width [Figs. 2(b) and 2(c)]. As in multifocal SIM [14,19] and ISIM [20], applying deconvolution further improved lateral resolution, resulting in microtubules with apparent width ~160 nm and allowing us to resolve microtubules spaced by as little as 120 nm (Fig. S7 in Supplement 1). We further quantified the resolution afforded by 2P ISIM by imaging 100-nm-diameter, yellow–green beads (Fig. S8 in Supplement 1), finding that 2P ISIM followed by deconvolution doubled the lateral resolution of the microscope when operated in diffraction-limited 2P excitation mode (from 311 ± 10 nm to 146 ± 5 nm, 30 beads). 2P ISIM did not improve the axial resolution of our system beyond the gains provided by deconvolution (predeconvolution, 668 ± 33 nm; postdeconvolution, 438 ± 22 nm, equivalent to the improvement obtained on images acquired in diffraction-limited 2P excitation mode). By “axial resolution,” we mean the axial FWHM of a subdiffractive bead as opposed to “sectioning,” which we define as the ability to distinguish two thin planes in z. We suspect the lack of improvement in z is due to aberrations in our 1P emission PSF that enlarge its axial extent compared to the diffraction-limited result predicted by theory. In this case, the recorded image is dominated by the axial extent of the 2P excitation PSF and the photon reassignment process does not result in any improvement in axial resolution.
Fig. 2.

Resolution enhancement in two-photon instant structured illumination microscopy (2P ISIM). (a) Immunolabeled microtubules in a fixed U2OS human osteosarcoma cell, as viewed in 2P ISIM, after deconvolution. (b) Higher-magnification view of the yellow rectangular region in (a), emphasizing resolution differences between images taken in 2P widefield (2P WF), 2P ISIM, and deconvolved 2P ISIM modes. (c) Line-outs of microtubules marked in green, red, and blue in (b). Scale bar: 10 μm in (a) and 3 μm in (b). See also Fig. S7 in Supplement 1.
Having verified that 2P ISIM doubled the lateral resolution of the 2P microscope, we next characterized its performance at depths further from the coverslip in phantom samples consisting of subdiffractive fluorescent beads embedded in a scattering matrix [5.0% agarose gel containing 2.5%, 62 nm diameter nonfluorescent polystyrene beads (Fig. 3)]. Compared to previous forms of multifocal structured illumination microscopy, 2P ISIM provided better imaging depth in scattering samples: we observed recognizable images of individual beads at depths up to ~125 μm [Fig. 3(a)], even though the images at depth deteriorated relative to those collected at the coverslip surface. In contrast, we were unable to observe recognizable beads at depths >50 μm from the coverslip surface when using 1P excitation (1P ISIM [18]). When setting the illumination conditions for similar SNR at the coverslip, both SNR and the signal-to-background ratio (SBR) degraded much faster with depth when using 1P rather than 2P illumination [Fig. 3(b)]. We also compared 2P ISIM to 2P multi-focal SIM (2P MSIM [19]), and found similar, albeit less dramatic, improvements in SBR and SNR when using 2P ISIM [Fig. 3(b)]. Next, we examined signal degradation and background levels independently (Fig. S9 in Supplement 1). In all three methods, signal deteriorated significantly as a function of depth, with 1P ISIM signal diminishing the fastest. Background remained essentially constant for 2P ISIM but increased significantly with 1P ISIM, and to a lesser extent with 2P MSIM. Consistent with these observations, when comparing the background rejection ability of all three techniques in a thick fluorescent lake (Fig. S10 in Supplement 1), 2P ISIM offered the best sectioning performance. We conclude that the superior performance of 2P ISIM at depth relative to other forms of point-based structured illumination microscopy results from (i) the use of 2P illumination (less scattering of excitation, and thus higher signal generation in thick samples) and (ii) the lack of excitation and emission crosstalk inherent to parallelized methods such as 1P ISIM and 2P MSIM (leading to lower levels of background in thick samples) [24].
Fig. 3.

Two-photon ISIM improves SNR and SBR relative to single-photon implementations. (a) Representative images of subdiffractive fluorescent beads in a scattering matrix, as observed in 2P ISIM, 2P MSIM, and 1P ISIM systems. All images are autoscaled independently, and “0 μm” corresponds to the coverslip surface. Scale bar: 500 nm. The limited range of the 2P MSIM piezo stage prevented us from comparing 2P ISIM and 2P MSIM at depths greater than 75 μm from the coverslip, and data are not shown for depths further than 50 μm from the coverslip for the 1P ISIM system due to low SBR. (b) Graphs indicating falloff in SNR and SBR as a function of depth from the coverslip surface. Means and standard deviations are indicated from measurements taken on 6 beads at each depth (see also Fig. S9 in Supplement 1). Note that these images were not deconvolved.
To demonstrate that the benefits of 2P ISIM also apply in thick, live samples, we imaged transgenic Caenorhabditis elegans embryos expressing GFP–H2B [Fig. 4(a), Media 1]. In these samples, scattering prevents super-resolution imaging at depths more than ~15 μm from the coverslip when using 1P illumination [14] (Fig. S11 and Media 2 in Supplement 1): subnuclear chromatin structure is evident close to the coverslip surface, but image quality rapidly worsens at depth so that nuclei eventually resemble featureless blobs. The loss in image resolution and contrast at depth that result from using 1P illumination cannot be compensated for simply by increasing the exposure time, which serves mainly to increase the background (Fig. S12 in Supplement 1). When using 2P ISIM, we observed only a slight degradation in signal at increasing depth, which was easily compensated for by applying a modest ramp in input intensity during volumetric acquisition (Methods). Unlike 1P ISIM, we were easily able to observe subnuclear chromatin structure throughout all planes of the imaging stack [Fig. 4(b)].
Fig. 4.
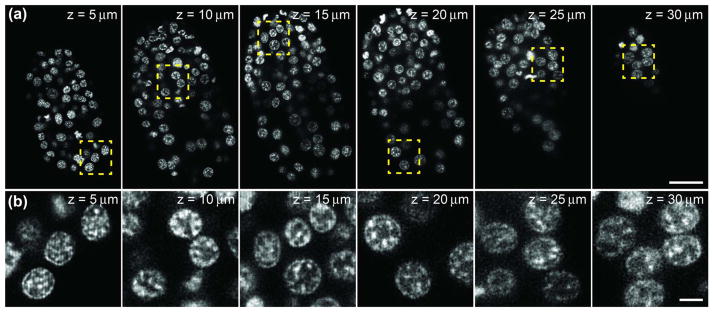
Two-photon ISIM enables visualization of subnuclear chromatin structure throughout nematode embryos. (a) Selected slices at indicated axial distance from the coverslip, through a live nematode embryo (bean stage). Scale bar: 10 μm. (b) Higher magnification views of yellow rectangular regions in (a), emphasizing subnuclear chromatin structure throughout the imaging volume. Scale bar: 2 μm. All images have been deconvolved. See also Media 1.
We then used 2P ISIM to image the nervous system in whole, live, anesthetized C. elegans larvae (Fig. 5) with the transcriptional reporter psax-3::GFP. SAX-3 is the C. elegans ROBO receptor homolog and is widely expressed throughout the nervous system [25]. By stitching together 10 fields of view, we reassembled an entire L2 stage larva, visualizing neurons and neurites throughout its ~350 μm length [Fig. 5(a), Media 3]. In addition to the GFP signal that highlighted these neuritis and cell bodies, we visualized autofluorescence in the 425–475 nm range from the larval body and gut structures (pseudo-red color, Fig. 5). This second color proved useful in defining the extent of the worm body, as well as internal regions such as the isthmus, terminal bulb of the pharynx, intestine, and intestinal lumen. Higher-magnification views within this dataset highlight the dorsal and ventral nerve cords [Fig. 5(b)], and resolved these processes [to an apparent width <200 nm, Fig. 5(c)], as well as the cell bodies and neurites of nerve ring neurons [Fig. 5(d)]. Our resolution and SNR were sufficiently high to distinguish closely spaced neuronal structures in both lateral [Fig. 5(d)] and axial [Figs. 5(e) and 5(f)] views of the nerve ring, and to capture transverse neurites that spanned the head of the larva [Fig. 5(e), in Media 3].
Fig. 5.

Two-color, 2P ISIM imaging in a live, anesthetized nematode larva. (a) Ten 2P ISIM volumes were acquired and stitched together to generate a two-color (green, GFP; red, blue-shifted autofluorescence) XY maximum intensity projection of an L2 nematode larva expressing transcriptional reporter psax-3::GFP, which is widely expressed throughout the nervous system. The head of the animal lies to the right, while the tail is located to the left. The yellow rectangle denotes the nerve ring and anterior portion of the nematode gut. Scale bar: 60 μm. (b) Higher-magnification view of the yellow rectangular region in (a), emphasizing nerve cords and nerve ring. Numerous head neurons and ventral cord motor neurons are visible in this view, as well as autofluorescent structures like the terminal bulb of the pharynx and the intestine. Scale bar: 20 μm. (c), (d) Higher-magnification views of yellow rectangular regions in (b). The yellow arrows show both the left and right fascicles of the ventral nerve cord, while the magenta arrow denotes the dorsal nerve cord. A neuronal process connecting the dorsal and ventral nerve cords is visible just anterior to the magenta arrow. In (d), neurons and neuronal processes in the nematode head can be resolved. Scale bar: 4 μm in (c), (d). The green colormap has been saturated in order to highlight dim neurites and subneuronal structures. (e), (f) Axial cuts through the imaging volume, corresponding to magenta and blue dashed lines in (b). Head neurons are visible in both views, while a neurite crossing the dorsal region of the head is denoted by a yellow arrow in (e). Scale bar: 5 μm. Neurites and fasciculating neurites denoted by yellow arrows in (c) and (e) have apparent lateral width <200 nm. All images were deconvolved. See also Media 3.
We also investigated the performance of 2P ISIM in thicker live samples. Our technique allowed us to visualize the microtubule network in a 38–40 h zebrafish eye in transgenic zebrafish engineered to have GFP-tagged microtubules, enabling inspection of the cytoskeleton of the developing lens beneath both the corneal and lens epithelial layers, at depths greater than 100 μm from the coverslip surface [Fig. 6(a), Media 4]. Upon close inspection of individual XY slices [Figs. 6(b)–6(f)] and a medial XZ slice [Fig. 6(g)], the rudimentary organization of the lens can be clearly observed, with multiple layers of microtubule networks forming clear, concentric spherical shells surrounding a central core. Although eventually the secondary lens fibers are elongated and well organized into symmetric layers [26], at this stage, microtubules in the lens are more disordered and 3D reconstruction revealed a surprisingly dense network of cytoskeletal fibers that appeared to curve tortuously throughout the volume of the lens [Figs. 6(a), 6(b), and 6(d); Media 5]. Despite our modest imaging rate of 1.67 frames per second (0.33 frames per second after 5× frame averaging), we were able to capture dividing cells in the lateral proliferative zone without significant motion blur [Fig. 6(a)]. Furthermore, although scattering degraded image quality farther into the sample, affecting both lateral and (to a greater extent) axial resolution [Fig. S13 in Supplement 1], we were still able to resolve thin microtubule bundles of apparent lateral width <200 nm throughout our 110 μm imaging volume [Figs. 6(b)–6(e)].
Fig. 6.

2P ISIM enables super-resolution imaging in volumes of ~100 μm thickness. (a) Rendering of ~60 × 60 × 110 μm volume (eye of 38–40 h old, live zebrafish embryo) captured with 2P ISIM. Single microtubules, bundles of microtubules, and dividing cells (magenta arrows) are visible in the volume. See also Media 4. (b), (d), (f) XY slices at indicated axial (Z) distance from the base of the stack. See also Media 5. Scale bar: 10 μm. (c), (e) Higher-magnification views of the yellow regions in (b), (d), emphasizing thin filaments (yellow arrows) with apparent width <200 nm. Scale bar: 5 μm. (g) XZ slice at indicated lateral (Y) distance from the origin of the stack, emphasizing concentric, circular cytoskeletal organization within the eye. Scale bar: 10 μm. All data presented in this figure were deconvolved.
The zebrafish samples also facilitated a comparison between 2P ISIM and conventional, point-scanning 2P imaging on a Leica SP5 microscope (an example of a modern, commercial 2P system used by biologists). We compared a lateral view of the midbrain–hindbrain boundary in a 36 h old zebrafish embryo on both systems [Fig. 7]. Although a rigorous comparison between the two systems is difficult due to differences in objectives, excitation power, and detection efficiency, at similar NA and acquisition speed, 2P ISIM delivered images with higher resolution and better SNR. For example, both microscopes revealed that microtubule filaments appeared to assemble into polygonal shapes throughout the volume [an example in Fig. 7(a) is shown ~20 μm from the coverslip]. While these structures appeared mostly continuous in the conventional 2P system, 2P ISIM clearly revealed the punctate nature of the individual bundles that made up the polygons [Fig. 7(b), Media 6]. The superior resolution of 2P ISIM also resolved fine filament bundles in axial views of the sample along the long axes of cells [Figs. 7(c) and 7(d), Media 7], which were otherwise obscured in the conventional 2P microscope.
Fig. 7.
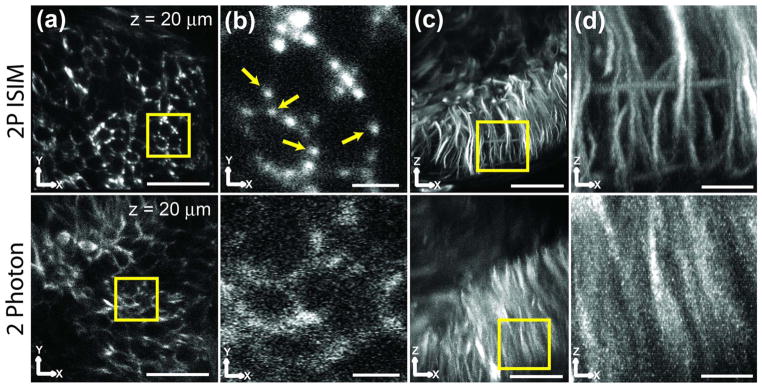
2P ISIM provides better resolution and SNR than conventional 2P microscopy. The same brain region in a zebrafish embryo was imaged in 2P ISIM (top row) and on a conventional, point-scanning 2P system (the Leica SP5, bottom row). (a) XY slices ~20 μm from the coverslip. Scale bar: 20 μm. (b) Higher-magnification views of region marked by the yellow square in (a). Scale bar: 3 μm. Yellow arrows indicate individual microtubule bundles. (c) XZ maximum intensity projections of the volumes. Scale bar: 20 μm. (d) Higher-magnification views of the region marked by the yellow square in (c). Scale bar: 5 μm. Images are raw, i.e., they have not been deconvolved. See also Media 6 and Media 7.
3. DISCUSSION
Our technique improves the spatial resolution of point-scanning 2P microscopy (2PM), requiring only simple modifications to the emission-side optical train of this workhorse system. Relative to the recent rescanning microscopy technique [21] (where a detection pinhole is needed to prevent out-of-focus emission from landing on the camera), 2P excitation eliminates the need for a detection pinhole in the emission path because out-of-focus emission does not occur (i.e., optical sectioning is inherent in the excitation process) [Note S1, 27]. This allows direct expansion of the illumination pattern without the need to first descan, thereby simplifying instrument design and alignment. Relative to our previous instant SIM implementation, 2P ISIM utilizes a single excitation focus instead of an array of foci, which also simplifies instrument design by eliminating the need for microlens arrays. 2P ISIM capability can thus be straightforwardly added to many existing 2P systems. Similar to our previous ISIM implementation [20], 2P ISIM approximately doubled the lateral spatial resolution of the conventional microscope that we modified, without any tradeoff in data acquisition speed or phototoxicity. Moreover, the longer wavelengths, 2P effect, and focused illumination employed offer better resolution, depth penetration, and inherent optical sectioning capability in thick fluorescent specimens relative to all previous SIM implementations. Since our technique requires an areal detector (camera) instead of a point detector (photomultiplier tube), it is more susceptible to scattering of emission than conventional 2PM. Nevertheless, combining 2P ISIM with chemical clearing methods [28,29] mitigates this issue and may prove immediately fruitful, likely enabling super-resolution imaging at unprecedented depth.
Many improvements to conventional 2PM would also benefit 2P ISIM, as the latter technology is based on the former. For example, excitation efficiency might be improved with dispersion compensation, which we did not incorporate here. Using adaptive optics on both the excitation [30] and emission side [31] would likely improve the SNR and resolution, especially axially and at increasing depths from the coverslip. Finally, the speed of the galvanometric mirrors limits our current full field of view (FOV) frame rate to 1.67 Hz (and typically less than 0.5 Hz, as we frame-averaged all datasets). Increasing the frame rate can be easily achieved by reducing the FOV along the slow galvo axis (e.g., by reducing our FOV to ~60 μm × 15 μm from ~60 μm × 60 μm, a frame rate of ~7 frames per second may be achieved). This flexibility contrasts with 2P MSIM [19], where the current scan pattern requires that 340 individual frames be collected to produce a single super-resolved image with a ~33 μm × 33 μm FOV, thereby preventing frame rates greater than ~0.3 Hz. Implementing resonant scanners would also improve our hardware speed, although in live biological samples we often found our frame rate more limited by signal than hardware, and synchronization of scanners may prove difficult. Finally, an alternative route to high-speed operation may be to parallelize the excitation. In this case, care must be taken when choosing the number and spacing of additional excitation foci to balance increases in speed against degradation in sectioning and to make best use of the available laser power.
4. METHODS
A. Optical System
The output from an 80 MHz Ti:sapphire oscillator capable of producing pulses of 140 fs duration (Coherent, Chameleon Ultra II) was used as the 2P excitation source. The laser beam was passed through a Glan-Laser calcite polarizer (Newport, 10GL08AR.16) and a half-wave plate (HWP, Newport, 10RP52-2), and the HWP was placed in a motorized rotation mount (Thorlabs, PRM1Z8E) for automated power control. After passing through the polarization optics, the beam was passed through a Pockels cell (Conoptics, 350-80-LA-02 KD*P Series E-O modulator and Driver 302M) and mechanical shutter (Thorlabs, SH05) for further intensity control. The Pockels cell provided rapid (on the μs scale) intensity modulation with incomplete extinction, whereas the mechanical shutter operated more slowly (on the ms scale), but provided complete extinction. Next, the beam was expanded 5× with two achromatic lenses (Thorlabs, f = 40 mm and f = 200 mm, AC254-040-B-ML, AC254-200-B-ML), passed through an iris (which served to reduce the beam diameter during alignment), and directed onto a two-dimensional galvanometric mirror assembly (Thorlabs, GVS012). The point midway between each mirror in the galvo assembly was imaged onto the back focal plane of a 60×, 1.2 NA objective (Olympus, UPLSAPO60XWPSF) using two achromatic lenses (Thorlabs, f = 250 mm and f = 200 mm, AC508-250-A-ML, AC254-200-A-ML) placed in a 4f telescope configuration between objective back focal plane and galvo assembly. Rotations of the galvanometric mirror assembly thus translated the excitation at the sample plane. The objective was housed in a microscope frame (Olympus, IX-70), and a reflective mirror (Chroma, 21010) was placed inside the filter turret in order to direct the beam into the objective. Samples were housed on an automated XY stage (ASI, S3192100FT) equipped with a 300 μm z axis piezo top plate (ASI, PZ-2300).
Fluorescence was collected through the same 60× objective lens, and passed through the same 200 mm/250 mm achromatic telescope mentioned above. A dichroic mirror (DC, Iridian, 6 mm thick and transmits 488 nm, 561 nm, 700–1100 nm, BJR000005) separated fluorescence from excitation, and was placed between the 250 mm achromat and a second, two-dimensional galvanometric mirror (Thorlabs, GVS012) assembly. The second galvo system was placed one focal length away from the 250 mm achromat, imaging the back focal plane of the objective lens onto the midpoint between galvo mirrors. Rotations of the galvo thus served to rescan fluorescence originating from the sample plane. A final achromatic lens (Thorlabs, f = 250 mm, AC508-250-A-ML) was placed one focal length after the galvo assembly, and served to focus the fluorescence image onto an electron-multiplying CCD (CAMERA, Andor, DU-885K-CS0-#VP, back-thinned with 1004 × 1002, 8 μm × 8 μm pixels and a CCI-23 control card). Two 680 nm short pass filters (Semrock, FF01-680/SP-25) were also included in the emission path to eliminate 2P illumination light reflected by the dichroic. These optics are shown in schematic form in (Fig. 1).
The optical magnification between sample and camera was (200/3 * 250/250) = 67 ×, resulting in a pixel size of 119.5 nm, which was confirmed using transmitted light images of a reticle. However, in SIM mode, the 2× expansion factor produced when the emission-side 2D galvo is scanned reduces the pixel size to 60 nm, which we confirmed by translating a sample of fluorescent beads a known distance with our automated stage.
B. Data Acquisition and Imaging Parameters
1. 2P ISIM
All 2P ISIM data were collected on a PC running 32 bit Windows XP SP3, with an Intel Xeon X3450 4 Core CPU, 2.67 GHz/core, and 3 GB RAM. Hardware components were controlled using custom control software written in LabVIEW (National Instruments, Version: 2012 SP1), and image acquisition was accomplished using manufacturer’s software (Andor, Solis Version: 4.12.30003.0) via external trigger mode. Acquisition of 3D data required integrated control of the mechanical shutter, Pockels cell, 2D galvos, camera, and piezo stage. All devices were controlled by external voltage using digital and analog outputs from a multichannel DAQ device (National Instruments, PCI-6733) and BNC connector block (National Instruments, BNC-2110). The mechanical shutter and camera were triggered via digital outputs, and the Pockels cell, 2D galvos, and piezo z stage were controlled by analog output. Due to its low impedance, we built a buffer amplifier to provide additional current to optimally drive the Pockels cell.
Rastering the illumination spot across the sample was accomplished by scanning one mirror from each 2D galvo assembly at 2 Hz and the other at 900 Hz. Note that in order to double the distance between adjacent emission foci before imaging, the amplitude and frequency of the waveforms delivered to excitation- and emission-side galvo assemblies was identical (this choice is justified in Note S2 in). Given the amplitude of our scan pattern, 900 Hz was the fastest scan speed at which we could maintain synchronization between the excitation- and emission-side galvos. During imaging, scanning of the galvos was initiated via the fire out signal from the camera. Figure S2 in Supplement 1 shows examples of the triangular and sinusoidal waveforms used to control the 2D galvo assemblies. Figure S3 in Supplement 1 shows examples of the control waveforms for the mechanical shutter, camera, and piezo stage for a 3D stack consisting of 4 image planes separated by 1 μm, with 2 frames acquired per plane.
As the fast-axis excitation galvo changes direction at the end of each sinusoidal line scan, it slows down, resulting in a significant increase in the total 2P illumination dose delivered to the sample near the edges of the FOV. The fluorescence signal rate F is proportional to I2t, where I is the instantaneous illumination intensity and t the dwell time of the excitation. A sinusoidal galvo scan implies that the dwell time of the galvo can be described by a secant waveform. We therefore fed the Pockels cell a √cosine waveform, which improved image uniformity (Figs. S3 and S4 in Supplement 1).
Andor Solis settings for all images were identical: the camera was cooled to −65°C, the exposure time was set to 500 ms, electron multiplication gain set to 300, the preamplifier gain to 3.8×, vertical pixel shift speed set to 3.72715 μs, and the horizontal pixel shift readout rate set to 35 MHz at 14 bits. Triggering was set to kinetic acquisition mode via external trigger, and data spooled to the hard disk as. tiffs.
Images for all figures were collected at 1.0–1.5 mW average power, as measured at the sample. All images were collected using 900 nm wavelength illumination except for Fig. 5, where GFP was excited at 950 nm and autofluorescence excited at 750 nm. For the images in Fig. 5, an additional 525 nm bandpass filter (Semrock, FF01-525/50-25) was included during GFP imaging, and a 442 nm bandpass filter (Semrock, FF01-442/46-25) was included during autofluorescence imaging. Details on frame averaging, total frame time, z step size, and total number of z slices for all 2P ISIM images can be found in Table S1 in Supplement 1. The average illumination intensity was held constant during acquisition of all 3D data sets except for the C. elegans embryo shown in Fig. 4, where the average illumination intensity per 2D slice was linearly ramped from 100% to 125% of its initial value during the 3D acquisition.
2. 2P MSIM
We used a previously described 2P MSIM system to acquire images [19], employing the same 1.2 NA objective used for 2P ISIM experiments. For images of fluorescent beads, the exposure time was set to 30 ms per image, and the z step size set to 200 nm. For each 2D slice, 340 images were acquired, resulting in an effective imaging rate of ~0.1 Hz. For images of the thick fluorescent lake, the exposure time was also 30 ms per image and the z step size set to 100 nm.
3. 1P ISIM
We used a previously described 1P ISIM system to acquire images [20], employing the same 1.2 NA objective used for 2P ISIM experiments. For all data presented here, the laser was set to maximum power. For the images of fluorescent beads in 3D gels, the exposure time was 80 ms per image, and the z step between images was 200 nm. For images of the thick fluorescent lake, the exposure time was 40 ms per image, and the z step was 100 nm. For images of C. elegans embryos in Fig. S11 in Supplement 1, the exposure time was 80 ms per image and the z step was 250 nm. For the C. elegans images presented in Fig. S12 in Supplement 1, the low-exposure images were taken at 80 ms exposure and the high-exposure images taken at 800 ms exposure. For both of these datasets, the z step size was 250 nm.
C. System Alignment
Alignment of most of the optics in our 2P ISIM is straightforward. Particular care was taken when aligning the relative scan patterns of the excitation- and emission-side 2D galvos. Figure S5 in Supplement 1 shows the effects of an initial rotary offset between the images produced by these two galvo assemblies. We corrected this offset by rotating the scan pattern of one of the galvo assemblies relative to the other assembly using the LabVIEW Cartesian Coordinate Rotation VI. In principle, the scan pattern of either galvo can be modified. We chose to modify the scan pattern of the excitation-side galvo assembly to match the emission-side galvo assembly. The optimal scan pattern correction was determined empirically by scanning identical regions in a thick fluorescent slide (Chroma, 92001) with either the excitation-side or emission-side galvos alone, and then examining the images as pseudo-colored overlays in Image J (NIH, version: 1.48c). By applying a rotation angle of 2.81°, we were able to minimize the offset until it was no longer discernible. We then oriented the camera to match the scan pattern of the emission-side 2D galvo assembly.
In addition to alignment of the galvo scan patterns, correct setting of the phase offset between the excitation- and emission-side fast galvos was critically important for optimal performance. Figure S6 in Supplement 1 shows the effects of small mismatches in the phase alignment of the two fast galvos. Mismatches of even 1° result in a distinctive doubling artifact. Mismatches of less than 1°, while not producing a perceptible doubling, greatly reduce the lateral resolution of the system along the fast galvo scan axis by smearing intensity across the camera. We found the best way to align the relative phases of the fast-scanning galvos was to image 100 nm, yellow–green fluorescent beads (Invitrogen, F8803) at different phase offsets and analyze their FWHM.
D. Conventional 2P Microscopy
A Leica SP5 inverted confocal microscope equipped with an ultrafast pulsed laser (SpectraPhysics, Mai Tai DeepSee) tuned to 900 nm at 2.18 W was used for conventional 2P experiments (Fig. 7). A bidirectional resonance scanner operating at 8000 Hz with 32 lines of scan averaging was used for image acquisition, resulting in a 2D acquisition time of ~4 s. Stacks were acquired using a 20× NA 1.0 objective lens (Leica, HCX APO 20X) at 11.6× zoom, with 0.25 μm steps between slices. Single slices measured 1024 × 1024 pixels for a field of 63.53 μm2 (62 nm pixel size).
E. Bead Samples
We prepared two kinds of bead samples in this manuscript: fluorescent bead layers for measuring PSFs at the coverslip surface and fluorescent beads embedded in 3D scattering gels for measuring PSF degradation at depth. Fluorescent bead layers were prepared by coating 24 × 50 mm #1.5 coverslips (VWR, 48393241) with 100 mg/ml poly-L lysine (Sigma, P8920), depositing 20–40 μL of 100 nm diameter yellow–green fluorescent beads (Invitrogen, F8803, 1:1000 dilution in water) on the coverslips, waiting for 10 min, and gently washing the coverslips in water to remove excess beads from the coverslip. Samples were then immersed in water for imaging. Fluorescent beads in 3D scattering gel samples were prepared by suspending 100 nm diameter yellow–green fluorescent beads at 1:250 dilution into a solution containing 5% agarose (Sigma, A9539) and 2.5% nonfluorescent 0.062 μm scattering polystyrene beads (Bangs Laboratories, PS02N). This mixture was vortexed vigorously and sonicated for 2 min before heating. After heating, the gelled mixture was deposited on a #1.5 coverglass bottomed dish (Matek, P35G-1.5-14-C), allowed to cool, and then immersed in water for imaging.
F. Quantification of SNR and SBR
Signal (for Figs. 3 and Fig. S9 in Supplement 1) was estimated by computing the average intensity of an 8 × 8 pixel box centered over the bead of interest. Background was estimated by computing the average intensity from an equally sized area containing no visible beads at the same depth. SNR was calculated as the ratio of the average signal from a bead over the standard deviation of the background region. SBR was calculated as the ratio of the average signal from a bead over the average background intensity. For all bead measurements, an averaged “dark current” (camera shutter open, no excitation light) image was subtracted from the bead image stack prior to analysis.
G. Quantification of Bead Size/Apparent Resolution
All measurements of bead FWHM, except the measurement of the 2P excitation PSF, were calculated using the ImageJ plugin “Plot FWHM” that fits a Gaussian function to vertical and horizontal cuts centered on the brightest point in the image (http://www.umanitoba.ca/faculties/science/astronomy/jwest/plugins.html courtesy of Jennifer West, University of Manitoba). The emission PSF was measured by exciting the bead sample with filtered light (Semrock, FF02-482/18-25) from a halogen lamp (i.e., using the transillumination pillar of our microscope to provide wide-field illumination), and collecting emission through a 525 bandpass (Semrock, FF01-525/50-25) filter. The 2P excitation PSF lateral FWHM was manually estimated from a plot of the average intensity of a 6 × 6 pixel region centered on a bead, as the excitation light was sequentially stepped across the field of view (note that the emission galvo was held stationary for this measurement; only the excitation galvos were scanned). Images were cropped prior to analysis.
H. Immunolabeled Cells
Microtubules in cultured U2OS human osteosarcoma cells (ATCC, HTB-96) were immunolabeled as follows: Cells were washed 3× with cytoskeletal buffer (CB) (10 mM PIPES, 138 mM KCl, 3 mM MgCl2, 2 mM EGTA, 0.01% NaN3, 160 mM sucrose, pH 6.8), fixed and permeabilized for 30 min at 37°C in a mixture of 0.5% glutaraldehyde, 3.7% formaldehyde, and 0.3% Triton X-100 (vol/vol) in CB, washed 5× with CB, and quenched 3× in ice-cold CB containing 100 mM Glycine. Cells were washed 1× with CB between each round of quenching. After quenching, cells were washed 3× with CB and blocked in antibody dilution buffer (Abdil, 150 mM NaCl, 20 mM Tris, 0.1% Triton X-100 (vol/vol), 0.1% NaN3, 4% BSA, pH 7.4) for 1 h, rocking them gently on an agitator (Lab Quake). After blocking, Abdil was aspirated and cells were incubated with 4 μg/mL monoclonal mouse anti-α-tubulin primary antibody (Sigma, T6199) in Abdil for 2 h at room temperature with rocking, washed 5× in Abdil, incubated in a 1:200 dilution of Alexa fluor 488 goat antimouse secondary antibody (Molecular probes, A11001) in Abdil for 3 h at room temp with rocking, washed 4× with Abdil, washed 4× in deionized water, and imaged.
I. Nematode Samples
Worm strains were raised at 20°C on NGM plates seeded with OP50 E. coli. Strain BV24 ([ltIs44 [pie-1p-mCherry:: PH(PLC1delta1) + unc-119(+)]; zuIs178 [(his-72p::HIS-72::GFP); unc-119(+)]V]) was used to image nuclei, and strain IC692 (quEx 162 sax-3p::GFP + pRF4 rol-6) was used to image neuronal cell bodies and neurites. BV24 embryos were obtained by cutting adult hermaphrodites in half with a razor blade, then transferring embryos to a well chamber coated with Poly-L-lysine for imaging via mouth pipette (Sigma-Aldrich, A5177). L2 stage worms of strain IC692 were immobilized with 50 mM levamisole and imaged on an agarose pad sandwiched between two #1.5 coverslips. Strain IC692 was obtained from the Caenorhabditis Genetics Center, and strain BV24 was kindly supplied by Dr. Zhirong Bao.
J. Zebrafish Samples
T g(X lEef 1a1:dclk2 – GFP)io008 zebrafish embryos [14] were collected by natural spawning and maintained at 28°C. Embryos were anesthetized in Tricaine (Sigma, E10521) at a concentration of 600 μM in embryo medium [60 mg RedSea Coral Pro Salt (Drs. Foster and Smith Pet Supplies) per liter ddH2O]. Anesthetized embryos were mounted in 1% low-melt agarose (Cambrex, 50080) in Lab-Tek II chamber slides (Nunc, 155379), covered with embryo medium, and imaged at room temperature.
Supplementary Material
Acknowledgments
FUNDING INFORMATION
National Institute of Biomedical Imaging and Bioengineering (NIBIB) (EB000074); National Institute of Child Health and Human Development (NICHD).
This work was supported by the Intramural Rsearch Programs of the U.S. NIBIB (to P.W.W., A.G.Y, M.I., R.C., G.H.P., and H.S.), and the Eunice Kennedy Shriver NICHD (to D.D.N. and A.C.). We thank Yicong Wu for help with LabVIEW programming and Christian Combs for help in rendering 3D datasets. We thank Victor Wang for help with stitching the 3D datasets. We thank students in the Marine Biology Laboratory Physiology course, especially Greg Alushin, Daria Bonazzi, and Alexis Lomakin, for help with construction of an early prototype of this device. We also thank Henry Eden for critical feedback on the manuscript. The NIH does not endorse or recommend any commercial products, processes, or services.
Footnotes
The views and opinions of authors expressed here do not necessarily state or reflect those of the U.S. Government and they may not be used for advertising or product endorsement purposes.
A. G. Y., G. H. P., and H. S. conceived the idea. P. W. W., A. G. Y., and H. S. designed the optical system. P. W. W. built the optical system and implemented data acquisition software with guidance from A. G. Y. and P. W. W., D. D. N., and M. I. acquired data. R. C., D. D. N., and A. C. provided guidance on biological experiments. P. W. W., R. C., and D. D. N. prepared the samples. D. D. N., M. I., G. H. P., and A. C. provided biological reagents or equipment. P. W. W., D. D. N., R. C., and H. S. analyzed the data. P. W. W. and H. S. wrote the paper with input from all authors. H. S. supervised the Research.
See Supplement 1 for supporting content.
References
- 1.Fischer RS, Wu Y, Kanchanawong P, Shroff H, Waterman CM. Microscopy in 3D: a biologist’s toolbox. Trends Cell Biol. 2011;21:682–691. doi: 10.1016/j.tcb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Y, Christensen R, Colon-Ramos D, Shroff H. Advanced optical imaging techniques for neurodevelopment. Curr Opin Neurobiol. 2013;23:1090–1097. doi: 10.1016/j.conb.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cox S, Jones GE. Imaging cells at the nanoscale. Int J Biochem Cell Biol. 2013;45:1669–1678. doi: 10.1016/j.biocel.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Hell SW, Reiner G, Cremer C, Stelzer EHK. Aberrations in confocal fluorescence microscopy induced by mismatches in refractive index. J Microsc. 1993;169:391–405. [Google Scholar]
- 5.Wilson BC, Jacques SL. Optical reflectance and transmittance of tissues: principles and applications. IEEE J Quantum Electron. 1990;26:2186–2199. [Google Scholar]
- 6.Berning S, Willig KI, Steffens H, Dibaj P, Hell SW. Nanoscopy in a living mouse brain. Science. 2012;335:551. doi: 10.1126/science.1215369. [DOI] [PubMed] [Google Scholar]
- 7.Gould TJ, Burke D, Bewersdorf J, Booth MJ. Adaptive optics enables 3D STED microscopy in aberrating specimens. Opt Express. 2012;20:20998–21009. doi: 10.1364/OE.20.020998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takasaki KT, Ding JB, Sabatini BL. Live-cell superresolution imaging by pulsed STED two-photon excitation microscopy. Biophys J. 2013;104:770–777. doi: 10.1016/j.bpj.2012.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.York AG, Ghitani A, Vaziri A, Davidson MW, Shroff H. Confined activation and subdiffractive localization enables whole-cell PALM with genetically expressed probes. Nat Methods. 2011;8:327–333. doi: 10.1038/nmeth.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zanacchi FC, Lavagnino Z, Donnorso MP, Del Bue A, Furia L, Faretta M, Diaspro A. Live-cell 3D super-resolution imaging in thick biological samples. Nat Methods. 2011;8:1047–1049. doi: 10.1038/nmeth.1744. [DOI] [PubMed] [Google Scholar]
- 12.Shao L, Kner P, Rego EH, Gustafsson MGL. Super-resolution 3D microscopy of live whole cells using structured illumination. Nat Methods. 2011;8:1044–1046. doi: 10.1038/nmeth.1734. [DOI] [PubMed] [Google Scholar]
- 13.Muller CB, Enderlein J. Image scanning microscopy. Phys Rev Lett. 2010;104:198101. doi: 10.1103/PhysRevLett.104.198101. [DOI] [PubMed] [Google Scholar]
- 14.York AG, Parekh SH, Nogare DD, Fischer RS, Temprine K, Mione M, Chitnis AB, Combs CA, Shroff H. Resolution doubling in live, multicellular organisms via multifocal structured illumination microscopy. Nat Methods. 2012;9:749–754. doi: 10.1038/nmeth.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schulz O, Pieper C, Clever M, Pfaff J, Ruhlandt A, Kehlenbach RH, Wouters FS, Großhans J, Bunt G, Enderlein J. Resolution doubling in fluorescence microscopy with confocal spinning-disk image scanning microscopy. Proc Natl Acad Sci USA. 2013;110:21000–21005. doi: 10.1073/pnas.1315858110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gustafsson MGL. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- 17.Heintzmann R, Gustafsson MGL. Subdiffraction resolution in continuous samples. Nat Photonics. 2009;3:362–364. [Google Scholar]
- 18.Ingaramo M, York AG, Hoogendoorn E, Postma M, Shroff H, Patterson GH. Richardson–Lucy deconvolution as a general tool for combining images with complementary strengths. Chem Phys Chem. 2014;15:794–800. doi: 10.1002/cphc.201300831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ingaramo M, York AG, Wawrzusin P, Milberg O, Hong A, Weigert R, Shroff H, Patterson GH. Two-photon excitation improves multifocal structured illumination microscopy in thick scattering tissue. Proc Natl Acad Sci USA. 2014;111:5254–5259. doi: 10.1073/pnas.1314447111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.York AG, Chandris P, Dalle Nogare D, Head J, Wawrzusin P, Fischer RS, Chitnis A, Shroff H. Instant super-resolution imaging in live cells and embryos via analog image processing. Nat Methods. 2013;10:1122–1126. doi: 10.1038/nmeth.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Luca GMR, Breedijk RMP, Brandt RAJ, Zeelenberg CHC, de Jong Babette E, Timmermans W, Nahidi Azir L, Hoebe RA, Stallinga S, Manders EMM. Re-scan confocal microscopy: scanning twice for better resolution. Biomed Opt Express. 2013;4:2644–2656. doi: 10.1364/BOE.4.002644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roth S, Sheppard CJR, Wicker K, Heintzmann R. Optical photon reassigment microscopy (OPRA) Opt Nanosc. 2013;2:1–6. [Google Scholar]
- 23.Sheppard CJR. Super-resolution in confocal Imaging. Optik. 1988;80:53–54. [Google Scholar]
- 24.Winter PW, Shroff H. Faster fluorescence microscopy: advances in high speed biological imaging. Curr Opin Chem Biol. 2014;20:46–53. doi: 10.1016/j.cbpa.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zallen JA, Yi BA, Bargmann CI. The conserved immunoglobulin superfamily member SAX-3/Robo directs multiple aspects of axon guidance in C. elegans. Cell. 1998;92:217–227. doi: 10.1016/s0092-8674(00)80916-2. [DOI] [PubMed] [Google Scholar]
- 26.Greiling TM, Clark JI. Early lens development in the zebrafish: a three-dimensional time-lapse analysis. Dev Dyn. 2009;238:2254–2265. doi: 10.1002/dvdy.21997. [DOI] [PubMed] [Google Scholar]
- 27.Gustafsson MGL, Shao L, Carlton PM, Wang CJR, Golubovskaya IN, Cande WZ, Agard DA, Sedat JW. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J. 2008;94:4957–4970. doi: 10.1529/biophysj.107.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hama H, Kurokawa H, Kawano H, Ando R, Shimogori T, Noda H, Fukami K, Sakaue-Sawano A, Miyawaki A. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat Neurosci. 2011;14:1481–1488. doi: 10.1038/nn.2928. [DOI] [PubMed] [Google Scholar]
- 29.Chung K, Wallace J, Kim SY, Kalyanasundaram S, Andalman AS, Davidson TJ, Mirzabekov JJ, Zalocusky KA, Mattis J, Denisin AK, Pak S, Bernstein H, Ramakrishnan C, Grosenick L, Gradinaru V, Deisseroth K. Structural and molecular interrogation of intact biological systems. Nature. 2013;497:332–337. doi: 10.1038/nature12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Debarre D, Botcherby EJ, Watanabe T, Srinivas S, Booth MJ, Wilson T. Image-based adaptive optics for two-photon microscopy. Opt Lett. 2009;34:2495–2497. doi: 10.1364/ol.34.002495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kner P, Sedat JW, Agard DA, Kam Z. High-resolution wide-field microscopy with adaptive optics for spherical aberration correction and motionless focusing. J Microsc. 2010;237:136–147. doi: 10.1111/j.1365-2818.2009.03315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.