

Abstract
Objective
Inflammation has been proposed as a key component in the development of hypertension and cardiac remodeling associated with different cardiovascular diseases. However, the role of the proinflammatory cytokine interleukin-6 in the chronic stage of hypertension is not well defined. Here, we tested the hypothesis that deletion of interleukin-6 protects against the development of hypertension, cardiac inflammation, fibrosis, remodeling and dysfunction induced by high salt diet and angiotensin II (Ang II).
Methods
Male C57BL/6J and interleukin-6-knock out (KO) mice were implanted with telemetry devices for blood pressure (BP) measurements, fed a 4% NaCl diet, and infused with either vehicle or Ang II (90 ng/min per mouse subcutaneously) for 8 weeks. We studied BP and cardiac function by echocardiography at baseline, 4 and 8 weeks.
Results
Myocyte cross-sectional area (MCSA), macrophage infiltration, and myocardial fibrosis were also assessed. BP increased similarly in both strains when treated with Ang II and high salt (Ang II-high salt); however, C57BL/6J mice developed a more severe decrease in left ventricle ejection fraction, fibrosis, and macrophage infiltration compared with interleukin-6-KO mice. No differences between strains were observed in MCSA, capillary density and MCSA to capillary density ratio.
Conclusion
In conclusion, absence of interleukin -6 did not alter the development of Ang II-high salt-induced hypertension and cardiac hypertrophy, but it prevented the development of cardiac dysfunction, myocardial inflammation, and fibrosis. This indicates that interleukin-6 plays an important role in hypertensive heart damage but not in the development of hypertension.
Keywords: angiotensin II, heart failure, hypertension, interleukin 6
INTRODUCTION
Inflammation has been proposed as a key component in the development of hypertension and cardiac remodeling associated with different cardiovascular diseases [1–3]. However, the role of specific cytokines in the development of hypertension and cardiac damage is not well defined.
Interleukin-6 is a cytokine with multiple physiological roles. In the innate immune system, interleukin-6 stimulates the production of neutrophils and the synthesis of acute-phase proteins by hepatocytes, thus contributing to the acute-phase response, whereas in the adaptive immunity, interleukin-6 stimulates the proliferation of B cells [4]. However, interleukin-6 has also been proposed to play a detrimental role in hypertension, in two ways. First, interleukin-6 may participate in the development of hypertension, and there is some evidence from human and animal studies to support this possibility. For example, in healthy human subjects, an acute angiotensin II (Ang II) infusion increased both interleukin-6 and blood pressure (BP) [5], and levels of circulating interleukin-6 showed a strong, positive correlation with BP [6]. Furthermore, in mice lacking interleukin-6, an attenuated pressor response to Ang II has been reported [7–9]. Also, interleukin-6 may participate in the development of hypertensive end-organ damage by causing cardiac and renal inflammation as well as extra-cellular matrix remodeling [7,9,10]. For example, infusion of interleukin-6 for 1 week induced a pattern of adverse cardiac remodeling similar to that observed in hypertensive hearts in the absence of changes in BP [10]. However, not all studies have shown a consistent detrimental effect of interleukin-6 on cardiovascular diseases. One study showed no effect of chronic infusion of interleukin-6 on pressure responsiveness to Ang II in vivo [11]. And another reported that deletion of interleukin-6 actually caused adverse cardiac remodeling, leading to myocardial fibrosis and failure [12], thus opening the possibility that the reported attenuation in Ang II-induced hypertension might be secondary to heart failure.
Thus, whether interleukin-6 is protective or detrimental to cardiac function and remodeling in hypertension-induced cardiac damage, and whether any effects seen are pressure-dependent or independent, remain unknown and need to be further explored.
We tested the hypothesis that deletion of interleukin-6 prevents the development of hypertension, cardiac inflammation, fibrosis, remodeling, and dysfunction induced by high salt and Ang II. We used a model of hypertension and end-organ damage induced by Ang II infusion in combination with a high-salt diet, the purpose of high salt being to exacerbate the phenotype induced by Ang II [8].
METHODS
Animal model, blood pressure measurement, and experimental protocol
All experiments were approved by the Henry Ford Health System Institutional Animal Care and Use Committee and were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Male C57BL/6J (C57) and interleukin-6knock out (KO) mice (B6.129S6-IL-6tm1/Kopf) were obtained from Jackson Laboratories (Bar Harbor, Maine, USA) at 9 weeks of age (20–24 g). Mice had access to regular chow (0.5% NaCl) and tap water ad libitum and were allowed to adjust to the environment for at least 1 week. Then, mice were implanted with telemetry devices to monitor BP and heart rate as previously described [13]. Briefly, mice were anesthetized with sodium pentobarbital (50 mg/kg intraperitoneally), the left carotid artery was isolated, and a telemetry catheter was then inserted through it and advanced to the aortic arch. The transmitting device was placed subcutaneously along the animal’s flank. Seven to 10 days later, the animals had recovered from surgery, displayed a normal BP circadian rhythm, and reached a weight of 27–30 g (12 weeks’ old). The regular chow was then replaced by a high-salt diet containing 4% NaCl (Teklad, Madison, Wisconsin, USA). One week later, mice were again anesthetized with sodium pentobarbital, and osmotic minipumps (Alzet, Cupertino, California, USA) were implanted under aseptic conditions to deliver either Ang II (90 ng/min per mouse; Bachem, Torrance, California, USA) or vehicle (0.01 N acetic acid in saline) as previously described [2]. The treatment groups were as follows: C57 + vehicle-high salt; interleukin-6 KO + vehicle-high salt; C57 + Ang II-high salt; and interleukin-6 KO + Ang II-high salt. Treatment was continued for 8 weeks.
Echocardiography
Echocardiography was performed at baseline, and then at 4 and 8 weeks in nonanesthetized mice as previously described [14]. Briefly, an Acuson Sequoia 512 system (Mountain View, California, USA) equipped with a 15-MHz linear transducer was used. Left ventricle remodeling was assessed by measuring left ventricle systolic and diastolic dimension areas (LVAs and LVAd), left ventricle dimensions (LVDs and LVDd), and posterior wall thickness (PWT). Systolic performance was assessed by measuring ejection fraction and shortening fraction.
Histopathological studies
Mice were sacrificed after 8 weeks of Ang II-high salt-induced hypertension. Hearts were stopped at diastole by injecting 15% KCl, then excised and weighed. Afterwards, the left ventricle was sectioned transversely into two slices, rapidly frozen in isopentane that was precooled in liquid nitrogen, and stored at −70°C. Sections measuring 6 μm were cut from each slice and double-stained with fluorescein-labeled peanut agglutinin [to outline the myocyte cross-sectional area (MCSA) and the interstitial space] and rhodamine-labeled Griffonia simplicifolia lectin I (to outline the capillaries). Radially oriented microscopic fields from each section were photographed at 400× using a microscope (IX81; Olympus America, Center Valley, Penn-sylvania, USA) and a digital camera (DP70; Olympus America), and analyzed with a computerized image analysis system (MicroSuite Biological imaging software; Olympus America). MCSA was measured by computer-based planimetry and averaged using the data obtained from all the photographs. The interstitial collagen fraction (ICF) was calculated as the ratio of the collagen area to the entire area of an individual section, which is the sum of the areas representing the myocytes and the interstitial space [15–17].
Immunohistochemistry
Frozen sections (6 μm) were immunostained with antirat antibody specific for CD68, a marker for macrophages (OX-38, BD Pharmingen, San Diego, California, USA). Vectastain Elite ABC-peroxidase kit (Vector Laboratories, Burlingame, California, USA) and AEC substrate 3-amino-9-ethylcarbazole (Invitrogen, Grand Island, New York, USA) were used to detect positive immunoreactivity. Negative controls were processed in a similar fashion except that they were not incubated with the primary antibody. Positive cells (reddish brown) were counted in 40× high-power fields in each section and expressed as number of CD68-positive cells per mm2.
Measurement of urinary albumin
Forty-eight hours prior to the end of the protocol, mice were acclimated to the metabolic cages (custom-made, specially designed for mice) 48 h prior to the 24-h urine collection, as described by Rhaleb et al. [18]. Urinary volume was measured and the urine was centrifuged twice at 1500g at 4°C for 10 min (Eppendorf centrifuge 5415R). The supernatant was passed through a 25-mm syringe filter (0.2 μm HT Tuffryn membrane; Gelman Laboratories, Ann Arbor, Michigan, USA) and stored at −20°C. Urinary albumin was measured with an enzyme-linked immunosorbent assay kit (GenWay, San Diego, California, USA) and expressed as micrograms albumin per 10 g body weight per 24 h.
Data analysis
Data are expressed as mean ± standard error. One-way analysis of variance was used followed by all pairwise testing of body weight, left ventricular weight/body weight, echocardiographic measurements of ventricular function, capillary density, MCSA, and the ratio between capillary density and MCSA. Two-sample Wilcoxon tests were used for heart weight/body weight, ICF, macrophage infiltration, and urinary albumin excretion, as the data were not normally distributed. Hochberg’s method was used to adjust for multiple testing. A P value less than 0.05 was considered significant.
RESULTS
Heart rate, mean blood pressure, and heart weight
There were no differences in heart rate among groups, irrespective of diet or Ang II treatment (Fig. 1a). There were no differences in mean BP among groups either at baseline or after 1 week of high-salt diet. BP was significantly increased in C57 mice after starting the infusion of Ang II and this increase was not attenuated in interleukin-6 KO mice, indicating that interleukin-6 is not necessary for Ang II-high salt-induced hypertension (Fig. 1b). Chronic treatment with Ang II-high salt increased heart weight similarly in both C57 and interleukin-6 KO mice (Table 1).
FIGURE 1.

Heart rate (a) and mean arterial pressure (b) as measured by telemetry at baseline, during high-salt diet (4% NaCl) and with the addition of either angiotensin (Ang) II or vehicle for 8 weeks (W) in C57 and interleukin-6 knock out (KO) mice. Heart rate and blood pressure were recorded during 24 h and then grouped in periods of 12 h of daytime (D) and night-time (N) for each day. Heart rate was not affected by either diet or Ang II treatment. The high-salt diet did not affect blood pressure. Ang II increased blood pressure in both C57 andinterleukin-6 KO mice (*P <0.05 C57 Ang II vs. C57 vehicle, #P <0.05 interleukin-6 KO Ang II vs. interleukin-6 KO vehicle). Deletion of interleukin-6 did not attenuate Ang II-high salt-induced hypertension in mice.
TABLE 1.
Body weight and hypertrophic index at 8 weeks of treatment
| Strain n |
High salt +vehicle
|
High salt +Ang II
|
||
|---|---|---|---|---|
| C57BL/6J 9 |
Interleukin-6 KO 9 |
C57BL/6J 8 |
Interleukin-6 KO 6 |
|
| Body weight (g) | 31.8 ± 0.5 | 31.4 ± 0.8 | 28.9 ± 1.8 | 30.1 ± 1.3 |
|
| ||||
| Heart weight/body weight (mg/g) | 4.0 ± 0.3 | 3.9 ± 0.2 | 6.9 ± 0.6* | 7.1 ± 0.6* |
|
| ||||
| LVW/body weight (mg/g) | 3.3 ± 0.3 | 3.3 ± 0.2 | 6.0 ± 0.5* | 6.1 ± 0.5* |
KO, knock out; LVW, left ventricular weight.
P <0.001 vs. same genotype infused with vehicle. There were no differences between genotypes.
Cardiac remodeling and function
At baseline, LVDs and LVDd and LVAs and LVAd, obtained from parasternal short-axis view, were similar among groups. None of these parameters were affected by high-salt diet in either genotype of mice when infused with vehicle (Table 2; Fig. 2a and 2c). Systolic measurements of the left ventricle chamber (LVDs and LVAs) were increased by Ang II-high salt in wild-type, but not in interleukin-6 KO mice (Table 2; Fig. 2b). However, no changes were observed in diastolic measurements of the left ventricle chamber (Table 2; Fig. 2d). Posterior wall thickness was increased by Ang II-high salt; however, no difference between genotypes was detected (Table 2).
TABLE 2.
Echocardiographic parameters at 8 weeks of treatment
| Strain n |
High salt +vehicle
|
High salt +Ang II
|
||
|---|---|---|---|---|
| C57BL/6J 9 |
Interleukin-6 KO 9 |
C57BL/6J 8 |
Interleukin-6 KO 6 |
|
| PWTs (mm) | 1.5 ± 0.08 | 1.49 ± 0.07 | 2.1 ± 0.08* | 2.02 ± 0.07* |
|
| ||||
| PWTd (mm) | 0.87 ± 0.01 | 0.95 ± 0.04 | 1.68 ± 0.073 | 1.65 ± 0.12* |
|
| ||||
| LVDs (mm) | 1.21 ± 0.05 | 1.22 ± 0.04 | 1.4 ± 0.08 | 1.07 ± 0.05# |
|
| ||||
| LVDd (mm) | 2.6 ± 0.09 | 2.6 ± 0.08 | 2.5 ± 0.1 | 2.4 ± 0.05 |
|
| ||||
| Shortening fraction (%) | 53 ± 1 | 53 ± 1 | 46 ± 2* | 56 32# |
d, diastole; LVA, left ventricular area; LVD, left ventricular dimension; PWT, posterior wall thickness; s, systole.
P <0.001 vs. same genotype infused with vehicle.
P <0.05 interelukin-6 KO-Ang II-high salt vs. C57-Ang II-high salt.
FIGURE 2.

Left ventricular area measured by echocardiography in systole (a and b) and diastole (c and d). Left ventricular areas were similar among groups at baseline and were not affected by high-salt diet in animals infused with vehicle. Ang II-high salt increased systolic left ventricular area in C57 mice (*P <0.01), but not in interleukin-6 KO mice, indicating that left ventricular dysfunction was prevented in interleukin-6 KO mice.
Left ventricle systolic function, evaluated by ejection fraction and shortening fraction, was not affected by high salt in either genotype of mice when infused with vehicle. Chronic treatment with Ang II-high salt depressed left ventricle systolic function in C57 mice, as evidenced by decreased ejection fraction and shortening fraction, whereas left ventricle function was well preserved in interleukin-6 KO mice (Table 2; Fig. 3). These results indicate that deletion of interleukin-6 prevents the progression of systolic dysfunction independently of changes in BP.
FIGURE 3.

Left ventricular ejection fraction in mice infused with vehicle (a) or Ang II (b). Ejection fraction was similar among groups at baseline and was not affected by high-salt diet in animals infused with vehicle. Ang II-salt decreased ejection fraction at 4 and 8 weeks in C57 mice relative to baseline (##P <0.01, ###P <0.001), but not in interleukin-6 KO mice. Consequently, ejection fraction was lower in C57 mice relative to interleukin-6 KO mice after 4–8 weeks of Ang II-high salt (***P <0.001), suggesting that left ventricular dysfunction in Ang II salt was partly mediated by interleukin-6.
Myocyte cross-sectional area and capillary density
In both C57 and intreleukin-6 KO mice, Ang II-high salt treatment significantly increased MCSA while decreasing capillary density and the ratio of capillary density to MCSA, an index of myocyte oxygenation. However, there were no differences between two genotypes when mice were treated with either vehicle-high salt or Ang II-high salt (Fig. 4), indicating that Ang II-high salt impairs myocardium oxygenation in an interleukin-6-independent manner.
FIGURE 4.

Myocyte cross-sectional area (MCSA; a), capillary density (b) and ratio of capillaries to MCSA (capillaries/MCSA; c). Deletion of interleukin-6 in mice did not prevent Ang II-high salt-induced changes in these parameters.
Myocardial macrophage infiltration and interstitial fibrosis
Quantitative analysis of myocardial macrophage infiltration and myocardial ICF revealed no differences between genotypes when mice were treated with vehicle-high salt. Ang II-high salt treatment increased the numbers of CD68-positive macrophages in the heart as compared with the hearts of vehicle-high salt-treated mice, but this increase was attenuated in interleukin-6 KO mice (Fig. 5). Likewise, Ang II-high salt increased ICF in C57 mice compared with vehicle-high salt, and this increase was also blunted in interleukin-6 KO mice (Fig. 6), indicating that interleukin-6 mediates Ang II-high salt-induced inflammation and fibrosis in the heart.
FIGURE 5.

Myocardial macrophage infiltration, representative images (top panels) and quantification (bottom). Ang II-salt increased myocardial macrophage infiltration, but this increase was attenuated in interleukin-6 KO. CD68 immunostaining, magnification 400×.
FIGURE 6.

Myocardial interstitial collagen fraction, representative images (top panels) and quantification (bottom). Ang II-salt increased myocardial fibrosis but this increase was attenuated in interleukin-6 KO mice. Fluorescein-labeled peanut agglutinin staining, magnification 400×.
Urinary albumin excretion
Urinary albumin excretion was not different between genotypes when mice were treated with vehicle-high salt. Ang II-high salt increased urinary albumin excretion compared with vehicle-high salt-treated animals of both genotypes. Urinary albumin excretion was nearly four times higher in the C57-Ang II-high salt group compared with the interleukin-6 KO-Ang II-high salt group; however, this comparison did not reach statistical significance. However, the difference between the two C57 groups (i.e., Ang II-high salt minus vehicle-high salt treated) was 969.5 ± 308 μg/ 24 h, which is significantly greater than the difference between the two interleukin-6 KO groups (236.4 ± 74 μg/ 24 h, P <0.05), suggesting that interleukin-6 contributes to hypertensive renal damage (Fig. 7).
FIGURE 7.
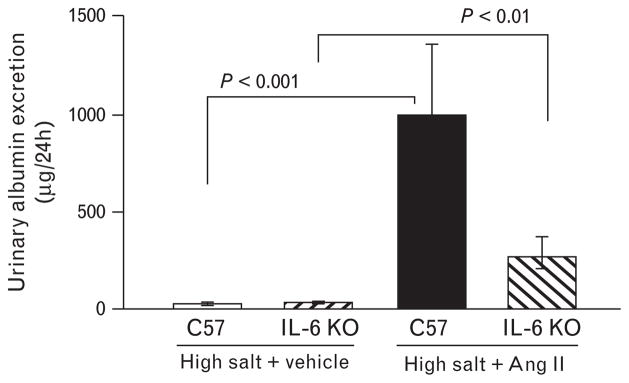
Urinary albumin excretion. Ang II-salt increased urinary albumin excretion, in both C57 and interleukin-6 KO mice.
DISCUSSION
In the present study, we investigated the effect of interleukin-6 deficiency on the development of hypertension and end-organ damage induced by chronic Ang II infusion and high-salt diet in mice. We found that deletion of interleukin-6 did not affect the development of Ang II-high salt-induced hypertension; however, it prevented myocardial inflammatory cell infiltration and fibrosis, preserved left ventricle ejection fraction, and reduced urinary album excretion. Lack of interleukin-6 did not prevent Ang II-high salt-induced myocyte hypertrophy or Ang II-high salt-induced decreases in the ratio of capillary density to myocyte area. Thus, our study is the first to show that interleukin-6 deficiency, independently of the development of hypertension and cardiac hypertrophy, prevents cardiac dysfunction, myocardial inflammation, and renal damage induced by chronic Ang II infusion and high salt feeding.
In the interest of elucidating the pathophysiological role of interleukin-6 in cardiovascular disease, we have taken advantage of a very unique tool, namely the interleukin-6 knockout mouse. Of the available mouse models of hypertension, the infusion of Ang II is widely used because it raises BP in a reliable manner [19], and because of the human relevance of the renin–angiotensin system, as demonstrated by the clinical benefits of ACE inhibitors and angiotensin II receptor type 1 (AT1) receptor blockers. Nevertheless, Ang II infusion by itself is a rather mild mouse model in terms of heart function. Whereas heart dysfunction is one important complication of hypertension in humans, Ang II-infused, hypertensive mice have little cardiac dysfunction. In fact, there are no mouse models of heart failure induced by systemic hypertension [20,21]. In the present study, we used a mouse model of Ang II-induced hypertension combined with a high-salt diet (4% NaCl in the chow). This model was first described in rats by Muirhead et al. [22], and is characterized by an enhanced pressor response and increased end-organ damage compared with treatment with Ang II and regular chow. Using this model over a period of 8 weeks, we achieved a mild–moderate degree of heart dysfunction in mice, but not overt heart failure. Thus, this model allowed us to test the hypothesis that interleukin-6 participates in Ang II/salt-induced hypertension and end-organ damage. Nevertheless, confirmatory studies in other animal models should be undertaken to validate the translational relevance of our findings before any clinical trials are proposed. For example, with the recent availability of knockout rats [23], it is possible that in the future interleukin-6 knockout rats could be used in rat models of hypertension and heart failure.
Our experimental design was similar to that of Lee et al. [8], who also used Ang II-high salt in interleukin-6 KO mice, but with two important differences: we extended the time of follow-up from 2 to 8 weeks, and we focused mainly on studying cardiac remodeling, inflammation, fibrosis, ventricular function, and BP by telemetry. To evaluate the role of interleukin-6 in the pathogenesis of hypertension, BP and heart rate were continuously monitored via subcutaneously implanted telemetric devices. Ang II in combination with high salt intake markedly increased BP in both C57 and interleukin-6 KO mice. However, interleukin-6 deletion had no influence on the development of hypertension. There is some controversy in the literature over whether or not interleukin-6 contributes to the pressor response in the commonly used mouse model of Ang II-induced hypertension. The above-mentioned study by Lee et al. [8] showed an attenuation of the pressor response to Ang II in mice fed 4% NaCl diet, and a blunted pressor response to Ang II was reported in mice fed regular chow as well [9,24]. However, Schrader et al. [25] found that C57 and interleukin-6 KO mice had similar increases in BP after 2 weeks of Ang II infusion. The reasons for this discrepancy are not entirely clear. To provide a fuller picture on this matter, we took the approach of extending our observations to 8 weeks, as opposed to previous studies that focused only on the early development of hypertension. Our results showed that interleukin-6 plays a role in hypertension-induced end-organ damage, rather than in the development of hypertension per se. In this regard, our findings are consistent with previous reports showing that interleukin-6 did not affect acute pressor responsiveness to Ang II in vivo [11] and that infusion of interleukin-6 for 7 days did not modify BP [10].
Whether pressure-dependent or not, the vast majority of studies have shown a detrimental effect of interleukin-6 on the cardiovascular system, and a protective effect of its deletion. However, this view is not unanimous, as Banerjee et al. [12] recently reported a significant reduction in ejection fraction in interleukin-6 KO mice compared with wild-type mice. We studied the effect of interleukin-6 deletion on cardiac function by echocardiography at baseline, that is, before the administration of Ang II infusion and high-salt diet. We found that interleukin-6 KO mice did not display adverse heart remodeling or dysfunction under basal conditions. Of note, we performed echocardiography in awake mice, as we have previously shown this method to be better for measuring mouse cardiac function compared to echo-cardiography under anesthesia [14].
In human studies, interleukin-6 has been shown to be associated with left ventricle hypertrophy, dilatation, dysfunction, and fibrosis [26–29]. In rats, Melendez et al. [10] reported that chronic interleukin-6 infusion did not increase BP, but caused myocardial hypertrophy and diastolic dysfunction. Likewise, Coles et al. [7] showed that myocardial hypertrophy induced by 1 week of Ang II infusion was attenuated by interleukin-6 deletion. However, Lai et al. [30] found no attenuation in myocardial hypertrophy in interleukin-6 KO mice after 4 weeks of aortic banding. We measured myocardial hypertrophy by three different indices, namely left ventricle weight, left ventricle mass (assessed by echocardiography), and MCSA, and found no attenuation of Ang II-high salt-induced cardiac hypertrophy in interleukin-6 KO mice. Thus, our results are in agreement with those of Lai et al. [30] and suggest that interleukin-6 does not contribute to the development of myocardial hypertrophy induced by chronic high-salt feeding and Ang II infusion. It is possible that, at least in this model, the development of myocardial hypertrophy is driven by pressure overload rather than by inflammation. Since we also found that interleukin-6 KO and C57 mice had a similar ratio of capillary density to MCSA, our results suggest that the cardioprotective effect of interleukin-6 deficiency in mice was independent not only of blood BP, but also of changes in myocardial oxygenation as well.
It is well known that untreated hypertension leads to cardiac failure. Ventricular dilatation and myocardial fibrosis are considered hallmarks of hypertensive heart disease and indicators of poor prognosis. Therefore, we also analyzed the effect of not only interleukin-6 deficiency on cardiac remodeling and dysfunction. We did not observe left ventricle dilatation after 8 weeks of treatment with Ang II-high salt (assessed by echocardiographic measurements of left ventricle diastolic dimension and area); thus, a different animal model would be necessary to study whether interleukin-6 plays a role in left ventricle dilatation. Nevertheless, Ang II-high salt-induced left ventricle systolic dysfunction was attenuated in interleukin-6 KO mice compared with wild–type mice, as evidenced by smaller left ventricle systolic chamber dimension and area and better ejection fraction in interleukin-6 KO mice. These results suggest that interleukin-6 contributes to the development of cardiac systolic dysfunction.
We and others have shown that sustained elevation of BP by Ang II is accompanied by myocardial macrophage infiltration and fibrosis [31–34]. Here we examined whether genetic deletion of interleukin-6 prevents cardiac inflammation and fibrosis. We found that myocardial macrophage infiltration increased with Ang II-high salt, but this was largely prevented in mice deficient in interleukin-6. Our data are in agreement with the study by Kobara et al. [35] who showed that a selective interleukin-6 antagonist reduced leukocyte and macrophage infiltration. We also found that Ang II-high salt increased myocardial collagen deposition in C57, but not interleukin-6 KO mice. These findings are congruent with those of Janssen et al. [36] who showed that cardiac fibrosis in an experimental model of myocardial infarction was accompanied by high levels of interleukin-6, and with the report of Kobara et al. [35] who demonstrated that inhibition of interleukin-6 reduced the left ventricle fibrosis after myocardial infarction. Furthermore, the attenuation in fibrosis that we observed is likely a consequence of the attenuation in macrophage infiltration, as Ma et al. [33] recently showed that during Ang II infusion, macrophages stimulate cardiac fibroblasts to produce interleukin-6, leading to TGFβ1 production, Smad3 phosphorylation, and fibrosis. In addition to its effect on macrophages, interleukin-6 has an important role in differentiation of T-cell subpopulations. For example, in the presence of transforming growth factor (TGF)β, naïve T cells will differentiate toward a regulatory (Treg) phenotype; however, addition of interleukin-6 to TGFβ will shift differentiation away from a Treg, toward a Th17 phenotype [37]. As Th17 are thought to participate in Ang II-induced damage [38], whereas Tregs are protective against it [39], our findings that interleukin-6 KO mice have decreased end-organ damage could be because of decreased Th17 and/or increased Treg cells. Of note, Kvakan et al. [39] reported that adoptive transfer of Treg cells ameliorates Ang II-induced end-organ damage in mice without affecting BP, which is similar to our findings in interleukin-6 KO mice.
Although we mainly focused on the role of interleukin-6 in the heart, we also tested whether interleukin-6 deletion prevented Ang II-high salt-induced albuminuria, a marker for renal damage. In humans, interleukin-6 accumulates in the glomeruli of patients with chronic kidney disease and hypertension [9]. And in mice, interleukin-6 participates in the development of albuminuria after 2 weeks of Ang II infusion [8,9]. Our work suggests that interleukin-6 may contribute to Ang II-induced albuminuria after 8 weeks of follow-up, independently of changes in BP.
In summary, our results indicate that in a model of hypertension and end-organ damage induced by Ang II-high salt, deletion of interleukin-6 does not prevent the development of hypertension, but can attenuate myocardial inflammation and fibrosis, leading to an improvement in cardiac function. These results suggest that interleukin-6 plays an important role in hypertensive heart disease. The present findings have important implications for understanding the role of interleukin-6 in the pathogenesis of hypertension and end-organ damage. In addition, although more research is needed, these findings provide support for novel therapeutic possibilities that target interleukin-6 [40].
Acknowledgments
This study was supported by Henry Ford Health System institutional funds (NER) and by National Institutes of Health grant HL-028982–31 (OAC).
Abbreviations
- Ang II
angiotensin II
- BP
blood pressure
- ICF
interstitial collagen fraction
- LVAd
left ventricle area diastolic
- LVAs
left ventricle area systolic
- LVDd
left ventricle dimension diastolic
- LVDs
left ventricle dimension systolic
- MCSA
myocyte cross-sectional area
Footnotes
Conflicts of interest
There are no conflicts of interest.
References
- 1.Harrison DG, Marvar PJ, Titze JM. Vascular inflammatory cells in hypertension. Front Physiol. 2012;3:128. doi: 10.3389/fphys.2012.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peng H, Yang XP, Carretero OA, Nakagawa P, D’Ambrosio M, Leung P, et al. Angiotensin II-induced dilated cardiomyopathy in Balb/c but not C57BL/6J mice. Exp Physiol. 2011;96:756–764. doi: 10.1113/expphysiol.2011.057612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu Q, Vazquez R, Zabadi S, Watson RR, Larson DF. T-lymphocytes mediate left ventricular fibrillar collagen cross-linking and diastolic dysfunction in mice. Matrix Biol. 2010;29:511–518. doi: 10.1016/j.matbio.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abbas AK, Lichtman AHH, Pillai S. Cellular and molecular immunology. Philadelphia, PA: Elsevier Health Sciences; 1994. [Google Scholar]
- 5.Luther JM, Gainer JV, Murphey LJ, Yu C, Vaughan DE, Morrow JD, et al. Angiotensin II induces interleukin-6 in humans through a mineralo-corticoid receptor-dependent mechanism. Hypertension. 2006;48:1050–1057. doi: 10.1161/01.HYP.0000248135.97380.76. [DOI] [PubMed] [Google Scholar]
- 6.Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension. 2001;38:399–403. doi: 10.1161/01.hyp.38.3.399. [DOI] [PubMed] [Google Scholar]
- 7.Coles B, Fielding CA, Rose-John S, Scheller J, Jones SA, O’Donnell VB. Classic interleukin-6 receptor signaling and interleukin-6 trans-signaling differentially control angiotensin II-dependent hypertension, cardiac signal transducer and activator of transcription-3 activation, and vascular hypertrophy in vivo. Am J Pathol. 2007;171:315–325. doi: 10.2353/ajpath.2007.061078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee DL, Sturgis LC, Labazi H, Osborne JB, Jr, Fleming C, Pollock JS, et al. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol. 2006;290:H935–H940. doi: 10.1152/ajpheart.00708.2005. [DOI] [PubMed] [Google Scholar]
- 9.Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C, et al. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension. 2012;59:136–144. doi: 10.1161/HYPERTENSIONAHA.111.173328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56:225–231. doi: 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boesen EI, Pollock DM. Effect of chronic IL-6 infusion on acute pressor responses to vasoconstrictors in mice. Am J Physiol Heart Circ Physiol. 2007;293:H1745–H1749. doi: 10.1152/ajpheart.00329.2007. [DOI] [PubMed] [Google Scholar]
- 12.Banerjee I, Fuseler JW, Intwala AR, Baudino TA. IL-6 loss causes ventricular dysfunction, fibrosis, reduced capillary density, and dramatically alters the cell populations of the developing and adult heart. Am J Physiol Heart Circ Physiol. 2009;296:H1694–H1704. doi: 10.1152/ajpheart.00908.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhaleb N-E, Yang XP, Nanba M, Shesely EG, Carretero OA. Effect of chronic blockade of the Kallikrein-Kinin system on the development of hypertension in rats. Hypertension. 2001;37:121–128. doi: 10.1161/01.hyp.37.1.121. [DOI] [PubMed] [Google Scholar]
- 14.Yang XP, Liu YH, Rhaleb N-E, Kurihara N, Kim HE, Carretero OA. Echocardiographic assessment of cardiac function in conscious and anesthetized mice. Am J Physiol Heart Circ Physiol. 1999;277 (5 Pt 2):H1967–H1974. doi: 10.1152/ajpheart.1999.277.5.H1967. [DOI] [PubMed] [Google Scholar]
- 15.Cavallero S, Gonzalez GE, Seropian IM, Cerrudo CS, Matorra F, Morales C, et al. Ventricular function and natriuretic peptides in sequentially combined models of hypertension. Am J Physiol Heart Circ Physiol. 2010;298:H1290–H1299. doi: 10.1152/ajpheart.00911.2009. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez GE, Seropian IM, Krieger ML, Palleiro J, Lopez Verrilli MA, Gironacci MM, et al. Effect of early versus late AT(1) receptor blockade with losartan on postmyocardial infarction ventricular remodeling in rabbits. Am J Physiol Heart Circ Physiol. 2009;297:H375–H386. doi: 10.1152/ajpheart.00498.2007. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y-H, Xu J, Yang X-P, Yang F, Shesely E, Carretero OA. Effect of ACE inhibitors and angiotensin II type 1 receptor antagonists on endothelial NO synthase knockout mice with heart failure. Hypertension. 2002;39 (2 Pt 2):375–381. doi: 10.1161/hy02t2.102796. [DOI] [PubMed] [Google Scholar]
- 18.Rhaleb N-E, Pokharel S, Sharma U, Carretero OA. Renal protective effects of N-acetyl-Ser-Asp-Lys-Pro in deoxycorticosterone acetate-salt hypertensive mice. J Hypertens. 2011;29:330–338. doi: 10.1097/HJH.0b013e32834103ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monassier L, Combe R, Fertak LE. Mouse models of hypertension. Drug Discov Today: Dis Models. 2006;3:273–281. [Google Scholar]
- 20.Breckenridge R. Heart failure and mouse models. Dis Model Mech. 2010;3:138–143. doi: 10.1242/dmm.005017. [DOI] [PubMed] [Google Scholar]
- 21.Patten RD, Hall-Porter MR. Small animal models of heart failure: development of novel therapies, past and present. Circ Heart Fail. 2009;2:138–144. doi: 10.1161/CIRCHEARTFAILURE.108.839761. [DOI] [PubMed] [Google Scholar]
- 22.Muirhead EE, Leach BE, Armstrong FB. Angiotensin-salt hypertension. Clin Sci Mol Med Suppl. 1973;45 (Suppl 1):257s–261s. doi: 10.1042/cs045257s. [DOI] [PubMed] [Google Scholar]
- 23.Enzler T, Bonizzi G, Silverman GJ, Otero DC, Widhopf GF, nzelon-Mills A, et al. Alternative and classical NF-kappa B signaling retain autoreactive B cells in the splenic marginal zone and result in lupus-like disease. Immunity. 2006;25:403–415. doi: 10.1016/j.immuni.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 24.Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension. 2010;56:879–884. doi: 10.1161/HYPERTENSIONAHA.110.158071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 deficiency protects against angiotensin II–induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol. 2007;27:2576–2581. doi: 10.1161/ATVBAHA.107.153080. [DOI] [PubMed] [Google Scholar]
- 26.Fisman EZ, Benderly M, Esper RJ, Behar S, Boyko V, Adler Y, et al. Interleukin-6 and the risk of future cardiovascular events in patients with angina pectoris and/or healed myocardial infarction. Am J Cardiol. 2006;98:14–18. doi: 10.1016/j.amjcard.2006.01.045. [DOI] [PubMed] [Google Scholar]
- 27.Hirota H, Izumi M, Hamaguchi T, Sugiyama S, Murakami E, Kunisada K, et al. Circulating interleukin-6 family cytokines and their receptors in patients with congestive heart failure. Heart Vessels. 2004;19:237–241. doi: 10.1007/s00380-004-0770-z. [DOI] [PubMed] [Google Scholar]
- 28.Ohtsuka T, Hamada M, Inoue K, Ohshima K, Sujzuki J, Matsunaka T, et al. Relation of circulating interleukin-6 to left ventricular remodeling in patients with reperfused anterior myocardial infarction. Clin Cardiol. 2004;27:417–420. doi: 10.1002/clc.4960270712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan AT, Yan RT, Cushman M, Redheuil A, Tracy RP, Arnett DK, et al. Relationship of interleukin-6 with regional and global left-ventricular function in asymptomatic individuals without clinical cardiovascular disease: insights from the Multi-Ethnic Study of Atherosclerosis. Eur Heart J. 2010;31:875–882. doi: 10.1093/eurheartj/ehp454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai NC, Gao MH, Tang E, Tang R, Guo T, Dalton ND, et al. Pressure overload-induced cardiac remodeling and dysfunction in the absence of interleukin 6 in mice. Lab Invest. 2012;92:1518–1526. doi: 10.1038/labinvest.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levick SP, Murray DB, Janicki JS, Brower GL. Sympathetic nervous system modulation of inflammation and remodeling in the hypertensive heart. Hypertension. 2010;55:270–276. doi: 10.1161/HYPERTENSIONAHA.109.142042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao TD, Yang XP, D’Ambrosio M, Zhang Y, Rhaleb N-E, Carretero OA. N-acetyl-seryl-aspartyl-lysyl-proline attenuates renal injury and dysfunction in hypertensive rats with reduced renal mass: Council for High Blood Pressure Research. Hypertension. 2010;55:459–467. doi: 10.1161/HYPERTENSIONAHA.109.144568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma F, Li Y, Jia L, Han Y, Cheng J, Li H, et al. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS One. 2012;7:e35144. doi: 10.1371/journal.pone.0035144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu J, Carretero OA, Liao TD, Peng H, Shesely EG, Xu J, et al. Local angiotensin II aggravates cardiac remodeling in hypertension. Am J Physiol Heart Circ Physiol. 2010;299:H1328–H1338. doi: 10.1152/ajpheart.00538.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobara M, Noda K, Kitamura M, Okamoto A, Shiraishi T, Toba H, et al. Antibody against interleukin-6 receptor attenuates left ventricular remodelling after myocardial infarction in mice. Cardiovasc Res. 2010;87:424–430. doi: 10.1093/cvr/cvq078. [DOI] [PubMed] [Google Scholar]
- 36.Janssen SP, Gayan-Ramirez G, Van den BA, Herijgers P, Maes K, Verbeken E, et al. Interleukin-6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation. 2005;111:996–1005. doi: 10.1161/01.CIR.0000156469.96135.0D. [DOI] [PubMed] [Google Scholar]
- 37.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 38.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, et al. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation. 2009;119:2904–2912. doi: 10.1161/CIRCULATIONAHA.108.832782. [DOI] [PubMed] [Google Scholar]
- 40.Seropian IM, Toldo S, Van Tassell BW, Abbate A. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. J Am Coll Cardiol. 2014;63:1593–1603. doi: 10.1016/j.jacc.2014.01.014. [DOI] [PubMed] [Google Scholar]