

Abstract
The goal of this study was to determine the impact of silk biomaterial structure (e.g., solution, hydrogel, film) on proteolytic susceptibility. In vitro enzymatic degradation of silk fibroin hydrogels and films was studied using a variety of proteases, including proteinase K, protease XIV, α-chymotrypsin, collagenase, matrix metalloproteinase-1 (MMP-1) and MMP-2. Hydrogels were used to assess bulk degradation while films were used to assess surface degradation. Weight loss, secondary structure determined by Fourier Transform Infrared (FT-IR) spectroscopy and degradation products analyzed via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) were used to evaluate degradation through five days. Silk films were significantly degraded by proteinase K, while silk hydrogels were degraded more extensively by protease XIV and proteinase K. Collagenase preferentially degraded the β-sheet content in hydrogels while protease XIV and α-chymotrypsin degraded the amorphous structures. MMP-1 and MMP-2 degraded silk fibroin in solution resulting in a decrease in peptide fragment sizes over time. The link between primary sequence mapping with protease susceptibility provides insight into the role of secondary structure in impacting proteolytic access by comparing solution vs. solid state proteolytic susceptibility.
Keywords: silk, protease, digestion, hydrogels, films
1. Introduction
Silk secreted from Bombyx mori silkworms has emerged as a useful protein polymer due to its biodegradability and utility in biomaterials for regenerative medicine and drug delivery [1–4]. B. mori silk is comprised of a number of proteins: fibroin (heavy chain, light chain and P25), the key structural components, and sericin, the glue-like outer layer that coats fibroin during fiber spinning and formation of cocoons [5]. The fibroin heavy chain can be divided into four different regions based on amino acid chemistry and sequence: the N-terminus, C-terminus, 11 spacer regions, and 12 large repeat bulk domains. The N-terminus, C-terminus and 11 spacer regions are hydrophilic and form the non-repetitive, amorphous regions of the assembled proteins. The 12 large bulk domains are hydrophobic and predominantly consist of the repeating hexapeptides GAGAGS and GAGAGY [6], which form the dominating crystalline β-sheet regions responsible for the strength and stability of silk biomaterials [7].
Silk fibroin can be formed into a variety of different biomaterials, such as hydrogels, sponges or films [8]. Hydrogels are water insoluble networks of polymer chains, and can be comprised of a number of synthetic (e.g. polyethylene glycol or polyvinyl alcohol) or naturally derived (e.g. collagen or hyaluronic acid) polymers [9]. To fully utilize a hydrogel in a biomedical context, an understanding of the degradation process is essential to determine the utility of the material for specific medical needs. For example, natural polymers, such as collagen and hyaluronic acid, have been used as filler materials for soft tissue augmentation. These materials work well as short term cosmetic fillers, but exhibit low volume persistence due to the presence of endogenous enzymes in the body [10]. An understanding of the enzyme kinetics associated with these materials is important in order to design crosslinking agents that inhibit their degradation and prolong their volume retention in the body [11,12].
Although silk fibers are defined by the US Pharmacopeia (USP) as non-degradable materials because they retain tensile integrity (>50%) after 60 days in vivo [13], recent studies have demonstrated biodegradation of silk fibroin when prepared without waxes and coatings, and with tunability of β-sheet (crystalline) content via augmentation of processing conditions [8,14–19]. Several silk fibroin degradation studies investigating porous sheets, yarns, powders and films have demonstrated a role for proteases (including protease XIV, α-chymotrypsin and collagenase) in the degradation process [20,21]. In addition, we have recently reported studies of silk fibroin degradation [16,22] which provide the foundation for the additional study of the process with other proteases, including matrix metalloproteinases. The results of these studies on fibers and films suggest that silk degrades via surface erosion, with little bulk degradation observed.
Matrix metalloproteinases (MMPs) are a family of naturally occurring enzymes that degrade extracellular matrix (ECM) proteins [23]. In their natural environment, they contribute to a range of physiological mechanisms useful in cell and blood vessel growth, cell death, reproduction and embryonic development, as well as tissue remodeling and wound healing [24]. MMPs are classified into different classes (i.e. collagenases, gelatinases, stromelysins) based on their in vitro substrate specificity [23]. Within each class, MMPs recognize specific peptide sequences common to that substrate. Some of these known recognition sequences are found within the amino acid profile of the silk fibroin heavy chain. Therefore, we would predict that MMPs would degrade silk fibroin materials. Understanding how MMPs interact with silk fibroin hydrogels and films will provide insight into silk biomaterials related to degradation in vitro and in vivo [25]. In the current work, the degradation of silk fibroin using both proteases and MMPs was studied, with a focus on how material format impacts access to proteolysis. The proteolytic degradation of silk fibroin materials was characterized using two types of enzymes: serine proteases (proteinase K, protease XIV, α-chymotrypsin and collagenase) and MMP’s (MMP-1, interstitial collagenase, and MMP-2, gelatinase A). Additionally, we sought to assess bulk degradation of silk through the use of hydrogels, due to the open porous structure which promotes transport and access of the enzymes throughout the material, in comparison to films where such access is limited. It was hypothesized that hydrogels would provide a useful biomaterial format to define the differences in surface versus bulk enzymatic degradation processes, while films would emphasize differences in silk proteolytic susceptibility with respect to surface erosion. Together, the findings provide a foundation towards predictive outcomes when these biomaterials are used in vivo.
2. Experimental Section
2.1 Silk Fibroin Solution
Silk fibroin solution was prepared as we have previously described [26]. Briefly, 30 grams of cocoons from B. mori silkworms were cut into fragments and boiled for 20 minutes in an aqueous solution of 0.04M Na2CO3 to remove the sericin coating from the fibroin fibers. Cocoons were then washed in deionized water to further remove sericin and Na2CO3. Remaining silk fibroin was left to dry overnight in a flow-hood. Next, the extracted silk fibroin was dissolved in a 9.3 M LiBr solution for 4 hours in a 60°C oven in order to breakdown and solubilize the protein in water, yielding a 20% (w/v) solution. This solution was dialyzed in 3,500 Da cut-off dialysis tubing for three days to remove LiBr. The final concentration of the silk fibroin solution generated was diluted to approximately 3% (w/v) and contained silk fibroin heavy chain, light chain and P25 proteins. The silk solution was not tested for active endogenous proteases or protease inhibitors remaining from the native silk cocoon. However, if present they would be in all the materials and accounted for in the control samples.
2.2 Preparation of Silk Hydrogels
Silk fibroin solution was sonicated with a Branson 450 Sonifier (Branson Ultrasonics, Danbury, CT, USA), consisting of the Model 450 power supply, converter, externally threaded disruptor horn, and 1/8″ (3.175 mm) diameter-tapered microtip, at 20% of the maximum amplitude for 5 seconds. Sonication physically induces β-sheet crosslinks via alteration in hydrophobic hydration of the protein chains and accelerated sol-gel transitions, as we have previously reported [15]. After sonication, 75 μL aliquots of solution were cast onto 4 mm diameter PDMS discs and allowed to sit on the bench-top overnight.
2.3 Preparation of Silk Films
A 75 μL aliquot of silk fibroin solution was cast onto a 4 mm diameter PDMS disc and allowed to sit at room temperature overnight. These films were water annealed by placing them inside a vacuum chamber containing a small water reservoir and pulling an 80 kPa vacuum for 1 hour. We have previously shown that water annealing increases the β-sheet content of the silk films and therefore makes them water insoluble [27]. Films were removed from the vacuum chamber and allowed to air dry for 12 hours on the bench-top.
2.4 Preparation of Enzymes
Proteinase K from Engyodontium album (specific activity: ≥30 units/mg protein), protease XIV from Streptomyces griseus (specific activity: ≥3.5 units/mg protein), α-chymotrypsin from bovine pancreas (specific activity: ≥40 units/mg protein) and collagenase from Clostridium histolyticum (specific activity: >125 collagen digestion units/mg solid) were all purchased from Sigma-Aldrich (St. Louis, MO, USA). Solutions of proteinase K, protease XIV, α-chymotrypsin and collagenase were freshly prepared by dissolving the enzyme powder in deionized water. An Amplite™ Universal Fluorimetric Protease Activity Assay kit (AAT Bioquest®, Inc., Sunnyvale, CA) was used to normalize digestion rate. From the derived data, enzyme concentration was determined based on equal enzymatic activity (160 RFU/min), proteinase K: 1.8 units/mL; protease XIV: 0.1 units/mL; α-chymotrypsin: 12 units/mL; collagenase: 75 units/mL.
MMP-1 (interstitial collagenase, E. coli expressed, ≥700 units/μg protein) and MMP-2 (gelatinase A, yeast expressed, ≥25 units/μg protein) were purchased from Enzo Life Sciences (Farmingdale, NY). MMP-1 and MMP-2 were reconstituted in assay buffer (50 mM HEPES, 10 mM CaCl2, 0.05% Brij-35, pH 7.5) and preliminary studies determined that concentrations of 8,750 units MMP-1/mL and 313 units MMP-2/mL were necessary to detect degradation of silk materials.
2.5 Degradation of Silk Materials by Serine Proteases and MMP’s
Silk fibroin hydrogels and films were removed from the PDMS discs and incubated in deionized water for 24 hours to leach out soluble peptides. Materials were then immersed in 40 μL of enzyme solution (or deionized water for control) and incubated at 37°C. Enzyme solution was removed and replaced with fresh enzyme once every 24 hours for up to 5 days. After removal, enzyme solution was heated to 100°C for 15 minutes to denature the enzymes per manufacturer’s instructions and stored at −80°C until analysis. At each time point, one set of silk materials was removed from incubation and washed with deionized water for 48 hours, with water changes every 12 hours, to remove adsorbed protein. Materials were then dried in an airflow hood for 3 days and stored dry at room temperature until analysis.
Silk films were also treated with EDTA inhibited MMP for 48 hours. For inhibition of MMP, EDTA at 10 mM was added to a 40 μL solution of MMP in assay buffer and incubated at 37°C for one hour. Films were placed in solutions of inhibited MMP enzyme and incubated at 37°C for 48 hours with refreshment at 24 hours. Films and supernatant were removed at the end of the study and analyzed for degradation.
To ascertain the effects of MMP degradation on aqueous silk fibroin proteins, a silk fibroin solution was diluted to 3% (w/v) and split into 50 μL aliquots. 350 units of MMP-1 or 12.5 units of MMP-2 were added to its appropriate treatment group and allowed to react for 6, 24 and 72 hours at 37°C. Enzyme was refreshed only once at 36 hours for appropriate groups. After treatment, samples were incubated at 100°C for 15 minutes to denature the enzymes and then stored at −80°C until analysis.
2.6 Analytical Techniques
After treatment of silk fibroin materials with enzyme, films, hydrogels and their supernatants were collected and analyzed for degradation. Weight loss was used for detecting degradation products and determining degradation rate. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate the degradation products and estimate molar mass of the degradation products. Secondary structure of the degraded silk materials was studied via Fourier Transform Infrared (FT-IR) spectroscopy. Light microscopy and scanning electron microscopy (SEM) were used to observe physical changes in film topography. Based on initial studies, measurements were taken at time points 1, 3 and 5 days for FT-IR as they were sufficient to determine trends in the data. Time points were measured daily for weight loss and SDS-PAGE because the additional data was needed to determine when the degradation of materials by different enzymes became significantly different.
2.6.1 Weight Loss
After drying, samples were weighted and percent weight loss was calculated by comparing each group to the control. Treatment of silk films with MMP’s resulted in aggregates forming on the surface of the films. To analyze these aggregates and to study the underlying topography of the films, a probe sonicator (Branson Ultrasonics, Danbury, CT, USA) was used to physically desorb the aggregates from the surface of the films after 48 and 120 hours. Treated films were placed in Eppendorf tubes containing 500 μL of deionized water, and sonication was performed at 20% of the max amplitude for 10 seconds. Films were then removed, and the supernatant containing deionized water and aggregate proteins was lyophilized, centrifuged at 4000 g for 5 minutes, and reconstituted in 100 μL deionized water before being analyzed.
2.6.2 Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE)
A bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Rockford, IL, USA) was used per manufacturer’s protocol to approximate equal protein loading between samples within a particular group. Samples were reduced with NuPAGE® lithium dodecyl sulfate (LDS) 4X sample buffer and NuPAGE ® Sample Reducing Agent (Invitrogen, Carlsbad, CA) at 80°C for 15 minutes, then loaded onto NuPAGE® 4–12% Bis-Tris gels in 1X MES running buffer (Invitrogen, Carlsbad, CA). SeeBlue® Pre-stained standards (Invitrogen, Carlsbad, CA) were run as the molecular weight markers from 3 to 188 kDa. An Invitrogen SilverXpress Silver Staining Kit (Invitrogen, Carlsbad, CA) was used to stain gels, and images were captured with a digital camera. Peptide molecular weight distribution was quantified using densitometric analysis along the length of the gel (ImageJ, NIH, Bethesda, MD).
2.6.3 Fourier Transform Infrared Spectroscopy (FT-IR)
Materials were extensively dried for 3 days in an air-flow hood to minimize the contribution of a water peak during FT-IR analysis [28,29]. For analysis of liberated surface aggregates, particles were resuspended in deuterium oxide and allowed to equilibrate for 24 hours before analysis [29]. Silk materials were analyzed via a JASCO FT-IR 6200 spectrometer (JASCO, Tokyo, Japan) combined with a MIRacle ™ attenuated total reflection (ATR) germanium crystal. Background measurements were recorded before sample reading and subtracted from the sample spectrum. For each measurement, spectral scans were run from 1550 to 1750 cm−1 at a resolution of 2 cm−1 for 32 scans per sample. Five samples were measured for each group, and each film or hydrogel was measured only once because samples were not recoverable after a single measurement.
Secondary structure was evaluated as in Hu et al. [30] Briefly, Fourier self-deconvolution (FSD) of the FT-IR spectra covering the amide I region (1590 –1710 cm−1) was performed using Opus 5.0 software. Spectra were deconvoluted using a Lorentzian line shape, a half-bandwidth between 25 and 26 cm−1, a noise reduction factor of 0.3, and apodization with a Blackman-Harris function. A baseline correction for the deconvoluted amide I region was subtracted before curve fitting. For curve fitting, Gaussian line shape profiles were input at positions (in cm−1) that correlated to the second derivative of the original spectra, and the input for peak width was kept constant at the software’s default value of 6 cm−1 for all peaks. Eleven fitted bands were chosen to define the secondary structure of the amide I. Assignments for each band were based off those made in Hu et al. (SUPPLEMENTAL FIGURE S1). Relative areas of the single bands were used to determine the fraction of the structural element. We assumed that the extinction coefficient was the same for each secondary structural component [30], and that any changes in the amide I absorptivity due to hydration of water (either within the materials or from the atmosphere) affected all secondary structure elements equally [31]. Reproducibility of the measurements was compared to that discussed in Hu et al. [30]
2.7 Statistics
Data are expressed as mean ± standard deviation with N = 5. One-way analysis of variance (ANOVA) and Tukey post-hoc analysis were used to determine statistical significance. Significance was accepted at the p < 0.05 level, and indicated in figures as * = p < 0.05, ┼ = p < 0.01, ╪ = p < 0.001.
3. Results
3.1. Quantitative Analysis
For silk fibroin films, proteinase K exhibited the most extensive degradation after 120 hours (FIGURE 1A, SUPPLEMENTAL TABLE S1). Silk fibroin hydrogels were degraded most by both protease XIV and proteinase K, but mass loss by protease XIV was significantly greater than proteinase K up to 72 hours (FIGURE 1B). From 72 to 120 hours, there was no significant difference between silk film mass losses due to protease XIV, α-chymotrypsin and collagenase, however, mass loss by protease XIV was significantly less than α-chymotrypsin and collagenase before 48 hours.
Figure 1.
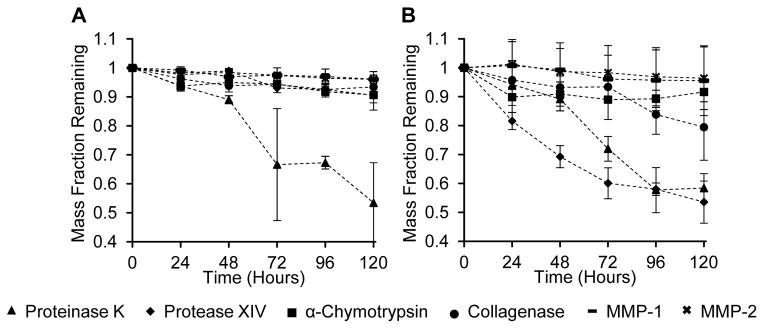
Weight loss from enzymatic degradation of silk fibroin materials. Films (Figure 1A) and hydrogels (Figure 1B) were prepared from a 3% (w/v) silk fibroin solution and immersed in proteinase K, protease XIV, α-chymotrypsin, collagenase, MMP-1, MMP-2, or deionized water (control). Remaining mass of degraded silk material was measured after each time point and compared against the control group. Values are the average± standard deviation of N=5.
3.2. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE)
Background controls for each group were enzyme solutions without silk materials incubated at 37°C for 24 hours, and controls for silk materials were incubated in deionized water (FIGURE 2A). The supernatant of silk films contained soluble protein fragments with a molecular weight of approximately 14 kDa on day 1. These fragments were detectable up to day 3, after which there were no detectable fragments. No light chain or P25 (both ~25 kDa) bands were detected at any time point. Silk hydrogels also showed a strong band at 14 kDa on day 1, along with a smear of proteins ranging from 20 to 38 kDa. The intensity of the smear and 14 kDa band diminished over 3 days and were no longer detectable at days 4 and 5.
Figure 2.

SDS-PAGE of degradation products released from silk fibroin materials. (A) deionized water only, (B) proteinase K, (C) protease XIV, (D) α-chymotrypsin, and (E) collagenase. Days are listed as 1–5; control lanes are enzyme solution incubated for 24 hours at 37°C without silk.
Degradation products from proteinase K treated films appeared with defined bands between 3–14 kDa on day 1, with a distinct band at 14 kDa. From days 2–5, these defined bands gradually become a smear of proteins between 3–14 kDa, and the band at 14 kDa was no longer detectable after day 3. Hydrogels treated with proteinase K showed a smear between 3–28 kDa which increased in intensity after each day of incubation (FIGURE 2B). Supernatant from silk films incubated with protease XIV did not contain detectable silk bands at day 1 and 2. A strong band at 45 kDa and several bands between 55–66 kDa were detectable between days 3–5. Supernatant from hydrogels had bands between 3–14 kDa that steadily grew in intensity from days 1 to 4. On day 5, the smear of peptides extended from 3–31 kDa. (FIGURE 2C).
For α-chymotrypsin, supernatant from silk films had no detectable bands at any time point (FIGURE 2D), while those degraded with collagenase had bands between 62–98 kDa present on day 1, a 49 kDa band present through day 2, and a band at 98 kDa, a smear of peptides between 38 and 62 kDa, and lower molecular weight bands at 10 kDa on days 3–5 (FIGURE 2E). Hydrogels degraded by α-chymotrypsin showed peptide bands from 36–70 kDa forming between days 2–4, which turned into a smear of peptides on day 5. Hydrogels degraded by collagenase show high molecular weight bands between 49 to 98 kDa, as well as a smear of mid and low molecular weight peptides ranging from 3 to 38 kDa appearing after day 2.
Silk films treated with MMP-1 and MMP-2 exhibited the formation of surface aggregates. The occurrence of these aggregates increased as a function of time (SUPPLEMENTAL FIGURE S2). To analyze the underlying topography and degradation, a probe sonicator was used to remove particles after days 2 and 5. Supernatant from untreated films after sonication had 14–28 kDa silk peptides which were similar in the EDTA-inhibited MMP groups. Post-sonication supernatant from 48 hours, MMP-1 exposed silk films had additional peptides between 28–38 kDa and 42–62 kDa. After sonication, the supernatant from MMP-2 treated films exhibited lower intensity peptide bands between 17–28 kDa compared to control and EDTA-inhibited MMP-2 lanes. The supernatant also exhibited lower molecular weight bands at ~6 kDa, which were not seen in other groups. Active MMP-2 is known to undergo enzyme autocatalysis [32,33] as was seen with bands at 28, 38 and 49 kDa, whereas EDTA inhibited MMP-2 did not show these bands.
Silk hydrogels treated with MMP-1 showed a band at 17 kDa, a smear from 20–36 kDa and a smear of peptides below 17 kDa on day 1. The intensities of all these peptides diminished each subsequent day. (FIGURE 3A). MMP-2 treated hydrogels also showed a band at 17 kDa on day 1 which persisted until day 5, as well as distinct bands between 4–14 kDa (FIGURE 3B).
Figure 3.
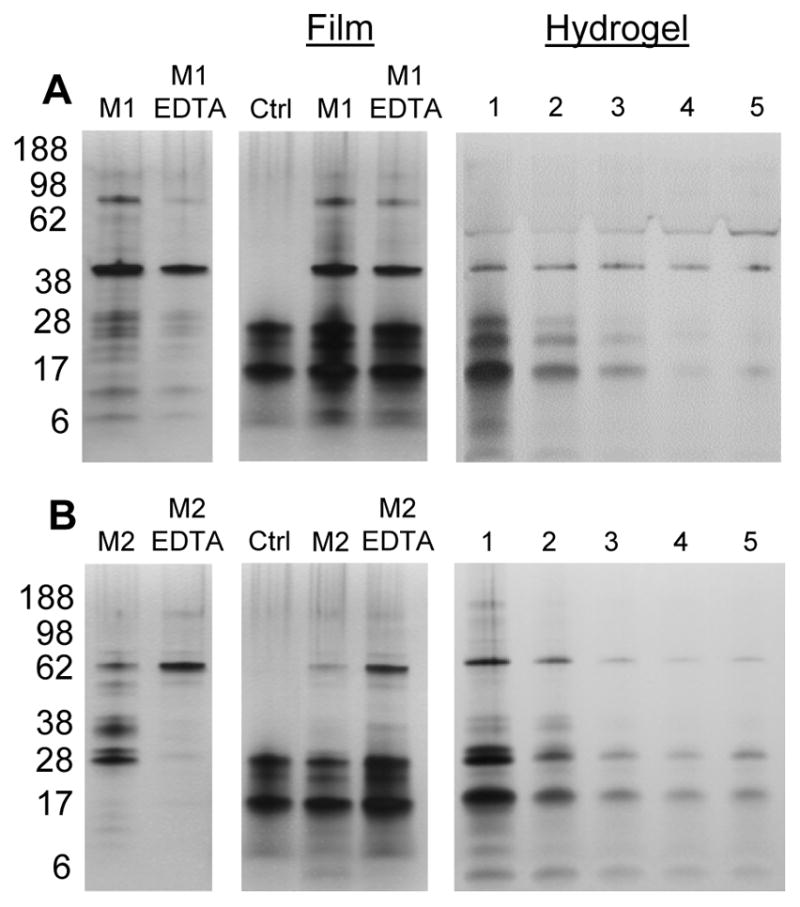
SDS-PAGE of degradation products after MMP degradation. Silk fibroin materials were incubated in MMP-1 (A) and MMP-2 (B) for 48 hours in 37°C incubation. Left-most lanes represent enzyme only without any silk material. “Film” lanes display the supernatant of an untreated film after sonication (“Ctrl”), a film treated with active MMP, and a film treated with EDTA-inhibited MMP. Film samples were sonicated after 48 hours of incubation in MMP solution. Days are listed for hydrogels as 1–5.
To determine if the aqueous soluble silk was susceptible to MMP cleavage, solutions were exposed to MMP1 and MMP2 separately (FIGURE 4). This experiment was not performed with serine proteases as this had been published previously [20–22]. In the non-degraded control sample, a smear of high molecular weight proteins between 100–200 kDa was seen. Densitometric analysis (SUPPLEMENTAL FIGURE S3) showed that the peak molecular weight of non-degraded aqueous silk was approximately 200 kDa. After degradation with MMP-1 and MMP-2, there was a decrease in smear intensity and peak molecular weight shifted to between 98–188 kDa. Peak molecular weight did not substantially change from 6 hours to 3 days of incubation in MMP, but a decline in the high molecular weight peptides (188+ kDa) was observed over time.
Figure 4.
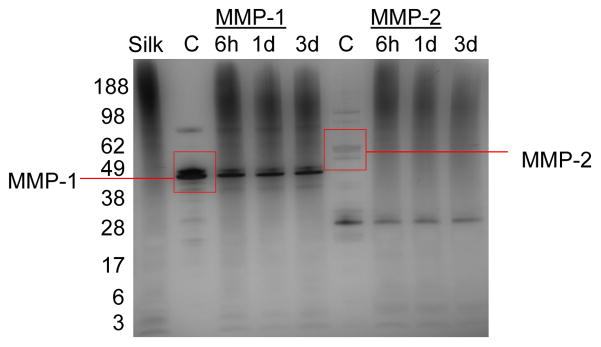
SDS-PAGE of aqueous silk fibroin after degradation by MMP-1 and MMP-2 for 3 days. Enzymes were refreshed only once at 36 hours, and all samples were incubated at 37°C. Silk lane is of aqueous silk fibroin in MMP buffer without enzyme. Control lanes are MMP enzyme incubated without silk.
3.3. Secondary Structure
Silk protein secondary structure was assessed by FT-IR (FIGURE 5, SUPPLEMENTAL TABLE S2 and FIGURE S4). For each of the four structures, statistical significance was analyzed by comparing the 72 and 120 hour time points to the 24 hour time point within each enzyme group. Only trends that are statistically significant are discussed. Control films incubated in water had a significant increase in α-helix content over time, but no change in other structures. Control hydrogels showed an increase in β-sheet structure at 120 hours. Proteinase K degradation of films reduced β-sheet content over time, while α-helix and random coil content increased after 120 hours. Hydrogels showed an initial increase in both α-helix content and turn structures after 72 hours, but α-helix decreased while turns remain unchanged after 120 hours. Random coil decreased across all time points and the β-sheet content slightly increased at 120 hours.
Figure 5.

Quantification of secondary structure in films and hydrogels after analysis by FT-IR. (A) β-sheet, (B) α-helix (C) random coil and (D) turns content after treatment with proteinase K, protease XIV, α-chymotrypsin, collagenase, MMP-1 and MMP-2 for 24, 72, and 120 hours. For controls, hydrogels and films were treated in de-ionized water only. Black bars represent secondary structure of materials before enzyme treatment. Number of samples per group = 5. Statistical significance is shown in Table S2.
Incubating silk films with protease XIV resulted in decreased turn content and increased β-sheet content over time, while α-helix structure increased after 72 hours. In hydrogels, both turns and β-sheet structures increased by 72 hours, and random coil decreased at 72 hours before showing a small increase at 120 hours. The α-helix content initially increased at 72 hours, but decreased after 120 hours.
Films treated with α-chymotrypsin showed an increase in β-sheet content only after 120 hours, an increase in random structure after 72 hours, and a large decrease in α-helix structure over time. Hydrogels showed a similar increase in β-sheet content after 120 hours and decreased α-helix content over time. Furthermore, the hydrogels had decreased random structure after 120 hours and increased turn structure over time. Collagenase degradation of films produced a decrease in random structure and an increase in β-sheet content after 120 hours. Hydrogels had a decrease in β-sheet and α-helix structure over time, and an increase in random coil structure after 120 hours. Finally, MMP-1 treatment of films resulted in decreased random content after 72 hours. Hydrogels showed a decrease in β-sheet structure after 72 hours. MMP-2 degradation produced a decrease in α-helix content after 120 hours in silk films, while hydrogels had decreased α-helix content after 120 hours.
4. Discussion
Cleavage sites within the silk fibroin heavy chain, light chain and P25 were predicted for each enzyme studied TABLE 1. The serine proteases (proteinase K, protease XIV, α-chymotrypsin) cleave amide bonds adjacent to aliphatic, aromatic or hydrophobic residues, while collagenase and the MMP’s cleave specific amino acid sequences found in collagen (MMP-1: Gly-Ile, Gly-Leu; MMP-2: Gly-Ile, Gly-Leu, Gly-Val, Gly-Phe, Gly-Asn, Gly-Ser). From the cocoon, the light chain and P25 glycoprotein combined account for approximately 5% of the amino acids present in silk fibroin [34]. Therefore, our discussion on degradation is focused on the heavy chain, the major constituent in silk fibroin. The recognition sites were used to calculate enzymatic cleavage sites within silk fibroin heavy chain without consideration for hindered accessibility due to secondary structure. The ability to correlate sequence chemistry of the silk fibroin heavy chain protein to degradation profiles found experimentally may help predict degradation kinetics and outcomes when silk is used for various biomaterials studies.
Table 1.
Summary of predicted protease cleavage sites for the silk fibroin proteins.
| Protease | Source | Cleavage Sites | # of Cleavage Sites | References | ||
|---|---|---|---|---|---|---|
| HC | LC | P25 | ||||
| Proteinase K | Engyodontium album | Adjacent to His, Phe, Trp, Tyr, Ala, Ile, Leu, Pro, Val, Met | 2050 | 134 | 114 | [40,41] |
| Protease XIV | Streptomyces griseus | Adjacent to His, Phe, Trp, Tyr, Lys, Arg | 348 | 41 | 56 | [45,46] |
| α-chymotrypsin | Bovine Pancreas | Adjacent to Tyr, Phe, Trp, Val, Ile, Leu | 434 | 81 | 76 | [20,45,47] |
| Collagenase a | Clostridium histolyticum | X-Gly-Pro b | 12 | 2 | 1 | [20,45] |
| MMP-1 a | E. coli expressed | Gly-Ile, Gly-Leu | 4 | 3 | 1 | [48,49] |
| MMP-2 a | Yeast expressed | Gly-Ile, Gly-Leu, Gly-Val, Gly-Phe, Gly-Asn, Gly-Ser | 590 | 10 | 3 | [48,50] |
HC = Heavy chain; LC = Light chain
Cleavage sites have been experimentally determined in collagen
X can represent any amino acid
For the control samples, some soluble peptides leached out and were detected in the supernatant by SDS-PAGE. For films, light chain and P25 proteins were not detected. The contribution from these proteins in the supernatant was likely too small to detect. Furthermore, the film density may have impeded leaching of light chain and P25 peptides from the material. In hydrogels, peptides were present at 25 kDa where the loose crosslink network allowed more soluble fragments to leach out. Again, no distinct bands were seen likely due to the low concentration of light chain and P25 peptides.
We predicted high mass loss by proteinase K because it hydrolyzes peptide bonds adjacent to aliphatic or aromatic amino acid residues, which account for ~39% of the silk fibroin heavy chain peptide bonds. Mass loss by proteinase K on both material formats were comparable, with approximately 10% weight loss after 48 hours. In comparison, protease XIV showed little degradation of films within the first 48 hours, while for hydrogels, approximately 30% of the material mass was lost in the first two days. Arai et al. made a comparison between protease degradation of silk films and fibers, and attributed faster degradation of films to better accessibility of the cleavage sites [20]. The present data agreed and the faster degradation of hydrogels by protease XIV was likely due to increased accessibility to recognition sites. We predict that proteinase K has a lower binding affinity or catalytic efficiency compared to protease XIV, accounting for slower degradation at early time points.
In terms of peptide fragment size, we expected to detect mostly small protein fragments for both films and hydrogels after proteinase K degradation. It was suggested that soluble protein fragments will continue to be enzymatically cleaved in solution [20], which is what was found based on the data. In films, the 14 kDa bands that appeared in days 1 and 2 vanished as they are most likely degraded into smaller peptides. Hydrogels also showed a continual increase in the amount of small molecular weight peptides.
It was unexpected that there would be a significant difference in mass loss between protease XIV and α-chymotrypsin given that they have a similar number of cleavage sites within the silk fibroin heavy chain sequence. One explanation is that protease XIV has an affinity towards β-sheet structures, while α-chymotrypsin preferentially digests the less crystalline regions and does not degrade β-sheets [21,22]. Unlike the loose network of hydrogels, the dense packing of fibroin chains within films may limit the ability of protease XIV to cleave the crystalline regions, while swelling of the amorphous regions in both material formats allowed α-chymotrypsin and collagenase access to their cleavage sites.
In films, the hydrophobic domains may be the only regions accessible to protease XIV. The present results have peptides with molecular weights of approximately 40 kDa releasing after day 3 correlating with an increase in mass loss at day 3, suggesting that protease XIV degrades surface residues allowing access to cleave off larger peptides. Alternatively, protease XIV degradation of hydrogels looked similar to proteinase K, with low molecular weight peptides steadily released over time. The loose network of hydrogels combined with the low specificity of protease XIV allowed for cleavage of peptides into small soluble fragments which were released into solution, accounting for the consistent mass loss over time. A-chymotrypsin, on the other hand, released larger soluble peptide fragments, which may be attributed to the higher specificity for non-crystalline regions in silk heavy chain. Silk peptides released from films degraded by α-chymotrypsin may have been smaller than 2.5 kDa or overlapped with the enzyme’s bands.
It was also unexpected that α-chymotrypsin and collagenase would show similar trends in degradation. Collagenase hydrolyzes the X-Gly bond in the sequence of X-Gly-Pro [20]. Even though glycine accounts for roughly 46% of the amino acid composition of fibroin heavy chain [35], the combination of X-Gly-Pro occurs only 12 times and exclusively within the amorphous regions. Peptides up to 98 kDa were found from both films and hydrogels after 24 hours of degradation, which agrees with prior studies [20,21], suggesting that collagenase degradation of the amorphous region produces higher molecular weight peptides when compared to protease XIV or α-chymotrypsin. Despite the scarcity of cleavage sites, Li et al. showed that collagenase degraded porous silk sheets more than α-chymotrypsin [21]. Based on the mass loss trends through 120 hours, longer enzymatic digestion time may have led to more collagenase-induced degradation of hydrogels and a significant difference compared to α-chymotrypsin. The greater incidence of high molecular weight bands in hydrogels compared to films is likely due to the accessibility within the hydrogel network.
Figures 4 and S3 illustrate that soluble silk fibroin degraded in the presence of MMP. Since the “Silk” lane represented the molecular weights of fibroin peptides present after incubation for 3 days without MMP, we assumed any change in molecular weight of the silk compared to the control was due to MMP degradation. The largest shift in peak molecular weight occurred within 6 hours for MMP-1 and MMP-2. There also appeared to be continuous degradation of high molecular weight proteins through the first day. MMP-1 and MMP-2 have a finite number of specific recognition sites in the silk heavy chain, which may explain why there is less change in molecular weight distribution between later time points.
Surface aggregates were observed on the films incubated with MMP-1 and MMP-2. These aggregates, which could not be desorbed by washing with water, made it difficult to observe surface topography and measure mass loss. Therefore, a probe sonicator was used to remove aggregates. After sonication, SEM revealed signs of erosion on the surface of MMP-1 treated films that were not apparent on control films (FIGURE 6). Hydrogels, which could not be sonicated showed no significant difference in mass loss compared to controls. Given the predicted number of cleavage sites within the silk fibroin heavy chain, we expected to see more degradation by MMP-2 than MMP-1, but again, degradation may have been limited by accessibility. The formation of surface aggregates on films could have blocked degradation at the surface. Frequent sonication was not feasible due to damaging the substrate.
Figure 6.
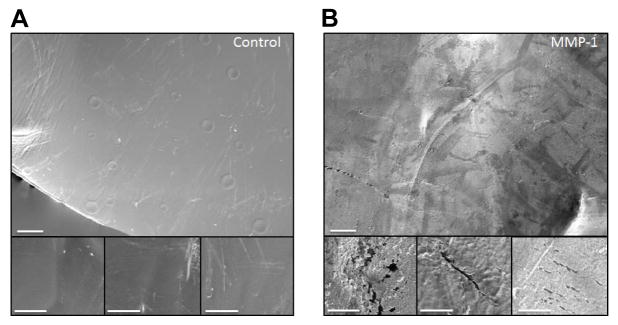
SEM images of film topography. Control (buffer only) and MMP-1 treated films after 48 hours of treatment. After sonication, control films (Figure A) show a mostly smooth topography with some marring due to contact with the sonicator probe. MMP-1 treated films (Figure B) show signs of crevices, pitting and degradation uniformly across the surface of the film. These topographical features could not be seen before sonication when aggregates were present and obscuring the surface details. Scale bar is 40 μm; inset is 10 μm.
To test whether these aggregates were adsorbed MMP enzyme or a byproduct of enzymatic degradation, we treated films with EDTA inhibited MMP enzyme. Films treated in inhibited MMP showed fewer surface aggregates (SUPPLEMENTAL FIGURE S5). After sonication, several peptide smears were observed which were not present in either assay buffer only or inhibited MMP groups. In the presence of MMP-2, the peptides between 17–28 kDa may be degraded to lower molecular weight – explaining the presence of peptides around ~6 kDa. The peptides between 17–28 kDa were soluble, since they appear in the film control lane, making them more accessible to enzymatic cleavage. Therefore, if MMP-2 has recognition sites within these soluble peptides, they will be susceptible to degradation. When MMP-2 was inhibited by EDTA, we again saw higher intensity peptide smears between 17–28 kDa and minimal peptides at ~6 kDa.
It is hypothesized that aggregates may have been silk-MMP complexes that formed on the surface as a result of degradation, but were not released into solution until sonication. These complexes may have stabilized MMP activity, which is why the hydrogels, which could not be sonicated, did not present any distinguishable bands for degraded peptides.
In terms of secondary structure, silk fibroin hydrogels are formed through physical crosslinking between protein chains forming into β-sheets, surrounded by the amorphous regions containing α-helices and random coil structures [15]. FT-IR results suggested that enzymes with low specificity (proteinase K and protease XIV) will primarily degrade the exposed amorphous regions (α-helix and random coil). These regions are more accessible in hydrogels compared to films, perhaps explaining why random coil content was reduced in hydrogels after degradation by these enzymes. Alternatively in films, amorphous regions are not readily accessible, therefore proteinase K degraded the β-sheet structures while protease XIV degraded turns. With more cleavage sites within the hydrophilic regions, proteinase K could degrade β-sheet structures, thus the large increase in mass loss.
Based on previous studies [21,22], we did not expect α-chymotrypsin to degrade β-sheet structure in either material format, which is reflected in the data. Instead, α-chymotrypsin degraded α-helix structures in both films and hydrogels, and degraded the random coil structures in hydrogels. These findings suggested that the specificity for non-crystalline regions made it difficult for α-chymotrypsin to access cleavage sites within the film bulk, whereas the open network of the hydrogels allowed for access and degradation of the cleavage sites.
Since collagenase cleavage sites are located strictly in the hydrophilic regions of the silk sequence, we expected a reduction in random coil, turn or α-helix structure with time, but instead saw degradation of the β-sheet structure. Cleavage at the hydrophilic regions by collagenase may lead to destabilization of the β-sheet, thus accounting for the decrease in β-sheet and increase in random coil structures. This destabilization may also have led to easier accessibility of more cleavage sites, thus allowing release of larger peptide fragments seen in the SDS-PAGE data.
Observable secondary structure changes by MMP degradation may have been obscured by the formation of aggregates on the film surface. Noting the difference in β-sheet content between control films (51.2%) and MMP-1 and MMP-2 treated films (40.2% and 42.5%, respectively), the ATR probe may have detected the adsorbed particles as well as the underlying film. To identify the composition of these aggregates, FT-IR analysis was performed on: (1) films pre and post-sonication, (2) desorbed aggregates in the supernatant post-sonication, and (3) MMP only (SUPPLEMENTAL FIGURE S6). The IR spectra did not sufficiently identify the composition of the aggregates, since MMPs have overlapping peaks within the amide I region (keeping in mind that deuteration of proteins causes a peak displacement of about 5–10 cm−1 within the amide I region [29]). This overlap may contribute to the secondary structures within the amide I for MMP treated films and hydrogels. Interestingly, we see strong peaks from 1150–1000 cm−1 for MMP-1 treated films pre-sonication that were diminished post-sonication. These peaks were not seen in the IR spectra of MMP-1 alone. These peaks most likely represent the adsorbed surface aggregates, but this was inconclusive. Cells secrete MMP-1 in both glycosylated and unglycosylated forms [36], and glycosylation of proteins has resulted in peaks between 1150–1000 cm−1 [37,38]. The peaks we observed could be a glycosylated form of adsorbed MMP-1. Similar peaks were seen in the pre-sonicated MMP-2 treated films, but at lower intensity.
It is interesting to compare the present results to prior studies with other enzymatically degradable biomaterials. For example, collagen gels and sponges degraded more rapidly than dense films when incubated with bacterial collagenase [39,40]. In UV-treated collagen, porous materials were significantly more degraded than dense films, regardless of the treatment time [39]. Similarly, collagenase resistance due to acyl azide crosslinking was structure dependent, as gels and sponges had significantly lower resistance to degradation than films [40]. In these experiments, degradation resistance was attributed to the level of crosslinking, which was inherently greater in dense films than in porous materials. A study focusing on the relationship between crosslink density and degradation in collagen films revealed that crosslinked films remained completely intact during a 7-week in vivo study, while non-crosslinked films were completely degraded [41]. Previous work with silk films has also shown degradation rates dependent on the amount of crosslinking [27,42], suggesting that physical crosslink density may also be a major contributor in the degradation profiles of silk materials as it is with collagen.
4. Conclusions
The goal of this study was to determine whether silk fibroin hydrogels exhibited different degradation profiles than films having high crystalline structure when exposed to proteases, thus clarifying the role of primary sequence versus secondary structure of silk materials on accessibility for enzymatic digestion. The degradation properties of a biomaterial are important because they provide insight into possible biomaterial fates in vivo. There are applications for biomaterials where rapid or slow rates of remodeling are desirable during tissue regeneration, thus insight into modes of action of different enzymes can be critical in predicting the degradation rate. The results of this study show that the ability of enzymes to break down a biomaterial are not apparent even if cleavage sites (based on primary sequence analysis) are present, as the structural state of the material (higher order structures vs. solution features) play a role. In the present study, silk fibroin heavy chain degradation was dependent upon material structure; for proteinase K, protease XIV and collagenase, degradation occurred more quickly in hydrogels than films. In hydrogels, proteinase K and protease XIV favored the degradation of non-β-sheet regions, but proteinase K degraded β-sheet regions in films. Collagenase, however, favored the degradation of β-sheet regions in hydrogels but not in films. MMPs showed signs of degradation in aqueous silk solutions, and aggregate formation on the surface of films for both MMP enzymes suggested that silk materials were susceptible to degradation by matrix metalloproteinases. These results provide the basis for predicting how silk biomaterial secondary structure will impact degradation during in vivo applications.
Supplementary Material
Example of deconvoluted spectra with underlying peak decomposition of the amide I region for secondary structure quantification. Black line depicts the deconvoluted spectra, red line represents the cumulative results of the curve fitting, and the 11 peak curves are below. Original peak positions were selected as seen in Hu et al. [30] β-sheet locations were between 1616–1637 and 1697–1703 cm−1; random coil locations were between 1638–1655 cm−1; α-helix locations were between 1656–1662 cm−1; and turn locations were between 1663–1695 cm−1. The table below contains the position, height and width at full width half max for each of the 11 peaks.
Macroscopic (left) and SEM images (right) of films treated with buffer only, MMP-1 and MMP-2. Macroscopic images show increased opacity over time with films treated with MMP. SEM images show the formation of aggregates on the film surface, while control films maintain a smooth topography. Macroscopic scale bar is 1 mm; SEM scale bar is 40 μm.
Densitometric analysis of aqueous silk degradation by MMP-1 and MMP-2. Vertical lines represent the peak molecular weight in each group. Top curve represents MMP enzyme only. Predominant MMP peaks have been demarked with arrows. The molecular weight ladder is given as reference.
Raw FTIR spectra for fibroin films and hydrogels. Spectra represent 24 hours (top), 72 hours (middle) and 120 hours (bottom) incubation in enzyme solution, or DI water for controls.
SEM images of films incubated in MMP enzyme with and without EDTA inactivation. Active MMP enzyme caused aggregates to form at the film surface. EDTA-inactivated MMP produced fewer aggregates. Scale bar is 20 μm; inset = 10 μm
Raw FTIR spectrum of MMP treated films and desorbed aggregates. (a) Deuterium Oxide, (b) MMP treated film, (c) sonicated film, (d) resuspended particles in supernatant after sonication, and (e) MMP enzyme only, or MMP buffer for control. Desorbed aggregates and MMP enzyme were resuspended in deuterium oxide, allowed to equilibrate for 24 hours, placed onto the FTIR crystal and dried before scanned. Control films were treated in DI water without MMP enzyme. Deuteration is known to cause a shift in the position of the amide I and II bands of about 5–10 cm−1 and 100 cm−1, respectively [29,43].
Quantification of mass loss results.
Quantification of FTIR results.
Acknowledgments
We thank the NIH for support of this research (P41 EB002520, U01 EB014976, R01 DE017207).
Footnotes
Conflicts of Interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Wang Y, Kim H-J, Vunjak-Novakovic G, Kaplan DL. Stem cell-based tissue engineering with silk biomaterials. Biomaterials. 2006;27(36):6064–82. doi: 10.1016/j.biomaterials.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Numata K, Kaplan DL. Silk-based gene carriers with cell membrane destabilizing peptides. Biomacromolecules. 2010;11:3189–95. doi: 10.1021/bm101055m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lammel AS, Hu X, Park S-H, Kaplan DL, Scheibel TR. Controlling silk fibroin particle features for drug delivery. Biomaterials. 2010;31(16):4583–91. doi: 10.1016/j.biomaterials.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wharram SE, Zhang X, Kaplan DL, McCarthy SP. Electrospun silk material systems for wound healing. Macromol Biosci. 2010;10(3):246–57. doi: 10.1002/mabi.200900274. [DOI] [PubMed] [Google Scholar]
- 5.Yamada H, Igarashi Y, Takasu Y, Saito H, Tsubouchi K. Identification of fibroin-derived peptides enhancing the proliferation of cultured human skin fibroblasts. Biomaterials. 2004;25(3):467–72. doi: 10.1016/s0142-9612(03)00540-4. [DOI] [PubMed] [Google Scholar]
- 6.Zhou CZ, Confalonieri F, Medina N, Zivanovic Y, Esnault C, Yang T, et al. Fine organization of Bombyx mori fibroin heavy chain gene. Nucleic Acids Res. 2000;28(12):2413–9. doi: 10.1093/nar/28.12.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou CZ, Confalonieri F, Jacquet M, Perasso R, Li ZG, Janin J. Silk fibroin: structural implications of a remarkable amino acid sequence. Proteins. 2001;44(2):119–22. doi: 10.1002/prot.1078. [DOI] [PubMed] [Google Scholar]
- 8.Vepari C, Kaplan D. Silk as a biomaterial. Prog Polym Sci. 2007;32(8–9):991–1007. doi: 10.1016/j.progpolymsci.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tibbitt MW, Anseth KS. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol Bioeng. 2009;103(4):655–63. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eppley BL, Dadvand B. Injectable soft-tissue fillers: clinical overview. Plast Reconstr Surg. 2006;118(4):98e–106e. doi: 10.1097/01.prs.0000232436.91409.30. [DOI] [PubMed] [Google Scholar]
- 11.Jackson JK, Zhao J, Wong W, Burt HM. The inhibition of collagenase induced degradation of collagen by the galloyl-containing polyphenols tannic acid, epigallocatechin gallate and epicatechin gallate. J Mater Sci: Mater Med. 2010;21(5):1435–43. doi: 10.1007/s10856-010-4019-3. [DOI] [PubMed] [Google Scholar]
- 12.Jones D, Tezel A, Borrell M. In vitro resistance to degradation of hyaluronic acid dermal fillers by ovine testicular hyaluronidase. Dermatol Surg. 2010;36:804–9. [Google Scholar]
- 13.Altman GH, Diaz F, Jakuba C, Calabro T, Horan RL, Chen J, et al. Silk-based biomaterials. Biomaterials. 2003;24:401–16. doi: 10.1016/s0142-9612(02)00353-8. [DOI] [PubMed] [Google Scholar]
- 14.Yucel T, Cebe P, Kaplan DL. Vortex-induced injectable silk fibroin hydrogels. Biophys J. 2009;97(7):2044–50. doi: 10.1016/j.bpj.2009.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Kluge J, Leisk GG, Kaplan DL. Sonication-induced gelation of silk fibroin for cell encapsulation. Biomaterials. 2008;29(8):1054–64. doi: 10.1016/j.biomaterials.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horan RL, Antle K, Collette AL, Wang Y, Huang J, Moreau JE, et al. In vitro degradation of silk fibroin. Biomaterials. 2005;26(17):3385–93. doi: 10.1016/j.biomaterials.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 17.Kim U-J, Park J, Li C, Jin H-J, Valluzzi R, Kaplan DL. Structure and properties of silk hydrogels. Biomacromolecules. 2004;5(3):786–92. doi: 10.1021/bm0345460. [DOI] [PubMed] [Google Scholar]
- 18.Fini M, Motta A, Torricelli P, Giavaresi G, Nicoli Aldini N, Tschon M, et al. The healing of confined critical size cancellous defects in the presence of silk fibroin hydrogel. Biomaterials. 2005;26(17):3527–36. doi: 10.1016/j.biomaterials.2004.09.040. [DOI] [PubMed] [Google Scholar]
- 19.Kim U-J, Park J, Kim HJ, Wada M, Kaplan DL. Three-dimensional aqueous-derived biomaterial scaffolds from silk fibroin. Biomaterials. 2005;26(15):2775–85. doi: 10.1016/j.biomaterials.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 20.Arai T, Freddi G, Innocenti R, Tsukada M. Biodegradation of Bombyx mori silk fibroin fibers and films. J Appl Polym Sci. 2003;91:2383–90. [Google Scholar]
- 21.Li M, Ogiso M, Minoura N. Enzymatic degradation behavior of porous silk fibroin sheets. Biomaterials. 2003;24(2):357–65. doi: 10.1016/s0142-9612(02)00326-5. [DOI] [PubMed] [Google Scholar]
- 22.Numata K, Cebe P, Kaplan DL. Mechanism of enzymatic degradation of beta-sheet crystals. Biomaterials. 2010;31(10):2926–33. doi: 10.1016/j.biomaterials.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Curran S, Murray G. Matrix metalloproteinases in tumour invasion and metastasis. J Pathol. 1999;189:300–8. doi: 10.1002/(SICI)1096-9896(199911)189:3<300::AID-PATH456>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 24.Nagase H. Matrix Metalloproteinases. J Biol Chem. 1999;274(31):21491–4. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 25.Talukdar S, Mandal M, Hutmacher DW, Russell PJ, Soekmadji C, Kundu SC. Engineered silk fibroin protein 3D matrices for in vitro tumor model. Biomaterials. 2011;32(8):2149–59. doi: 10.1016/j.biomaterials.2010.11.052. [DOI] [PubMed] [Google Scholar]
- 26.Sofia S, McCarthy MB, Gronowicz G, Kaplan DL. Functionalized silk-based biomaterials for bone formation. J Biomed Mater Res. 2001;54(1):139–48. doi: 10.1002/1097-4636(200101)54:1<139::aid-jbm17>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 27.Lu Q, Hu X, Wang X, Kluge J, Lu S, Cebe P, et al. Water-insoluble silk films with silk I structure. Acta Biomater. 2010;6(4):1380–7. doi: 10.1016/j.actbio.2009.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong A, Huang P, Caughey WS. Protein secondary structures in water from second-derivative amide I infrared spectra. Biochemistry. 1990;29(13):3303–8. doi: 10.1021/bi00465a022. [DOI] [PubMed] [Google Scholar]
- 29.Byler DM, Susi H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers. 1986;25(3):469–87. doi: 10.1002/bip.360250307. [DOI] [PubMed] [Google Scholar]
- 30.Hu X, Kaplan D, Cebe P. Determining beta-sheet crystallinity in fibrous proteins by thermal analysis and infrared spectroscopy. Macromolecules. 2006;39(18):6161–70. [Google Scholar]
- 31.De Jongh HH, Goormaghtigh E, Ruysschaert JM. The different molar absorptivities of the secondary structure types in the amide I region: an attenuated total reflection infrared study on globular proteins. Anal Biochem. 1996;242(1):95–103. doi: 10.1006/abio.1996.0434. [DOI] [PubMed] [Google Scholar]
- 32.Turpeenniemi-Hujanen T. Gelatinases (MMP-2 and -9) and their natural inhibitors as prognostic indicators in solid cancers. Biochimie. 2005;87(3–4):287–97. doi: 10.1016/j.biochi.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 33.Bello L, Lucini V, Carrabba G, Giussani C, Machluf M, Pluderi M, et al. Simultaneous inhibition of glioma angiogenesis, cell proliferation, and invasion by a naturally occurring fragment of human metalloproteinase-2. Cancer Res. 2001;61:8730–6. [PubMed] [Google Scholar]
- 34.Inoue S, Tanaka K, Arisaka F, Kimura S, Ohtomo K, Mizuno S. Silk fibroin of Bombyx mori is secreted, assembling a high molecular mass elementary unit consisting of H-chain, L-chain, and P25, with a 6:6:1 molar ratio. J Biol Chem. 2000;275(51):40517–28. doi: 10.1074/jbc.M006897200. [DOI] [PubMed] [Google Scholar]
- 35.Wray LS, Hu X, Gallego J, Georgakoudi I, Omenetto FG, Schmidt D, et al. Effect of processing on silk-based biomaterials: reproducibility and biocompatibility. J Biomed Mater Res Part B. 2011;99(1):89–101. doi: 10.1002/jbm.b.31875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saarinen J, Welgus HG, Flizar C, Kalkkinen N, Helin J. N-glycan structures of matrix metalloproteinase-1 derived from human fibroblasts and from HT-1080 fibrosarcoma cells. Eur J Biochem. 1999;259(3):829–40. doi: 10.1046/j.1432-1327.1999.00105.x. [DOI] [PubMed] [Google Scholar]
- 37.Petibois C, Cazorla G, Cassaigne A, Déléris G. Plasma protein contents determined by Fourier-transform infrared spectrometry. Clin Chem. 2001;47(4):730–8. [PubMed] [Google Scholar]
- 38.Petibois C, Gionnet K, Gonçalves M, Perromat A, Moenner M, Déléris G. Analytical performances of FT-IR spectrometry and imaging for concentration measurements within biological fluids, cells, and tissues. Analyst. 2006;131(5):640–7. doi: 10.1039/b518076g. [DOI] [PubMed] [Google Scholar]
- 39.Lee J-E, Park J-C, Hwang Y-S, Kim JK, Kim J-G, Suh H. Characterization of UV-irradiated dense/porous collagen membranes: morphology, enzymatic degradation, and mechanical properties. Yonsei Med J. 2001;42(2):172–9. doi: 10.3349/ymj.2001.42.2.172. [DOI] [PubMed] [Google Scholar]
- 40.Rault I, Frei V, Herbage D, Abdul-Malak N, Huc A. Evaluation of different chemical methods for cross-linking collagen gel, films and sponges. J Mater Sci Mater Med. 1996;7(4):215–21. [Google Scholar]
- 41.Boon ME, Ruijgrok JM, Vardaxis MJ. Collagen implants remain supple not allowing fibroblast ingrowth. Biomaterials. 1995;16(14):1089–93. doi: 10.1016/0142-9612(95)98905-t. [DOI] [PubMed] [Google Scholar]
- 42.Jin H-J, Park J, Karageorgiou V, Kim U-J, Valluzzi R, Cebe P, et al. Water-stable silk films with reduced β-sheet content. Adv Funct Mater. 2005;15(8):1241–7. [Google Scholar]
- 43.Kong J, Yu S. Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim Biophys Sin. 2007;39(8):549–59. doi: 10.1111/j.1745-7270.2007.00320.x. [DOI] [PubMed] [Google Scholar]
- 44.Ebeling W, Hennrich N, Klockow M, Metz H, Orth HD, Lang H. Proteinase K from Tritirachium album Limber. Eur J Biochem. 1974;47(1):91–7. doi: 10.1111/j.1432-1033.1974.tb03671.x. [DOI] [PubMed] [Google Scholar]
- 45.Sweeney PJ, Walker JM. Proteinase K. In: Burrell Michael., editor. Enzymes of Molecular Biology. Humana Press; Totwa, NJ: 1993. pp. 305–11. [Google Scholar]
- 46.Bauer C-A. Active Centers of Streptomyces griseus Protease 1, Streptomyces griseus Protease 3, and α-Chymotrypsin: Enzyme-Substrate Interactions. Am Chem Soc. 1978;17(2):375–80. doi: 10.1021/bi00595a028. [DOI] [PubMed] [Google Scholar]
- 47.Appel W. Chymotrypsin: molecular and catalytic properties. Clin Biochem. 1986;19(6):317–22. doi: 10.1016/s0009-9120(86)80002-9. [DOI] [PubMed] [Google Scholar]
- 48.Yu AE, Murphy AN, Stetler-Stevenson WG. 72-kDa Gelatinase (Gelatinase A): structure, activation, regulation, and substrate specificity. In: Parks W, Mecham R, editors. Matrix Metalloproteinases. Academic P; 1998. pp. 85–113. [Google Scholar]
- 49.Pardo A, Selman M. MMP-1: the elder of the family. Int J Biochem Cell Biol. 2005;37(2):283–8. doi: 10.1016/j.biocel.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 50.Fernandez-Patron C, Stewart KG, Zhang Y, Koivunen E, Radomski MW, Davidge ST. Vascular matrix metalloproteinase-2-dependent cleavage of calcitonin gene-related peptide Promotes Vasoconstriction. Circ Res. 2000;87(8):670–6. doi: 10.1161/01.res.87.8.670. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Example of deconvoluted spectra with underlying peak decomposition of the amide I region for secondary structure quantification. Black line depicts the deconvoluted spectra, red line represents the cumulative results of the curve fitting, and the 11 peak curves are below. Original peak positions were selected as seen in Hu et al. [30] β-sheet locations were between 1616–1637 and 1697–1703 cm−1; random coil locations were between 1638–1655 cm−1; α-helix locations were between 1656–1662 cm−1; and turn locations were between 1663–1695 cm−1. The table below contains the position, height and width at full width half max for each of the 11 peaks.
Macroscopic (left) and SEM images (right) of films treated with buffer only, MMP-1 and MMP-2. Macroscopic images show increased opacity over time with films treated with MMP. SEM images show the formation of aggregates on the film surface, while control films maintain a smooth topography. Macroscopic scale bar is 1 mm; SEM scale bar is 40 μm.
Densitometric analysis of aqueous silk degradation by MMP-1 and MMP-2. Vertical lines represent the peak molecular weight in each group. Top curve represents MMP enzyme only. Predominant MMP peaks have been demarked with arrows. The molecular weight ladder is given as reference.
Raw FTIR spectra for fibroin films and hydrogels. Spectra represent 24 hours (top), 72 hours (middle) and 120 hours (bottom) incubation in enzyme solution, or DI water for controls.
SEM images of films incubated in MMP enzyme with and without EDTA inactivation. Active MMP enzyme caused aggregates to form at the film surface. EDTA-inactivated MMP produced fewer aggregates. Scale bar is 20 μm; inset = 10 μm
Raw FTIR spectrum of MMP treated films and desorbed aggregates. (a) Deuterium Oxide, (b) MMP treated film, (c) sonicated film, (d) resuspended particles in supernatant after sonication, and (e) MMP enzyme only, or MMP buffer for control. Desorbed aggregates and MMP enzyme were resuspended in deuterium oxide, allowed to equilibrate for 24 hours, placed onto the FTIR crystal and dried before scanned. Control films were treated in DI water without MMP enzyme. Deuteration is known to cause a shift in the position of the amide I and II bands of about 5–10 cm−1 and 100 cm−1, respectively [29,43].
Quantification of mass loss results.
Quantification of FTIR results.