

Background: Prostaglandin glycerol esters are rapidly hydrolyzed in biological systems.
Results: Complementary approaches demonstrated that lysophospholipase A2 hydrolyzes prostaglandin glycerol esters.
Conclusion: Lysophospholipase A2 is a major prostaglandin glycerol ester-specific hydrolase in human cancer cells.
Significance: Perturbation of lysophospholipase A2 provides a means to understand prostaglandin glycerol ester function in vivo.
Keywords: Cancer, Chemical Biology, Enzyme Kinetics, Lipid Metabolism, Prostaglandin, Activity-based Protein Profiling, Endocannabinoids, Prostaglandin Glycerol Esters, Serine Hydrolase
Abstract
Prostaglandin glycerol esters (PG-Gs) are produced as a result of the oxygenation of the endocannabinoid, 2-arachidonoylglycerol, by cyclooxygenase 2. Understanding the role that PG-Gs play in a biological setting has been difficult because of their sensitivity to enzymatic hydrolysis. By comparing PG-G hydrolysis across human cancer cell lines to serine hydrolase activities determined by activity-based protein profiling, we identified lysophospholipase A2 (LYPLA2) as a major enzyme responsible for PG-G hydrolysis. The principal role played by LYPLA2 in PGE2-G hydrolysis was confirmed by siRNA knockdown. Purified recombinant LYPLA2 hydrolyzed PG-Gs in the following order of activity: PGE2-G > PGF2α-G > PGD2-G; LYPLA2 hydrolyzed 1- but not 2-arachidonoylglycerol or arachidonoylethanolamide. Chemical inhibition of LYPLA2 in the mouse macrophage-like cell line, RAW264.7, elicited an increase in PG-G production. Our data indicate that LYPLA2 serves as a major PG-G hydrolase in human cells. Perturbation of this enzyme should enable selective modulation of PG-Gs without alterations in endocannabinoids, thereby providing a means to decipher the unique functions of PG-Gs in biology and disease.
Introduction
Endocannabinoids are a class of arachidonic acid (AA)2-containing bioactive lipids that have myriad physiological functions (1–7). Key endocannabinoids include 2-arachidonoylglycerol (2-AG) and arachidonoylethanolamide (AEA), which are produced from membrane phospholipids and initiate cellular responses through interactions with the cannabinoid receptors, CB1 and CB2 (5–7). Their effects are mitigated through metabolism by the serine hydrolases, monoacylglycerol lipase (MAGL), α/β-hydrolase domain-containing proteins 6 and 12 (ABHD6 and ABHD12), and fatty acid amide hydrolase (8–12). In addition to these lipases, endocannabinoids have been shown to be selective substrates for cyclooxygenase enzymes, particularly cyclooxygenase-2 (COX-2). Oxidation of 2-AG by COX-2, followed by metabolism by prostaglandin synthases, results in the production of prostaglandin glycerol esters (PG-Gs, Fig. 1). These lipids are of growing interest because they elicit a wide array of cellular responses, including activation of calcium mobilization, modulation of synaptic transmission, induction of hyperalgesia, exacerbation of neurotoxicity and neuroinflammation, and stimulation of anti-inflammatory effects upon lipopolysaccharide stimulation (13–19).
FIGURE 1.

Biosynthesis and metabolism of 2-AG. Following production of 2-AG, lipases (MAG lipase, ABHD6 and -12, CES1, and PPT1) can hydrolyze 2-AG to AA, which can be oxidized to form prostaglandins (PGE2 shown here). Additionally, 2-AG is oxidized by COX-2 to form PG-Gs (PGE2-G shown here). PG-Gs can be hydrolyzed by CES1, PPT1, and/or other enzymes to form the corresponding free acid prostaglandins. DAG, diacylglycerol.
Establishing the physiological relevance of PG-Gs in vivo has been a significant challenge due to their sensitivity to enzymatic hydrolysis to PGs (20). PG-Gs are hydrolyzed in vitro by MAGL (21, 22), α,β-hydrolase-6 (ABHD6) (23), α,β-hydrolase-12 (ABHD12) (23), carboxylesterase-1 (CES1), and palmitoyl-protein thioesterase-1 (PPT1) (24). CES1 and PPT1 have been shown to metabolize PG-Gs in human THP1 cells (24). CES1, a xenobiotic-metabolizing enzyme that is expressed in high amounts in the liver, hydrolyzes a wide array of substrates, ranging from ester and amide-containing xenobiotics (25) to long chain fatty acid esters and thioesters (26) and cholesteryl esters from lipid droplets (26, 27). Similarly, PPT1, a lysosomal hydrolase, has multiple substrates; however, it is predominantly responsible for the depalmitoylation of a number of proteins as well as hydrolysis of palmitoyl-CoA and palmitoyl thioglucoside (28, 29). Consistent with the wide substrate acceptance exhibited by CES1 and PPT1, both enzymes are capable of hydrolyzing PG-Gs and 2-AG (24, 30, 31). In THP1 monocytes, the hydrolysis of 2-AG is almost entirely attributed to CES1, with minor involvement of PPT1 (24, 30, 31). Kinetic analysis of both enzymes showed almost 2-fold greater catalytic turnover for 2-AG than for PG-Gs (31).
We chose to investigate the hydrolase responsible for PG-G metabolism in human cancer cell lines because of the high PGE2-G hydrolytic activity detected in preliminary experiments, the ease of cell maintenance, and the potential for straightforward biochemical and genetic manipulation. The various enzymes described above are serine hydrolases, so we explored the possibility that the PGE2-G hydrolase(s) in human cancer cells is(are) a member of this superfamily. Serine hydrolases are a diverse class of enzymes that include lipases, proteases, and esterases (32, 33), and many class members are involved in lipid biosynthesis and metabolism (9–12). A unifying feature of the serine hydrolase family is a catalytic mechanism that involves the activation of a serine nucleophile for attack on substrates containing esters, amides, or thioester bonds (33). This conserved mechanism has enabled the development of irreversible fluorophosphonate probes that can covalently modify the active site serine and render the enzyme catalytically inactive (32). Nomura et al. (34, 35) coupled fluorophosphonate probe binding with mass spectrometric proteomics techniques, known as activity-based protein profiling with multidimension protein identification technology, to determine the relative activity levels of serine hydrolases across different cancer cell lines. Utilizing these inventories and comparing the relative activities of individual serine hydrolases to PGE2-G hydrolase activities has allowed us to identify lysophospholipase A2 (LYPLA2) as a principal hydrolase responsible for PG-G metabolism in human cells.
Lysophospholipases compose an important class of serine hydrolases that metabolize lysophospholipids to form free fatty acid and the glycerol phosphate-containing head group (36). Thus, we have identified a novel function and substrate for LYPLA2. Specifically, we identify LYPLA2 as the serine hydrolase responsible for hydrolysis of PG-Gs across a number of different cancer cell lines. siRNA knockdown and cDNA overexpression validated the involvement of LYPLA2 in PG-G hydrolysis. Active enzyme was expressed and purified in Escherichia coli, which allowed for kinetic evaluation of an array of different substrates. In contrast to other PG-G-hydrolyzing enzymes, we found that LYPLA2 exerted no action on 2-AG or AEA but did hydrolyze 1-AG.
EXPERIMENTAL PROCEDURES
Chemicals, Cells, and Reagents
2-AG, AEA, PG-Gs (PGE2-glycerol ester, PGD2-glycerol ester, and PGF2α-glycerol ester), prostaglandin serinol amide (PGE2-SA), deuterated prostaglandins and PG-Gs (PGE2-d4 and PGE2-G-d5), and deuterated AA (AA-d8) were purchased from Cayman Chemicals (Ann Arbor, MI). All lysophospholipids and plasmalogens were purchased from Avanti Polar Lipids (Alabaster, AL). LC-MS solvents were from Fisher. DharmaFECT 1 and methoxy fluorophosphonate coupled to tetramethylrhodamine (FP-TAMRA) were acquired from Thermo Scientific (Pittsburgh, PA). Lipofectamine 2000, Lipofectamine RNAiMAX, and all siRNAs were from Invitrogen. Human breast cancer adenocarcinoma cell line, MDA-MB-231 and MCF7, prostate cancer cell lines PC3 and LNCaP, human embryonic kidney cells HEK293, and mouse macrophage-like RAW264.7 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). All cell culture media were from Invitrogen. Recombinant LYPLA1 cDNA, recombinant LYPLA2 cDNA, and TurboFect were purchased from OriGene Technologies (Rockville, MD). Fetal bovine serum (FBS) was from Atlas Biologicals (Fort Collins, CO). HIS®-Select nickel affinity beads were from Sigma. HiPrep 16/60 Sephacryl S-200 HR was from GE Healthcare. The Luna liquid chromatography reverse phase C18 column was from Phenomenex (Torrance, CA). The Thermo liquid chromatography reverse phase C4 column was from Thermo Scientific. The Zorbax Eclipse XDB reverse phase C18 column was from Agilent (Santa Clara, CA).
Culture Conditions
MDA-MB-231, MCF7, LNCaP, and HEK293 cells were maintained as adherent cultures in RPMI 1640 medium supplemented with 10% (v/v) FBS at 37 °C and 5% CO2. PC3 cells were grown in DMEM/F-12 supplemented with 10% (v/v) FBS. RAW264.7 mouse macrophage cells were maintained in high glucose DMEM supplemented with 10% FBS. All cells were grown to no more than 75% confluence.
siRNA Knockdown of LYPLA1 and LYPLA2 in Multiple Cancer Cell Types
The siRNAs for LYPLA2 were as follows: siRNA, sense 5′-GCUCCGGACUGUUGUCACAtt and antisense 5′-UGUGACAACAGUCCGGAtt; and siRNA2, sense 5′-GGGUCCAGUUCAAGACAUAtt and antisense 5′-UAUGUCUUGAACUGGACCCtt. The siRNAs for LYPLA1 were sense 5′-GGGUUUUCUCAGGGAGGAGtt and antisense 5′-CUCCUCCCUGAGAAAACCCtt. MDA-MB-231 cells were plated at 1.0 × 106 cells per 100 mm2 and allowed to incubate for 24 h to 40–60% confluence. siRNA was introduced into the cells as a complex with DharmaFECT 1 according to the manufacturer's recommendations. Briefly, siRNA-DharmaFECT complexes were formed for 20 min in serum-free Opti-MEM I. RPMI 1640 medium from each plate was removed and replaced with 5 ml of serum-free Opti-MEM I medium followed by addition of 1 ml of transfection solution. Cells were cultured for 24 h at 37 °C. The medium was subsequently replaced with RPMI 1640 medium containing 10% FBS and incubated at 37 °C for an additional 48 h before harvesting.
PC3 cells were plated at 1.0 × 106 cells per 100 mm2 and allowed to incubate for 24 h to give 40–60% confluence. Complex formation with siRNA and Lipofectamine 2000 was performed according to the manufacturer's recommendations. Briefly, siRNA-Lipofectamine complexes were formed for 20 min in serum-free Opti-MEM I. The medium in each plate was removed and replaced with serum-free Opti-MEM I medium followed by addition of 1 ml of transfection solution. The cells were cultured for 8 h at 37 °C. The medium was subsequently replaced with DMEM/F-12 supplemented with 10% (v/v) FBS. Cells were incubated at 37 °C for an additional 48 h before harvesting.
LNCaP cells were plated at 1.0 × 106 cells per 100 mm2 and allowed to incubate for 24 h to 40–60% confluence. Complex formation with siRNA and Lipofectamine RNAiMAX was performed according to the manufacturer's recommendations. Briefly, siRNA-Lipofectamine complexes were formed for 5 min in serum-free Opti-MEM I. One ml of transfection solution was added to each plate, and the cells were cultured for 72 h at 37 °C before harvesting.
LYPLA1 and LYPLA2 cDNA Overexpression in HEK293 Cells
HEK293 cells were plated at a density of 3 × 105 cells per well in a 6-well plate in RPMI 1640 medium supplemented with 10% FBS and incubated for 24 h at 37 °C. LYPLA1 or LYPLA2 cDNA was prepared by adding 45 μl of TurboFect to 250 μl of Opti-MEM I followed by 15 μl (1.5 μg) of cDNA. The cells were incubated at room temperature for 30 min. The cDNA solution was added to each well dropwise, and cells were incubated at 37 °C for an additional 48 h before harvesting.
Preparation of Cell Lysates
Cells were harvested by scraping, pelleted by centrifugation, and washed once with phosphate-buffered saline. The cells were resuspended in 500 μl of ice-cold 25 mm Tris buffer (pH 7.5) containing 0.1 mm of both EDTA and DTT. Cells were lysed by sonication (Virsonic Cell Disrupter model 16-850, 10 × 10-s pulses at relative output of 0.5, on ice), and the cytosolic fractions were separated by centrifugation (100,000 × g for 1 h). Protein concentrations were determined using the BCA reagent kit according to the manufacturer's instruction (Pierce).
PG-G Hydrolase Assay
Hydrolytic activity was determined by adding 10 nmol of PGE2-G to 100 μl of cell lysates (250 μg/ml total protein) at 37 °C. Reactions were quenched after 2 h by addition of 1 ml of ethyl acetate containing deuterated internal standard (PGE2-d4). The organic layer was removed, evaporated to near dryness under nitrogen, and reconstituted with 50% methanol. Samples were analyzed by LC-MS/MS for the hydrolytic product, PGE2, which was quantified against the internal standard using stable isotope dilution.
Serine Hydrolase Inhibition
The general serine hydrolase probe, FP-TAMRA, was used to determine the involvement of serine hydrolases in the metabolism of PG-Gs. FP-TAMRA (100 nm) or DMSO was added to 100 μl of MDA-MB-231 cytosol and allowed to incubate for 30 min at 37 °C. PG-G hydrolase activity was determined as described above, except that samples were incubated from 0 to 60 min to obtain a full hydrolysis time course.
Western Blot Analysis
Protein expression was determined by Western blot analysis. Samples were separated by SDS-PAGE. The proteins were then transferred to a nitrocellulose membrane and probed with either rabbit anti-LYPLA2 (1:500 v/v, Thermo Scientific), rabbit anti-LYPLA1 (1:500 v/v, Thermo Scientific), or anti-COX-2 (1:1000 v/v, Cell Signaling) and goat anti-β-actin (1:5000 v/v, Santa Cruz Biotechnology) overnight at 4 °C. Membranes were washed and incubated with IR-visible anti-rabbit or anti-goat secondary antibodies (1:5000 v/v, LI-COR). Blots were visualized using an Odyssey IR Imager.
Recombinant Human LYPLA1, LYPLA2, and His-tagged LYPLA2 E. coli Expression
Full-length LYPLA2 was cloned into an untagged (pC6H) or a hexahistidine-tagged (p6Hb) vector using overlap PCR and isothermal assembly (37). These LYPLA2 constructs were subsequently transformed into BL21 Rosetta E. coli cells (catalog no. 71402, EMD Millipore) for protein expression. Large scale expression was carried out in 10 liters of autoinduction medium (38) at 37 °C overnight.
His-tagged LYPLA2 E. coli Purification
All purification was performed at 4 °C. E. coli cell pellets were resuspended in 20 mm sodium phosphate buffer (pH 7.4) containing 500 mm NaCl, 20 mm imidazole, and 0.1 mm DTT. Cells were lysed by sonication (10 × 10-s pulses at relative output of 0.5, on ice), and the cytosolic fraction was separated by centrifugation (100,000 × g for 1 h). HIS-Select® nickel affinity beads pre-equilibrated with Buffer A (20 mm sodium phosphate buffer (pH 7.4), 500 mm NaCl, 20 mm imidazole, and 0.1 mm DTT) were added to the cytosolic fraction, and protein was bound overnight. Beads were packed into a column and washed with 4 column volumes of Buffer A. His-tagged protein was eluted by linear gradient from 0 to 100% Buffer B (20 mm sodium phosphate buffer (pH 7.4), 500 mm NaCl, and 500 mm imidazole). Eluted protein was collected and concentrated using a Millipore 3000 molecular weight cutoff centrifugal filter. Protein was loaded onto a 120-ml HiPrep 16/60 Sephacryl S-200 HR size exclusion column pre-equilibrated with 2 column volumes of running buffer (Tris buffer (pH 7.5), 0.1 mm EDTA, 0.1 mm DTT). The column was eluted with 1 column volume of running buffer at a flow rate of 0.5 ml/min; protein was collected, and purity was validated by SDS-PAGE.
LYPLA2 Kinetic Analysis
Hydrolysis reactions with recombinant LYPLA2 (100 nm protein) were performed in 25 mm Tris buffer (pH 7.5) with 0.1 mm EDTA and 0.1 mm DTT. Substrates dissolved in either 50% ethanol (PG-Gs), methanol (lysophospholipids), or 2-propanol (plasmalogens) were added at varying concentrations, ranging from 0 to 200 μm. 1-AG was prepared by diluting 2-AG stock solution in PBS to give final concentrations of 20 mm AG, 0.65 mm potassium chloride, 33.55 mm sodium chloride, 0.36 mm monobasic potassium phosphate, and 1.96 mm dibasic sodium phosphate. This solution was incubated at 4 °C overnight to induce isomerization of 2-AG to 1-AG, giving a final solution of ∼85% 1-AG and 15% 2-AG. After preincubation of LYPLA2 for 5 min at 37 °C, reactions were initiated with the addition of substrate. Reactions were quenched after 5 min with 1 ml of ethyl acetate containing 20 ng/ml of either PGE2-d4 (for PGE2-G and PGD2-G), PGF2α-d4 (for PGF2α-G), AA-d8 (for 2-AG, 1-AG, and AEA), or with 150 μl of ethanol containing 20 ng/ml of 17:0 lysophosphatidylcholine (lyso-PC) (for stearoyl, palmitoyl, and oleoyl lyso-PC), 17:0 lysophosphatidic acid (lyso-PA) (for palmitoyl lyso-PA), 17:1 lysophosphatidic acid (lyso-PS) (for palmitoyl lyso-PS and oleoyl lysophosphatidylethanolamine (lyso-PE) or 18:0(plasm)/18:1 PC plasmalogen (for 18:0(plasm)/20:4 plasmalogen). The optimal time for determining kinetic parameters was 5 min. The organic layer was collected and dried to completion under nitrogen. Samples were reconstituted in 50% methanol and analyzed by LC-MS/MS. Kinetic parameters were determined by performing nonlinear regression analysis using a Michaelis-Menten equation with Prism GraphPad version 5.0d.
Hydrolytic Activity of LYPLA2 Following Small Molecule Inhibition
Inhibition reactions were conducted in 25 mm Tris buffer (pH 7.5) with 0.1 mm EDTA and 0.1 mm DTT containing 100 nm recombinant LYPLA2. 100 μl of protein was preincubated with 0–100 μm of either Compound 1 or, 21 (39), the PGE2-G structural analog (prostaglandin E2 serinol amide, PGE2-SA), or 0–1 mm JZL184 for 5 min. Reactions were initiated by addition of 5 μm of PGE2-G and then quenched after 5 min with 1 ml of ethyl acetate containing 20 ng/ml of PGE2-d4. The organic layer was evaporated to dryness under nitrogen, reconstituted in 50% methanol, and analyzed by LC-MS/MS for PGE2.
LYPLA2 Inhibition in RAW264.7 Murine Macrophage-like Cells
The effects of LYPLA2 inhibition on PG-G production were tested by exposure of cells to Compound 1. RAW264.7 cells were plated at 3 × 106 cells per 100-mm2 plate and incubated for 24 h in DMEM supplemented with 10% FBS. Medium was replaced with serum-free DMEM containing either 1 μg/ml of lipopolysaccharide (LPS) (Sigma), 10 μm Compound 1, or a combination of both, and allowed to incubate for 6 h. Cells treated with LPS were then treated with 5 μm ionomycin (Calbiochem) for 45 min. Ionomycin stimulates the production of 2-AG and AA from endogenous sources (40). Lipids were extracted from the cell medium by addition of 2 volumes of ethyl acetate containing PGE2-d4, PGE2-G-d5, and AA-d8 internal standards. The organic layer was dried under nitrogen, and the residue was reconstituted in 50% methanol for analysis of PGs, PG-Gs, and AA by LC-MS/MS.
LC-MS Analytical Procedures
Analysis of prostanoids was accomplished by reverse phase chromatography followed by mass spectrometric detection by selected reaction monitoring (SRM) using a Shimadzu LC-20 HPLC system coupled to an Applied Biosystems 3200 QTRAP mass spectrometer. Separation of PGs and lyso-PCs was achieved by gradient elution of a Phenomenex Luna C18 column (50 × 2.0 mm, 3 μm particle size). Solvent A was HPLC-grade water with 0.1% formic acid, and solvent B was acetonitrile (ACN) with 0.1% formic acid. Samples were injected onto the column with a starting condition of 80% solvent A and 20% solvent B at a flow rate of 0.5 ml/min. A linear gradient of increasing solvent B to 98% was run over 2 min and then held for 1.5 min. SRM transitions were as follows: PGE2 transition m/z 351.3 → 271.2 and PGE2-d4 m/z 355.3 → 275.2.
Separation of PGs and PG-Gs was accomplished with a gradient formed between solvent A, HPLC-grade water with 5 mm ammonium acetate (pH 3.6), and solvent B, ACN supplemented with 6% (v/v) of solvent A. Samples were injected onto the column with a starting condition of 70% solvent A and 30% solvent B at a flow rate of 0.6 ml/min. A linear gradient of increasing solvent B to 100% was run over 3.1 min and then held for 1.8 min. SRM transitions were as follows: PGE2-G transition m/z 444.3 → 391.3; PGE2-G-d5 m/z 449.3 → 396.3; PGE2 m/z 370.3 → 317.2; and PGE2-d4 m/z 374.3 → 321.2.
Separation of AA was achieved by gradient elution. Solvent A was HPLC-grade water with 80 μm silver acetate, and solvent B was methanol with 118 μm silver acetate. Samples were injected onto the column with a starting condition of 80% solvent A and 20% solvent B at a flow rate of 0.4 ml/min. A linear gradient of increasing solvent B to 100% was run over 1 min and then held for 2 min. SRM transition was as follows: AA m/z 518.9 → 411.1 and AA-d8 m/z 526.9 → 419.1.
Resolution of AG isomers was achieved by gradient elution using an Agilent Eclipse XDB-C18 column (150 × 2.1 mm, 3.5 μm particle size). Solvent A was HPLC-grade water with 2 mm ammonium acetate, and solvent B was methanol with 10 mm ammonium acetate. Samples were injected onto the column with a starting condition of 15% solvent A and 85% solvent B at a flow rate of 0.325 ml/min. A linear gradient of increasing solvent B to 95% was run over 9.5 min, followed by a sharp increase of B to 100% over 0.5 min and then held at 100% for an additional 3 min. SRM transitions were as follows: 1-AG and 2-AG transition m/z 396.2 → 287.1 and 2-AG-d8 m/z 404.2 → 295.1.
Separation of choline-containing lysophospholipids was achieved by gradient elution using a Thermo C4 reverse phase column. Solvent A was HPLC-grade water with 0.1% formic acid, and solvent B was ACN with 0.1% formic acid. Samples were injected onto the column with a starting condition of 30% solvent A and 70% solvent B at a flow rate of 0.5 ml/min. A linear gradient of increasing solvent B to 99% was run over 2 min and then held for 1 min. RSM transitions were as follows: 18:0 lyso-PC m/z 524.3 → 184.3; 18:1 lyso-PC m/z 524.3 → 184.3; 17:0 lyso-PC m/z 510.3 → 184.3; and 16:0 lyso-PC m/z 496.3 → 184.3.
Separation of all other lysophospholipids was achieved by gradient elution using a Thermo C4 reverse phase column. Solvent A was HPLC-grade water with 0.1% formic acid, and solvent B was ACN with 0.1% formic acid. Samples were injected onto the column with a starting condition of 50% solvent A and 50% solvent B at a flow rate of 0.5 ml/min. A linear gradient of increasing solvent B to 99% was run over 4 min and then held for 2.5 min. SRM transitions were as follows: 16:0 lyso-PA m/z 409.2 → 152.8; 17:0 lyso-PA m/z 423.4 → 152.8; 16:0 lyso-PS m/z 496.4 → 153.0; 17:1 lyso-PS m/z 508.4 → 153.0, and 18:1 lyso-PE m/z 478.3 → 281.2.
Resolution of plasmalogens was achieved by gradient elution using a Thermo C4 reverse phase column. Solvent A was HPLC-grade water with 0.1% formic acid, and solvent B was a 2:1 ratio of 2-propanol to ACN with 0.1% formic acid. Samples were injected onto the column with a starting condition of 50% solvent A and 50% solvent B at a flow rate of 0.5 ml/min. A linear gradient of increasing solvent B to 100% was run over 2 min and then held for 5 min. SRM transitions were as follows: 18:0(plasm)/18:1 PC plasmalogen m/z 773.5 → 184.3 and 18:0(plasm)/20:4 PC plasmalogen m/z 795.3 → 184.3.
RESULTS
Characterization of PGE2-G Hydrolysis in Cancer Cells
MDA-MB-231 breast cancer cells were initially tested for hydrolytic activity by quantitatively monitoring the formation of PGE2 from exogenously provided PGE2-G by LC-MS/MS. Cytosolic fractions demonstrated PGE2-G hydrolase activity that was linear with time for up to 60 min (Fig. 2A). Initial studies demonstrated that the majority of hydrolytic activity was present in the cytosol, with no activity detectable in the membrane fraction (data not shown). Importantly, serine hydrolase inventories have been identified and published for multiple cancer cell lines, including MDA-MB-231. This led us to explore whether a member of the serine hydrolase family was responsible for PGE2-G hydrolysis. MDA-MB-231 cytosol was preincubated with an irreversible serine hydrolase inhibitor, FP-TAMRA, and assessed for hydrolytic activity following incubation. As shown in Fig. 2A, fluorophosphonate treatment abolished hydrolysis of PGE2-G. This supported the hypothesis that a serine hydrolase is responsible for PGE2-G hydrolysis in MDA-MB-231 cells.
FIGURE 2.
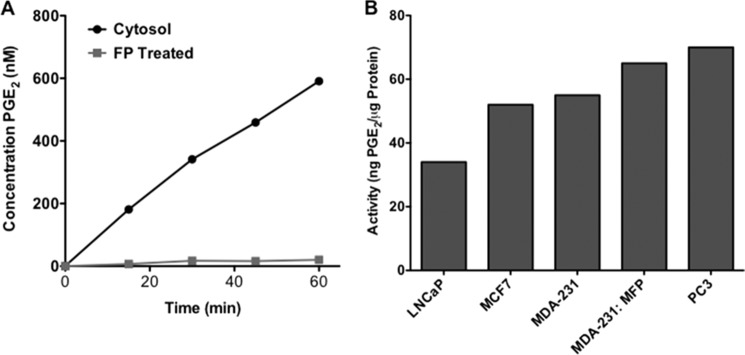
PGE2-G hydrolase activities in human cancer cells. A, MDA-MB-231 cells displayed PGE2-G hydrolase activity that was abolished by pretreatment with an irreversible fluorophosphonate inhibitor, FP-TAMRA. B, PGE2-G hydrolytic activity was assessed across multiple human cancer cell types.
Nomura et al. (34, 35) recently profiled serine hydrolases in a series of human cancer cell lines. The serine hydrolase proteome was enriched by covalently labeling the enzymes with fluorophosphonate molecules bound to biotin followed by avidin chromatography. The purified serine hydrolases were identified by multidimensional liquid chromatography mass spectrometry-based proteomic analysis of tryptic digests. Spectral counting revealed the relative levels of serine hydrolase activities across a number of aggressive and nonaggressive cancer cell types (34, 35). Utilizing the cancer cell types investigated in these serine hydrolase inventories, we quantified PGE2-G hydrolysis by the cytosolic fractions of two breast cancer cell lines, MDA-MB-231 and MCF7, and two prostate cancer cell lines, PC3 and LNCaP. Fig. 2B demonstrates that LNCaP cells exhibited the lowest hydrolytic activity, and PC3 cells displayed the highest rate of PGE2-G hydrolysis. All breast cancer cell lines tested, including MCF7, MDA-MB-231, and MDA-MB-231 cells passaged through a mouse fat pad, exhibited intermediate levels of activity. By comparing the PGE2-G hydrolytic activity across the cell lines to published inventories of over 60 cytosolic serine hydrolases (supplemental Table 1) (34, 35), we were able to identify one enzyme, acyl-protein thioesterase 2 (APT2), also referred to lysophospholipase A2 (LYPLA2), that correlated across all data sets.
LYPLA2 Involvement in PGE2-G Hydrolysis
To investigate the role LYPLA2 plays in PGE2-G hydrolysis, siRNA knockdown of enzyme expression was initially conducted in MDA-MB-231 cells using two distinct siRNAs. The level of LYPLA2 protein was markedly reduced (95 and 69%) in MDA-MB-231 cells upon transfection with siRNA and siRNA2, respectively, as determined by Western blot analysis (Fig. 3, C and D). Cytosolic fractions obtained from the siRNA- and siRNA2-treated MDA-MB-231 cells displayed a respective 60 and 50% reduction in hydrolysis of PGE2-G compared with control cells (Fig. 3A). PC3 and LNCaP cell lines transfected with the LYPLA2-directed siRNA also displayed significant decreases in the levels of LYPLA2 (90 and 86%, respectively) as compared with the cells transfected with the control siRNA (Fig. 3, B and E). Comparisons of PGE2-G hydrolytic activity from cytosol obtained from control and LYPLA2 knockdown cells (Fig. 3A) demonstrated a reduction in hydrolysis of 60% in PC3 cells and 25% in LNCaP cells. The minor decrease in LNCaP hydrolytic activity may be due to lower expression levels of LYPLA2 (34, 35), which also correlate with a smaller total amount of PGE2-G hydrolytic activity in this cell line. In contrast, cells that demonstrated high levels of PGE2-G hydrolytic activity were the most affected by siRNA knockdown of LYPLA2, with decreases in activity ranging from 60 to 80%. Taken together, siRNA knockdown across multiple cell lines supported the hypothesis that LYPLA2 constitutes a major PG-G hydrolytic enzyme in human cancer cells.
FIGURE 3.

LYPLA2 siRNA knockdown in PC3, MDA-MB-231, and LNCaP cells. A, PGE2-G hydrolytic activity of cytosol obtained from PC3, MDA-MB-231, and LNCaP control (scrambled) or LYPLA2-deficient (siRNA) cells. Western blot analysis of LYPLA2 in control (scrambled) and LYPLA2-depleted (siRNA) PC3 (B), MDA-MB-231 (C and D), or LNCaP (E) cells. β-Actin Western blotting verified equaling protein loading (5 μg per lane). Data are presented as the mean ± S.D. of triplicate analyses. ****, p < 0.001; **, p < 0.01.
To further validate LYPLA2 involvement in PGE2-G hydrolysis, the cDNA for human LYPLA2 was transfected into HEK293 cells. Untransfected HEK293 cells express no detectable levels of LYPLA2 by Western blot (Fig. 4A) and had low levels of PGE2-G hydrolase activity (Fig. 4B). In contrast, HEK293 cells transfected with LYPLA2 showed high expression of LYPLA2 by Western blotting (Fig. 4A) and a substantial increase in PGE2-G hydrolytic activity compared with fractions from untransfected cells (Fig. 4B). Combined with our siRNA knockdown data, these overexpression data confirm LYPLA2 as a major PGE2-G hydrolytic enzyme in human cells.
FIGURE 4.
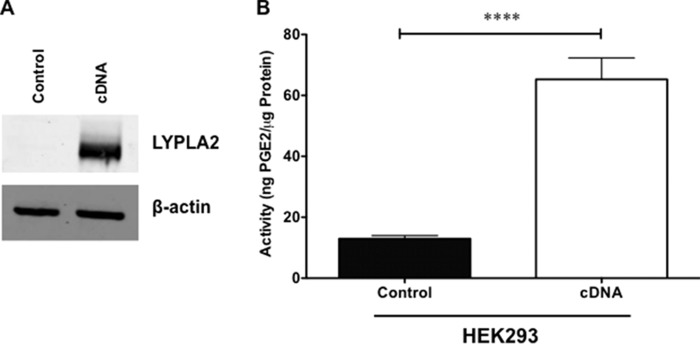
LYPLA2 overexpression in HEK293. A, Western blot analysis of LYPLA2 in control HEK293 cells (control) and LYPLA2-overexpressing HEK293 cells (cDNA). β-Actin Western blotting verified equaling protein loading (5 μg per lane). B, PGE2-G hydrolytic activity of cytosol obtained from HEK293- (control) or LYPLA2-overexpressing (cDNA) cells. Data are presented as the mean ± S.D. of n = 6 analyses. ****, p < 0.001 by t test.
LYPLA1 Involvement in PGE2-G Hydrolysis
LYPLA2 is a member of the serine hydrolase family responsible for lysophospholipid metabolism. A second isoform, lysophospholipase A1 (LYPLA1), is also present in all tested cancer cell lines. Similarly to LYPLA2, LYPLA1 is responsible for hydrolysis of lysophospholipids, and the two enzymes share 60% sequence homology. To investigate the role LYPLA1 plays in PGE2-G hydrolysis, siRNA knockdown of LYPLA1 was conducted in MDA-MB-231 cells. The levels of LYPLA1 protein were markedly reduced (70%) in MDA-MB-231 cells upon transfection with siRNA as determined by Western blot analysis (Fig. 5A). Cytosolic fractions obtained from MDA-MB-231 cells following LYPLA1 knockdown displayed a small but significant reduction in hydrolysis of PGE2-G compared with that of control cells (Fig. 5B).
FIGURE 5.

LYPLA1 siRNA knockdown in MDA-MB-231 cells and LYPLA1 overexpression in HEK293. A, Western blot analysis of LYPLA1 in control MDA-MB-231 cells (scrambled) and LYPLA1-depleted cells (siRNA). β-Actin Western blotting verified uniform protein loading (5 μg per lane). B, PGE2-G hydrolytic activity of cytosol obtained from MDA-MB-231 control cells (scrambled) or LYPLA1-deficient MDA-MB-231 cells (siRNA). C, Western blot analysis of LYPLA1 in control HEK293 cells (control) and LYPLA2-overexpressing HEK293 cells (cDNA). β-Actin Western blotting verified equaling protein loading (5 μg per lane). D, PGE2-G hydrolytic activity of cytosol obtained from HEK293 (control) or LYPLA1-overexpressing (cDNA) cells. Data are presented as the mean ± S.D. of triplicate analyses. **, p < 0.01 by t test. N/S indicates no significance.
To further test LYPLA1 involvement in PGE2-G hydrolysis, cDNA for human LYPLA1 was transfected into HEK293 cells. The cytosolic fraction from the transfected cells demonstrated high expression of LYPLA1 compared with that of control cells as indicated by Western blot (Fig. 5C). Importantly, upon overexpression of LYPLA1, HEK293 cytosolic fractions showed no significant increase in PGE2-G hydrolytic activity when compared with those from untransfected cells (Fig. 5D). Thus, unlike the siRNA knockdown data, the overexpression data do not confirm LYPLA1 as a serine hydrolase responsible for PGE2-G hydrolysis in highly hydrolytically active cancer cell lines.
Expression and Activity of Recombinant Human LYPLA1 and LYPLA2
Recombinant LYPLA1 and LYPLA2 were successfully expressed in E. coli as determined by Coomassie staining (Fig. 6B). Expression of human LYPLA1 in E. coli caused no significant increase in PGE2-G hydrolysis, whereas expression of human LYPLA2 in E. coli was accompanied by a significant increase in PGE2-G hydrolytic activity (Fig. 6A). E. coli expression data in combination with the siRNA knockdown and cDNA overexpression data in HEK cells all confirm that LYPLA2 is the major hydrolase responsible for PGE2-G hydrolysis in human cancer cells, while essentially ruling out a significant role for LYPLA1.
FIGURE 6.

Production and hydrolytic activity of recombinant human LYPLA1 and LYPLA2 by overexpression in E. coli. A, PGE2-G hydrolytic activity of cytosol obtained from wild-type E. coli and E. coli overexpressing recombinant human LYPLA1 and recombinant human LYPLA2. B, Coomassie Blue staining of recombinant LYPLA2 (left) and recombinant LYPLA1 (right) by E. coli compared with control. C, PGE2-G hydrolytic activity of cytosol obtained from wild-type E. coli and E. coli overexpressing recombinant human LYPLA2 or recombinant His-tagged human LYPLA2. D, Coomassie Blue staining (left) and Western blot analysis of LYPLA2 (center) and His tag (right) of purified, recombinant His-tagged LYPLA2 produced by E. coli.
Expression, Isolation, and Activity of Recombinant Human His-tagged LYPLA2
Having demonstrated that LYPLA2 is involved in PGE2-G hydrolysis, we next aimed to elucidate the biochemical activities of this serine hydrolase utilizing expressed and purified His-tagged recombinant human enzyme (hLYPLA2). Expression of LYPLA2 or His-tagged LYPLA2 in E. coli significantly increased PGE2-G hydrolytic activity as compared with the activity of control E. coli cells (Fig. 6C). Consistent with the predicted molecular weight of LYPLA2, the His-tagged hLYPLA2 was purified as a single 25-kDa band (Fig. 6D). On the basis of Coomassie Blue staining, the expressed His-tagged hLYPLA2 was >95% pure.
Substrate Specificity of Recombinant LYPLA2
We evaluated the kinetics of LYPLA2 hydrolysis against an array of substrates, including multiple PG-Gs (PGE2-G, PGD2-G, and PGF2α-G), endocannabinoids (2-AG, 1-AG, and AEA), and lysophospholipids (palmitoyl (16:0), stearoyl-(18:0), and oleoyl- (18:1) lyso-PC, palmitoyl-lyso-PS, and palmitoyl-lyso-PA). Analysis of substrate concentration-velocity plots (Fig. 7) yielded steady-state Michaelis-Menten kinetic parameters (Table 1) for LYPLA2 hydrolytic activity against all substrates tested. Initially, we determined activity toward lysophospholipid substrates and found that the catalytic efficiencies of LYPLA2 for palmitoyl and stearoyl lyso-PC, palmitoyl lyso-PA, and palmitoyl lyso-PS (kcat/Km = 4.0, 5.6, 3.1, and 3.3 min−1 μm−1, respectively) were comparable with published values (41). Interestingly, addition of a single site of unsaturation in the lipid (18:1 lyso-PC) significantly reduced the catalytic efficiency of the enzyme (Kcat/Km =0.6 min−1 μm−1), suggesting that unsaturated lipids hinder hydrolysis. LYPLA2 did not display any catalytic activity against plasmalogens.
FIGURE 7.
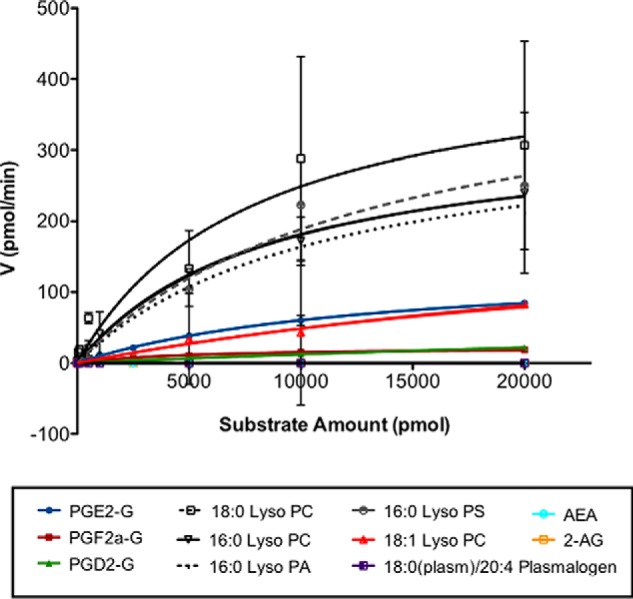
LYPLA2 hydrolytic activity against multiple lipid substrates. Substrate concentration (pmol) versus reaction velocity (pmol/min) plots for recombinant His-tagged LYPLA2. Data are presented as the mean ± S.D. of triplicate analyses.
TABLE 1.
Kinetic parameters for hydrolysis of multiple lipid substrates by recombinant human LYPLA2
| Substrate | kcat | Km | kcat/Km |
|---|---|---|---|
| min−1 | μm | min−1 μm−1 | |
| PGE2-G | 14.1 ± 0.4 | 13 ± 1.1 | 1.08 |
| PGF2α-G | 2.2 ± 0.3 | 5 ± 1.1 | 0.44 |
| PGD2-G | 9.5 ± 1.6 | 67 ± 14 | 0.14 |
| Lyso-PC (16:0) | 33.3 ± 1.7 | 8.3 ± 1.0 | 4.0 |
| Lyso-PC (18:0) | 44.3 ± 10.2 | 7.8 ± 4.4 | 5.6 |
| Lyso-PC (18:1) | 24.8 ± 9.1 | 40.7 ± 20.6 | 0.6 |
| Lyso-PA (16:0) | 34.6 ± 4.7 | 11.1 ± 3.2 | 3.1 |
| Lyso-PS (16:0) | 44.1 ± 10.4 | 13.3 ± 6.2 | 3.3 |
| 18:0(plasm)/20:4 plasmalogen | NAa | NA | NA |
| 1-AG | 1.7 ± 0.07 | 7.6 ± 1.2 | 0.23 |
| 2-AG | NA | NA | NA |
| AEA | NA | NA | NA |
a NA means no activity.
LYPLA2 had lower hydrolytic efficiency toward PG-Gs relative to its activity with saturated lysophospholipids. LYPLA2 showed a greater catalytic efficiency for PGE2-G (Kcat/Km = 1.1 min−1 μm−1) than for PGF2α-G and PGD2-G (kcat/Km = 0.44 and 0.14 min−1 μm−1, respectively).
Commercially available glyceryl prostaglandins are an equilibrium mixture of the 2-glyceryl ester (minor) and the 1(3)-glyceryl ester. To evaluate the regiochemistry of hydrolysis, we modified the HPLC conditions to enable separation of the 2- and the 1(3)-isomers of PGE2-G. HPLC analysis of the incubation mixtures of PGE2-G and LYPLA2 demonstrated that the 1(3)-glyceryl ester was selectively hydrolyzed; no hydrolysis of the 2-glyceryl ester was observed (data not shown).
Neither of the endocannabinoids, 2-AG or AEA, was hydrolyzed by LYPLA2. Because commercial 2-AG is nearly pure 2-glyceryl ester, we incubated 2-AG in PBS overnight to allow it to equilibrate with the 1(3)-AG isomer. Addition of LYPLA2 to this equilibrium mixture demonstrated hydrolysis of the 1(3)-AG but no hydrolysis of 2-AG. The kcat/Km value for hydrolysis of 1-AG was lower than that of PGE2-G (Table 1). Consideration of the substrate specificity data summarized in Table 1 suggests that LYPLA2 only hydrolyzes 1(3)-glyceryl esters.
Bovine serum albumin (BSA) increases the rate of PGD2-G hydrolysis by human MAGL (23), so we determined the relative activity of LYPLA2 to a series of substrates in the presence of 0.5% BSA. The activity of LYPLA2 toward PGE2-G increased, whereas the activity toward 1-AG, 16:0-LPC, and 18:0-LPC decreased (Fig. 8). In fact, PGE2-G is the preferred substrate of LYPLA2 in the presence of BSA.
FIGURE 8.

Effect of BSA on LYPLA2 activity toward a series of substrates. LYPLA2 hydrolytic activity against multiple substrates in the absence (solid bars) or presence (open bars) of bovine serum albumin. Data are presented as the mean ± S.D. of triplicate analyses. ****, p < 0.001; ns, no significance.
Inhibition of Recombinant LYPLA2 Results in Decreased Hydrolysis of PG-Gs
Selective inhibitors of LYPLA2 and the related enzyme LYPLA1 have recently been discovered (39). We assessed the effects of these inhibitors, named Compound 1 (LYPLA2-specific inhibitor) and Compound 21 (LYPLA1-specific inhibitor) (Fig. 9A), on the hydrolytic activity of LYPLA2. Compound 1 inhibited the PGE2-G hydrolysis activity of purified LYPLA2 with an IC50 of 904 nm (Fig. 9B), comparable with previously reported values (510 nm) for the inhibition of hydrolysis of a fluorescent substrate, resorufin acetate (39). Compound 21, however, did not affect LYPLA2 activity. An amidated PGE2-G structural analog, PGE2-SA, was also assessed for LYPLA2 inhibition. PGE2-SA had no effect on the hydrolysis of PGE2-G, indicating the enzyme may have a very tight requirement for substrate binding. The MAGL inhibitor JZL-184 inhibited LYPLA2 at a concentration of 29 μm, which is ∼500-fold higher than the concentrations at which it inhibits MAGL (Fig. 9) (42).
FIGURE 9.

Inhibition of LYPLA2 by small molecule inhibitors. A, chemical structures and IC50 values for Compound 1 (LYPLA2 inhibitor), Compound 21 (LYPLA1 inhibitor), JZl-184 (MAGL inhibitor), and PGE2-SA (structural analog of PGE2-G). B, inhibition curves of LYPLA2-mediated hydrolysis of PGE2-G by Compound 1 (■), Compound 21 (●), and PGE2-SA (▴). Data are presented as the mean ± S.D. of triplicate analyses.
LYPLA2 Inhibition Increases PG-G Levels in RAW264.7 Murine Macrophage-like Cells
To assess the physiological relevance of PG-G hydrolysis by LYPLA2, murine RAW264.7 macrophage-like cells were stimulated to produce PGE2-G and PGD2-G by priming cells with LPS (1 μg/ml) to induce expression of COX-2 (Fig. 10) followed by treatment with the calcium ionophore, ionomycin (5 μm), to promote release of 2-AG/AA. We determined whether inhibition of LYPLA2 by Compound 1 could affect the amount of PG-Gs produced in stimulated RAW264.7 cells. Upon simulation, RAW264.7 cells produce high levels of PGD2 and PGE2 and lower levels of PGD2-G and PGE2-G, all of which are secreted into the culture medium, as reported previously (Fig. 10) (20). Pretreatment of stimulated RAW264.7 cells with 10 μm compound 1 prior to addition of ionomycin, increased the levels of PGD2/PGE2-G in the medium compared with those in the medium of uninhibited cells (40 versus 26 pmol). A concomitant decrease in PGD2/PGE2 was not observable, likely because of the large excess of prostaglandins to PG-Gs in stimulated RAW cells.
FIGURE 10.

LYPLA2 inhibition significantly increases endogenous levels of PGE2-G/PGD2-G formed in RAW264.7 murine macrophage-like cells. Cells were treated with LPS (1 μg/ml) in the presence or absence of an LYPLA2-specific inhibitor (10 μm) for 6 h in serum-free medium followed by treatment with ionomycin (5 μm) for 45 min. The culture medium was removed, and lipids were extracted for LC-MS/MS analysis for PGE2-G/PGD2-G (A) and PGE2/PGD2 (B). C, Western blot analysis of untreated and treated RAW264.7 macrophages confirmed the expression of COX-2 and LYPLA2 in cells. β-Actin Western blot verified equal protein loading (10 μg per lane). Data are presented as the mean ± S.D. of triplicate analyses. **, p < 0.01
DISCUSSION
Endocannabinoids are lipid mediators that elicit a variety of physiological effects, including analgesia and suppression of inflammation (6, 7, 43). COX-2 oxygenates 2-AG to form PG-Gs, which display effects that are frequently opposite those of endocannabinoids, e.g. hyperalgesia and neuroinflammation (13–19). However, complete elucidation of the effects of PG-Gs in vivo has been challenging due to their instability to enzymatic hydrolysis (20). These studies have identified a serine hydrolase, LYPLA2, as a major enzyme responsible for PGE2-G hydrolysis in human cancer cells.
LYPLA2 was expressed in E. coli and purified to apparent homogeneity to determine its specific activity against a range of substrates. LYPLA2 exhibited higher catalytic efficiency against lysophospholipids than against PG-Gs when assays were conducted in the absence of albumin. However, the inclusion of albumin in the assay buffer led to a reversal of substrate specificity such that PGE2-G was hydrolyzed more rapidly than other substrates, including lysophospholipids. Among the PG-Gs tested, PGE2-G was the preferred substrate, followed by PGF2α-G and PGD2-G. The PG-Gs assayed as substrates were an equilibrium mixture of the 1(3)- and 2-glycerol esters; the 1(3)-glycerol ester composes 85% of the mixture. Monitoring the isomeric composition in the presence of LYPLA2 under conditions in which hydrolysis was faster than isomerization revealed that LYPLA2 selectively hydrolyzed the 1(3)- but not the 2-isomer. LYPLA2 also displayed no activity against 2-AG or AEA but did hydrolyze 1(3)-AG at rates lower than PGE2-G. In fact, in the presence of albumin, PGE2-G was hydrolyzed nearly 10-fold faster than 1(3)-AG. Fig. 11 summarizes the lipid substrates for LYPLA2.
FIGURE 11.
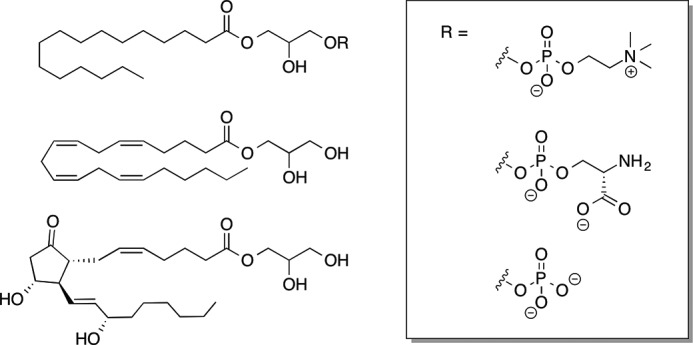
Lipid substrates for hydrolysis by LYPLA2.
The preference of LYPLA2 for 1(3)-glycerol esters over 2-glycerol esters is reminiscent of ABHD6 and ABHD12, which hydrolyze 1(3)-AG faster than 2-AG but is different from MAGL, which hydrolyzes 1(3)-AG and 2-AG at comparable rates (23, 44). The relative activity of ABHD6, ABHD12, and MAGL for the 2- or 1(3)-isomers of PG-Gs is not known, but MAGL appears to hydrolyze the 2-glycerol isomer of the PGD2-G dehydration product, 15-deoxy-PGJ2, quite efficiently. In fact, MAGL exhibited higher activity against the 2-glycerol ester of 15-deoxy-PGJ2 than the 1(3)-isomers of PGE2-G or PGD2-G (23). Whether this is due to the more polar nature of PGE2-G and PGD2-G compared with 15-deoxy-PGJ2 or to the isomeric differences in the glycerol ester moiety is unknown.
LYPLA2 is more active against PG-Gs than the three enzymes previously reported to exhibit PGE2-G hydrolase activity. LYPLA2's catalytic activity for PGE2-G is 2-, 17-, and 52-fold greater than those reported for CES1, MAGL, and PPT1, respectively (21, 31). However, inclusion of albumin in the buffer solutions increases the rates of PGE2-G hydrolysis by LYPLA2 and MAGL. Furthermore, unlike LYPLA2, which does not hydrolyze 2-AG, CES1, MAGL, and PPT1 are all more active against 2-AG than against PGE2-G (24, 30, 31).
LYPLAs are widely distributed in multiple human tissues (36). Interestingly, LYPLA2 has a narrower range of lysophospholipid substrates than LYPLA1. The latter enzyme, which is 60% identical to LYPLA2 in sequence, hydrolyzes a range of lysophospholipids, including lyso-PC, lyso-, lyso-PS, lysophosphatidylglycerol, and lysophosphatidylinositol. In contrast, LYPLA2 hydrolyzes only lyso-PC, and lyso-PS. Yet, LYPLA2 hydrolyzes PGE2-G and other PG-Gs, whereas LYPLA1 does not. LYPLA1 and LYPLA2 also catalyze depalmitoylation of proteins that associate with membranes by virtue of the presence of palmitoyl-cysteine residues in the proteins. LYPLA1 removes the palmitate group from G protein α subunits and H-Ras, whereas LYPLA2 acts on GAP-43 (45, 46). Because of their acyl protein hydrolase activities, LYPLA1 and LYPLA2 are also known as APT1 and APT2, respectively.
Because multiple enzymes hydrolyze PG-Gs with different rates and under different conditions, selective inhibitors or genetic manipulations will be important in dissecting the contributions of individual hydrolases to PG-G hydrolysis in intact cells or tissues. Compound 1, which had been previously reported to inhibit LYPLA2 (39), exhibited 32-fold selectivity for inhibition of PGE2-G hydrolysis compared with the MAGL inhibitor JZL-184, and greater than 100-fold selectivity compared with the LYPLA1 inhibitor compound 21. JZL-184 is a potent MAGL inhibitor but is frequently used in vitro and in vivo at concentrations much higher than its stated IC50 for MAGL of 50 nm (42). Compound 1 significantly reduced PGE2-G hydrolysis by LYPLA2 in vitro and increased endogenous levels of PGD2-G/PGE2-G in LPS-activated RAW macrophages. This is the first demonstration of the elevation of a lipid substrate by inhibition of LYPLA2 in intact cells.
The specificity of LYPLA2 for 1(3)-glycerol ester substrates suggests that isomerization of the initially formed 2-glycerol esters of PGE2-G/PGD2-G occurred following LPS treatment of the RAW cells. The half-life to spontaneous acyl migration of 2-glycerol esters to 1(3)-glycerol esters is ∼10 min in serum-free medium but reduces to ∼3 min in the presence of serum (47). Because the synthesis of PGE2-G/PGD2-G took place over 45 min following addition of ionomycin to trigger 2-AG release, there was ample time for isomerization to occur. The possibility also exists that isomerization of 2-AG to 1(3)-AG occurred during this time course and that addition of compound 1 inhibited 1(3)-AG hydrolysis by LYPLA2 leading to elevated levels of PGE2-G/PGD2-G following COX-2 oxygenation. This possibility seems less likely than compound 1 inhibition of LYPLA2 hydrolysis of PGE2-G/PGD2-G because 1(3)-AG is an inferior substrate for LYPLA2 compared with glyceryl prostaglandins and because 1(3)-AG is a poorer substrate than 2-AG for oxygenation by COX-2.
The biological effects of PG-Gs that have been documented in the literature have all been recorded with commercial material, which are primarily the 1(3)-glycerol esters. It is not known whether the 2-glycerol esters of PG-Gs have more potent or different effects in the same assays. Therefore, it seems likely that LYPLA2 plays an important role in controlling the biological effects of endocannabinoid-derived prostaglandin glycerol esters. This study demonstrates that it is a major and perhaps the major glyceryl prostaglandin hydrolase in human cancer cells. However, the multiplicity of enzymes that hydrolyze these compounds, either the 2- or the 1(3)- isomers, suggests that different enzymes will play significant roles in controlling the activities of glyceryl prostaglandins depending on the cell type and physiological state. It will be exciting to dissect the contributions of individual enzymes to PG-G hydrolysis using a combination of chemical biological and genetic approaches.
Supplementary Material
Acknowledgments
We are grateful to the Vanderbilt Antibody and Protein Resource Core. We especially thank Erin Gribben for assistance with E. coli growth and protein expression. We are also thankful to Carol Rouzer for help in reviewing and preparing the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants GM15431 (to L. J. M.) and DK033760 (to B. F. C.) and Training Grant GM65086 (to J. D. M.).

This article contains supplemental Table S1.
- AA
- arachidonic acid
- 2-AG
- 2-arachidonoyl glycerol
- AEA
- arachidonylethanolamine
- MAGL
- monoacylglycerol lipase
- ABHD
- α/β hydrolase domain containing
- CES
- carboxylesterase
- LYPLA2
- lysophospholipase A2
- LYPLA1
- lysophospholipase A1
- PG
- prostaglandins
- PG-G
- prostaglandin glycerol esters
- SRM
- selected reaction monitoring
- FP-TAMRA
- methoxy fluorophosphonate coupled to tetramethylrhodamine
- ACN
- acetonitrile
- lyso-PS
- lysophosphatidic acid
- lyso-PA
- lysophosphatidic acid
- lyso-PC
- lysophosphatidylcholine
- lyso-PE
- lysophosphatidylethanolamine.
REFERENCES
- 1. Walker J. M., Huang S. M. (2002) Cannabinoid analgesia. Pharmacol. Ther. 95, 127–135 [DOI] [PubMed] [Google Scholar]
- 2. Pertwee R. G. (2001) Cannabinoid receptors and pain. Prog. Neurobiol. 63, 569–611 [DOI] [PubMed] [Google Scholar]
- 3. Valenzano K. J., Tafesse L., Lee G., Harrison J. E., Boulet J. M., Gottshall S. L., Mark L., Pearson M. S., Miller W., Shan S., Rabadi L., Rotshteyn Y., Chaffer S. M., Turchin P. I., Elsemore D. A., Toth M., Koetzner L., Whiteside G. T. (2005) Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology 48, 658–672 [DOI] [PubMed] [Google Scholar]
- 4. Whiteside G. T., Gottshall S. L., Boulet J. M., Chaffer S. M., Harrison J. E., Pearson M. S., Turchin P. I., Mark L., Garrison A. E., Valenzano K. J. (2005) A role for cannabinoid receptors, but not endogenous opioids, in the antinociceptive activity of the CB2-selective agonist, GW405833. Eur. J. Pharmacol. 528, 65–72 [DOI] [PubMed] [Google Scholar]
- 5. Di Marzo V., Bisogno T., De Petrocellis L. (2007) Endocannabinoids and related compounds: walking back and forth between plant natural products and animal physiology. Chem. Biol. 14, 741–756 [DOI] [PubMed] [Google Scholar]
- 6. Kogan N. M., Mechoulam R. (2006) The chemistry of endocannabinoids. J. Endocrinol. Invest. 29, 3–14 [PubMed] [Google Scholar]
- 7. Piomelli D. (2003) The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 4, 873–884 [DOI] [PubMed] [Google Scholar]
- 8. Natarajan V., Schmid P. C., Reddy P. V., Schmid H. H. (1984) Catabolism of N-acylethanolamine phospholipids by dog brain preparations. J. Neurochem. 42, 1613–1619 [DOI] [PubMed] [Google Scholar]
- 9. Schmid P. C., Zuzarte-Augustin M. L., Schmid H. H. (1985) Properties of rat liver N-acylethanolamine amidohydrolase. J. Biol. Chem. 260, 14145–14149 [PubMed] [Google Scholar]
- 10. Maccarrone M., van der Stelt M., Rossi A., Veldink G. A., Vliegenthart J. F., Agrò A. F. (1998) Anandamide hydrolysis by human cells in culture and brain. J. Biol. Chem. 273, 32332–32339 [DOI] [PubMed] [Google Scholar]
- 11. Lichtman A. H., Hawkins E. G., Griffin G., Cravatt B. F. (2002) Pharmacological activity of fatty acid amides is regulated, but not mediated, by fatty acid amide hydrolase in vivo. J. Pharmacol. Exp. Ther. 302, 73–79 [DOI] [PubMed] [Google Scholar]
- 12. Blankman J. L., Simon G. M., Cravatt B. F. (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 14, 1347–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nirodi C. S., Crews B. C., Kozak K. R., Morrow J. D., Marnett L. J. (2004) The glyceryl ester of prostaglandin E2 mobilizes calcium and activates signal transduction in RAW264.7 cells. Proc. Natl. Acad. Sci. U.S.A. 101, 1840–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sang N., Zhang J., Chen C. (2006) PGE2 glycerol ester, a COX-2 oxidative metabolite of 2-arachidonoyl glycerol, modulates inhibitory synaptic transmission in mouse hippocampal neurons. J. Physiol. 572, 735–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sang N., Zhang J., Chen C. (2007) COX-2 oxidative metabolite of endocannabinoid 2-AG enhances excitatory glutamatergic synaptic transmission and induces neurotoxicity. J. Neurochem. 102, 1966–1977 [DOI] [PubMed] [Google Scholar]
- 16. Hu S. S., Bradshaw H. B., Chen J. S., Tan B., Walker J. M. (2008) Prostaglandin E2 glycerol ester, an endogenous COX-2 metabolite of 2-arachidonoylglycerol, induces hyperalgesia and modulates NFκB activity. Br. J. Pharmacol. 153, 1538–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Richie-Jannetta R., Nirodi C. S., Crews B. C., Woodward D. F., Wang J. W., Duff P. T., Marnett L. J. (2010) Structural determinants for calcium mobilization by prostaglandin E2 and prostaglandin F2α glyceryl esters in RAW264.7 cells and H1819 cells. Prostaglandins Other Lipid Mediat. 92, 19–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Valdeolivas S., Pazos M. R., Bisogno T., Piscitelli F., Iannotti F. A., Allarà M., Sagredo O., Di Marzo V., Fernández-Ruiz J. (2013) The inhibition of 2-arachidonoyl-glycerol (2-AG) biosynthesis, rather than enhancing striatal damage, protects striatal neurons from malonate-induced death: a potential role of cyclooxygenase-2-dependent metabolism of 2-AG. Cell Death Dis. 4, e862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alhouayek M., Masquelier J., Cani P. D., Lambert D. M., Muccioli G. G. (2013) Implication of the anti-inflammatory bioactive lipid prostaglandin D2-glycerol ester in the control of macrophage activation and inflammation by ABHD6. Proc. Natl. Acad. Sci. U.S.A. 110, 17558–17563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kozak K. R., Crews B. C., Ray J. L., Tai H. H., Morrow J. D., Marnett L. J. (2001) Metabolism of prostaglandin glycerol esters and prostaglandin ethanolamides in vitro and in vivo. J. Biol. Chem. 276, 36993–36998 [DOI] [PubMed] [Google Scholar]
- 21. Vila A., Rosengarth A., Piomelli D., Cravatt B., Marnett L. J. (2007) Hydrolysis of prostaglandin glycerol esters by the endocannabinoid-hydrolyzing enzymes, monoacylglycerol lipase and fatty acid amide hydrolase. Biochemistry 46, 9578–9585 [DOI] [PubMed] [Google Scholar]
- 22. Laitinen T., Navia-Paldanius D., Rytilahti R., Marjamaa J. J., Kařízková J., Parkkari T., Pantsar T., Poso A., Laitinen J. T., Savinainen J. R. (2014) Mutation of Cys242 of human monoacylglycerol lipase disrupts balanced hydrolysis of 1- and 2-monoacylglycerols and selectively impairs inhibitor potency. Mol. Pharmacol. 85, 510–519 [DOI] [PubMed] [Google Scholar]
- 23. Savinainen J. R., Kansanen E., Pantsar T., Navia-Paldanius D., Parkkari T., Lehtonen M., Laitinen T., Nevalainen T., Poso A., Levonen A. L., Laitinen J. T. (2014) Robust hydrolysis of prostaglandin glycerol esters by human monoacylglycerol lipase (MAGL). Mol. Pharmacol. 86, 522–535 [DOI] [PubMed] [Google Scholar]
- 24. Xie S., Borazjani A., Hatfield M. J., Edwards C. C., Potter P. M., Ross M. K. (2010) Inactivation of lipid glyceryl ester metabolism in human THP1 monocytes/macrophages by activated organophosphorus insecticides: role of carboxylesterases 1 and 2. Chem. Res. Toxicol. 23, 1890–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ross M. K., Borazjani A., Wang R., Crow J. A., Xie S. (2012) Examination of the carboxylesterase phenotype in human liver. Arch. Biochem. Biophys. 522, 44–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quiroga A. D., Lehner R. (2011) Role of endoplasmic reticulum neutral lipid hydrolases. Trends Endocrinol. Metab. 22, 218–225 [DOI] [PubMed] [Google Scholar]
- 27. Ghosh S. (2000) Cholesteryl ester hydrolase in human monocyte/macrophage: cloning, sequencing, and expression of full-length cDNA. Physiol. Genomics 2, 1–8 [DOI] [PubMed] [Google Scholar]
- 28. Lu J. Y., Hofmann S. L. (2006) Thematic review series: lipid posttranslational modifications. Lysosomal metabolism of lipid-modified proteins. J. Lipid Res. 47, 1352–1357 [DOI] [PubMed] [Google Scholar]
- 29. Lu J. Y., Hofmann S. L. (2006) Inefficient cleavage of palmitoyl-protein thioesterase (PPT) substrates by aminothiols: implications for treatment of infantile neuronal ceroid lipofuscinosis. J. Inherit. Metab. Dis. 29, 119–126 [DOI] [PubMed] [Google Scholar]
- 30. Crow J. A., Bittles V., Herring K. L., Borazjani A., Potter P. M., Ross M. K. (2012) Inhibition of recombinant human carboxylesterase 1 and 2 and monoacylglycerol lipase by chlorpyrifos oxon, paraoxon and methyl paraoxon. Toxicol. Appl. Pharmacol. 258, 145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang R., Borazjani A., Matthews A. T., Mangum L. C., Edelmann M. J., Ross M. K. (2013) Identification of palmitoyl protein thioesterase 1 in human THP1 monocytes and macrophages and characterization of unique biochemical activities for this enzyme. Biochemistry 52, 7559–7574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Simon G. M., Cravatt B. F. (2010) Activity-based proteomics of enzyme superfamilies: serine hydrolases as a case study. J. Biol. Chem. 285, 11051–11055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Long J. Z., Cravatt B. F. (2011) The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem. Rev. 111, 6022–6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nomura D. K., Lombardi D. P., Chang J. W., Niessen S., Ward A. M., Long J. Z., Hoover H. H., Cravatt B. F. (2011) Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. 18, 846–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nomura D. K., Long J. Z., Niessen S., Hoover H. S., Ng S. W., Cravatt B. F. (2010) Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang A., Dennis E. A. (1999) Mammalian lysophospholipases. Biochim. Biophys. Acta 1439, 1–16 [DOI] [PubMed] [Google Scholar]
- 37. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A., 3rd, Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 38. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 39. Adibekian A., Martin B. R., Chang J. W., Hsu K. L., Tsuboi K., Bachovchin D. A., Speers A. E., Brown S. J., Spicer T., Fernandez-Vega V., Ferguson J., Hodder P. S., Rosen H., Cravatt B. F. (2012) Confirming target engagement for reversible inhibitors in vivo by kinetically tuned activity-based probes. J. Am. Chem. Soc. 134, 10345–10348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Di Marzo V., Bisogno T., De Petrocellis L., Melck D., Orlando P., Wagner J. A., Kunos G. (1999) Biosynthesis and inactivation of the endocannabinoid 2-arachidonoylglycerol in circulating and tumoral macrophages. Eur. J. Biochem. 264, 258–267 [DOI] [PubMed] [Google Scholar]
- 41. Wang A., Yang H. C., Friedman P., Johnson C. A., Dennis E. A. (1999) A specific human lysophospholipase: cDNA cloning, tissue distribution and kinetic characterization. Biochim. Biophys. Acta 1437, 157–169 [DOI] [PubMed] [Google Scholar]
- 42. Long J. Z., Li W., Booker L., Burston J. J., Kinsey S. G., Schlosburg J. E., Pavón F. J., Serrano A. M., Selley D. E., Parsons L. H., Lichtman A. H., Cravatt B. F. (2009) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 5, 37–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Di Marzo V., De Petrocellis L., Bisogno T. (2005) The biosynthesis, fate and pharmacological properties of endocannabinoids. Handb. Exp. Pharmacol. 168, 147–185 [DOI] [PubMed] [Google Scholar]
- 44. Navia-Paldanius D., Savinainen J. R., Laitinen J. T. (2012) Biochemical and pharmacological characterization of human α/β-hydrolase domain containing 6 (ABHD6) and 12 (ABHD12). J. Lipid Res. 53, 2413–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Duncan J. A., Gilman A. G. (1998) A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein α subunits and p21(RAS). J. Biol. Chem. 273, 15830–15837 [DOI] [PubMed] [Google Scholar]
- 46. Tomatis V. M., Trenchi A., Gomez G. A., Daniotti J. L. (2010) Acyl-protein thioesterase 2 catalyzes the deacylation of peripheral membrane-associated GAP-43. PLoS One 5, e15045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rouzer C. A., Ghebreselasie K., Marnett L. J. (2002) Chemical stability of 2-arachidonylglycerol under biological conditions. Chem. Phys. Lipids 119, 69–82 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.