

Background: Serine racemase produces d-serine, which is required for optimal NMDA receptor activity.
Results: FBXO22 interacts with and activates serine racemase by preventing its targeting to membranes.
Conclusion: The data suggest an atypical role of FBXO22 in regulating d-serine synthesis unrelated to its effects on the ubiquitin system.
Significance: The results provide a new mechanism affecting d-serine synthesis with implications for the regulation of NMDA receptors.
Keywords: Glutamate Receptor, Neurotransmitter, Neurotransmitter Receptor, Ubiquitin, Ubiquitylation (Ubiquitination), d-Serine, F-box Proteins, FBXO22, NMDA Receptor, Serine Racemase
Abstract
d-Serine is a physiological activator of NMDA receptors (NMDARs) in the nervous system that mediates several NMDAR-mediated processes ranging from normal neurotransmission to neurodegeneration. d-Serine is synthesized from l-serine by serine racemase (SR), a brain-enriched enzyme. However, little is known about the regulation of d-serine synthesis. We now demonstrate that the F-box only protein 22 (FBXO22) interacts with SR and is required for optimal d-serine synthesis in cells. Although FBXO22 is classically associated with the ubiquitin system and is recruited to the Skip1-Cul1-F-box E3 complex, SR interacts preferentially with free FBXO22 species. In vivo ubiquitination and SR half-life determination indicate that FBXO22 does not target SR to the proteasome system. FBXO22 primarily affects SR subcellular localization and seems to increase d-serine synthesis by preventing the association of SR to intracellular membranes. Our data highlight an atypical role of FBXO22 in enhancing d-serine synthesis that is unrelated to its classical effects as a component of the ubiquitin-proteasome degradation pathway.
Introduction
The NMDA receptor (NMDAR)2 is a main excitatory receptor in the nervous system and is involved in a wide array of processes, including synaptic plasticity, learning, and memory, and also in neurodegenerative diseases (1). NMDARs display unique regulatory mechanisms requiring binding of glutamate along with a coagonist (glycine or d-serine) for the receptor/channel activation (2). Accumulating evidence demonstrates that d-serine, a d-amino acid present in the mammalian brain, is the major ligand at the coagonist site of the receptors and mediates several NMDAR-dependent processes (3–9).
d-serine is synthesized from l-serine by the enzyme serine racemase (SR) (10, 11). In addition to producing d-serine, SR generates pyruvate by catalyzing the α,β-elimination of water from l-serine (12, 13). SR KO mice display about 90% decrease in brain d-serine and exhibit deficits in NMDAR-dependent synaptic plasticity and spatial learning (14–16). These mice are also resistant to α,β-mediated neurotoxicity in vivo and are less susceptible to stroke damage (17, 18), indicating that SR may be involved in neurodegeneration. In this framework, SR inhibitors may provide a novel neuroprotective strategy in neurodegenerative conditions.
Until recently, d-serine was thought to be exclusively released by exocytosis from astrocytes, a type of glia cells that ensheath synapses (4, 6, 19). However, recent data demonstrate that neurons are a main site of d-serine production and storage in the brain and that they regulate their own NMDARs by releasing d-serine (5, 8, 20–24). Astrocytic SR is activated by interaction with a number of proteins, including Grip-1 (25), Pick-1 (26), and Disc-1 (27). Disc-1 binds to and stabilizes SR in glia cells but has no effect on neuronal SR (27). Astrocytic SR is inhibited by interaction with inositol phospholipids and also by S-nitrosylation (28, 29). In neurons, NMDAR stimulation promotes translocation of SR from the cytosol (where it normally resides) to the membrane, a process that appears to involve atypical palmitoylation of the enzyme (30). NMDAR-stimulated translocation to the membrane inactivates SR, providing a feedback inhibition of neuronal d-serine synthesis that may work as a failsafe mechanism to prevent NMDAR overactivation in vicinal neurons or synapses (30).
Here we sought to identify new SR-interacting proteins that might regulate SR. We found that FBXO22, an F-box motif-containing protein (31), interacts with SR and regulates its subcellular localization and activity in cells. The FBXO22 effect on d-serine dynamics is unrelated to its role as a component of the SCF ubiquitin ligase complex. Our data imply a non-canonical role of FBXO22 in regulating d-serine synthesis via changes in SR intracellular targeting.
EXPERIMENTAL PROCEDURES
Materials
DMEM, glutathione-agarose beads, anti-Myc-agarose matrix, red anti-FLAG M2 affinity gel, red anti-HA affinity gel, MG132, rabbit anti c-Myc (catalog no. C3956), mouse anti-c-Myc (catalog no. M4439), mouse anti-β-tubulin (catalog no. T7816), rabbit anti-KDM4A (catalog no. HPA007610), mouse anti-FBXO22 (catalog no. WH0026263M1), and mouse and rabbit anti-FLAG antibodies (catalog nos. F1804 and F7425, respectively) were purchased from Sigma-Aldrich. The A172 glioblastoma cell line was obtained from the ATCC. Mini-complete protease inhibitor mixture was purchased from Roche. [35S]methionine/cysteine was purchased from PerkinElmer Life Sciences. Mouse anti-FBXO22 (catalog no. sc-100736), rabbit anti Cul1 (catalog no. H-213), rabbit anti-HA (catalog no. sc-805), and mouse anti-KDEL (catalog no. sc-58774) were purchased from Santa Cruz Biotechnology. Mouse anti-β-actin (catalog no. 691001) was purchased from MP Biomedicals. Anti-rabbit and anti-mouse peroxidase-conjugated antibodies, anti-rabbit Cy3 or Cy2, anti-mouse Cy3 or Cy2, and normal goat serum were obtained from Jackson ImmunoResearch Laboratories. Mouse anti-HA (catalog no. MMS-101P) was purchased from Covance. Mouse anti-Cul1 (catalog no. C32620) and mouse anti-Skp1 (catalog no. 610530) were from BD Biosciences. Mouse anti-histone H4 (catalog no. ab17036) was obtained from Abcam. Rabbit anti-H3K9me3 (catalog no. 491008) was purchased from Invitrogen.
Basal medium Eagle, minimum essential medium, FBS, penicillin/streptomycin, penicillin/streptomycin/amphotericin, trypsin, and soybean trypsin inhibitor were obtained from Biological Industries (Kibbutz Beit Haemek, Israel). Lipofectamine 2000 was purchased from Invitrogen.
Protein Identification by Mass Spectrometry
Immunoprecipitation of HA-tagged mouse SR (HA-SR) from transfected SH-SY5Y neuroblastoma cells and mass spectrometric analysis of coimmunoprecipitated proteins was performed as described previously (30). Briefly, HA-SR-transfected SH-SY5Y cells were lysed by sonication with 20 mm Tris-HCl (pH 7.4), 1 mm EDTA, 50 mm KCl, protease inhibitor mixture, 2 mm pyrophosphate, 1 mm NaF, and 1 mm orthovanadate. Then Triton X-100 (0.3%) was added to the samples and kept under rotation for 10 min at 4 °C. After removing cell debris by a 10-min centrifugation at 1400 × g, the suspension was centrifuged at 200,000 × g for 1 h to obtain cytosolic and membrane fractions. Immunoprecipitation of HA-SR from the cytosolic fraction was carried out with anti-HA affinity matrix. The immunoprecipitate was washed extensively for 2 h by seven changes of high-stringency buffer consisting of 50 mm Tris-HCl (pH 7.4), 0.5 m NaCl, 1% Triton X-100, and 0.1% SDS. Following SDS-PAGE, the bands were excised at the 36- to 42-kDa range, and proteins were reduced, carbamidomethylated, and digested in-gel with trypsin (modified sequencing grade from Promega). The tryptic peptides were extracted from the gel and desalted using C18 STAGE tips. Capillary LC-MS/MS analysis was performed on an UltiMate 3000 LC system (Dionex) coupled to an electrospray ionization (ESI)-Q-TOF mass spectrometer (Q-Tof micro, Waters) using a custom-made reverse-phase analytical column. The mass spectrometer was operated in an automated data-dependent acquisition mode where each MS scan (m/z 350–1500, 1-s scan time) was followed by three MS/MS scans (m/z 50–1500, 1-s scan time) of the most intense peptide ions (z = 2–4). The MassLynx raw files from the Q-TOF micro were processed using ProteinLynx Global Server 2.0.5 (Waters) and exported in Micromass pkl format for automated peptide identification using an in-house Mascot server v2.2.03 (Matrix Sciences). The search was performed against the taxonomy-filtered (Mammalia) SwissProt database (version 56.0, 63,150 sequences after taxonomy filter) and a corresponding decoy database. The following parameters were applied: enzyme, trypsin; maximum missed cleavages, 3; fixed modifications, carbamidomethyl (C); variable modifications, oxidation (M), Phospho_STY (STY), and PhosphoIntact (STY); peptide mass tolerance, 0.25 Da; fragment mass tolerance, 0.6 Da; mass values, monoisotopic; instrument type, ESI-QUAD-TOF. With default Mascot search parameters (significance threshold, p < 0.05; ion score cutoff, 15) the estimated false discovery rate was 0.0% (number of matches above identity threshold in search of real/decoy database, 115/0). Using these parameters, we identified the doubly charged tryptic peptide VVAEELENVR (m/z 579.32) covering position 87–96 and the doubly charged tryptic peptide STFVLSNLAEVVER (m/z 782.45) covering position 19–32 of human FBXO22. The given peptides were assigned to the spectra with Mascot ion scores of 60 and 77, respectively, and the identification was verified by manual de novo sequencing and homology search.
Recombinant Proteins
Sequence-verified constructs of FBXO22a and b were subcloned into the pGEX4T-2 vector. The GST-FBXO22 fusion constructs were introduced in codon plus BL21 bacteria and induced by isopropyl 1-thio-β-d-galactopyranoside (0.3 mm) at 30 °C for 3 h. Bacteria were pelleted by centrifugation for 10 min at 5000 × g at 4 °C. The pellet was resuspended in PBS supplemented with 100 mm NaCl, 2 mm DTT, and 0.4 mm PMSF, and cells were disrupted by sonication. After addition of Triton X-100 to 1%, insoluble material was removed by centrifugation at 40,000 × g. Then GST-FBXO22 fusion proteins were purified by binding to glutathione-agarose beads, followed by extensive washes with cold PBS. Proteins were eluted and dialyzed against PBS plus 6% glycerol. GST-α-synuclein and GST-CHIP (C terminus of Hsc70-interacting protein) constructs (received from Prof. S. Engelender, Technion Institute of Technology) were produced in the same fashion. His-SR was purified from BL21 bacteria as described previously (13).
In Vitro Binding Experiments
Purified His-SR (0.4 μg/ml) was incubated with GST recombinant proteins bound to glutathione-Sepharose beads (0.8 μg/ml GST-FBXO22a, 1.5 μg/ml GST FBXO22b, or 30 μg/ml GST-α-synuclein). The binding buffer consisted of 20 mm Tris-HCl (pH 7.4), 0.2% Triton X-100, 100 μg/ml bovine serum albumin, 200 mm NaCl, and protease inhibitor mixture. After 2 h under rotation at 4 °C, the samples were washed five times with PBS supplemented with 0.2% Triton X-100. Bound SR was analyzed by SDS-Page and Western blot analysis with a rabbit anti-SR serum (1:1000) characterized previously (8).
Preparation of FBXO22 Deletion Mutants
To delete the F-box region (ΔFBXO22 construct), we carried out PCR with PfuUltra high fidelity (HF) polymerase (Agilent) using primer pairs flanking the F-box region (between amino acids 20 and 68). The primers were as follows: primer 1, 5′ CGCGGCTCCTCCGTAGACCCGCGGAGCACCTGGATCTCCGCAGGC 3′, corresponding to amino acids 11–20 fused with amino acids 68–74; primer 2, 5′ ATAAGAATGCGGCCGCTTATTTAGATGACCCCAG 3′, complementary to the C-terminal region of FBXO22b with a restriction site for the NotI enzyme. After gel purification, the Sal1 site and amino acids 1–10 were inserted by PCR with primer 3 (5′ ACGCGTCGACCATGGAGCCGGTAGGCTGCGGCGAGTGCCGCGGCTCCTCCGTA 3′) and primer 2. The sequence was verified by double-stranded DNA sequence, and the desired product was inserted into the appropriate pRK5 plasmids.
Cell Culture and Transfection
HEK293, SH-SY5Y, or A172 cells were cultured in DMEM containing 10% FBS, antibiotics (penicillin, 100 units/ml; streptomycin, 0.1 mg/ml; and amphotericin B, 0.25 μg/ml) and 4 mm glutamine. Cells were plated on 6-well tissue culture plates (Nunc) and transfected after 24 h at 70–90% confluence using Lipofectamine 2000 (Invitrogen) with the indicated DNAs. Transfection of A172 cells was carried out without FBS or antibiotics. Cells were harvested 48–72 h post-transfection. Transfection of siRNA was carried out with 100 nm siRNA using Lipofectamine 2000 according to the specifications of the manufacturer, and cells were harvested 72 h after transfection. FBXO22 siRNA (Sigma), referred to as siRNA-988, consisted of 5′ GCCAUAAGAGAGCAAGGAA 3′.
Coimmunoprecipitations and Western Blot Analysis
HEK293A cells were cotransfected with Myc-FBXO22a and either HA-SR or HA-glucosamine 6-phosphate deaminase (GNPDA). Thirty-six hours after transfection, cells were lysed by gentle sonication in 20 mm Tris-HCl (pH 7.4), 1 mm EDTA, 30 μm MG132, 300 mm NaCl, and protease inhibitor mixture. After sonication, Triton X-100 was added to 1% final concentration, and the suspension was cleared by centrifugation at 16,000 × g for 10 min. Myc-FBXO22a was immunoprecipitated by adding anti-Myc matrix (Sigma) for 2 h under rotation at 4 °C. After washing six times with high stringency buffer consisting of 20 mm Tris-HCl (pH 7.4), 300 mm NaCl, and 1% Triton X-100, the immunoprecipitates were analyzed by SDS-PAGE and Western blot using mouse anti-HA 1:1000 (Sigma) and rabbit anti-Myc 1:500 (Sigma). Coimmunoprecipitation of SR and FBXO22b was carried out essentially as described above, except we employed overnight incubation with anti-HA matrix and 400 mm NaCl instead of 300 mm in the washing steps to prevent nonspecific binding.
To investigate the interaction of FBXO22 with the SCF complex, HEK293 cells were transfected with Myc-FBXO22b, Myc-FBXO22b Δ20–67, or Myc-FBXO22a. Thirty-six hours after transfection, MG132 (30 μm) was added for 12 h, and cells were lysed by gentle sonication in buffer containing 50 mm Tris-HCl (pH 7.4), 1 mm EDTA, 10% glycerol, 30 μm MG132, 140 mm NaCl, and protease inhibitor mixture. Then Nonidet P-40 was added to 1% final concentration, and the suspension was cleared by centrifugation for 10 min at 16,000 × g at 4 °C. Immunoprecipitation was performed with anti-Myc matrix (Sigma) for 3 h at 4 °C. The beads were washed six times with 50 mm Tris-HCl (pH 7.4), 10% glycerol, 140 mm NaCl, and 1% Nonidet P-40. The Western blot was probed using mouse anti-Cul1 1:500 (BD Biosciences), mouse anti-Skp1 1:500 (BD Biosciences), and mouse anti-Myc 1:2500 (Sigma).
For the endogenous interaction of SR with FBXO22, rat brains were homogenized using a glass homogenizer with 5 volumes of 50 mm Tris-HCl (pH 7.4), 140 mm NaCl, 1% CHAPS, 30 μm MG132, and protease inhibitor mixture. The homogenate was cleared by centrifugation at 30,000 × g for 20 min at 4 °C. 10 μg of purified anti-SR and rabbit normal IgG was coupled to protein G-agarose using dimethylpimelimidate (Sigma) as described previously (32) and incubated for 12 h with rat brain homogenate at 1.5 mg/ml. The immunoprecipitate was washed six times with 50 mm Tris-HCl (pH 7.4), 140 mm NaCl, and 0.5% CHAPS, and the coimmunoprecipitation was revealed with mouse anti-FBXO22 (1:100, Santa Cruz Biotechnology) and rabbit anti-SR serum (1:1000).
To investigate whether SR associates with SCFhFBXO22a, we employed conditions as described previously (33), with slight modifications. HEK293 cells were cotransfected with HA-hFBXO22a and Myc-KDM4A or Myc-SR. Forty-eight hours post-transfection, cells were lysed for 30 min on ice in buffer containing 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 0.2% Nonidet P-40, and protease inhibitor mixture (Mini Complete, Roche Diagnostics). The lysate was cleared by centrifugation at 14,000 × g for 10 min, and the immunoprecipitation was carried out using anti-Myc matrix (Sigma). After washing six times with lysis buffer, the samples were analyzed by SDS-PAGE and Western blot. In some experiments, HA-SR of HA-FBXO22a was cotransfected with either mouse Cul1 or mouse Skp1a in the pCMV-Sport6 vector (GE Healthcare), and coimmunoprecipitations were carried out as described above. To quantify the Western blots, the densitometry of the bands were calculated at the linear phase and divided by β-tubulin in each lane.
In Vivo Ubiquitination
HEK293 cells were transfected with FLAG-ubiquitin, HA-SR, and either GFP or Myc-hFBXO22a in the pRK5 plasmid. Twenty-four hours after transfection, cells were incubated with 30 μm MG132 for 12 h. Immunoprecipitation was carried out as described previously (34), with some modifications. Briefly, the cells were lysed by sonication in medium containing 10 mm Tris-HCl (pH 7.4), 1 mm EDTA, 20 mm N-ethylmaleimide, and protease inhibitor mixture. Subsequently, SDS was added to 1% final concentration, and the samples were boiled for 5 min. This was followed by a 10-fold dilution in buffer containing 20 mm Tris-HCl (pH 7.4), 2% Triton X-100, 0.5% deoxycholate, 1 mm EDTA, 100 mm NaCl, and protease inhibitor mixture. After centrifugation at 16,000 × g for 10 min to remove any insoluble material, the immunoprecipitation was carried out with anti-HA affinity matrix, and the immunoprecipitates were washed six times with 20 mm Tris-HCl (pH 7.4), 200 mm NaCl, 0.5% deoxycholate, 0.1% SDS, and 1% Triton X-100. Subsequently, FLAG-ubiquitin-SR conjugates were revealed with rabbit anti-FLAG (1:1000), and total immunoprecipitated SR was monitored with mouse anti-HA (1:1000).
Pulse-Chase Experiments
Half-life experiments were carried out as described previously (35), with the following modifications. HEK293 cells were transfected with HA-SR and hFBXO22a, hFBXO22b, or GFP at a SR/hFBXO22 cDNA ratio of 1:14. Forty-eight hours after transfection, the medium was replaced by DMEM lacking methionine/cysteine (Sigma) for 1 h. Then the cells were pulsed with methionine/cysteine-free medium containing 100 μCi of [35S]methionine/cysteine (PerkinElmer Life Sciences) for 70 min and subsequently chased in complete DMEM. At the specified times, the cells were harvested and lysed in buffer containing 20 mm Tris-HCl (pH 7.4), 0.3 m NaCl, 1% Triton X-100, 30 μm MG132, and protease inhibitors (Mini-complete, Roche Diagnostics). Immunoprecipitation of HA-SR was carried out with anti-HA affinity matrix (Covance), and the immunoprecipitates were washed six times with radioimmune precipitation assay buffer. The samples were resolved on SDS-PAGE gels, transferred to nitrocellulose membranes, and quantified by PhosphorImager analysis. The amount of immunoprecipitated HA-SR was checked by Western blot analysis, followed by densitometry of the chemiluminescent signal.
Brain Subcellular Fractionation
Brains of SR-KO mice (23) and WT controls were homogenized using a glass homogenizer with 5 volumes of 50 mm Tris-HCl (pH 7.4), 140 mm NaCl, 0.2% Triton X-100, 0.32 m sucrose, 1 mm MgCl2, 1 mm potassium Pi, and Mini Complete (Roche Diagnostics). The homogenate was centrifuged at 1250 × g for 10 min at 4 °C to yield crude nuclear fraction (P1) and supernatant 1 (S1). After two washes by centrifugation in homogenization buffer, P1 was suspended in 2 m sucrose, layered over a 2.4 m sucrose cushion, and centrifuged at 53,000 × g for 75 min at 4 °C to yield a purified nuclear pellet (36). For obtaining the cytosolic fraction, the S1 fraction was centrifuged at 200,000 × g for 30 min to remove membranes.
For subcellular fractionation of A172 cultures, the cells were lysed by three freeze and thaw cycles in buffer containing 20 mm Tris-HCl (pH 7.4), 100 mm NaCl, and protease inhibitor mixture. An aliquot of the homogenate was put aside, and the remaining lysate was centrifuged at 1500 × g for 10 min (4 °C) to give a supernatant and crude nuclear pellet. P1 was washed twice by centrifugation and purified further to produce a purified nuclear fraction as described previously (36, 37), with the following modifications. P1 was suspended in 10 mm Tris-HCl (pH 7.4), 0.25% Triton X-100, 1 mm potassium Pi (pH 6.5), 1 mm MgCl2, and 1.32 m sucrose supplemented with protease inhibitors and homogenized by six to seven strokes with a glass homogenizer. Subsequently, P1 was layered over a 1.7 m sucrose cushion and centrifuged at 53,000 × g for 75 min at 4 °C to obtain purified nuclei. Cytosolic and membrane fractions were obtained by centrifugation of S1 at 200,000 × g for 30 min. The membrane fraction was washed twice by centrifugation with lysis buffer.
Primary Cultures
Pregnant Sprague-Dawley rats were killed by quick decapitation following isoflurane anesthesia with the approval of the Committee for Supervision of Animal experiments (Technion-Israel Institute of Technology). Serum-free neuronal cultures from the cerebral cortex were prepared from E16–18 as described previously (24, 38) with the following modifications. The culture medium consisted of Neurobasal supplemented with B-27, 0.4 mm glutamine, and penicillin/streptomycin (Neurobasal + B-27). On DIV1, the cultured medium was changed to fresh Neurobasal + B27. Afterward, half of the culture media was replaced every 3 days to fresh Neurobasal + B27. Biochemical experiments were carried out on DIV10–12.
Primary astrocytic cultures were carried out with P0-P1 Sprague-Dawley rat pups as described previously (24). The cells were maintained in basal medium Eagle supplemented with 10% FBS, 0.4 mm glutamine, and penicillin/streptomycin and used at DIV14.
Lentivirus Production and shRNA-mediated Knockdown
A lentivirus harboring shRNA was produced by cotransfection of the pGIPZ vector (13 μg), pCMV-dR8.74 packing (8.7 μg), and VSVG envelope pMD2G (4.6 μg) in HEK293T cells (39). Medium containing virus was collected after 24 and 48 h post-transfection. The virus was concentrated by centrifugation at 116,000 × g for 2 h at 4 °C and resuspended in Neurobasal medium. Aliquots were stored at −70 °C until use. Primary neuronal cultures were infected on DIV4 with a multiplicity of infection of 30–50. Levels of FBXO22 were monitored by Western blot analysis with mouse anti-FBXO22 antibody (Sigma). Only cultures exhibiting at least 30–40% infection efficiency on DIV12–13 monitored by GFP fluorescence were employed. The pGIPZ construct containing FBXO22 shRNA (GE Healthcare) encompassed a mature antisense consisting of TATTCCTTCAATTTGAGGG (catalog no. RHS4430–98894301, referred to as 9889). Controls were carried out with pGIPZ non-silencing shRNA (catalog no. RHS4346, GE Healthcare).
Astrocytes were transduced at DIV10 with lentivirus harboring FBXO22-silencing or non-silencing shRNA (multiplicity of infection 20). The astrocytes were selected for 7 days with 2 μg/ml puromycin before use.
Immunocytochemistry
A172 cells were seeded on glass coverslips coated with poly-d-lysine (0.2–0.5 mg/ml) in 12-well plates at a concentration of 0.75–0.9 × 106 cells/well. After 72 h, cells were fixed for 20 min with fresh 4% paraformaldehyde. After washing with PBS, the cells were blocked and permeabilized at room temperature for 90 min with PBS supplemented with 8% normal goat serum, 80 mm NaCl, 2 mg/ml IgG-free BSA, and 0.1% Triton X-100. Colocalization was revealed by the use of serum anti-SR at 1:400 (8) and antibodies against FBXO22 (1:100) and KDEL (1:100) (Santa Cruz Biotechnology). Following primary antibody incubation for 16–20 h at 4 °C, the cells were blocked at room temperature with 6% normal goat serum (NGS) and 0.1% Triton X-100 in PBS. After washing the slides, anti-rabbit Cy3 and anti-mouse Cy2 were added to 4% NGS and 0.1% Triton X-100 in PBS. Afterward, the cells were washed, and DAPI was added for 30 s at a final concentration of 0.1 μg/ml for nuclear labeling, followed by three additional washes. Images were obtained using laser-scanning confocal microscopy (LSM 510 Meta laser-scanning confocal system (Zeiss)) with a ×63 oil objective. Optical sections of 2–3 μm were used at 1024 × 1024 pixel resolution. Final picture editing was done with 510 LSM software.
d-Serine Synthesis
For endogenous d-serine production, A172 cells were plated in 24-well plates at 0.1 × 106 cells/well and transfected with siRNA to FBXO22 or control siRNA as described in the previous section. Seventy-two hours after transfection, the cells were washed with basal medium Eagle containing 10% FCS and 4 mm glutamine. Then the medium was supplemented with 10 mm l-serine that was rendered free of contaminant d-serine as described previously (40). Synthesis of d-serine was monitored by HPLC of the culture media collected after 72 h and normalized by the SR expression levels in each well monitored by Western blot analysis and quantification using the Odissey system (LI-COR Biosciences). The identity of d-serine was confirmed by treating the media with recombinant d-serine deaminase from E. coli prior to HPLC analysis (40). In some experiments, d-serine was quantified by an enzymatic assay using recombinant yeast d-serine deaminase and Amplex Red (Invitrogen) amplification as described previously (41).
Serine Racemase Activity in Vitro
Recombinant His-SR (0.1 μg) was preincubated in PBS containing 3% glycerol and 5 μg of soluble GST-FBXO22a, GST-α-synuclein, or GST-CHIP (a 30- to 40 fold-excess of GST fusion proteins to SR monomer) for 1 h at 4 °C in silanized 1.5-ml polypropylene tubes. Subsequently, the samples were diluted 20-fold into reaction medium containing 20 mm Tris-HCl (pH 7.4), 1 mm MgCl2, 100 μm ATP, 30 μm pyridoxal-5′-phosphate, 0.3 mm NADH, and 0.5 units/ml lactate dehydrogenase. SR activity was started by addition of 10 mm l-serine Ο-sulfate, an efficient SR substrate generating pyruvate, sulfate, and NH4. Pyruvate was quantified by monitoring NADH absorbance as described previously (42).
RESULTS
To identify new SR interactors that regulate d-serine synthesis in cells, we carried out immunoprecipitation of HA-SR from SH-SY5Y neuroblastoma cells, followed by LC-MS analysis. The immunoprecipitate was washed extensively under high stringency conditions that favored the detection of strong interactors. Under these conditions, human FBXO22 (hFBXO22) was the only additional protein identified in the 36–42 kDa range of HA-SR gel bands, indicating the possible presence of a SR-FBXO22 complex (Fig. 1A).
FIGURE 1.

Association of SR with FBXO22 in vitro and in vivo. A, schematic of the FBXO22a and b isoforms. FBXO22b is a predicted smaller isoform that lacks the FIST C domain and whose last 11 amino acids at the C terminus are different (gray box). B, purified His-SR binds to GST-FBXO22 in vitro. His-SR (0.4 μg/ml) was incubated with glutathione-agarose beads containing 0.8 μg/ml GST-FBXO22a, 1.5 μg/ml GST-FBXO22b, or 30 μg/ml GST-α-synuclein. SR bound to the beads was monitored with anti-SR serum (1:1000) (top panel). Bottom panel, the GST-fusion proteins used in the binding experiments stained by Ponceau S. C, SR coimmunoprecipitates with FBXO22a. HEK 293 cells were cotransfected with Myc-FBXO22a and either HA-SR or HA-GNPDA (control protein), and immunoprecipitation (IP) was carried out with anti-Myc matrix. HA-SR or HA-GNPDA in the immunoprecipitate were detected with mouse anti-HA (1:1000) (top panel). Immunoprecipitated Myc-FBXO22a was detected with mouse anti-Myc (1:5000) (center panel). The bottom panel corresponds to the input (5%) probed with anti-HA. D, SR/FBXO22 interaction does not depend on the F-box domain. Myc-FBXO22b or Myc-FBXO22b Δ20–67 (lacking the F-box domain) coimmunoprecipitate with HA-SR but not with HA-FKBP12 control protein (top panel). The center panel corresponds to HA-SR and HA-FKBP12 immunoprecipitate probed with anti-HA. Levels of Myc-FBXO22a and Myc-FBXO22b in the input were monitored with polyclonal anti-Myc (1:1000) (bottom panel). The blots are representative of at least three experiments. E, coimmunoprecipitation of Myc-FBXO22a, Myc-FBXO22b, or Myc-FBXO22b Δ20–67 with endogenous Cul1 and Skp1 from HEK293 cells. Myc-FBXO22a interacts with both Skp1 and Cul1, which are the core components of the SCF complex. A much weaker interaction was observed with Myc-FBXO22b, whereas no binding was detectable with Myc-FBXO22b lacking the F-box region (Δ20–67). Top panel, coimmunoprecipitation with Skp1 monitored mouse anti-Skp1 (1:500). Center panel, coimmunoprecipitation with Cul1 monitored with mouse anti-Cul1 (1:500). F, SR and FBXO22A interact in vivo. Immunoprecipitation from rat brain homogenate with anti-SR demonstrates coimmunoprecipitation of SR to FBXO22 but not when rabbit IgG was used (B). The coimmunoprecipitation was checked using anti-FBXO22 (1:100, top panel). SR presence was verified using anti-SR serum (1:1000, bottom panel). The blots are representative of at least three experiments.
Human FBXO22 protein contains an N-terminal F-box domain that mediates its interaction with the Skp1 component of the Skp1·Cullin·F-box (SCF) ubiquitin-ligase complex (43, 44). Analysis of the expressed sequence tag database indicates the existence of two FBXO22 isoforms, 22a and 22b (Fig. 1A). FBXO22a controls the degradation of KDM4A, a histone demethylase (33), and seems to be involved in the inflammatory response during Salmonella infection (45). In its C-terminal region, FBXO22a has an F-box intracellular transduction C-terminal domain (FIST C), presumably involved in amino acid sensing and signal transduction (46). The shorter and previously uncharacterized FBXO22b isoform lacks the FIST C region and differs in its 11 C-terminal amino acids because of alternative splicing (Fig. 1A). A search in the expressed sequence tags database revealed only two expressed sequence tags containing the shorter FBXO22b isoform compared with more than 100 expressed sequence tags containing FBXO22a, indicating that the latter is the dominant isoform.
To verify whether SR interacts directly with hFBXO22, we monitored the in vitro binding of purified His-tagged SR protein with purified GST-hFBXO22a and hFBXO22b fusion proteins in the presence of excess bovine serum albumin to prevent nonspecific binding (Fig. 1B). His-SR binds to hFBXO22 isoforms but not to GST-α-synuclein control protein, even when present at large excess, indicating that hFBXO22 directly interacts with SR (Fig. 1B).
To confirm that SR and hFBXO22 interact, we carried out coimmunoprecipitation from transfected HEK-293 cells. Myc-hFBXO22a coimmunoprecipitated with HA-SR (Fig. 1C) but not with unrelated control protein HA-GNPDA (Fig. 1C). A construct that mimics the smaller FBXO22 isoform, Myc-hFBXO22b (Fig. 1D), also coimmunoprecipitated with HA-SR but not with HA-FKBP12 (Fig. 1D). Immunoprecipitation of SR with the smaller isoform (hFBXO22b) that lacks the FIST C domain indicates that this region is not required for the interaction (Fig. 1D).
In contrast to hFBXO22a, the smaller hFBXO22b isoform displays only minimal binding to the Skp1 and Cul1 components of the SCF complex (Fig. 1E, compare lanes 1 and 3). Deletion of the F-Box domain (Myc-hFBXO22b Δ20–67 mutant) abrogates the association of hFBXO22 with the SCF complex, inferred from the lack of coimmunoprecipitation with Skp1 and Cul1 (Fig. 1E, lane 2). However, the Myc-hFBXO22b Δ20–67 mutant still coimmunoprecipitates with HA-SR (Fig. 1D, lane 2), indicating that the SFC complex is not required for SR/FBXO22 interaction.
SR and rat FBXO22a coimmunoprecipitate from rat brain homogenates, indicating that the proteins appear to interact in vivo (Fig. 1F). We did not detect any band corresponding to the smaller isoform FBXO22b in brain or cultured cells, likely because of very low levels of expression. Therefore, most subsequent experiments were carried out with the FBXO22a isoform, a more likely candidate to regulate SR. Indeed, in situ hybridization of mouse brain shows that FBXO22a is nearly ubiquitously expressed in all easily defined brain cell types and all brain regions (47) (available from the Allen Brain Atlas).
Harper and co-workers (33) reported that the SCFhFBXO22a complex ubiquitinates and regulates the steady-state levels of KDM4A, which demethylates histone H3 lysine 9 and 36. We wondered whether hFBXO22a is also required for ubiquitination and degradation of SR by the ubiquitin-proteasomal system. We found that cotransfection of hFBXO22a with SR does not promote SR ubiquitination either with or without a proteasomal inhibitor (Fig. 2A). Levels of SR ubiquitination with FBXO22a were even somewhat lower than the control levels (Fig. 2A, compare lanes 1 and 3). Overexpression of hFBXO22a or hFBXO22b does not affect the [S35]SR half-life in pulse-chase experiments (Fig. 2B), indicating the lack of a significant effect on SR turnover.
FIGURE 2.

hFBXO22a does not affect SR turnover or targeting to the proteasome. A, SR ubiquitination (Ubiq) is not increased when cotransfected with FBXO22a. HA-SR was cotransfected with Myc-hFBXO22a or GFP and incubated with or without MG132 (a proteasome inhibitor). HA immunoprecipitate (IP) was probed with mouse anti-FLAG (1:500, Sigma) to reveal ubiquitin conjugates (top panel). Non-ubiquitinated SR was revealed with mouse anti-HA (1:1000, bottom panel). B, SR turnover is unaffected by hFBXO22 overexpression. HA-SR was cotransfected with hFBXO22a, hFBXO22b, or GFP (n = 3). Cells were incubated in a medium containing [35S]methionine/cysteine and chased for the indicated times (top panel). Bottom panel, PhosphorImager analysis of [35S]SR. C, endogenous FBXO22a knockdown does not affect SR ubiquitination. HEK293 cells were cotransfected with HA-SR, FLAG-ubiquitin, siRNA to hFBXO22a, or control (Ctl) siRNA, and ubiquitination was revealed as in Fig. 2A after 12 h incubation with 30 μm MG132. Bottom panel (input), successful knockdown of endogenous hFBXO22a expression revealed with mouse anti-FBXO22 (1:1000). D, knockdown of endogenous hFBXO22a does not affect endogenous SR steady-state levels in A172 glioblastoma cells (top panel). Transfection was performed with siRNA against hFBXO22a, which down-regulated hFBXO22a expression (center panel). Bottom panel, β-actin loading control. The graph shows the lack of change in the steady-state levels of endogenous SR from seven independent experiments. a.u., arbitrary units; ns, not significant. E, SR preferentially interacts with free FBXO22a species. Myc-SR or Myc-KDM4A was cotransfected with HA-FBXO22a and immunoprecipitated with anti-Myc. Cul1 was present in the Myc-KDM4A/HA-FBXO22a coimmunoprecipitate but absent from the Myc-SR·HA-FBXO22a complex (top panel). HA-FBXO22a was revealed with mouse anti-HA (1:1000, center panel), whereas immunoprecipitated Myc-KDM4A or Myc-SR was revealed with mouse anti-Myc (1:1000, Sigma, bottom panel). F and G, HA-FBXO22a, but not HA-SR, interacts with ectopically expressed Cul1 (F) or Skp1a (G). H, FBXO22a, KDM4A, and H3K9Me3 levels are unchanged in brain homogenates of SR-KO mice. Cyt, brain cytosolic fraction; PN, brain purified nuclear fraction. The blots are representative of at least three different experiments.
Conceivably, ectopically expressed FBXO22a may generate artifacts because of its competition with the endogenous FBXO22. To more definitely exclude a role of FBXO22a in SR ubiquitination, we employed siRNA-mediated FBXO22a knockdown strategy. We found no changes in SR ubiquitination when levels of endogenous FBXO22 were decreased efficiently (Fig. 2C, compare lanes 1 and 2 with lanes 3 and 4).
To investigate whether endogenous hFBXO22a affects SR targeting to the proteasome, we tested the effect of hFBXO22a knockdown on steady-state levels of endogenous SR in human brain glioblastoma A172 cells (Fig. 2D). Endogenous SR expression was unaffected by FBXO22 siRNA, which efficiently knocked down endogenous hFBXO22a expression (Fig. 2D). These data are consistent with the notion that hFBXO22a does not play a role in SR degradation by the ubiquitin system.
Because hFBXO22a coimmunoprecipitates with components of the SCF complex (Fig. 1E), we investigated whether the pool of hFBXO22a interacting with SR is associated with the SCF complex. We found that Cul1 is absent from the SR/hFBXO22 coimmunoprecipitate, indicating that SR interacts preferentially with a free hFBXO22a pool (Fig. 2E). As a control, we confirmed that the SCFhFBXO22 complex substrate KDM4A coimmunoprecipitates with both hFBXO22a and Cul1 under the same experimental conditions (Fig. 2E) (33). Furthermore, FBXO22a, but not SR, coimmunoprecipitated with ectopically expressed Cul1 (Fig. 2F) or Skp1a (Fig. 2G). These data are consistent with the notion that SR does not interact with the SCFhFBXO22 complex.
Does SR affect FBXO22a function? To address this question, we monitored the levels of FBXO22a and the SCFFBXO22a substrate KDM4A in brain homogenates from wild-type and SR-KO mice (Fig. 2H). We found that levels of both FBXO22a and KDM4A were unchanged in SR-KO mice, indicating that the function of SCFFBXO22a is not changed globally in SR-KO mice. Likewise, methylated histone 3, which is demethylated by KDM4A, was unaffected, excluding a significant effect of SR on FBXO22a targets (Fig. 2H).
We next examined whether FBXO22 regulates SR activity. SR binds avidly to membrane lipids, especially of the phosphoinositol type (29). This leads to inhibition of SR activity. Accordingly, NMDAR-elicited translocation of SR from the cytosol to the membrane completely inactivates d-serine synthesis in neurons (30). However, despite the apparent high affinity of SR to lipids, most of the protein is soluble in the cytosol under basal conditions. We found that FBXO22a seems to be important in keeping SR in the cytosol (Fig. 3, A and B). siRNA-mediated hFBXO22a knockdown increased severalfold the levels of endogenous SR associated to the membrane fraction of glioblastoma cells (Fig. 3A). Levels of endogenous SR in homogenate (Fig. 3A, Hom) were unchanged, indicating that FBXO22a does not affect SR expression. Knockdown of endogenous hFBXO22a was monitored in the homogenate (Fig. 3A, Hom). As expected, the levels of the SCFFBXO22a substrate KDM4A increased in the purified nucleus and membrane fraction, confirming the role of hFBXO22a in regulating KDM4A levels (33).
FIGURE 3.

FBXO22a primarily affects SR subcellular localization. A, siRNA-mediated knockdown of endogenous hFBXO22 expression in A172 glioblastoma cells increases endogenous SR levels in the membrane fraction. Levels of endogenous SR in the membrane fraction (Mem) increased upon hFBXO22a knockdown, whereas β-tubulin (β-tub) levels were unchanged. Knockdown of endogenous hFBXO22a monitored in the homogenate fraction (Hom) did not affect the total expression of SR or GAPDH revealed with rabbit anti-SR (1:1000) or mouse anti-GAPDH (1: 200). siRNA to hFBXO22a increased in the levels of its substrate KDM4A both in the purified nuclei fraction (KDM4 pn) and in the cytosolic fraction (KDM4 cyt), as revealed by rabbit anti-KDM4A (1:500). The graph depicts the increase in membrane-bound SR in three experiments. a.u., arbitrary units; Ctl, control. B, knockdown of endogenous rat FBXO22 expression in primary cortical neuron cultures increases membrane-bound SR levels. Primary cortical neuron cultures were infected with a lentivirus harboring shRNA to hFBXO22 or non-silencing (NS) shRNA and compared with uninfected cultures (Ctl). Subcellular fractionation reveals an increase in the levels of endogenous membrane-bound SR with no change in the β-tubulin loading control. Total levels of SR and β-actin in homogenate (Hom) were unaffected, whereas shRNA to rat FBXO22 was associated with a decrease in its expression (Hom, bottom panel). The graph depicts the increase in membrane-bound SR in three experiments. C–H, siRNA to hFBXO22 increases the extent of colocalization of endogenous SR (red) with the endoplasmic reticulum marker KDEL (green) in A172 glioblastoma cells, as analyzed by confocal laser microscopy. Control (C–E) and siRNA to hFBXO22 (F–H) were used. Scale bar = 50 μm. The panels are representative of at least three different experiments. *, p < 0.05; **, p < 0.01.
Similar to that seen in glioblastoma cells, infection of primary cortical neuronal cultures with shRNA to rat FBXO22a increases the levels of membrane-bound SR without changing β-tubulin levels (Fig. 3B). Total expression of SR was unaffected by shRNA to FBXO22a but significantly decreased the levels of endogenous FBXO22a (Fig. 3B, Hom).
Immunocytochemical experiments confirmed the biochemical findings. hFBXO22a knockdown appears to increase levels of SR that colocalizes with intracellular membranes, as seen by increased colocalization with KDEL, an endoplasmic reticulum marker (Fig. 4, C–H).
FIGURE 4.
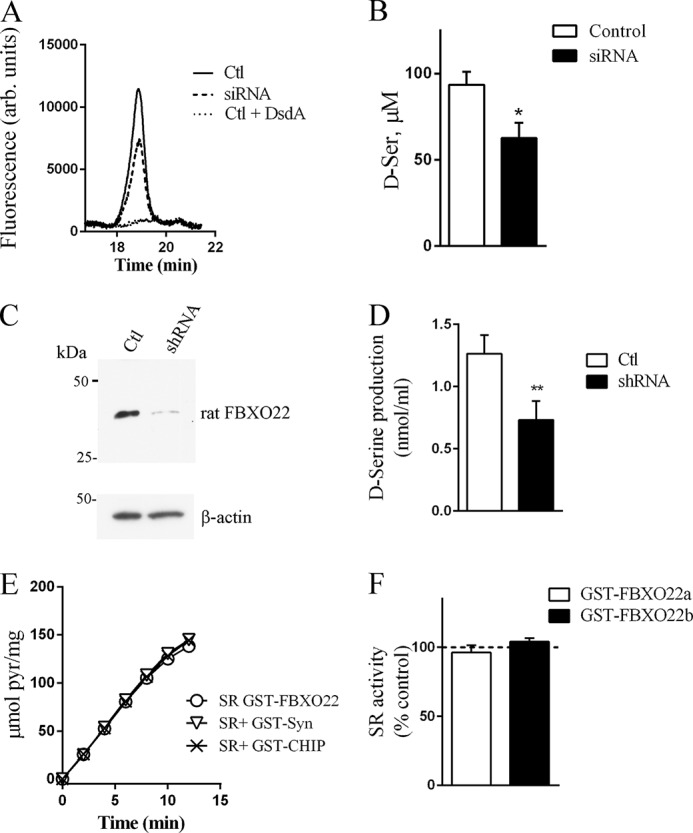
FBXO22a is required for optimal SR activity in cells. A, HPLC analysis of endogenous d-serine production in A172 glioblastoma cells revealed a discrete d-serine peak in the culture media of cells transfected with non-silencing siRNA (Ctl), which decreased in cells transfected with siRNA to hFBXO22. Treatment with d-serine deaminase enzyme (DsdA) prior to HPLC analysis abolished the peak of d-serine, indicating that it corresponds to authentic d-serine. arb. units, arbitrary units. B, quantification of d-serine (d-Ser) in culture media of cells transfected with non-silencing siRNA (Ctl) or siRNA to hFBXO22. Values represent the mean ± S.E. of four experiments with different cell cultures. C, knockdown of endogenous rat FBXO22 in primary astrocyte cultures infected with lentivirus containing FBXO22 shRNA when compared with non-silencing shRNA. D, FBXO22 knockdown decreases d-serine production in primary astrocyte cultures. E and F, lack of effect of hFBXO22a (E and F) or hFBXO22b (F) on SR activity in vitro. A 30-fold molar excess of GST-hFBXO22 isoforms did not affect His-SR activity when compared with GST-α-synuclein or GST-CHIP controls. Values represent the mean ± S.E. of at least three experiments with different protein preparations. *, p < 0.05.
Because SR is inactivated upon binding to membranes (29, 30), we next examined whether the effects of hFBXO22a on SR subcellular localization may influence d-serine production. We found that siRNA-mediated knockdown of endogenous hFBXO22a in glioblastoma cells decreases endogenous d-serine production by about 40% (Fig. 4, A and B), indicating that hFBXO22a is required for optimal d-serine synthesis in the cellular environment. Similarly, lentivirus-mediated shRNA knockdown of FBXO22 in rat primary astrocytes (Fig. 4C) decreased d-serine production by 40% (Fig. 4D). Experiments employing recombinant proteins demonstrate that a 30- to 40-fold molar excess of hFBXO22a or hFBXO22b does not directly affect the activity of the soluble recombinant SR measured under standard conditions in vitro (Fig. 4, E and F). The data are consistent with the notion that changes in subcellular localization of SR (Fig. 3), rather than a direct effect on SR activity at the cytosol, mediate FBXO22 effect in d-serine synthesis observed in cells (Fig. 4, A–D).
DISCUSSION
We described a new interaction of SR with FBXO22a, which is required for optimal d-serine synthesis. We confirmed the interaction by biochemical methods and showed that knockdown of FBXO22a decreases d-serine synthesis in cells. This was associated with higher levels of SR in the membrane fraction of glioblastoma cells and primary neurons, suggesting that hFBXO22a is important for proper targeting of SR. Furthermore, changes in SR subcellular localization by FBXO22 are observed in both glia and neuronal cells, indicating that this may be a general regulatory mechanism. Except for Golga3, a protein that affects SR half-life (48), no other SR interacting protein was demonstrated to affect its function in both glial and neuronal cells.
F-Box proteins are generally thought to play a role in the ubiquitin-proteasome system (43, 44, 49, 50). The SCFFBXO22a ubiquitin ligase complex regulates the degradation of KDM4A and, ultimately, the levels of histone 3 methylation and transcription of KDM4A target genes (33). The interaction of FBXO22a with KDM4A is mediated via its FIST domain, whereas the F-Box motif is required for binding to SKP1 at the SCF complex (33). Conversely, we found that SR still interacts with an FBXO22 mutant that lacks both FIST and the F-Box motif (hFBXO22b Δ20–67), indicating that both of these domains are not required for the SR-FBXO22 interaction.
Our data indicate that FBXO22a has a non-canonical role in regulating SR activity by primarily affecting its intracellular localization. A lack of effect on SR ubiquitination with either ectopic expression of FBXO22a or siRNA knockdown indicates that SR is not a substrate of SCFFBXO22a. Furthermore, FXBO22a does not change the SR half-life or steady-state levels. On the other hand, we found that FBXO22a is required for SR activity in cells by preventing the accumulation of membrane-bound SR species, which is inactive toward d-serine synthesis (30). This effect appears to be mediated by free FBXO22a species without the participation of the SCFFBXO22a complex.
A number of F-box-containing proteins play additional roles that are not related to their ubiquitination activity as components of the SCF ubiquitin-ligase complex (51–53). Emi1/Rca1 is an F-box protein that inhibits the anaphase-promoting complex/cyclosome. The anaphase-promoting complex/cyclosome is regulated by cyclin-B, whose destruction is blocked by Emi1, but does not require its F-box region (54, 55). Fbxo7 associates with Cdk6 and functions as an assembly scaffold without affecting the ubiquitination of the subunits (56). Another F-box-containing protein, Fbs1, functions as a chaperone and prevents aggregation of glycoproteins (57).
How does FBXO22a modulate SR localization and d-serine production? SR displays lipid binding motifs that mediate its binding to inositol phospholipids (29). Conceivably, FBXO22a may function as a SR chaperone and occlude its lipid-binding region. This will prevent SR interaction with membranes, known to be deleterious for SR activity (29, 30). We found that knockdown of endogenous FBXO22 led to an increase in the colocalization of SR with KDEL, suggesting the presence of SR in the endoplasmic reticulum network. Proteins departing from the endoplasmic reticulum are known to undergo different posttranslational modifications that may also contribute to SR inhibition.
FBXO22a contains a C-terminal FIST-C domain (Fig. 1A) involved in signal transduction and may bind small ligand molecules, possibly amino acids or their derivatives (46). Even though FBXO22-SR interaction was not mediated by the FIST domain, it is conceivable that this region provides another layer of regulation of d-serine dynamics.
In sum, our data disclose an atypical role of FBXO22 unrelated to the ubiquitin system. FBXO22 enhances SR activity primarily by preventing its binding to membranes. Small molecules that disrupt SR-FBXO22 interaction provide a new strategy to inhibit d-serine production and prevent the overactivation of NMDARs that occurs following stroke and neurodegenerative conditions.
Acknowledgments
We thank Dr. Dina Rosenberg, Goren Kolodney, and Hazem Safory for help with primary culture preparations, Dr. Elena Dumin for advice, and Edith Suss-Toby for confocal microscopy assistance.
This work was supported by the Israel Science Foundation, by the Legacy Heritage Fund, by the Allen and Jewel Prince Center for Neurodegenerative Disorders of the Brain, and by the Herbert N. Goldman Medical Research Fund (to H. W.).
- NMDAR
- NMDA receptor
- SR
- serine racemase
- SCF
- Skp1·Cullin·F-box
- GNPDA
- glucosamine 6-phosphate deaminase
- P1
- crude nuclear fraction
- E16
- embryonic day 16
- DIV
- day(s) in vitro
- FIST C
- F-box intracellular transduction C-terminal domain
- CHIP
- C terminus of Hsc70-interacting protein.
REFERENCES
- 1. Traynelis S. F., Wollmuth L. P., McBain C. J., Menniti F. S., Vance K. M., Ogden K. K., Hansen K. B., Yuan H., Myers S. J., Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnson J. W., Ascher P. (1987) Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325, 529–531 [DOI] [PubMed] [Google Scholar]
- 3. Mothet J. P., Parent A. T., Wolosker H., Brady R. O., Jr., Linden D. J., Ferris C. D., Rogawski M. A., Snyder S. H. (2000) d-Serine is an endogenous ligand for the glycine site of the N-methyl-d-aspartate receptor. Proc. Natl. Acad. Sci. U.S.A. 97, 4926–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Panatier A., Theodosis D. T., Mothet J. P., Touquet B., Pollegioni L., Poulain D. A., Oliet S. H. (2006) Glia-derived d-serine controls NMDA receptor activity and synaptic memory. Cell 125, 775–784 [DOI] [PubMed] [Google Scholar]
- 5. Rosenberg D., Artoul S., Segal A. C., Kolodney G., Radzishevsky I., Dikopoltsev E., Foltyn V. N., Inoue R., Mori H., Billard J. M., Wolosker H. (2013) Neuronal d-serine and glycine release via the Asc-1 transporter regulates NMDA receptor-dependent synaptic activity. J. Neurosci. 33, 3533–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Henneberger C., Papouin T., Oliet S. H., Rusakov D. A. (2010) Long-term potentiation depends on release of d-serine from astrocytes. Nature 463, 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mothet J. P., Rouaud E., Sinet P. M., Potier B., Jouvenceau A., Dutar P., Videau C., Epelbaum J., Billard J. M. (2006) A critical role for the glial-derived neuromodulator d-serine in the age-related deficits of cellular mechanisms of learning and memory. Aging Cell 5, 267–274 [DOI] [PubMed] [Google Scholar]
- 8. Kartvelishvily E., Shleper M., Balan L., Dumin E., Wolosker H. (2006) Neuron-derived d-serine release provides a novel means to activate N-methyl-d-aspartate receptors. J. Biol. Chem. 281, 14151–14162 [DOI] [PubMed] [Google Scholar]
- 9. Shleper M., Kartvelishvily E., Wolosker H. (2005) D-serine is the dominant endogenous coagonist for NMDA receptor neurotoxicity in organotypic hippocampal slices. J. Neurosci. 25, 9413–9417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wolosker H., Blackshaw S., Snyder S. H. (1999) Serine racemase: a glial enzyme synthesizing d-serine to regulate glutamate-N-methyl-d-aspartate neurotransmission. Proc. Natl. Acad. Sci. U.S.A. 96, 13409–13414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wolosker H., Sheth K. N., Takahashi M., Mothet J. P., Brady R. O., Jr., Ferris C. D., Snyder S. H. (1999) Purification of serine racemase: biosynthesis of the neuromodulator d-serine. Proc. Natl. Acad. Sci. U.S.A. 96, 721–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Miranda J., Panizzutti R., Foltyn V. N., Wolosker H. (2002) Cofactors of serine racemase that physiologically stimulate the synthesis of the N-methyl-d-aspartate (NMDA) receptor coagonist d-serine. Proc. Natl. Acad. Sci. U.S.A. 99, 14542–14547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Foltyn V. N., Bendikov I., De Miranda J., Panizzutti R., Dumin E., Shleper M., Li P., Toney M. D., Kartvelishvily E., Wolosker H. (2005) Serine racemase modulates intracellular d-serine levels through an α,β-elimination activity. J. Biol. Chem. 280, 1754–1763 [DOI] [PubMed] [Google Scholar]
- 14. Balu D. T., Li Y., Puhl M. D., Benneyworth M. A., Basu A. C., Takagi S., Bolshakov V. Y., Coyle J. T. (2013) Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc. Natl. Acad. Sci. U.S.A. 110, E2400–E2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Basu A. C., Tsai G. E., Ma C. L., Ehmsen J. T., Mustafa A. K., Han L., Jiang Z. I., Benneyworth M. A., Froimowitz M. P., Lange N., Snyder S. H., Bergeron R., Coyle J. T. (2009) Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol. Psychiatry 14, 719–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Labrie V., Fukumura R., Rastogi A., Fick L. J., Wang W., Boutros P. C., Kennedy J. L., Semeralul M. O., Lee F. H., Baker G. B., Belsham D. D., Barger S. W., Gondo Y., Wong A. H., Roder J. C. (2009) Serine racemase is associated with schizophrenia susceptibility in humans and in a mouse model. Hum. Mol. Genet. 18, 3227–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Inoue R., Hashimoto K., Harai T., Mori H. (2008) NMDA- and β-amyloid1–42-induced neurotoxicity is attenuated in serine racemase knock-out mice. J. Neurosci. 28, 14486–14491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mustafa A. K., Ahmad A. S., Zeynalov E., Gazi S. K., Sikka G., Ehmsen J. T., Barrow R. K., Coyle J. T., Snyder S. H., Doré S. (2010) Serine racemase deletion protects against cerebral ischemia and excitotoxicity. J. Neurosci. 30, 1413–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mothet J. P., Pollegioni L., Ouanounou G., Martineau M., Fossier P., Baux G. (2005) Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter d-serine. Proc. Natl. Acad. Sci. U.S.A. 102, 5606–5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Balu D. T., Takagi S., Puhl M. D., Benneyworth M. A., Coyle J. T. (2014) d-Serine and serine racemase are localized to neurons in the adult mouse and human forebrain. Cell Mol. Neurobiol. 34, 419–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benneyworth M. A., Li Y., Basu A. C., Bolshakov V. Y., Coyle J. T. (2012) Cell selective conditional null mutations of serine racemase demonstrate a predominate localization in cortical glutamatergic neurons. Cell Mol. Neurobiol. 32, 613–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ehmsen J. T., Ma T. M., Sason H., Rosenberg D., Ogo T., Furuya S., Snyder S. H., Wolosker H. (2013) d-Serine in glia and neurons derives from 3-phosphoglycerate dehydrogenase. J. Neurosci. 33, 12464–12469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miya K., Inoue R., Takata Y., Abe M., Natsume R., Sakimura K., Hongou K., Miyawaki T., Mori H. (2008) Serine racemase is predominantly localized in neurons in mouse brain. J. Comp. Neurol. 510, 641–654 [DOI] [PubMed] [Google Scholar]
- 24. Rosenberg D., Kartvelishvily E., Shleper M., Klinker C. M., Bowser M. T., Wolosker H. (2010) Neuronal release of d-serine: a physiological pathway controlling extracellular d-serine concentration. FASEB J. 24, 2951–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim P. M., Aizawa H., Kim P. S., Huang A. S., Wickramasinghe S. R., Kashani A. H., Barrow R. K., Huganir R. L., Ghosh A., Snyder S. H. (2005) Serine racemase: activation by glutamate neurotransmission via glutamate receptor interacting protein and mediation of neuronal migration. Proc. Natl. Acad. Sci. U.S.A. 102, 2105–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fujii K., Maeda K., Hikida T., Mustafa A. K., Balkissoon R., Xia J., Yamada T., Ozeki Y., Kawahara R., Okawa M., Huganir R. L., Ujike H., Snyder S. H., Sawa A. (2006) Serine racemase binds to PICK1: potential relevance to schizophrenia. Mol. Psychiatry 11, 150–157 [DOI] [PubMed] [Google Scholar]
- 27. Ma T. M., Abazyan S., Abazyan B., Nomura J., Yang C., Seshadri S., Sawa A., Snyder S. H., Pletnikov M. V. (2013) Pathogenic disruption of DISC1-serine racemase binding elicits schizophrenia-like behavior via d-serine depletion. Mol. Psychiatry 18, 557–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mustafa A. K., Kumar M., Selvakumar B., Ho G. P., Ehmsen J. T., Barrow R. K., Amzel L. M., Snyder S. H. (2007) Nitric oxide S-nitrosylates serine racemase, mediating feedback inhibition of d-serine formation. Proc. Natl. Acad. Sci. U.S.A. 104, 2950–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mustafa A. K., van Rossum D. B., Patterson R. L., Maag D., Ehmsen J. T., Gazi S. K., Chakraborty A., Barrow R. K., Amzel L. M., Snyder S. H. (2009) Glutamatergic regulation of serine racemase via reversal of PIP2 inhibition. Proc. Natl. Acad. Sci. U.S.A. 106, 2921–2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Balan L., Foltyn V. N., Zehl M., Dumin E., Dikopoltsev E., Knoh D., Ohno Y., Kihara A., Jensen O. N., Radzishevsky I. S., Wolosker H. (2009) Feedback inactivation of d-serine synthesis by NMDA receptor-elicited translocation of serine racemase to the membrane. Proc. Natl. Acad. Sci. U.S.A. 106, 7589–7594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jin J., Cardozo T., Lovering R. C., Elledge S. J., Pagano M., Harper J. W. (2004) Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 18, 2573–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harlow E., Lane D. (1988) Antibodies: a Laboratory Manual, pp. 343–345, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 33. Tan M. K., Lim H. J., Harper J. W. (2011) SCF(FBXO22) regulates histone H3 lysine 9 and 36 methylation levels by targeting histone demethylase KDM4A for ubiquitin-mediated proteasomal degradation. Mol. Cell. Biol. 31, 3687–3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haskin J., Szargel R., Shani V., Mekies L. N., Rott R., Lim G. G., Lim K. L., Bandopadhyay R., Wolosker H., Engelender S. (2013) AF-6 is a positive modulator of the PINK1/parkin pathway and is deficient in Parkinson's disease. Hum. Mol. Genet. 22, 2083–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rott R., Szargel R., Haskin J., Bandopadhyay R., Lees A. J., Shani V., Engelender S. (2011) α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. U.S.A. 108, 18666–18671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mullen R. J., Buck C. R., Smith A. M. (1992) NeuN, a neuronal specific nuclear protein in vertebrates. Development 116, 201–211 [DOI] [PubMed] [Google Scholar]
- 37. Hara M. R., Agrawal N., Kim S. F., Cascio M. B., Fujimuro M., Ozeki Y., Takahashi M., Cheah J. H., Tankou S. K., Hester L. D., Ferris C. D., Hayward S. D., Snyder S. H., Sawa A. (2005) S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7, 665–674 [DOI] [PubMed] [Google Scholar]
- 38. Brewer G. J., Torricelli J. R., Evege E. K., Price P. J. (1993) Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res. 35, 567–576 [DOI] [PubMed] [Google Scholar]
- 39. Zufferey R., Nagy D., Mandel R. J., Naldini L., Trono D. (1997) Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 15, 871–875 [DOI] [PubMed] [Google Scholar]
- 40. Radzishevsky I., Wolosker H. (2012) An enzymatic-HPLC assay to monitor endogenous d-serine release from neuronal cultures. Methods Mol. Biol. 794, 291–297 [DOI] [PubMed] [Google Scholar]
- 41. Ito T., Takahashi K., Naka T., Hemmi H., Yoshimura T. (2007) Enzymatic assay of d-serine using d-serine dehydratase from Saccharomyces cerevisiae. Anal. Biochem. 371, 167–172 [DOI] [PubMed] [Google Scholar]
- 42. Panizzutti R., De Miranda J., Ribeiro C. S., Engelender S., Wolosker H. (2001) A new strategy to decrease N-methyl-d-aspartate (NMDA) receptor coactivation: inhibition of d-serine synthesis by converting serine racemase into an eliminase. Proc. Natl. Acad. Sci. U.S.A. 98, 5294–5299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bai C., Sen P., Hofmann K., Ma L., Goebl M., Harper J. W., Elledge S. J. (1996) SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 86, 263–274 [DOI] [PubMed] [Google Scholar]
- 44. Cardozo T., Pagano M. (2004) The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 5, 739–751 [DOI] [PubMed] [Google Scholar]
- 45. Pilar A. V., Reid-Yu S. A., Cooper C. A., Mulder D. T., Coombes B. K. (2012) GogB is an anti-inflammatory effector that limits tissue damage during Salmonella infection through interaction with human FBXO22 and Skp1. PLoS Pathog. 8, e1002773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Borziak K., Zhulin I. B. (2007) FIST: a sensory domain for diverse signal transduction pathways in prokaryotes and ubiquitin signaling in eukaryotes. Bioinformatics 23, 2518–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lein E. S., Hawrylycz M. J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A. F., Boguski M. S., Brockway K. S., Byrnes E. J., Chen L., Chen T. M., Chin M. C., Chong J., Crook B. E., Czaplinska A., Dang C. N., Datta S., Dee N. R., Desaki A. L., Desta T., Diep E., Dolbeare T. A., Donelan M. J., Dong H. W., Dougherty J. G., Duncan B. J., Ebbert A. J., Eichele G., Estin L. K., Faber C., Facer B. A., Fields R., Fischer S. R., Fliss T. P., Frensley C., Gates S. N., Glattfelder K. J., Halverson K. R., Hart M. R., Hohmann J. G., Howell M. P., Jeung D. P., Johnson R. A., Karr P. T., Kawal R., Kidney J. M., Knapik R. H., Kuan C. L., Lake J. H., Laramee A. R., Larsen K. D., Lau C., Lemon T. A., Liang A. J., Liu Y., Luong L. T., Michaels J., Morgan J. J., Morgan R. J., Mortrud M. T., Mosqueda N. F., Ng L. L., Ng R., Orta G. J., Overly C. C., Pak T. H., Parry S. E., Pathak S. D., Pearson O. C., Puchalski R. B., Riley Z. L., Rockett H. R., Rowland S. A., Royall J. J., Ruiz M. J., Sarno N. R., Schaffnit K., Shapovalova N. V., Sivisay T., Slaughterbeck C. R., Smith S. C., Smith K. A., Smith B. I., Sodt A. J., Stewart N. N., Stumpf K. R., Sunkin S. M., Sutram M., Tam A., Teemer C. D., Thaller C., Thompson C. L., Varnam L. R., Visel A., Whitlock R. M., Wohnoutka P. E., Wolkey C. K., Wong V. Y., Wood M., Yaylaoglu M. B., Young R. C., Youngstrom B. L., Yuan X. F., Zhang B., Zwingman T. A., Jones A. R. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 [DOI] [PubMed] [Google Scholar]
- 48. Dumin E., Bendikov I., Foltyn V. N., Misumi Y., Ikehara Y., Kartvelishvily E., Wolosker H. (2006) Modulation of d-serine levels via ubiquitin-dependent proteasomal degradation of serine racemase. J. Biol. Chem. 281, 20291–20302 [DOI] [PubMed] [Google Scholar]
- 49. Kipreos E. T., Pagano M. (2000) The F-box protein family. Genome Biol. 1, reviews s3002.1–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ho M. S., Ou C., Chan Y. R., Chien C. T., Pi H. (2008) The utility F-box for protein destruction. Cell Mol. Life Sci. 65, 1977–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nelson D. E., Randle S. J., Laman H. Beyond ubiquitination: the atypical functions of Fbxo7 and other F-box proteins. Open Biol. 3:130131, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jonkers W., Rep M. (2009) Lessons from fungal F-box proteins. Eukaryot. Cell 8, 677–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hermand D. (2006) F-box proteins: more than baits for the SCF? Cell Div. 1, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Reimann J. D., Freed E., Hsu J. Y., Kramer E. R., Peters J. M., Jackson P. K. (2001) Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell 105, 645–655 [DOI] [PubMed] [Google Scholar]
- 55. Zielke N., Querings S., Grosskortenhaus R., Reis T., Sprenger F. (2006) Molecular dissection of the APC/C inhibitor Rca1 shows a novel F-box-dependent function. EMBO Rep. 7, 1266–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Laman H., Funes J. M., Ye H., Henderson S., Galinanes-Garcia L., Hara E., Knowles P., McDonald N., Boshoff C. (2005) Transforming activity of Fbxo7 is mediated specifically through regulation of cyclin D/cdk6. EMBO J. 24, 3104–3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yoshida Y., Murakami A., Iwai K., Tanaka K. (2007) A neural-specific F-box protein Fbs1 functions as a chaperone suppressing glycoprotein aggregation. J. Biol. Chem. 282, 7137–7144 [DOI] [PubMed] [Google Scholar]