

Background: CRABP2 delivers RA to its cognate nuclear receptor RAR and regulates gene expression by cooperating with HuR in stabilizing mRNAs.
Results: In conjunction with HuR, CRABP2 regulates the expression of multiple cancer-related genes and suppresses tumor growth.
Conclusion: The anticarcinogenic activities of CRABP2 are mediated by both HuR and RAR.
Significance: The data demonstrate a novel mechanism through which CRABP2 inhibits tumorigenesis.
Keywords: Cancer Biology, Nucleus, Retinoic Acid, Retinoid-binding Protein, RNA-binding Protein, HuR, Nuclear Import/Export
Abstract
Cellular retinoic acid-binding protein 2 (CRABP2) potently suppresses the growth of various carcinomas, but the mechanism(s) that underlies this activity remains incompletely understood. CRABP2 displays two distinct functions. The classical function of this protein is to directly deliver retinoic acid (RA) to RA receptor (RAR), a nuclear receptor activated by this hormone, in turn inducing the expression of multiple antiproliferative genes. The other function of the protein is exerted in the absence of RA and mediated by the RNA-binding and stabilizing protein HuR. CRABP2 directly binds to HuR, markedly strengthens its interactions with target mRNAs, and thus increases their stability and up-regulates their expression. Here we show that the anticarcinogenic activities of CRABP2 are mediated by both of its functions. Transcriptome analyses revealed that, in the absence of RA, a large cohort of transcripts is regulated in common by CRABP2 and HuR, and many of these are involved in regulation of oncogenic properties. Furthermore, both in cultured cells and in vivo, CRABP2 or a CRABP2 mutant defective in its ability to cooperate with RAR but competent in interactions with HuR suppressed carcinoma growth and did so in the absence of RA. Hence, transcript stabilization by the CRABP2-HuR complex significantly contributes to the ability of CRABP2 to inhibit tumorigenesis. Surprisingly, the observations also revealed that HuR regulates the expression of multiple genes involved in nuclear pore formation and is required for nuclear import of CRABP2 and for transcriptional activation by RAR. The data thus point at a novel function for this important protein.
Introduction
The vitamin A metabolite retinoic acid (RA)3 regulates gene transcription by activating several members of the nuclear receptor family of transcription factors: the classical RA receptors (RARs) (1, 2) and peroxisome proliferator-activated receptor β/δ (PPARβ/δ) (3, 4). The partitioning of RA between these receptors is regulated by two intracellular lipid-binding proteins: cellular retinoic acid-binding protein 2 (CRABP2), which has a high affinity for the hormone and shuttles it to RARs, and fatty acid-binding protein 5 (FABP5), which has a lower affinity for RA and delivers it to PPARβ/δ. CRABP2 and FABP5 are cytosolic in the absence of their ligand, but upon binding of RA, they undergo a conformational change that activates their nuclear localization signals and results in their mobilization to the nucleus (5–8). In the nucleus, these binding proteins associate with their cognate receptors to form a complex through which RA is directly “channeled” to the receptor (9). CRABP2 and FABP5 thus markedly enhance the transcriptional activities of RAR and PPARβ/δ, respectively (3, 5, 7, 10, 11). Consequently, RA activates RARs in cells that highly express CRABP2 but functions through PPARβ/δ when FABP5 predominates. As RAR and PPARβ/δ regulate the expression of distinct cohorts of genes, RA displays different and sometimes opposing biological activities in cells where due to a high CRABP2/FABP5 ratio it activates RAR, and in cells where this ratio is low, it results in activation of PPARβ/δ. For example, in many carcinoma cells, RAR up-regulates genes that trigger differentiation, apoptosis, and cell cycle arrest (12–18), whereas PPARβ/δ induces the expression of genes that promote proliferation, angiogenesis, and survival (3, 19–22). Consequently, RA inhibits the growth of carcinoma cells that express CRABP2 (11–13, 23, 24) but promotes oncogenic activities in FABP5-expressing cells (3, 25, 26).
Although it is well established that CRABP2 suppresses carcinoma cell growth by delivering RA to RAR, it was noted previously that this binding protein also exerts biological activities independently of either RA or its receptor (27). It was thus reported that although expression of apoptotic peptidase-activating factor 1 (Apaf-1), the major protein in the apoptosome, is not controlled by either RA or RAR, ectopic expression of CRABP2 increases its level both in cultured carcinoma cells and in vivo (12, 24, 27). It was shown further that expression of CRABP2 in mammary carcinoma cells cultured in the absence of RA enhances the cleavage of several caspases, demonstrating that the protein exerts proapoptotic activities in the absence of its ligand (12, 27). These observations raise the possibility that the tumor-suppressive activities of CRABP2 may stem not only from its ability to activate RAR but also from an additional, RA- and RAR-independent function.
It was reported recently that CRABP2 devoid of RA (apo-CRABP2) functions in conjunction with HuR, one of the best characterized proteins involved in post-transcriptional regulation of gene expression in animals (28). HuR regulates various biological functions including RNA splicing, nuclear export, and transcript stabilization. It exerts the latter activity by binding to AU-rich elements in 3′-UTRs of target mRNAs, thereby protecting them against degradation and up-regulating their expression (29–32). CRABP2 cooperates with HuR in stabilization of certain mRNAs. It was thus shown that the binding protein directly interacts with HuR both in solution and when associated with some target transcripts and that it markedly increases the affinity of HuR for such transcripts. CRABP2 thus enhances the stability and increases the expression levels of such transcripts including mRNAs for the proapoptotic genes Apaf-1 and Casp7 and for HuR itself. Indeed, it was shown that CRABP2 can enhance apoptotic responses through its cooperation with HuR (27). The current work was undertaken to investigate whether its cooperation with HuR is involved in the anticarcinogenic activities of CRABP2 and to assess the relative contributions of CRABP2/RAR and CRABP2/HuR pathways in mediating these activities.
EXPERIMENTAL PROCEDURES
Cells
The M-2−/− cell line was generated from tumors that arose in murine mammary tumor virus-neu/CRABP2-null mice (24). MCF-7 cells were purchased from ATCC (Manassas, VA). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/liter glucose, 4.5 g/liter l-glutamine, 10% fetal bovine serum (Invitrogen), 100 IU/ml penicillin, and 100 μg/ml streptomycin.
Reagents
RA was purchased from Calbiochem. Antibodies against HuR (3A2; sc-5261), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 6C5; sc-32233), actin (I-19; sc-1616), and RARβ (C-19; sc-552) were from Santa Cruz Biotechnology, Inc. Antibodies against Apaf-1 (8723) and poly(ADP-ribose) polymerase (9542) were from Cell Signaling Technology, Inc. Antibody against CRABP2 was a gift from Cecile Rochette-Egly (Institut Génétique Biologie Moléculaire Cellulaire, Strasbourg, France). Transfections were carried out using PolyFect (Qiagen).
Vectors
Mammalian expression vectors harboring cDNA encoding wild type (WT) or hCRABP2ΔNLS N-terminally tagged with EGFP (pEGFP-C2 vector) and vector encoding FLAG-tagged CRABP2 were described previously (27).
Lentiviral shRNA Production
pLKO.1 vectors harboring shRNAs (Elavl1, TRCN0000112088; ELAVL1, TRCN0000017275; CRABP2, TRCN0000021373) were from Open Biosystems. pLKO.1 vectors harboring luciferase shRNA (SHC007) or non-targeting shRNA (SHC002) were from Sigma-Aldrich. Using pCMV packaging vector and pMD2.G envelope vector, lentiviruses were produced in HEK293T cells, and target cells were transduced according to standard protocols. Expression of Elavl1 and ELAVL1, encoding mouse HuR and human HuR, respectively, was reduced using respective shRNAs.
Transcriptome Analyses
MCF-7 cells were transduced with lentiviruses harboring the indicated shRNAs. 4 days post-transduction, cells were harvested, and RNA was extracted using RNeasy columns (Qiagen). Samples were amplified, labeled, and hybridized on Affymetrix® Human Gene 2.1 ST Arrays (Affymetrix) by the Gene Expression and Genotyping Facility of the Case Comprehensive Cancer Center of Case Western Reserve University. Raw data files were analyzed using Affymetrix Expression Console and Transcriptome Analysis Console. Signal intensities were normalized using the robust multichip average method. t test analyses were used to select genes differentially expressed in cells in which either ELAVL1 or CRABP2 was knocked down versus luciferase with -fold change and p value cutoffs fixed at 1.2 and 0.01, respectively. Venn analysis was used to identify the overlapping genes between the 2 groups. The list of overlapping genes was further analyzed for known functions and pathways using Ingenuity Pathway Analysis (Ingenuity Systems).
Real time quantitative PCR (qPCR)
Real time qPCR was performed using a StepOnePlus Real Time PCR System with TaqMan probes: Apaf1, Mm01223702_m1; HuR/Elavl1, Mm00516012_m1; Rarb, Mm01319677_m1; Brca1, Mm0129840_m1; Brca2, Mm01218747_m1; Casp7, Mm00432324_m1; Casp9, Mm00516563_m1; Btg2, Mm00476162_m1; BRCA1, Hs01556193_m1; BRCA2, Hs00609073_m1; CASP7, Hs00169152_m1; HuR/ELAVL1, Hs00171309_m1; CRABP2, Hs00275636_m1; 18 S, 4352930E (Applied Biosystems). Levels of mRNAs were normalized to 18 S ribosomal RNA using the ΔΔCt method (Applied Biosystems Technical Bulletin Number 2).
Transactivation Assays
Transactivation assays were carried out as described previously (10). Briefly, cells were cultured in delipidated medium for 48 h and co-transfected with a luciferase reporter driven by a DR-5 RAR response element and a vector encoding β-galactosidase used as a transfection control. Cells were treated with RA (1 μm) for 16 h and lysed, and expression of luciferase was measured and corrected for encoding β-galactosidase.
Confocal Fluorescence Microscopy
M-2−/− cells cultured in DMEM containing 10% charcoal-treated FBS were transfected with pCMV-3Tag-1 encoding FLAG-CRABP2. Cells were fixed in 4% paraformaldehyde, PBS; blocked; and permeabilized with PBS containing 0.2% Triton X-100 and 1% BSA (room temperature for 1 h). FLAG-tagged CRABP2 was visualized by immunostaining using antibodies against FLAG (Sigma-Aldrich, F1804). Nuclei were visualized by DAPI staining. Cells were mounted with Fluoromount-G (SouthernBiotech) and imaged using a LSM510 confocal microscope (Leica).
Animal Studies
9-week-old NCrnu/nu nude female mice were purchased from the Athymic Animal and Xenograft Core Facility of the Case Comprehensive Cancer Center and housed at the Case Western Reserve University School of Medicine Animal Facility in accordance with the regulations of the American Association for the Accreditation of Laboratory Animal Care. 3 × 106 cells in 100 μl of serum-free DMEM were injected subcutaneously. Tumor growth was measured with calipers, and tumor volumes were calculated using the following formula: (length × width2)/2.
Histology
Tumors were excised, fixed in 4% paraformaldehyde, embedded in paraffin, sectioned, mounted on glass slides, and stained with hematoxylin and eosin (H&E) by the Tissue Procurement, Histology, and Immunochemistry Core Facility of the Case Comprehensive Cancer Center. Immunohistochemistry was performed using the EXPOSE rabbit-specific HRP/diaminobenzidine detection immunohistochemistry IHC kit (Abcam, ab80437). Antigen retrieval was achieved by boiling slides in 10 mm sodium citrate, pH 6.0 for 10 min. Sections were incubated with antibody against phosphorylated histone H3 (Cell Signaling Technology, 9701) at a 1:200 dilution (16 h at 4 °C). Slides were imaged using a Leica DM6000 and Volocity Acquisition software at the Imaging Core Facility of the Department of Genetics and Genome Sciences at Case Western Reserve University.
RESULTS
CRABP2 and HuR Regulate a Common Cohort of Cancer-related Genes
In some carcinoma cells that express CRABP2, RA inhibits proliferation by activating RAR and thereby inducing the expression of antiproliferative RAR target genes (11–13, 23, 24, 33). In accordance, treatment with RA inhibited the growth of MCF-7 mammary carcinoma cells, which highly express CRABP2 (33), and decreased the expression of CRABP2 (Fig. 1A), diminishing the antiproliferative activity of RA (Fig. 1B). Interestingly, however, decreasing the expression of CRABP2 in these cells promoted cell proliferation even in the absence of RA (Fig. 1B). These observations suggest that, in addition to delivering RA to nuclear RAR, CRABP2 suppresses cell growth by an additional, RA-independent mechanism. In regard to this possibility, it was recently reported that apo-CRABP2 directly binds to the RNA-binding protein HuR, increases its affinity for some target transcripts, and thereby enhances the stability of these mRNAs and increases their expression (27). Transcriptome analyses were carried out to begin to examine whether the RA-independent growth-suppressive activity of CRABP2 may stem from its cooperation with HuR. The expression levels of CRABP2 or HuR in MCF-7 cells were reduced using respective shRNAs (Fig. 1, C and D). Cells were depleted of retinoids by culturing them in charcoal-treated medium, and transcriptome analyses were carried out using Affymetrix Human Gene 2.1 ST Arrays. Decreasing the expression of CRABP2 or HuR altered the expression of 607 or 1678 mRNAs, respectively. Of these, 135 transcripts were found to be regulated in common by CRABP2 and HuR (Fig. 1E and supplemental Table S1). Array data were deposited in the Gene Expression Omnibus (GEO) database of the NCBI under accession number GSE62291. Notably, commonly regulated mRNAs were predominantly regulated in the same fashion: 93 mRNAs were down-regulated and 38 mRNAs were up-regulated in response to reducing the expression of either protein (Fig. 1, F and G). Only four mRNAs were regulated by HuR and CRABP2 in opposite directions (Fig. 1, F and G). Hence, a significant subset of HuR-regulated mRNAs is also targeted by CRABP2. Ingenuity Pathway Analysis revealed that many genes commonly regulated by CRABP2 and HuR are involved in regulation of oncogenic properties including cell proliferation and survival, migration, invasion, and death with about 90 clustering as cancer genes (Fig. 2A). Validation of three genes identified by the transcriptome analysis by real time qPCR showed that mRNA for the apoptotic gene CASP7, which was shown previously to be controlled by the CRABP2-HuR complex (27), and the tumor suppressor genes BRCA1 and BRCA2 (34) were markedly down-regulated in cells with decreased expression of either CRABP2 (Fig. 2B) or HuR (Fig. 2C).
FIGURE 1.

CRABP2 and HuR regulate a common subset of genes. A, MCF-7 cells were transduced with lentiviral particles harboring a non-targeting shRNA (shCtrl) or shRNA targeting CRABP2 (shCRABP2). Cells were selected with puromycin to generate cell lines stably expressing the respective shRNAs. Immunoblots demonstrate reduced expression of CRABP2. B, cells were cultured in delipidated medium for 48 h, treated with vehicle or RA (1 μm) for 4 days, and counted. Data are mean ± S.E. (n = 3). C and D, MCF-7 cells were transduced with lentiviral particles containing shRNAs targeting luciferase (shCtrl), CRABP2 (shCRABP2; C), or HuR (shHuR; D). Levels of mRNA for CRABP2 (C) or HuR (D) were assayed by qPCR. Data are mean ± S.E. (n = 3). *, p ≤ 0.01 by two-tailed Student's t test. E, Venn diagram depicting changes in gene expression in cells with reduced expression of CRABP2 and HuR. 135 genes were found to be regulated by both CRABP2 and HuR. F and G, expression profiles of genes commonly regulated by CRABP2 and HuR represented in a heat map clustering (F) or plotted as log2(-fold change) of genes regulated by CRABP2 versus HuR (G). Error bars represent S.E.
FIGURE 2.
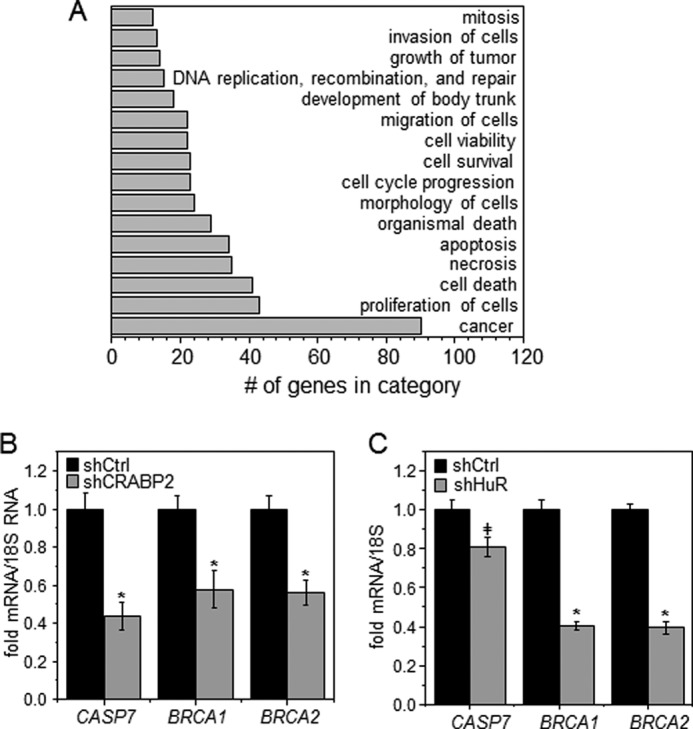
CRABP2 and HuR co-regulate cancer-related genes. A, top 16 biological functions and diseases found to be significantly represented in the set of genes commonly regulated by CRABP2 and HuR. B and C, levels of denoted mRNAs in MCF-7 cells expressing shRNAs targeting luciferase (shCtrl), CRABP2 (shCRABP2; B), or HuR (shHuR; C) measured by qPCR. Data are mean ± S.E. (C, n = 3; D, n = 4). *, p ≤ 0.01; ‡, p = 0.02 by two-tailed Student's t test. Error bars represent S.E.
CRABP2 Inhibits Mammary Carcinoma Cell Growth by Two Distinct Mechanisms
A nuclear localization-defective CRABP2 mutant was used to assess the relative contributions of the two functions of the protein to its ability to inhibit carcinoma cell growth. This mutant, CRABP2-K20A/R29A/K30A (CRABP2ΔNLS), binds RA with native affinity but lacks the nuclear localization signal of the protein and thus does not undergo RA-induced nuclear translocation and does not enhance the transcriptional activity of RAR (7, 27). CRABP2ΔNLS nevertheless retains a high affinity for HuR and is indistinguishable from the WT protein in its ability to cooperate with HuR in enhancing mRNA stability (27). M-2−/− mammary carcinoma cells, a line derived from mammary tumors that arose in the murine mammary tumor virus-neu mouse model of breast cancer bred with Crabp2-null mice (24), were used. These cells do not express CRABP2, providing a clean background for examining effects of CRABP2 on cell growth. M-2−/− cells lines that stably overexpress a control vector encoding EGFP, EGFP-tagged CRABP2, or EGFP-CRABP2ΔNLS were generated (Fig. 3A). Ectopic expression of CRABP2 suppressed the growth of M-2−/− cells in the absence of RA, and notably CRABP2ΔNLS exerted a similar effect (Fig. 3B). Hence, in accordance with its activity in MCF-7 cells (Fig. 1B), CRABP2 can suppress cell growth by an RA- and RAR-independent mechanism. Treatment of these cells with RA markedly facilitated their growth (Fig. 3B). This response reflects that, as these cells lack CRABP2 but express FABP5, RA is directed to PPARβ/δ and thus exerts proliferative activities (3, 24). Indeed, expression of CRABP2 converted RA from a proproliferative to a growth-suppressing agent (Fig. 3B). CRABP2ΔNLS also inhibited cell growth in the presence of RA, but it did so less efficiently than the WT protein. Interestingly, the rate of proliferation of CRABP2ΔNLS-expressing cells in the presence of RA was similar to that of CRABP2-expressing cells devoid of RA.
FIGURE 3.

CRABP2 inhibits mammary carcinoma cell growth by two distinct mechanisms. A, M-2−/− cells stably expressing EGFP (Ctrl), EGFP-CRABP2 (2), or EGFP-2ΔNLS (2ΔNLS). Immunoblotting demonstrates similar expression of WT and mutant CRABP2. GAPDH was used as a loading control. B, cells were cultured in delipidated medium for 48 h, treated with vehicle or RA (200 nm) for 4 days, and counted. C, M-2−/− cells (3 × 106) that stably overexpress EGFP (Ctrl), EGFP-CRABP2 (2), or EGFP-CRABP2ΔNLS (2ΔNLS) were injected subcutaneously into female NCrnu/nu mice, and tumor growth was monitored. Data are mean ± S.E. (mice: control, n = 20; CRABP2 and CRABP2ΔNLS, n = 10 each). *, p ≤ 0.05 versus control; ‡, p ≤ 0.05 using two-tailed Student's t test. D–H, levels of denoted mRNAs (D and F) and proteins (E, G, and H) in tumors expressing EGFP (Ctrl), CRABP2 (2), or CRABP2ΔNLS (2ΔNLS). D and F, levels of mRNAs assessed by qPCR. Data are mean ± S.E. (n = 3–5). *, p ≤ 0.05 versus control using two-tailed Student's t test. E, G, and H, left, immunoblots using denoted antibodies. Right, quantification of immunoblots. Data are mean ± S.E. (E and G, n = 8; H, n = 4). *, p ≤ 0.05 versus control using two-tailed Student's t test. Error bars represent S.E.
To further examine the involvement of the two functions of CRABP2 in regulation of carcinoma cell growth, M-2−/− cells that express CRABP2 or CRABP2ΔNLS were subcutaneously injected into female NCrnu/nu athymic mice, and tumor growth was monitored. To minimize variability between animals, each mouse was injected with the M-2−/− cells stably expressing a control vector into one flank, and with M-2−/− cells that stably express either CRABP2 or CRABP2ΔNLS were injected into the opposite flank. Tumors that arose at sites injected with CRABP2-expressing cells developed at a slower rate than those that arose from control cells (Fig. 3C). Similarly to their behavior in cultured cells, CRABP2ΔNLS-expressing cells developed tumors at an intermediate rate, displaying growth that was slower than that displayed by control cells but faster than that observed by cells that express WT-CRABP2 (Fig. 3C). Reflecting activation of RAR, expression of CRABP2 resulted in an increase in mRNA and protein of three established RAR target genes: Rarb, Casp9, and Btg2 (Fig. 3, D and E). In accordance with its inability to cooperate with RAR, CRABP2ΔNLS did not affect the levels of these RAR targets (Fig. 3, D and E). However, both CRABP2 and CRABP2ΔNLS up-regulated Apaf1, Elavl1, Casp7, Brca1, and Brca2, genes that are controlled by CRABP2 in conjunction with HuR (Fig. 3, F–H and Ref. 28). Taken together, these observations indicate that CRABP2 exerts anticarcinogenic activities through two distinct mechanisms, that one of these mechanisms is mediated through the ability of the protein to enhance RA-induced activation of RAR, and that the other mechanism likely emanates from up-regulation of antiproliferative genes brought about through the cooperation with HuR.
Histological analyses revealed that, although the general morphology of all tumors was similar (Fig. 4A), tumors that arose from cells expressing CRABP2 had fewer nuclei that were positively stained for the proliferation marker phosphorylated histone H3, whereas tumors from cells expressing CRABP2ΔNLS displayed an intermediate number of positive nuclei (Fig. 4A). Tumors that arose from cells that express either CRABP2 or CRABP2ΔNLS similarly displayed a marked increase in cleavage of the apoptotic protein poly(ADP-ribose) polymerase (Fig. 4B). The data thus suggest that suppression of cell growth by CRABP2 is mediated both by RAR and by HuR, whereas proapoptotic activities of the protein are exerted primarily through its cooperation with HuR.
FIGURE 4.

Cell proliferation and apoptosis in tumors expressing CRABP2 or CRABP2ΔNLS. A, histological analyses of tumors that arose from M-2−/− cells that stably overexpress EGFP (Ctrl), EGFP-CRABP2 (2), or EGFP-CRABP2ΔNLS (2ΔNLS). Top, H&E staining. Bottom, immunohistochemistry demonstrating expression of phosphorylated histone H3 (phospho-histone H3). Scale bars, 100 μm. B, left, immunoblots of full-length and cleaved poly(ADP-ribose) polymerase (PARP) in tumors. GAPDH was used as a loading control. Right, quantitation of immunoblots. *, p ≤ 0.05 versus control using two-tailed Student's t test. Error bars represent S.E.
HuR Is Required for CRABP2-mediated Activation of RAR
Depletion of retinoids and the use of CRABP2ΔNLS allowed for dissection between the two functions of CRABP2 by negating its cooperation with RAR. To further examine the relative contributions of these activities, M-2−/− cell lines that express different levels of HuR in the absence or presence of ectopically expressed CRABP2 were generated (Fig. 5A). Cells were cultured in the presence of 200 nm RA, and cell growth was monitored (Fig. 5B). Ectopic expression of CRABP2 markedly suppressed proliferation. In agreement with previous reports that HuR displays antiproliferative activities (27, 35–37), decreasing the expression level of this protein enhanced cell growth. Surprisingly, however, despite the presence of RA, decreasing the expression of HuR negated the ability of CRABP2 to inhibit cell growth. Cells with reduced expression of HuR and counterparts that express CRABP2 were then injected into NCrnu/nu athymic mice, and tumor growth was monitored (Fig. 5C). Similarly to the behavior of cultured cells, ectopic expression of CRABP2 in M-2−/− cells failed to suppress tumor development from cells with a reduced level of HuR (Fig. 5C). These observations surprisingly suggest that HuR not only directly cooperates with CRABP2 in mediating growth inhibition but that its presence is also necessary for enabling CRABP2 to inhibit proliferation in conjunction with RAR. In support of this conclusion, ectopic expression of CRABP2 had no effect on the RAR target genes Casp9 and Btg2 in tumors that arose from cells with reduced expression of HuR (Fig. 5D). Transcriptional activation assays were carried out to directly examine whether HuR affects the transcriptional activity of the CRABP2/RAR pathway. Cells that stably express different levels of CRABP2 and HuR (Fig. 5A) were transfected with a luciferase reporter driven by an RAR response element and treated with RA, and luciferase activity was measured (Fig. 5E). In control cells, RA activated the reporter, and CRABP2 enhanced the response. However, although expression of HuR was reduced in these cells by only 40–50% (Fig. 5A), the decrease inhibited RA-induced reporter activation both in the absence and presence of CRABP2.
FIGURE 5.

HuR is required for activation of RAR and suppression of tumor development by CRABP2. A, M-2−/− cell lines stably expressing EGFP (Ctrl) or EGFP-CRABP2 (CRABP2) (Fig. 3A) were transduced with lentiviral particles encoding a non-targeting shRNA (shCtrl) or shRNA targeting HuR (shHuR). Cells were selected with puromycin to generate cell lines stably expressing the respective shRNAs. Top, overexpression of CRABP2 and reduced expression of HuR demonstrated by immunoblotting. Bottom, quantitation of HuR immunoblots. B, cells were cultured in delipidated medium for 48 h, treated with vehicle or RA (200 nm) for 3 days, and counted. Data are mean ± S.E. (n = 3). *, p ≤ 0.01. C, 3 × 106 M-2−/− cells expressing reduced levels of HuR and ectopically expressing EGFP (Ctrl) or EGFP-CRABP2 (CRABP2) (A) were injected subcutaneously into the opposite flanks of female NCrnu/nu mice. Tumor growth was monitored. Data are mean ± S.E. (n = 10). D, levels of denoted mRNAs in tumors were assessed by qPCR. Data are mean ± S.E. (n = 3). E, cells with varying expression levels of CRABP2 and HuR (A) were cultured in delipidated medium for 48 h and then co-transfected with an RAR response element (RARE)-driven luciferase reporter gene and a vector encoding β-galactosidase. Cells were treated with vehicle or RA (20 nm for 16 h), and luciferase activity was measured and normalized to β-galactosidase (βgal) activity. Data are mean ± S.D. (n = 3). *, p ≤ 0.01 versus RA-treated control cells. Error bars represent S.E.
HuR Is Required for the RA-induced Nuclear Import of CRABP2
Upon binding RA, CRABP2 undergoes relocalization to the nucleus where it delivers the ligand to RAR. Possible involvement of HuR in the nuclear translocation of CRABP2 was thus examined. Control M-2−/− cells, which stably express shRNA against luciferase or HuR, were generated (Fig. 6A). Cells were transfected with a vector encoding FLAG-tagged CRABP2 and immunostained using FLAG antibodies, and the protein was visualized by confocal fluorescence microscopy (Fig. 6B). In control cells, CRABP2 was predominantly cytosolic in the absence of RA and mobilized to the nucleus 30 min following RA treatment. Strikingly, in cells with reduced expression of HuR, RA induced a discernable shift in the subcellular localization of CRABP2, but this shift did not culminate in nuclear import. Instead, 30 min following RA treatment, CRABP2 accumulated around the nucleus and did not enter this compartment (Fig. 6B). The observations thus show that HuR expression is critical for enabling the nuclear import of CRABP2. Interestingly, examination of data emerging from transcriptome analysis revealed that reducing the expression of HuR significantly decreased the expression levels of multiple proteins involved in nuclear pore formation and nuclear import/export (Fig. 6C).
FIGURE 6.

HuR is required for RA-induced nuclear translocation of CRABP2. A, M-2−/− cells were transduced with lentiviral particles encoding shRNAs targeting luciferase (shCtrl) or HuR (shHuR) and selected with puromycin to generate cell lines stably expressing the shRNAs. Immunoblotting demonstrates down-regulation of HuR in the cell line stably expressing shRNA targeting HuR. B, cells were cultured in delipidated medium and transfected with vector encoding FLAG-CRABP2. FLAG-CRABP2 was immunostained in untreated cells and in cells treated with RA for 30 min and visualized using confocal microscopy. DAPI was used to visualize nuclei. Scale bars, 20 μm. C, genes involved in nuclear import/export that were found by transcriptome analysis (Fig. 1) to be down-regulated upon decreasing the expression of HuR.
DISCUSSION
CRABP2 suppresses the growth of various carcinomas, and it has been established that this activity is exerted at least in part by CRABP2-mediated direct delivery of RA to RAR, leading to induction of antiproliferative RAR target genes (11–13, 23, 24). The observations described here show that CRABP2 also exerts anticarcinogenic activities through its ability to cooperate with HuR. Transcriptome analyses revealed that, in the absence of RA, a large cohort of transcripts is regulated in common by CRABP2 and HuR (Fig. 1, E–G) and that many of these are involved in regulation of oncogenic properties (Fig. 2A). Notably, the analyses failed to identify some proapoptotic transcripts known to be regulated by the cooperation of CRABP2 and HuR such as APAF1 and CASP7 (Ref. 27 and Fig. 2B), reflecting the sensitivity limit of the method. The complete spectrum of transcripts co-regulated by CRABP2 and HuR and their involvement in cancer cell biology remain to be elucidated.
CRABP2 cooperates with HuR in the absence of RA as well as in the absence of the nuclear localization signal of the protein that is essential for enabling it to deliver RA to RAR (27). In contrast, the RAR-mediated activities of CRABP2 strictly depend on the presence of RA and on an intact ability to undergo RA-induced nuclear localization. Consequently, CRABP2 and its nuclear localization-defective mutant similarly inhibited cell growth in the absence of retinoids (Fig. 3B), whereas in the presence of RA CRABP2 was more effective. In accordance, ectopic expression of CRABP2ΔNLS inhibited tumor growth in a xenograft mouse model, but CRABP2 was more efficient in this capacity (Fig. 3C), reflecting the additional growth-suppressing activity of RAR. Indeed, although both CRABP2 and CRABPΔNLS increased the expression of HuR target genes, only the WT protein activated RAR (Fig. 3, D–H). The data thus indicate that CRABP2 inhibits tumorigenesis both by cooperating with RAR and by enhancing HuR-mediated transcript stabilization. Notably, the data indicate that the contribution of the CRABP2/HuR pathway to the growth-inhibitory activities of CRABP2 is more substantial than that of CRABP2/RAR arm (Fig. 3B, 4B).
Surprisingly, down-regulation of HuR inhibited the transcriptional activity of RAR (Fig. 5E) and abolished the ability of CRABP2 to inhibit carcinoma cell growth (Fig. 5, B and C). The observations that HuR is critical for enabling the nuclear import of CRABP2 (Fig. 6B) and that down-regulation of this protein results in decreased expression of multiple genes involved in nuclear pore formation and in nuclear import and export (Fig. 6C) suggest a mechanism by which HuR is involved in regulating transcriptional activities. Taken together with the observations that HuR is necessary for the transcriptional activity of RAR even in the absence of CRABP2 (Fig. 5E), the data indicate that HuR does not specifically regulate the nuclear import of CRABP2 but is generally involved in regulating nuclear pore formation and nuclear entry and exit. The observations thus point at a novel function for this important protein.
Supplementary Material
Acknowledgments
We thank Cecile Rochette-Egly (Institut Génétique Biologie Moléculaire Cellulaire, Strasbourg, France) for CRABP2 antibodies. The core facilities of the Case Comprehensive Cancer Center are supported by National Institutes of Health Grant P30CA43703. The Imaging Core Facility, Department of Genetics and Genome Sciences, Case Western Reserve University, is supported by the National Institutes of Health Office of Research Infrastructure Programs under Award S10RR021228.
This work was supported, in whole or in part, by National Institutes of Health Grants DK060684 and NCI166955 (to N. N.), 5T32GM008803-09 (to A. C. V.), and RO1CA166955.
Array data have been deposited in the Gene Expression Omnibus (GEO) database of the NCBI under accession number GSE62291.

This article contains supplemental Table S1.
- RA
- retinoic acid
- CRABP2
- cellular retinoic acid-binding protein 2
- RAR
- RA receptor
- PPARβ/δ
- peroxisome proliferator-activated receptor β/δ
- FABP5
- fatty acid-binding protein 5
- Apaf-1
- apoptotic peptidase-activating factor 1
- EGFP
- enhanced GFP
- qPCR
- quantitative PCR.
REFERENCES
- 1. Chambon P. (1996) A decade of molecular biology of retinoic acid receptors. FASEB J. 10, 940–954 [PubMed] [Google Scholar]
- 2. Germain P., Chambon P., Eichele G., Evans R. M., Lazar M. A., Leid M., De Lera A. R., Lotan R., Mangelsdorf D. J., Gronemeyer H. (2006) International Union of Pharmacology. LX. Retinoic acid receptors. Pharmacol. Rev. 58, 712–725 [DOI] [PubMed] [Google Scholar]
- 3. Schug T. T., Berry D. C., Shaw N. S., Travis S. N., Noy N. (2007) Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 129, 723–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shaw N., Elholm M., Noy N. (2003) Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor β/δ. J. Biol. Chem. 278, 41589–41592 [DOI] [PubMed] [Google Scholar]
- 5. Tan N. S., Shaw N. S., Vinckenbosch N., Liu P., Yasmin R., Desvergne B., Wahli W., Noy N. (2002) Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol. Cell. Biol. 22, 5114–5127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gillilan R. E., Ayers S. D., Noy N. (2007) Structural basis for activation of fatty acid-binding protein 4. J. Mol. Biol. 372, 1246–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sessler R. J., Noy N. (2005) A ligand-activated nuclear localization signal in cellular retinoic acid binding protein-II. Mol. Cell 18, 343–353 [DOI] [PubMed] [Google Scholar]
- 8. Armstrong E. H., Goswami D., Griffin P. R., Noy N., Ortlund E. A. (2014) Structural basis for ligand regulation of the fatty acid-binding protein 5, peroxisome proliferator-activated receptor β/δ (FABP5-PPARβ/δ) signaling pathway. J. Biol. Chem. 289, 14941–14954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dong D., Ruuska S. E., Levinthal D. J., Noy N. (1999) Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J. Biol. Chem. 274, 23695–23698 [DOI] [PubMed] [Google Scholar]
- 10. Budhu A., Gillilan R., Noy N. (2001) Localization of the RAR interaction domain of cellular retinoic acid binding protein-II. J. Mol. Biol. 305, 939–949 [DOI] [PubMed] [Google Scholar]
- 11. Budhu A. S., Noy N. (2002) Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Mol. Cell. Biol. 22, 2632–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donato L. J., Noy N. (2005) Suppression of mammary carcinoma growth by retinoic acid: proapoptotic genes are targets for retinoic acid receptor and cellular retinoic acid-binding protein II signaling. Cancer Res. 65, 8193–8199 [DOI] [PubMed] [Google Scholar]
- 13. Donato L. J., Suh J. H., Noy N. (2007) Suppression of mammary carcinoma cell growth by retinoic acid: the cell cycle control gene Btg2 is a direct target for retinoic acid receptor signaling. Cancer Res. 67, 609–615 [DOI] [PubMed] [Google Scholar]
- 14. Noy N. (2010) Between death and survival: retinoic acid in regulation of apoptosis. Annu. Rev. Nutr. 30, 201–217 [DOI] [PubMed] [Google Scholar]
- 15. Afonja O., Juste D., Das S., Matsuhashi S., Samuels H. H. (2004) Induction of PDCD4 tumor suppressor gene expression by RAR agonists, antiestrogen and HER-2/neu antagonist in breast cancer cells. Evidence for a role in apoptosis. Oncogene 23, 8135–8145 [DOI] [PubMed] [Google Scholar]
- 16. Afonja O., Raaka B. M., Huang A., Das S., Zhao X., Helmer E., Juste D., Samuels H. H. (2002) RAR agonists stimulate SOX9 gene expression in breast cancer cell lines: evidence for a role in retinoid-mediated growth inhibition. Oncogene 21, 7850–7860 [DOI] [PubMed] [Google Scholar]
- 17. Zacheis D., Dhar A., Lu S., Madler M. M., Klucik J., Brown C. W., Liu S., Clement F., Subramanian S., Weerasekare G. M., Berlin K. D., Gold M. A., Houck J. R., Jr., Fountain K. R., Benbrook D. M. (1999) Heteroarotinoids inhibit head and neck cancer cell lines in vitro and in vivo through both RAR and RXR retinoic acid receptors. J. Med. Chem. 42, 4434–4445 [DOI] [PubMed] [Google Scholar]
- 18. Soprano D. R., Qin P., Soprano K. J. (2004) Retinoic acid receptors and cancers. Annu. Rev. Nutr. 24, 201–221 [DOI] [PubMed] [Google Scholar]
- 19. Di-Poï N., Michalik L., Tan N. S., Desvergne B., Wahli W. (2003) The anti-apoptotic role of PPARβ contributes to efficient skin wound healing. J. Steroid Biochem. Mol. Biol. 85, 257–265 [DOI] [PubMed] [Google Scholar]
- 20. Di-Poï N., Tan N. S., Michalik L., Wahli W., Desvergne B. (2002) Antiapoptotic role of PPARβ in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol. Cell 10, 721–733 [DOI] [PubMed] [Google Scholar]
- 21. Aggarwal B. B., Sethi G., Ahn K. S., Sandur S. K., Pandey M. K., Kunnumakkara A. B., Sung B., Ichikawa H. (2006) Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: modern target but ancient solution. Ann. N.Y. Acad. Sci. 1091, 151–169 [DOI] [PubMed] [Google Scholar]
- 22. Montagner A., Delgado M. B., Tallichet-Blanc C., Chan J. S., Sng M. K., Mottaz H., Degueurce G., Lippi Y., Moret C., Baruchet M., Antsiferova M., Werner S., Hohl D., Saati T. A., Farmer P. J., Tan N. S., Michalik L., Wahli W. (2014) Src is activated by the nuclear receptor peroxisome proliferator-activated receptor β/δ in ultraviolet radiation-induced skin cancer. EMBO Mol. Med. 6, 80–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Manor D., Shmidt E. N., Budhu A., Flesken-Nikitin A., Zgola M., Page R., Nikitin A. Y., Noy N. (2003) Mammary carcinoma suppression by cellular retinoic acid binding protein-II. Cancer Res. 63, 4426–4433 [PubMed] [Google Scholar]
- 24. Schug T. T., Berry D. C., Toshkov I. A., Cheng L., Nikitin A. Y., Noy N. (2008) Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARβ/δ to RAR. Proc. Natl. Acad. Sci. U.S.A. 105, 7546–7551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Levi L., Lobo G., Doud M. K., von Lintig J., Seachrist D., Tochtrop G. P., Noy N. (2013) Genetic ablation of the fatty acid-binding protein FABP5 suppresses HER2-induced mammary tumorigenesis. Cancer Res. 73, 4770–4780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morgan E., Kannan-Thulasiraman P., Noy N. (2010) Involvement of fatty acid binding protein 5 and PPARβ/δ in prostate cancer cell growth. PPAR Res. 2010, 234629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vreeland A. C., Yu S., Levi L., de Barros Rossetto D., Noy N. (2014) Transcript stabilization by the RNA-binding protein HuR is regulated by cellular retinoic acid-binding protein 2. Mol. Cell. Biol. 34, 2135–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hinman M. N., Lou H. (2008) Diverse molecular functions of Hu proteins. Cell. Mol. Life Sci. 65, 3168–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Myer V. E., Fan X. C., Steitz J. A. (1997) Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. EMBO J. 16, 2130–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brennan C. M., Steitz J. A. (2001) HuR and mRNA stability. Cell Mol. Life Sci. 58, 266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen C. Y., Xu N., Shyu A. B. (2002) Highly selective actions of HuR in antagonizing AU-rich element-mediated mRNA destabilization. Mol. Cell. Biol. 22, 7268–7278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. López de Silanes I., Zhan M., Lal A., Yang X., Gorospe M. (2004) Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl. Acad. Sci. U.S.A. 101, 2987–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jing Y., Waxman S., Mira-y-Lopez R. (1997) The cellular retinoic acid binding protein II is a positive regulator of retinoic acid signaling in breast cancer cells. Cancer Res. 57, 1668–1672 [PubMed] [Google Scholar]
- 34. Venkitaraman A. R. (2014) Tumour suppressor mechanisms in the control of chromosome stability: insights from BRCA2. Mol. Cells 37, 95–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Latorre E., Tebaldi T., Viero G., Spartà A. M., Quattrone A., Provenzani A. (2012) Downregulation of HuR as a new mechanism of doxorubicin resistance in breast cancer cells. Mol. Cancer 11, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gubin M. M., Calaluce R., Davis J. W., Magee J. D., Strouse C. S., Shaw D. P., Ma L., Brown A., Hoffman T., Rold T. L., Atasoy U. (2010) Overexpression of the RNA binding protein HuR impairs tumor growth in triple negative breast cancer associated with deficient angiogenesis. Cell Cycle 9, 3337–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mazroui R., Di Marco S., Clair E., von Roretz C., Tenenbaum S. A., Keene J. D., Saleh M., Gallouzi I. E. (2008) Caspase-mediated cleavage of HuR in the cytoplasm contributes to pp32/PHAP-I regulation of apoptosis. J. Cell Biol. 180, 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.