

Background: Bile acid-induced hepatocyte injury causes cholestatic liver disease.
Results: Inhibiting mitochondrial fission prevents bile acid-induced hepatocyte death, and liver-specific decrease of mitochondrial fission in vivo limits bile duct ligation-induced liver injury and fibrosis.
Conclusion: Controlling mitochondrial morphology is an effective strategy to decrease bile acid-induced liver injury.
Significance: Mitochondrial fission is a new target to control cholestatic liver disease.
Keywords: Bile Acid, Cell Death, Liver, Mitochondria, Reactive Oxygen Species (ROS), DLP1, Drp1, Cholestasis, Mitochondrial Fission, Mitochondrial Fusion
Abstract
Mitochondria frequently change their shape through fission and fusion in response to physiological stimuli as well as pathological insults. Disrupted mitochondrial morphology has been observed in cholestatic liver disease. However, the role of mitochondrial shape change in cholestasis is not defined. In this study, using in vitro and in vivo models of bile acid-induced liver injury, we investigated the contribution of mitochondrial morphology to the pathogenesis of cholestatic liver disease. We found that the toxic bile salt glycochenodeoxycholate (GCDC) rapidly fragmented mitochondria, both in primary mouse hepatocytes and in the bile transporter-expressing hepatic cell line McNtcp.24, leading to a significant increase in cell death. GCDC-induced mitochondrial fragmentation was associated with an increase in reactive oxygen species (ROS) levels. We found that preventing mitochondrial fragmentation in GCDC by inhibiting mitochondrial fission significantly decreased not only ROS levels but also cell death. We also induced cholestasis in mouse livers via common bile duct ligation. Using a transgenic mouse model inducibly expressing a dominant-negative fission mutant specifically in the liver, we demonstrated that decreasing mitochondrial fission substantially diminished ROS levels, liver injury, and fibrosis under cholestatic conditions. Taken together, our results provide new evidence that controlling mitochondrial fission is an effective strategy for ameliorating cholestatic liver injury.
Introduction
Cholestasis is characterized by impaired bile flow, resulting in the accumulation of bile acids within hepatocytes and in the serum. Toxic bile acids induce hepatocellular injury, followed by inflammation, liver fibrosis, and cirrhosis, leading to portal hypertension and liver failure. Pharmacological therapy is limited, and patients with end-stage cholestasis usually require liver transplantation.
Early evidence has suggested that toxic bile acids induce hepatocyte apoptosis through the extrinsic death receptor-mediated pathway or the intrinsic mitochondria-mediated pathway (1). However, recent studies have suggested that oncotic necrosis might be the main type of cell death in the rodent model of cholestasis (2–4). It is likely that both apoptotic and necrotic cell death are present in chronic cholestatic diseases, depending on the length, cause, and complications of the disease (1, 5). Nevertheless, both necrosis and apoptosis involve mitochondria; mitochondrial dysfunction and overproduction of reactive oxygen species (ROS)2 play a key role in the progression of chronic liver diseases. Toxic bile salts directly impair mitochondrial bioenergetics (6, 7), or induce mitochondrial outer membrane permeabilization (MOMP) (8, 9), leading to ATP depletion and subsequent necrotic cell death. MOMP also releases pro-apoptotic proteins such as cytochrome c to the cytosol, activating downstream caspases and apoptotic cell death (10).
Mitochondria are dynamic organelles that frequently change their shape and location. Fission and fusion are the main processes determining mitochondrial morphology. Membrane-remodeling large GTPases belonging to the dynamin family are the main proteins mediating mitochondrial fission and fusion. In mammals, these proteins are dynamin-like/related protein 1 (DLP1/Drp1) for mitochondrial fission, and mitofusin isoforms (Mfn1 and Mfn2) and optic atrophy 1 (OPA1) for mitochondrial outer and inner membrane fusion, respectively. DLP1 is a cytosolic protein that is recruited to mitochondria for fission. Apoptotic stimuli translocate DLP1 to the mitochondria and induce mitochondrial fragmentation (11). It has been shown that mitochondrial fission sites in apoptosis are enriched with the pore-forming pro-apoptotic protein Bax that induces MOMP and cytochrome c release (12). Necrosis is also accompanied by activation of DLP1 and mitochondrial fragmentation (13). Inhibition of mitochondrial fission prevents or delays both apoptosis and necrosis (11, 13, 14), indicating its importance in the cell death progression.
Aberrant mitochondrial morphology has been implicated in many human diseases. While mutations in Mfn2 and OPA1 cause genetic diseases Charcot Marie Tooth type 2A and autosomal dominant optic atrophy, respectively (15–17), a more severe newborn death is associated with a mutation in DLP1 (18). In addition, many pathological conditions such as neurodegeneration, metabolic disorders, and cardiovascular diseases exhibit mitochondrial dysfunction and altered mitochondrial morphology (19, 20), suggesting a close form-function relationship. Although mitochondrial deformation has been observed in cholestasis (21), the mechanism by which this altered mitochondrial morphology plays a role in the disease progression is unknown.
In this study, we found that a toxic bile salt induces mitochondrial fragmentation through the function of DLP1, which is accompanied by ROS increase and cell death. Importantly, we demonstrate that inhibition of mitochondrial fission prevents harmful downstream consequences of toxic bile salt exposure. Using transgenic mice expressing a dominant-negative form of DLP1 specifically in the liver, we showed that decreasing mitochondrial fission significantly diminished the liver injury and fibrosis after bile duct ligation. Our data provide new in vivo evidence that limiting mitochondrial fission is an effective strategy for ameliorating cholestatic liver injury.
EXPERIMENTAL PROCEDURES
Cell Isolation and Culture
All the procedures involving animals conform to the US National Institutes of Health regulations (NIH) and were approved by the Institutional Animal Care and Use Committee of Georgia Regents University. Mouse primary hepatocytes were prepared as described previously (22). Cells were maintained in DMEM on collagen-coated 35-mm dishes supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin, and used within 48 h after isolation. McNtcp.24 cells (a rat hepatocyte-derived cell line that stably expresses the sodium-dependent taurocholate cotransporting polypeptide and efficiently transports bile salts) were cultured in DMEM containing 10% fetal bovine serum, 10% bovine calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 200 μg/ml G418. For overexpression of the dominant-negative mutant DLP1-K38A, cells were either transfected with green fluorescent protein (GFP)-tagged DLP1-K38A (GFP-DLP1-K38A) by using Lipofectamine (Invitrogen) per manufacturer's instruction, or infected with an adenovirus carrying DLP1-K38A (Ad-DLP1-K38A) as described previously (23, 24).
Mitochondrial Morphology Analyses
Mitochondria were visualized by expressing mitochondria-targeted GFP or by MitoTracker red CMXRos (Invitrogen) staining. Quantitative analyses of mitochondrial morphology were performed using NIH-developed ImageJ software as described previously (22, 25, 26).
Fluorescence Microscopy
Fluorescence images were viewed with an Olympus IX71 epifluorescence microscope. Images were acquired with an Evolution QEi camera (Mediacybernetics, Inc.) driven by IPLab imaging software (Scanalytics, Inc.). Excitation/emission wavelengths were 480/535 nm for green fluorescence (GFP, annexin V-FITC, and Alexa 488), and 555/613 nm for red fluorescence (MitoTracker red, TMRE, propidium iodide, ethidium, and Alexa 594).
ROS Assessment
ROS levels were detected using the fluorescent probe dihydroethidium (DHE; Invitrogen) as described previously (22, 23, 25). Cultured cells or fresh frozen sections of mouse liver were loaded with 5 μm DHE at 37 °C for 30 min.
Cell Death Assay
Cell death was detected by using an annexin V-FITC apoptosis detection kit (BioVision) per manufacturer's instruction. To detect cell death in situ, a Terminal deoxynucleotidyl transferase dUTP Nick End Labeling (TUNEL, Biotium) assay was used to label apoptotic nuclei in fresh-frozen mouse liver sections per manufacturer's instruction. For quantification, TUNEL-positive hepatocytes were counted in 20 high power fields (400× magnification) for each animal.
Isolation of Mitochondria
Mitochondria were isolated by differential centrifugation. Cells were suspended in cold isolation buffer (10 mm Hepes pH 7.2, 1 mm EDTA, 320 mm sucrose) containing protease inhibitor and homogenized in a Dounce homogenizer. The homogenate was centrifuged at 700 × g for 8 min. The supernatant was saved, and the pellet was homogenized and centrifuged again. The supernatant was pooled and centrifuged at 17,000 × g for 15 min to obtain the mitochondrial pellet (27).
Immunoblotting and Immunofluorescence
For immunoblotting, the rabbit polyclonal anti-DLP-N (28), mouse monoclonal anti-cytochrome c (BD Bioscience Pharmingen), or rabbit polyclonal anti-VDAC (Rockland) antibodies were used for primary antibodies. For secondary antibodies, horseradish peroxidase-conjugated anti-rabbit, and anti-mouse antibodies were used. For indirect immunofluorescence, cells grown on the coverslip or mouse liver fresh-frozen sections were fixed in 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. Mouse anti-DLP1 (BD Biosciences) and rabbit anti-GFP antibodies (Invitrogen) were used for primary antibodies, and Alexa 488 or 594-conjugated antibodies (Invitrogen) for secondary antibodies. Fluorescence images were acquired and adjusted using Adobe Photoshop (Adobe Systems Inc.) software.
Generation of Triple Transgenic Mice Expressing DLP1-K38A
We generated a transgenic mouse line carrying DLP1-K38A and EGFP under the control of a bidirectional tet-responsive element (Tg[DLP1-K38A]) (22). Another transgenic line carrying R26STOPrtTA, which contains a transcription stop sequence (STOP) floxed in front of the rtTA, was obtained from Dr. Wei Hsu (29). We first generated a double transgenic line by crossing R26STOPrtTA with transgenic mice carrying Alb-Cre (Jax mice no. 003574). The double transgenic mice (dTg[Alb-Cre/R26STOPrtTA]) were then crossed with Tg[DLP1-K38A] to generate the triple transgenic mice tTg[Alb-Cre/R26STOPrtTA/DLP1-K38A] that express DLP1-K38A and EGFP only in the liver upon Dox induction. Transgenic mice were genotyped using PCR primers as follows: 5′-GGAACTCAGAGCAGTGGAGCG-3′ and 5′-CCTCTCTGGAAATCTTAAGTGCCTCTGAC-3′ for DLP1-K38A; 5′-GGAACAGGAGCATCAAGTAGC-3′ and 5′-GCGTCAGCAGGCAGCATATC-3′ for rtTA; 5′-ACCTGAAGATGTTCGCGATTATCT-3′ and 5′-ACCGTCAGTACGTGAGATATCTT-3′ for Cre; and 5′-TTGCTACTTGATGCTCCTGTTC-3′ and 5′-ACTCTTCGCGGTCTTTCCAGT-3′ for detecting STOP excision. For transgene expression, doxycycline was continuously supplied ad libitum in chow at 200 mg/kg (Bioserv).
RT-PCR
Total RNA from homogenized liver tissue was DNase-digested and reverse-transcribed using RQ1 DNase and GoScript Reverse Transcription System (Promega). RT-PCR primers were: 5′-GGAACTCAGAGCAGTGGAGCG-3′ and 5′-CCTCTCTGGAAATCTTAAGTGCCTCTGAC-3′ for DLP1-K38A, 5′-AACTTTGGCATTGTGGAAGGGCTC-3′ and 5′-TTGTCATTGAGAGCAATGCCAGCC-3′ for GAPDH, and 5′-TCAAGGACGACGGCAACTACAAGA-3′ and 5′-AGGACCATGTGATCGCGCTTCTC-3′ for EGFP. PCR cycle conditions were 95 °C for 5 min, then 35 cycles of 95 °C for 45 s; 60 °C, 57 °C, and 54 °C for 45 s for EGFP, DLP1-K38A, and GAPDH, respectively, and 72 °C for 1 min 15 s.
Bile Duct Ligation
8–10-week-old male triple transgenic tTg[Alb-Cre/R26STOPrtTA/DLP1-K38A] mice and the age- and sex-matched littermate control dTg[Alb-Cre/R26STOPrtTA] mice were used in these experiments. All mice were fed Dox 2 days before the operation to ensure DLP1-K38A expression. Mice were anesthetized with an intraperitoneal injection of a ketamine (100 mg/kg)/xylazine (10 mg/kg) mixture. The common bile duct was double ligated and cut between the ligatures. Sham operation mice underwent the same midline laparotomy and common bile duct exposure, without the ligation. On the day of evaluation, blood samples were collected before the mice were sacrificed. Livers were harvested and cut into pieces, immediately frozen in OCT compound, or fixed with 4% formaldehyde in PBS for further processing.
Serum Total Bilirubin Levels and Alanine Transaminase (ALT) Measurement
Serum total bilirubin levels and serum ALT were measured by commercial assay kits per manufacturers' instructions (Bilirubin Assay Kit, Abnova; Alanine Transaminase Activity Assay Kit, Cayman Chemical).
Liver Histology
Formaldehyde-fixed and paraffin-embedded mouse liver tissues were sectioned at the Histology Core Laboratory at Medical College of Georgia. H&E and trichrome staining were performed following the standard protocol.
Statistical Analysis
Error bars in all graphs represent the S.E. Student's t test (two-tailed, unpaired) was used to compare the two groups, and p < 0.05 was considered statistically significant.
RESULTS
Toxic Bile Salt GCDC Causes Cell Death and Induces Mitochondrial Fragmentation in Primary Hepatocytes
Impaired bile flow during cholestasis causes the accumulation of bile acids in hepatocytes and leads to cell death (1). Glycochenodeoxycholate (GCDC) is the major toxic bile acid accumulated in cholestasis. We treated primary mouse hepatocytes with GCDC and evaluated cell death using annexin V and propidium iodide (PI) staining. Positive staining for annexin V indicates early-stage apoptosis, whereas PI-positive cells are considered necrotic (30). We found that 50 μm GCDC significantly increased cell death in primary hepatocytes (Fig. 1, A and B), confirming previous observations (7, 9, 31–33). Significantly more cells were PI-positive after 4 h of GCDC treatment, illustrating the progression to necrotic cell death in GCDC insult (Fig. 1B). Studies indicate that GCDC and bile-duct ligation initiate cell death through the death receptor Fas as well as via nuclear proteases (33–37). In addition, the mitochondrial permeability transition has been shown to occur in bile acid-induced apoptosis (38), suggesting the involvement of mitochondria-dependent apoptosis. Because mitochondria often become fragmented during apoptosis (11), we examined mitochondrial morphology in primary hepatocytes treated with GCDC. Mitochondria in primary hepatocytes were densely packed throughout the cytoplasm. Unlike the filamentous mitochondria commonly seen in other cell types, untreated hepatocyte mitochondria were mostly spherical/ovoid shapes of varying sizes with occasional short rods (Fig. 1C). On the other hand, mitochondria in cells treated with GCDC were smaller and of more uniform size compared with those in control cells (Fig. 1E), as assessed by mitochondrial size distribution analyses (Fig. 1, D and F). In addition, the number of mitochondria per cell increased by a ∼2-fold in GCDC-treated cells. The formation of smaller mitochondria along with an increase of mitochondrial number upon treatment with GCDC is consistent with a fragmented phenotype. This morphological change indicates that despite their mostly spherical morphology, hepatic mitochondria undergo fragmentation during apoptosis induced by a toxic bile salt.
FIGURE 1.

Toxic bile salt GCDC induces cell death and mitochondrial fragmentation in primary mouse hepatocytes. A and B, primary mouse hepatocytes were incubated with 50 μm GCDC for the indicated time, and cell death was evaluated using annexin V (A) and PI (B) staining. 100–200 cells were counted in each treatment; the experiment was repeated three times. Error bars represent S.E. *, p < 0.05. C—F, GCDC-induced mitochondrial fragmentation. Spherical/ovoid mitochondria in primary hepatocytes under control conditions (C) became smaller after GCDC treatment (E). Scale bar: 10 μm. Insets: 2× magnified. Images were acquired after 2 h of GCDC incubation. Frequency plots for mitochondrial size show a broader distribution of different sizes of mitochondria under control conditions (D) versus smaller and more uniform mitochondria with an ∼2-fold increase in number after GCDC treatment (F).
GCDC Induces Mitochondrial Fragmentation and Cell Death in the Bile Transporter-expressing Hepatic Cell Line McNtcp.24
To further verify GCDC-induced mitochondrial fragmentation in cells containing more conventional tubular mitochondria, we used a hepatic cell line, McNtcp.24. McNtcp.24 stably expresses the bile salt transporter and responses to bile salt insult in a manner similar to primary hepatocytes (34). Mitochondria in untreated McNtcp.24 cells are filamentous and often interconnected to form networks (Fig. 2A). In contrast, cells treated with GCDC exhibit short and fragmented mitochondria (Fig. 2B). The number of cells with fragmented mitochondria increased to about 80% after 15 min of GCDC treatment and remained high with both 100 and 400 μm GCDC treatment (Fig. 2C). Time-lapse imaging revealed that shortened mitochondria started to appear at 8–10 min, and mitochondria became completely fragmented by 15 min after GCDC addition (Fig. 2F). We also evaluated the mitochondrial membrane potential using the potentiometric probe tetramethylrhodamine ethyl ester (TMRE) during GCDC incubation. Fluorescence quantification indicated a significant decrease of the membrane potential at 30-min or longer GCDC incubation, while no change was found with 15-min GCDC incubation (Fig. 2D). Because mitochondrial fragmentation was prevalent at 15-min GCDC treatment, these data indicate that mitochondrial fragmentation precedes the functional decrease of mitochondria during GCDC incubation.
FIGURE 2.

GCDC induces mitochondrial fragmentation in the bile transporter-expressing hepatic cell line McNtcp.24. A–C, mitochondria in McNtcp.24 are tubular in control conditions (A) and fragmented after GCDC treatment (B). Quantitative data show the numbers of cells with fragmented mitochondria increased to about 80% at 15 min and remained high with both 100 and 400 μm GCDC treatments (C). 100–200 cells were counted in each treatment; the experiment was repeated 3 times. Error bars are S.E. D, quantification of TMRE fluorescence shows a decrease in mitochondrial inner membrane potential at 30 min or longer treatments with GCDC. Error bars represent S.E. *, p < 0.05; ****, p < 0.0001. E and F, time-lapse imaging of mitochondrial in control cells (E) and GCDC-treated cells (F). Mitochondria remained tubular under control conditions (E) whereas mitochondrial tubules became shorter at 10 min and fragmented by 15 min after GCDC addition (F). Scale bar: 10 μm.
As in primary hepatocytes, GCDC treatment increased cell death in McNtcp.24 cells. The increase in cell death judged by annexin V and PI-positive staining in GCDC treatments was time and concentration dependent (Fig. 3, A and B). A prominent increase of PI-positive cells was observed with high concentration GCDC treatments (200 and 400 μm). Under these conditions, annexin V-positive cells were also PI positives, indicating necrosis (Fig. 3, B and C). In contrast, annexin V-positive cells in lower concentration GCDC treatments (50 and 100 μm) were PI negative (Fig. 3C), suggesting that this cell death was mostly apoptotic. In addition, the level of cytochrome c in the mitochondrial fraction was significantly diminished upon GCDC treatment (Fig. 3D), indicating its release from mitochondria during apoptosis. Taken together, our data show that hepatocyte mitochondria are fragmented during bile salt-induced cell injury.
FIGURE 3.

GCDC induces cell death in McNtcp.24 cells. A and B, GCDC-induced cell death is time- and concentration-dependent as measured by annexin V (A) and PI (B). ∼400 cells were counted in each treatment; the experiment was repeated three times. Error bars are S.E. C, images of annexin V and PI staining. Cells positive for annexin V are PI-negative at a low concentration of GCDC (100 μm) whereas annexin V-positive cells are PI-positive at a high-concentration of GCDC (400 μm). D, immunoblot of the mitochondrial fraction shows a significantly diminished cytochrome c level after 100 μm GCDC treatment for 2 h.
Inhibition of Mitochondrial Fission Prevents GCDC-induced Mitochondrial Fragmentation and Cell Death
In steady state, normal mitochondrial morphology is maintained by a balanced frequency of fission and fusion. Mitochondrial fragmentation induced by GCDC suggests a shift of the fission/fusion balance toward mitochondrial fission. Therefore, we examined the cellular distribution of the mitochondrial fission protein DLP1. Mitochondria were isolated from cells treated with GCDC for different periods and analyzed by immunoblotting. In both primary hepatocytes and McNtcp.24 cells, we found increased DLP1 levels in the mitochondrial fraction with concomitant decreases in the cytosolic fraction from cells treated with GCDC for 15 min or longer (Fig. 4A).
FIGURE 4.

Inhibition of mitochondrial fission prevents GCDC-induced mitochondrial fragmentation and cell death. A, immunoblot of the mitochondrial and cytosolic fractions from GCDC-treated primary hepatocytes and McNtcp.24 cells shows increased DLP1 levels in mitochondria and decreased DLP1 levels in the cytosol. B, McNtcp.24 cells expressing DLP1-K38A maintain elongated tubular mitochondria in GCDC treatment (100 μm, 30 min). 50–80 cells were counted in each treatment; the experiment was repeated four times. Error bars represent S.E. C, D, and D′, inhibition of mitochondrial fission in primary mouse hepatocytes induces mitochondrial enlargement and interconnection (D). Fission inhibition prevents the formation of small fragmented mitochondria in GCDC treatment, maintaining the enlarged and interconnected phenotype (D′). Insets: 2.5× magnified images of the boxed areas. Scale bar: 20 μm. E and F, inhibition of mitochondrial fission prevents apoptotic cell death. The number of annexin V-positive cells was significantly lower in DLP1-K38A-expressing McNtcp.24 (E) and primary hepatocytes (F) in 2-h GCDC treatment, compared with control cells. 300–500 cells were counted in each treatment; the experiment was repeated three times. Error bars represent S.E. *, p < 0.05; **, p < 0.01. G, fission inhibition blocks cytochrome c release from mitochondria. Immunofluorescence of McNtcp.24 cells treated with 100 μm GCDC for 2 h shows that cytochrome c is diffuse and its level is decreased in untransfected cell (U, right panel). The neighboring GFF-DLP1-K38A-expressing cell retains cytochrome c in the elongated mitochondrial tubules (T, right panel).
To directly test whether the mitochondrial fragmentation induced by GCDC requires DLP1-mediated fission, we inhibited mitochondrial fission by expression of the dominant-negative mutant DLP1-K38A and examined mitochondrial morphology after GCDC treatment. DLP1-K38A expression was achieved through adenoviral infection (Ad-DLP1-K38A) (23). Adenovirus carrying GFP (Ad-GFP) was used as infection control. McNtcp.24 cells expressing DLP1-K38A showed elongated tubular networks of mitochondria and, after GCDC treatment, still maintained long filamentous mitochondria (Fig. 4B). In primary hepatocytes, as we reported previously, small round mitochondria became enlarged and interconnected upon DLP1-K38A expression (22) (Fig. 4, C and D). As in McNtcp.24, fission inhibition in primary hepatocytes prevented GCDC-induced fragmentation, maintaining enlarged and connected mitochondria after GCDC treatment (Fig. 4, C′ and D′). These observations demonstrate that the mitochondrial fission protein DLP1 is required for bile salt-induced mitochondrial fragmentation.
Our observations indicate that GCDC-induced mitochondrial fragmentation occurs earlier than cell death, suggesting that mitochondrial fragmentation is a preceding process necessary for bile salt-induced cell death. To test this, we assessed the GCDC-induced cell death under fission inhibitory conditions. We found that fission inhibition by DLP1-K38A significantly decreased the number of annexin V-positive cells both in McNtcp.24 and in primary hepatocytes treated with GCDC for 2 h (Fig. 4, E and F). We also transfected cells with GFP-DLP1-K38A and evaluated the cytochrome c distribution. Consistent with the annexin V data, we found that fission inhibition blocked cytochrome c release from mitochondria. Cells expressing DLP1-K38A show characteristic DLP1-containing aggregates in the cytoplasm (39, 40) (Fig. 4G). Cytochrome c staining in untransfected cells is decreased in fragmented mitochondria and appeared dim and diffuse in GCDC-treated cells. However, in cells transfected with GFP-DLP1-K38A, cytochrome c was retained in the elongated mitochondrial tubules, indicating that fission inhibition prevented cytochrome c release (Fig. 4G). These data show that DLP1-mediated mitochondrial fission is required for bile salt-induced cell death and that inhibiting mitochondrial fission prevents that end.
Inhibition of Mitochondrial Fission Prevents GCDC-induced ROS Increase
Oxidative stress occurs in livers of patients with cholestasis and has been suggested to play a critical role in the pathogenesis of this disease (41). Therefore, we examined the role of ROS in GCDC-induced mitochondrial fragmentation and cell death. GCDC treatment significantly increased ROS levels in McNtcp.24 cells as judged by an increase of ethidium fluorescence (Fig. 5, A and B). Treating cells with manganese(III) tetrakis(1-methyl-4-pyridyl)porphyrin (MnTMPyP), a cell permeable manganese superoxide dismutase mimetic, completely blocked the GCDC-induced ROS increase (Fig. 5A). The mitochondria-targeted antioxidant Mito-TEMPO also eliminated the ROS increase in GCDC incubation, suggesting a mitochondrial origin of the ROS (Fig. 5C). In addition, in the presence of MnTMPyP, mitochondria were still fragmented after the GCDC treatment, indicating that ROS are not the cause of mitochondrial fragmentation (Fig. 5D).
FIGURE 5.

Inhibition of mitochondrial fission prevents GCDC-induced ROS increase. A–C, GCDC treatment increases ROS production from mitochondria. GCDC treatment (100 μm, 15 min) increases ethidium fluorescence in McNtcp.24 cells (B, middle panel), and MnTMPyP normalized the ROS level (B, right panel). Quantification of ethidium fluorescence (A). Mito-TEMPO also normalized GCDC-induced ROS increase, indicating mitochondrial origin of ROS (C). ****, p < 0.0001. D, inhibition of ROS increase by MnTMPyP does not prevent GCDC-induced mitochondrial fragmentation. ∼300 cells were counted in each treatment; the experiment was repeated three times. Error bars represent S.E. E and F, inhibition of mitochondrial fission prevents GCDC-induced ROS increase. In control McNtcp.24 cells, ROS levels were low in both untransfected and GFP-DLP1-K38A-expressing cells (outlined). In GCDC-treated cells (100 μm, 15 min), ROS in DLP1-K38A cells (outlined) remained low, whereas untransfected cells showed significantly increased ROS levels. Quantification of ethidium fluorescence (F). n = 40, ****, p < 0.0001. G, preventing ROS increase by MnTMPyP does not block cell death in GCDC treatment.
We found that inhibiting mitochondrial fission prevented the ROS increase in GCDC incubation. In untreated control conditions, McNtcp.24 cells transfected with GFP-DLP1-K38A showed a low level of ROS similar to untransfected cells (Fig. 5, E and F). After GCDC incubation, however, ROS in DLP1-K38A cells remained low whereas neighboring untransfected cells showed significantly increased ROS levels (Fig. 5, E and F). These data demonstrate that DLP1-mediated mitochondrial fragmentation from GCDC treatment is a causal factor for ROS increase. Interestingly, however, MnTMPyP did not block cell death in GCDC treatment (Fig. 5G), suggesting that an increased ROS level alone is not sufficient to cause cell death. Our in vitro results demonstrate that inhibition of mitochondrial fission prevents ROS increase and cell death from GCDC insult. Importantly, these findings predict that controlling mitochondrial fission may limit cholestatic liver injury in vivo.
Transgenic Mice Expressing DLP1-K38A in the Liver
To test our hypothesis that decreasing mitochondrial fission can ameliorate cholestatic liver injury, we generated a transgenic mouse model expressing DLP1-K38A specifically in the liver. To avoid a potential alteration in liver function from permanent loss of fission, we used a doxycycline (Dox)-inducible expression of the dominant-negative DLP1-K38A in the liver. In this system, a transcription stop sequence (STOP) was inserted between the ROSA26 promoter and the reverse tetracycline-controlled transactivator (rtTA) via loxP sequences (R26STOPrtTA) (29) (Fig. 6A). Crossing the mice carrying R26STOPrtTA with another transgenic line expressing the cre reombinase only in the liver (Alb-Cre: cre recombinase under the control of albumin promoter) produced the double transgenic mice in which the STOP sequence was excised out only in the liver, resulting in the liver-specific expression of rtTA. These double transgenic mice (dTg[Alb-Cre/R26STOPrtTA]) were crossed with a third transgenic line which carried DLP1-K38A and EGFP under the control of the bidirectional tet-responsive element (TRE) (22). The resulting triple transgenic mice tTg[Alb-Cre/R26STOPrtTA/DLP1-K38A] expressed DLP1-K38A only in the liver upon Dox induction (Fig. 6A). We confirmed the liver-specific excision of the STOP sequence in our triple transgenic mice using two different PCR primer sets. One primer set anneals within the rtTA coding region, amplifying the 270-base pair DNA fragment, whereas the other primer set binds upstream and downstream of the STOP sequence, amplifying the 650-base pair DNA fragment under our PCR conditions only after the STOP sequence excision (Fig. 6B). We found the amplification of the 650-base pair fragment only in the liver, indicating the liver-specific excision of the STOP sequence (Fig. 6C). We verified the expression of DLP1-K38A by RT-PCR and immunohistochemistry in the livers of Dox-fed triple transgenic mice (Fig. 6, D and E). The expression of DLP1-K38A in the triple transgenic mice was similar to that found in the double transgenic mice dTg[rtTA/DLP1-K38A], which express DLP1-K38A in whole body including the liver (22).
FIGURE 6.

Transgenic mice expressing DLP1-K38A in the liver. A, Cre-mediated excision of the transcription STOP sequence in the liver allows Dox-induced, liver-specific expression of DLP1-K38A in triple transgenic mice. B and C, detection of liver specific excision of the STOP sequence. B, primer 1 (Pr #1) binds upstream and downstream of the STOP sequence amplifying the 650-base pair DNA fragment only after STOP sequence excision. Primer 2 (Pr #2) anneals within the rtTA coding region amplifying a 270-base pair DNA fragment. Genotyping by PCR in different tissues from triple transgenic mice shows liver-specific excision of the STOP sequence. D and E, expression of transgene. RT-PCR of the livers from 2-days Dox-fed mice shows expression of DLP1-K38A and GFP in tTg (lane 2), not in dTg (lane 1) (D). Immunohistochemistry of the liver sections from 2-day Dox-fed mice shows increased levels of DLP1 and GFP in the triple transgenic mouse (lower panels) (E).
DLP1-K38A Expression Limits Cholestatic Liver Injury in Mice
Cholestasis was induced in mice by common bile duct ligation (BDL), a widely used rodent model that mimics human cholestatic liver disease. Immunoblotting analyses of mitochondrial fission and fusion proteins in the mouse livers at 3 days post-BDL showed a significantly increased level of the fission protein DLP1 and a marked decrease in the fusion protein Mfn2 (Fig. 7, A and B), suggesting a shift of the fission/fusion balance to fission, similar to in vitro observations. Consistent with these results, decreased Mfn2 levels have been observed in the livers of patients with cholestasis (42). No significant changes were found in the inner membrane fusion protein OPA1 or the fission protein Mff (Fig. 7, A and B). It has been shown that two splice variants of Mff are predominantly expressed out of nine possible variants (43, 44). While there appeared to be some alterations in the levels of small versus large Mff variants among different livers (Fig. 7A), the significance of such variations is unknown. Regardless, these results indicate that increased mitochondrial fission accompanies BDL-induced liver injury in vivo.
FIGURE 7.

Mitochondrial morphology in the livers after bile duct ligation. A and B, fission and fusion proteins in livers from sham- and BDL-operated mice (5 mice each) were analyzed by immunoblotting (A). Quantification indicates a significant increase of DLP1 and a marked decrease of Mfn2 in BDL mice (B). C, increased serum total bilirubin in both control and DLP1-K38A livers after BDL. Error bars represent S.E. *, p < 0.05; **, p < 0.01. D, electron micrographs of livers from control and tTg (DLP1-K38A) at 3 days post BDL. Mitochondria are elongated in sham-operated livers (Control sham, DLP1-K38A sham), whereas they appear fragmented in BDL livers (Control BDL, DLP1-K38A BDL).
To test the effect of decreasing mitochondrial fission on cholestatic liver injury, BDL was performed in mice expressing DLP1-K38A in the liver. We crossed heterozygous Tg[DLP1-K38A] mice with homozygous dTg[Alb-Cre/R26STOPrtTA] to produce the experimental tTg[Alb-Cre/R26STOPrtTA/DLP1-K38A] mice and the littermate control dTg[Alb-Cre/R26STOPrtTA] mice. Mice were fed Dox 2 days before the operation to ensure transgene expression of DLP1-K38A, and sacrificed 3 days after BDL when acute hepatocellular injury peaked (32). Jaundice developed in both control and experimental mice after BDL, as evidenced by yellow discoloration of the skin and confirmed by increased serum total bilirubin (Fig. 7C). Electron microscopy showed that DLP1-K38A-expressing livers contained more elongated and branched mitochondria compared with control livers (Fig. 7D, Control versus DLP1-K38A sham), suggesting decreased fission. In BDL livers, round mitochondria were prevalent, indicative of fragmentation (Fig. 7D, Control BDL). In addition, larger appearance of the round mitochondria suggests a potential swelling. This morphological change of mitochondria is consistent with original observations made in rat livers after BDL (21). No significant differences in mitochondrial morphology were observed after BDL of DLP1-K38A-expressing livers, compared with BDL livers from control mice (Fig. 7D, control versus DLP1-K38A BDL). The expression level of DLP1-K38A in these transgenic mice is modest (22). It is possible that the severe toxic insult caused by BDL is sufficient to mask the incomplete penetrance of DLP1-K38A in these mice.
Despite insufficient recovery in mitochondrial morphology, we found a drastic difference between control and DLP1-K38A mice in their appearance after the BDL. All control BDL mice appeared sick, showing hunched abnormal posture, ruffled hair, loss of groom behavior, and were stationary often huddled in the corner. Remarkably, the DLP1-K38A tTg mice undergone BDL appeared less sick and showed near normal behavior, suggesting the beneficial effect of DLP1-K38A expression. Consistent with this healthier phenotype observed in the tTg mice, hematoxylin and eosin staining of liver sections revealed that DLP1-K38A provided remarkable protection from BDL-induced liver injury. Portal edema and neutrophil infiltration were evident in control BDL mice. However, this phenotype was significantly decreased in DLP1-K38A BDL mice, indicating reduced liver injury. Most notably, large areas of biliary infarcts representing clusters of injured hepatocytes were found in control BDL mice (Fig. 8B). Importantly, we found that DLP1-K38A expression substantially decreased the number and area of infarct following the BDL (Fig. 8D). Quantification of biliary infarct demonstrated that both the area and number of infarcts were decreased by more than 3-fold in DLP1-K38A BDL mice compared with control BDL mice (Fig. 8, E and F), indicating that DLP1-K38A expression protects hepatocytes from cholestatic injury.
FIGURE 8.
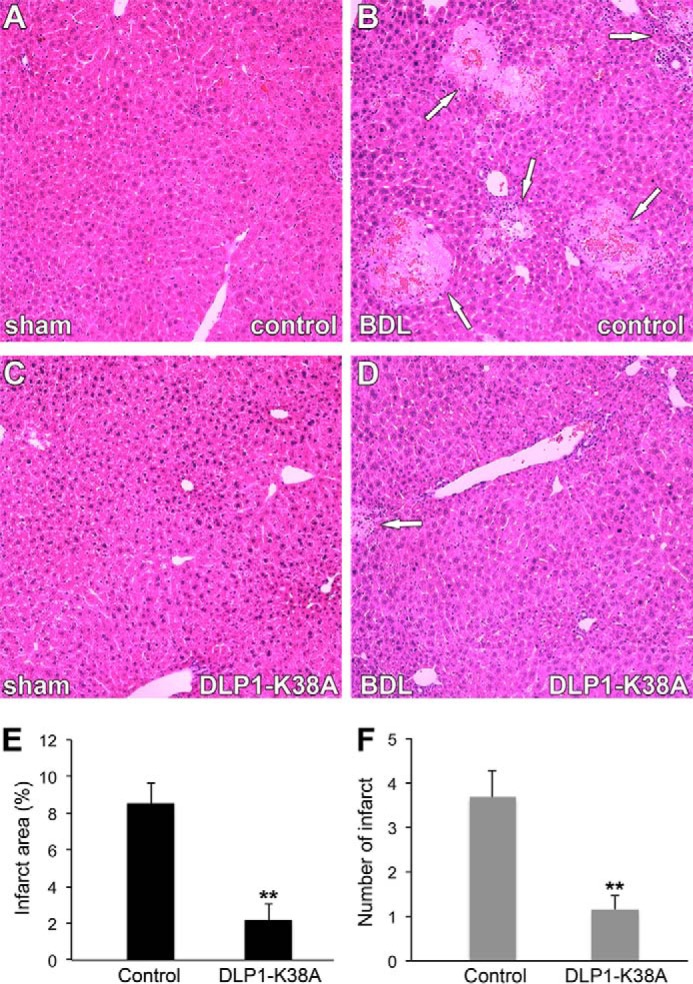
DLP1-K38A expression limits cholestatic liver injury in mice. A–D, representative images of H&E-stained liver sections from sham-operated or BDL mice. Large areas of biliary infarct (arrows) are present in control mice after BDL (B). DLP1-K38A substantially decreases the infarcts following the BDL (D). E and F, quantification shows decreases of infarct area (E) and number of infarcts (F) in DLP1-K38A BDL mice (n = 5) compared with control BDL mice (n = 5). Error bars represent S.E. **, p < 0.01.
DLP1-K38A Expression Decreases ROS Levels, Apoptosis, and Fibrosis in Cholestatic Livers
Because our in vitro studies indicated that inhibiting mitochondrial fission by DLP1-K38A diminished bile salt-induced ROS increase and apoptosis, we assessed the ROS levels and apoptosis in BDL livers from control and DLP1-K38A mice. DHE staining of the frozen liver sections showed increased ROS levels in 3-day BDL livers of control mice. In contrast, consistent with in vitro observations, no ROS increase was detected in the livers from DLP1-K38A BDL mice (Fig. 9, A and B). Likewise, TUNEL assays showed a significantly decreased number of apoptotic cells in DLP1-K38A BDL livers compared with that in control BDL livers (Fig. 9, C and D). TUNEL-positive cells could be necrotic due to potential DNA degradation by infiltrating neutrophils and other cells. However, representative images show TUNEL-positive cells to be dispersed, not confined within the necrotic infarcts (Fig. 9D), suggesting that they are apoptotic. As predicted from the BDL-induced liver injury, the serum alanine aminotransferase (ALT) level, an indicator of liver dysfunction, was increased in control BDL mice. However, DLP1-K38A BDL mice showed a notably lower serum ALT than control BDL, suggesting that DLP1-K38A expression significantly ameliorated the cholestasis-induced liver dysfunction (Fig. 9E). Finally, trichrome staining was performed to evaluate liver fibrosis, which can be detected by blue-colored staining for collagen accumulation (Fig. 9F). We found that control BDL livers developed significant fibrosis at 3 days post-BDL mostly in the infarct areas, which became more pronounced by 3 weeks post-BDL. Remarkably, DLP1-K38A expression in tTg livers resulted in significantly reduced fibrosis at both 3 days and 3 weeks post-BDL (Fig. 9, F and G).
FIGURE 9.

DLP1-K38A expression decreases ROS levels, apoptosis, and fibrosis in cholestatic livers. A–E, BDL for 3 days increases ROS levels (A, B), apoptosis (C, D, TUNEL), and liver injury (E, ALT) in control mice (n = 5). Livers from DLP1-K38A mice 3 days after BDL show significant decreases in these parameters (n = 5). *, p < 0.05; ****, p < 0.0001. F, trichrome-stained sections from 3 days and 3 weeks of BDL liver. Control mice show a significant increase in fibrosis (bluish fibrous staining, arrows) after 3 days, which is more pronounced after 3 weeks. DLP1-K38A expression significantly reduces fibrosis at both 3 days and 3 weeks post-BDL. G, quantification of liver fibrosis. At 3 days post BDL, n = 5 for both control and DLP1-K38A mice; at 3 weeks post-BDL, n = 3 for control mice and n = 5 for DLP1-K38A mice. *, p < 0.05; ***, p < 0.0001.
Our data demonstrate that expression of DLP1-K38A in the liver ameliorates hepatocellular injury and liver dysfunction through decreasing ROS and apoptosis. Our findings that controlling mitochondrial fission can limit cholestatic liver injury suggest that mitochondrial fission can be a new therapeutic target.
DISCUSSION
In this study, we demonstrated both in vitro and in vivo that decreasing mitochondrial fission has an ameliorating effect on bile acid-induced liver injury. It has been shown that cholestasis is associated with hepatic mitochondrial dysfunction, which includes loss of membrane potential and ATP production, reduced fatty acid oxidation, impaired respiratory complex activities, and decreased respiration, (6, 7). In addition, a decrease in mitochondrial biogenesis was reported in cholestasis along with impairment of autophagy (45). Mitochondrial deformation suggestive of increased division and swelling was reported in cholestatic livers decades ago (21). However, the contribution of mitochondrial morphology in cholestatic liver injury is still not well understood. Our study identified mitochondrial fission as a new factor that contributes to ROS production, hepatocellular injury, and fibrosis in cholestasis.
We observed that GCDC induced rapid fragmentation of mitochondria. This fragmentation was mediated by DLP1 that became associated with mitochondria in GCDC treatment. Mitochondrial translocation of DLP1 is regulated by phosphorylation at two different serine residues of DLP1. Phosphorylation at the upstream serine residue (corresponding to serine 616 in human DLP1) increases mitochondrial fission (46–49) whereas phosphorylation at the downstream serine (serine 637) increases or decreases fission depending on phosphorylating kinases (50–53). Cyclic AMP-dependent protein kinase (PKA) phosphorylates DLP1 at serine 637 to prevent its binding to mitochondria whereas the Ca2+-dependent phosphatase calcineurin dephosphorylates DLP1 to promote its translocation to mitochondria (50, 51, 54). Since toxic bile acids have been shown to increase cytosolic Ca2+ (55–57), it is likely that cellular Ca2+ increase by GCDC induces calcineurin-mediated DLP1 dephosphorylation and mitochondrial translocation, resulting in fission. Furthermore, it has been shown that cAMP protects hepatocytes from GCDC-induced apoptosis (58). It is possible that cAMP activates PKA to prevent DLP1-mediated mitochondrial fragmentation and apoptosis.
We found that GCDC-induced mitochondrial fragmentation was rapid and sustained. In previous studies, we reported that glucose or calcium stimulation also induced rapid mitochondrial fragmentation (25, 26, 49). However, unlike GCDC treatment, mitochondrial fragmentation under these conditions was reversible, recovering tubular morphology after an hour of sustained stimulations (25, 26). With glucose stimulation, ROS levels increase and decrease in the same time frame as mitochondrial fragmentation occurs and reverses (25). With GCDC treatment, ROS increase was also accompanied by mitochondrial fragmentation and remained high as mitochondrial fragmentation persisted. These observations suggest that there is a close relationship between mitochondrial fragmentation and ROS production. We showed that the mitochondrial MnSOD mimetic MnTMPyP was unable to prevent GCDC-induced mitochondrial fragmentation, whereas fission inhibition decreased ROS levels, indicating that mitochondrial fission is causal for the ROS increase.
Our data show that Mito-TEMPO normalized the ROS level in GCDC treatment, indicating mitochondria as a source of ROS production, which is consistent with previous findings (38, 59, 60). Bile acids have been shown to increase ROS production by mitochondria to activate the MAP kinase signaling cascade (61). This signaling activation was prevented by cyclosporine A or bongkrekic acid, suggesting that bile acids cause mitochondrial permeability transition (MPT) to induce ROS production (61). Based on our finding that mitochondrial fission is an upstream event to ROS increase in bile acids treatment, it is likely that increased mitochondrial fission induces the MPT to increase ROS levels in GCDC treatment. In support of this notion, we showed previously that inhibition of mitochondrial fission blocks MPT in metabolic insult-induced cell death (23). High levels of ROS also induce MPT (62). Once increased ROS generation is established in bile acids insult, the MPT and ROS would form a vicious cycle of ROS overproduction and mitochondrial dysfunction, leading to hepatic injury.
Interestingly, we observed that ROS suppression by MnTMPyP was not sufficient to block cell death in GCDC treatment, whereas fission inhibition not only normalized ROS levels but also prevented cell death. Bile acids induce apoptosis through activation of the death receptor Fas (34, 37). Also, bile acids can induce the MPT (61), thereby increasing ROS levels. However, Fas activation was reported to be independent of ROS production (63). In another study, bile salts-induced ROS/oxidative stress activates Fas, which was suppressed by inhibiting ROS production from NAPDH oxidases (64). It is possible that signals from ROS and Fas converge at the mitochondria, amplifying cell injury (64). Our data showed that decreasing mitochondrial fission prevented ROS increase in both GCDC-treated cells and BDL livers. In addition, it is well known that inhibition of mitochondrial fission blocks or delays the progression of intrinsic apoptosis (65). Therefore, fission inhibition can block both the early increase of ROS and the late progression of apoptosis in bile acid treatment. Our results showing in vitro and in vivo benefits of decreasing mitochondrial fission in bile acid-induced cell death demonstrate that controlling mitochondrial fission can be an effective therapeutic strategy for ameliorating cholestatic liver injury.
For in vivo evaluation of decreased fission in cholestatic liver injury, we have generated transgenic mice expressing DLP1-K38A specifically in the liver. As we showed previously with non tissue-specific transgenic mice, the level of transgene expression was modest in this mouse model (22). However, this level of DLP1-K38A expression was sufficient to decrease ROS levels and apoptosis, protecting livers from cholestatic injury. Our findings provide the proof-of-concept that mitochondrial fission can be a novel therapeutic target in cholestatic liver disease, and the new transgenic mice, which inducibly express DLP1-K38A specifically in the liver, will be useful for studying the role of mitochondrial fission in additional liver pathology including alcoholic and nonalcoholic liver diseases, as well as associated liver fibrosis and cirrhosis.
Acknowledgment
We thank Wei Hsu (University of Rochester) for providing R26STOPrtTA mice.
This study was supported by National Institutes of Health Grants DK061991 (to Y. Y.) and DK41876 (to G. J. G.).
- ROS
- reactive oxygen species
- MOMP
- mitochondrial outer membrane permeabilization
- TUNEL
- Terminal deoxynucleotidyl transferase dUTP Nick End Labeling
- DLP
- dynamin-like/related protein
- GCDC
- glycochenodeoxycholate
- PI
- propidium iodide
- TMRE
- tetramethylrhodamine ethyl ester
- MnTMPyP
- manganese(III) tetrakis(1-methyl-4-pyridyl)porphyrin
- Dox
- doxycycline
- MPT
- mitochondrial permeability transition.
REFERENCES
- 1. Malhi H., Gores G. J. (2008) Cellular and molecular mechanisms of liver injury. Gastroenterology 134, 1641–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gujral J. S., Liu J., Farhood A., Jaeschke H. (2004) Reduced oncotic necrosis in Fas receptor-deficient C57BL/6J-lpr mice after bile duct ligation. Hepatology 40, 998–1007 [DOI] [PubMed] [Google Scholar]
- 3. Fickert P., Trauner M., Fuchsbichler A., Zollner G., Wagner M., Marschall H. U., Zatloukal K., Denk H. (2005) Oncosis represents the main type of cell death in mouse models of cholestasis. J. Hepatol. 42, 378–385 [DOI] [PubMed] [Google Scholar]
- 4. Mitchell C., Mahrouf-Yorgov M., Mayeuf A., Robin M. A., Mansouri A., Fromenty B., Gilgenkrantz H. (2011) Overexpression of Bcl-2 in hepatocytes protects against injury but does not attenuate fibrosis in a mouse model of chronic cholestatic liver disease. Lab. Investig. 91, 273–282 [DOI] [PubMed] [Google Scholar]
- 5. Rehman H., Ramshesh V. K., Theruvath T. P., Kim I., Currin R. T., Giri S., Lemasters J. J., Zhong Z. (2008) NIM811 (N-methyl-4-isoleucine cyclosporine), a mitochondrial permeability transition inhibitor, attenuates cholestatic liver injury but not fibrosis in mice. J. Pharmacol. Exp. Therap. 327, 699–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rolo A. P., Oliveira P. J., Moreno A. J., Palmeira C. M. (2000) Bile acids affect liver mitochondrial bioenergetics: possible relevance for cholestasis therapy. Toxicol. Sci. 57, 177–185 [DOI] [PubMed] [Google Scholar]
- 7. Spivey J. R., Bronk S. F., Gores G. J. (1993) Glycochenodeoxycholate-induced lethal hepatocellular injury in rat hepatocytes. Role of ATP depletion and cytosolic free calcium. J. Clin. Invest. 92, 17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sokol R. J., Winklhofer-Roob B. M., Devereaux M. W., McKim J. M., Jr. (1995) Generation of hydroperoxides in isolated rat hepatocytes and hepatic mitochondria exposed to hydrophobic bile acids. Gastroenterology 109, 1249–1256 [DOI] [PubMed] [Google Scholar]
- 9. Sokol R. J., Straka M. S., Dahl R., Devereaux M. W., Yerushalmi B., Gumpricht E., Elkins N., Everson G. (2001) Role of oxidant stress in the permeability transition induced in rat hepatic mitochondria by hydrophobic bile acids. Pediatr. Res. 49, 519–531 [DOI] [PubMed] [Google Scholar]
- 10. Green D. R., Kroemer G. (2004) The pathophysiology of mitochondrial cell death. Science 305, 626–629 [DOI] [PubMed] [Google Scholar]
- 11. Frank S., Gaume B., Bergmann-Leitner E. S., Leitner W. W., Robert E. G., Catez F., Smith C. L., Youle R. J. (2001) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 1, 515–525 [DOI] [PubMed] [Google Scholar]
- 12. Karbowski M., Lee Y. J., Gaume B., Jeong S. Y., Frank S., Nechushtan A., Santel A., Fuller M., Smith C. L., Youle R. J. (2002) Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 159, 931–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Z., Jiang H., Chen S., Du F., Wang X. (2012) The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243 [DOI] [PubMed] [Google Scholar]
- 14. Lee Y. J., Jeong S. Y., Karbowski M., Smith C. L., Youle R. J. (2004) Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 15, 5001–5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Züchner S., Mersiyanova I. V., Muglia M., Bissar-Tadmouri N., Rochelle J., Dadali E. L., Zappia M., Nelis E., Patitucci A., Senderek J., Parman Y., Evgrafov O., Jonghe P. D., Takahashi Y., Tsuji S., Pericak-Vance M. A., Quattrone A., Battologlu E., Polyakov A. V., Timmerman V., Schröder J. M., Vance J. M. (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 36, 449–451 [DOI] [PubMed] [Google Scholar]
- 16. Delettre C., Lenaers G., Griffoin J. M., Gigarel N., Lorenzo C., Belenguer P., Pelloquin L., Grosgeorge J., Turc-Carel C., Perret E., Astarie-Dequeker C., Lasquellec L., Arnaud B., Ducommun B., Kaplan J., Hamel C. P. (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 26, 207–210 [DOI] [PubMed] [Google Scholar]
- 17. Alexander C., Votruba M., Pesch U. E., Thiselton D. L., Mayer S., Moore A., Rodriguez M., Kellner U., Leo-Kottler B., Auburger G., Bhattacharya S. S., Wissinger B. (2000) OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215 [DOI] [PubMed] [Google Scholar]
- 18. Waterham H. R., Koster J., van Roermund C. W., Mooyer P. A., Wanders R. J., Leonard J. V. (2007) A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 356, 1736–1741 [DOI] [PubMed] [Google Scholar]
- 19. Yoon Y., Galloway C. A., Jhun B. S., Yu T. (2011) Mitochondrial dynamics in diabetes. Antioxid. Redox Signal 14, 439–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Archer S. L. (2013) Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 369, 2236–2251 [DOI] [PubMed] [Google Scholar]
- 21. Schaffner F., Bacchin P. G., Hutterer F., Scharnbeck H. H., Sarkozi L. L., Denk H., Popper H. (1971) Mechanism of cholestasis. 4. Structural and biochemical changes in the liver and serum in rats after bile duct ligation. Gastroenterology 60, 888–897 [PubMed] [Google Scholar]
- 22. Galloway C. A., Lee H., Nejjar S., Jhun B. S., Yu T., Hsu W., Yoon Y. (2012) Transgenic control of mitochondrial fission induces mitochondrial uncoupling and relieves diabetic oxidative stress. Diabetes 61, 2093–2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu T., Sheu S. S., Robotham J. L., Yoon Y. (2008) Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 79, 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jhun B. S., Lee H., Jin Z. G., Yoon Y. (2013) Glucose stimulation induces dynamic change of mitochondrial morphology to promote insulin secretion in the insulinoma cell line INS-1E. PLoS ONE 8, e60810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu T., Robotham J. L., Yoon Y. (2006) Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. U.S.A. 103, 2653–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hom J. R., Gewandter J. S., Michael L., Sheu S. S., Yoon Y. (2007) Thapsigargin induces biphasic fragmentation of mitochondria through calcium-mediated mitochondrial fission and apoptosis. J. Cell. Physiol. 212, 498–508 [DOI] [PubMed] [Google Scholar]
- 27. Yu T., Fox R. J., Burwell L. S., Yoon Y. (2005) Regulation of mitochondrial fission and apoptosis by the mitochondrial outer membrane protein hFis1. J. Cell Sci. 118, 4141–4151 [DOI] [PubMed] [Google Scholar]
- 28. Yoon Y., Pitts K. R., Dahan S., McNiven M. A. (1998) A novel dynamin-like protein associates with cytoplasmic vesicles and tubules of the endoplasmic reticulum in mammalian cells. J. Cell Biol. 140, 779–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu H. M., Liu B., Chiu S. Y., Costantini F., Hsu W. (2005) Development of a unique system for spatiotemporal and lineage-specific gene expression in mice. Proc. Natl. Acad. Sci. U.S.A. 102, 8615–8620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vermes I., Haanen C., Steffens-Nakken H., Reutelingsperger C. (1995) A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 184, 39–51 [DOI] [PubMed] [Google Scholar]
- 31. Canbay A., Higuchi H., Bronk S. F., Taniai M., Sebo T. J., Gores G. J. (2002) Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology 123, 1323–1330 [DOI] [PubMed] [Google Scholar]
- 32. Georgiev P., Jochum W., Heinrich S., Jang J. H., Nocito A., Dahm F., Clavien P. A. (2008) Characterization of time-related changes after experimental bile duct ligation. Br. J. Surg. 95, 646–656 [DOI] [PubMed] [Google Scholar]
- 33. Miyoshi H., Rust C., Roberts P. J., Burgart L. J., Gores G. J. (1999) Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology 117, 669–677 [DOI] [PubMed] [Google Scholar]
- 34. Faubion W. A., Guicciardi M. E., Miyoshi H., Bronk S. F., Roberts P. J., Svingen P. A., Kaufmann S. H., Gores G. J. (1999) Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J. Clin. Invest. 103, 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kwo P., Patel T., Bronk S. F., Gores G. J. (1995) Nuclear serine protease activity contributes to bile acid-induced apoptosis in hepatocytes. Am. J. Physiol. 268, G613–G621 [DOI] [PubMed] [Google Scholar]
- 36. Roberts L. R., Kurosawa H., Bronk S. F., Fesmier P. J., Agellon L. B., Leung W. Y., Mao F., Gores G. J. (1997) Cathepsin B contributes to bile salt-induced apoptosis of rat hepatocytes. Gastroenterology 113, 1714–1726 [DOI] [PubMed] [Google Scholar]
- 37. Reinehr R., Graf D., Häussinger D. (2003) Bile salt-induced hepatocyte apoptosis involves epidermal growth factor receptor-dependent CD95 tyrosine phosphorylation. Gastroenterology 125, 839–853 [DOI] [PubMed] [Google Scholar]
- 38. Yerushalmi B., Dahl R., Devereaux M. W., Gumpricht E., Sokol R. J. (2001) Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology 33, 616–626 [DOI] [PubMed] [Google Scholar]
- 39. Pitts K. R., Yoon Y., Krueger E. W., McNiven M. A. (1999) The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol. Biol. Cell 10, 4403–4417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yoon Y., Pitts K. R., McNiven M. A. (2001) Mammalian dynamin-like protein DLP1 tubulates membranes. Mol. Biol. Cell 12, 2894–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Copple B. L., Jaeschke H., Klaassen C. D. (2010) Oxidative stress and the pathogenesis of cholestasis. Semin. Liver Dis. 30, 195–204 [DOI] [PubMed] [Google Scholar]
- 42. Chen Y., Lv L., Jiang Z., Yang H., Li S., Jiang Y. (2013) Mitofusin 2 protects hepatocyte mitochondrial function from damage induced by GCDCA. PLoS ONE 8, e65455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Otera H., Wang C., Cleland M. M., Setoguchi K., Yokota S., Youle R. J., Mihara K. (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 191, 1141–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gandre-Babbe S., van der Bliek A. M. (2008) The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 19, 2402–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin T. K., Huang L. T., Huang Y. H., Tiao M. M., Tang K. S., Liou C. W. (2012) The effect of the red wine polyphenol resveratrol on a rat model of biliary obstructed cholestasis: involvement of anti-apoptotic signalling, mitochondrial biogenesis and the induction of autophagy. Apoptosis 17, 871–879 [DOI] [PubMed] [Google Scholar]
- 46. Taguchi N., Ishihara N., Jofuku A., Oka T., Mihara K. (2007) Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529 [DOI] [PubMed] [Google Scholar]
- 47. Qi X., Disatnik M. H., Shen N., Sobel R. A., Mochly-Rosen D. (2011) Aberrant mitochondrial fission in neurons induced by protein kinase C{δ} under oxidative stress conditions in vivo. Mol. Biol. Cell 22, 256–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Strack S., Wilson T. J., Cribbs J. T. (2013) Cyclin-dependent kinases regulate splice-specific targeting of dynamin-related protein 1 to microtubules. J. Cell Biol. 201, 1037–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu T., Jhun B. S., Yoon Y. (2011) High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid. Redox Signal. 14, 425–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chang C. R., Blackstone C. (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 282, 21583–21587 [DOI] [PubMed] [Google Scholar]
- 51. Cribbs J. T., Strack S. (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Han X. J., Lu Y. F., Li S. A., Kaitsuka T., Sato Y., Tomizawa K., Nairn A. C., Takei K., Matsui H., Matsushita M. (2008) CaM kinase I α-induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 182, 573–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang W., Wang Y., Long J., Wang J., Haudek S. B., Overbeek P., Chang B. H., Schumacker P. T., Danesh F. R. (2012) Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 15, 186–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cereghetti G. M., Stangherlin A., Martins de Brito O., Chang C. R., Blackstone C., Bernardi P., Scorrano L. (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 105, 15803–15808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anwer M. S., Engelking L. R., Nolan K., Sullivan D., Zimniak P., Lester R. (1988) Hepatotoxic bile acids increase cytosolic Ca++ activity of isolated rat hepatocytes. Hepatology 8, 887–891 [DOI] [PubMed] [Google Scholar]
- 56. Beuers U., Nathanson M. H., Boyer J. L. (1993) Effects of tauroursodeoxycholic acid on cytosolic Ca2+ signals in isolated rat hepatocytes. Gastroenterology 104, 604–612 [DOI] [PubMed] [Google Scholar]
- 57. Thibault N., Ballet F. (1993) Effect of bile acids on intracellular calcium in isolated rat hepatocyte couplets. Biochem. Pharmacol. 45, 289–293 [DOI] [PubMed] [Google Scholar]
- 58. Webster C. R., Anwer M. S. (1998) Cyclic adenosine monophosphate-mediated protection against bile acid-induced apoptosis in cultured rat hepatocytes. Hepatology 27, 1324–1331 [DOI] [PubMed] [Google Scholar]
- 59. Serviddio G., Pereda J., Pallardó F. V., Carretero J., Borras C., Cutrin J., Vendemiale G., Poli G., Viña J., Sastre J. (2004) Ursodeoxycholic acid protects against secondary biliary cirrhosis in rats by preventing mitochondrial oxidative stress. Hepatology 39, 711–720 [DOI] [PubMed] [Google Scholar]
- 60. Rodrigues C. M., Fan G., Wong P. Y., Kren B. T., Steer C. J. (1998) Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol. Med. 4, 165–178 [PMC free article] [PubMed] [Google Scholar]
- 61. Fang Y., Han S. I., Mitchell C., Gupta S., Studer E., Grant S., Hylemon P. B., Dent P. (2004) Bile acids induce mitochondrial ROS, which promote activation of receptor tyrosine kinases and signaling pathways in rat hepatocytes. Hepatology 40, 961–971 [DOI] [PubMed] [Google Scholar]
- 62. Kowaltowski A. J., Castilho R. F., Vercesi A. E. (2001) Mitochondrial permeability transition and oxidative stress. FEBS Lett. 495, 12–15 [DOI] [PubMed] [Google Scholar]
- 63. Gupta S., Natarajan R., Payne S. G., Studer E. J., Spiegel S., Dent P., Hylemon P. B. (2004) Deoxycholic acid activates the c-Jun N-terminal kinase pathway via FAS receptor activation in primary hepatocytes. Role of acidic sphingomyelinase-mediated ceramide generation in FAS receptor activation. J. Biol. Chem. 279, 5821–5828 [DOI] [PubMed] [Google Scholar]
- 64. Reinehr R., Becker S., Keitel V., Eberle A., Grether-Beck S., Häussinger D. (2005) Bile salt-induced apoptosis involves NADPH oxidase isoform activation. Gastroenterology 129, 2009–2031 [DOI] [PubMed] [Google Scholar]
- 65. Youle R. J., van der Bliek A. M. (2012) Mitochondrial fission, fusion, and stress. Science 337, 1062–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]