

Background: IpaH bacterial ubiquitin ligases show no homology with eukaryotic ligases, and their mechanism is speculative.
Results: IpaH9.8 functions as a cooperative allosteric dimer with two Ubc5∼ubiquitin binding sites per subunit.
Conclusion: Kinetic parallels between IpaH and eukaryotic HECT ligases suggest convergent catalytic cycle evolution.
Significance: These are the first mechanistic details of the IpaH enzyme catalytic mechanism.
Keywords: Cooperativity, E3 Ubiquitin Ligase, Enzyme Mechanism, Polyubiquitin Chain, Protein Degradation, Virulence Factor, IpaH, Ubc5, Conjugation, Oligomer
Abstract
The human pathogen Shigella flexneri subverts host function and defenses by deploying a cohort of effector proteins via a type III secretion system. The IpaH family of 10 such effectors mimics ubiquitin ligases but bears no sequence or structural homology to their eukaryotic counterpoints. Using rates of 125I-polyubiquitin chain formation as a functional read out, IpaH9.8 displays V-type positive cooperativity with respect to varying concentrations of its Ubc5B∼125I-ubiquitin thioester co-substrate in the nanomolar range ([S]½ = 140 ± 32 nm; n = 1.8 ± 0.1) and cooperative substrate inhibition at micromolar concentrations ([S]½ = 740 ± 240 nm; n = 1.7 ± 0.2), requiring ordered binding to two functionally distinct sites per subunit. The isosteric substrate analog Ubc5BC85S-ubiquitin oxyester acts as a competitive inhibitor of wild-type Ubc5B∼125I-ubiquitin thioester (Ki = 117 ± 29 nm), whereas a Ubc5BC85A product analog shows noncompetitive inhibition (Ki = 2.2 ± 0.5 μm), consistent with the two-site model. Re-evaluation of a related IpaH3 crystal structure (PDB entry 3CVR) identifies a symmetric dimer consistent with the observed cooperativity. Genetic disruption of the predicted IpaH9.8 dimer interface reduces the solution molecular weight and significantly ablates the kcat but not [S]½ for polyubiquitin chain formation. Other studies demonstrate that cooperativity requires the N-terminal leucine-rich repeat-targeting domain and is transduced through Phe395. Additionally, these mechanistic features are conserved in a distantly related SspH2 Salmonella enterica ligase. Kinetic parallels between IpaH9.8 and the recently revised mechanism for E6AP/UBE3A (Ronchi, V. P., Klein, J. M., and Haas, A. L. (2013) E6AP/UBE3A ubiquitin ligase harbors two E2∼ubiquitin binding sites. J. Biol. Chem. 288, 10349–10360) suggest convergent evolution of the catalytic mechanisms for prokaryotic and eukaryotic ligases.
Introduction
Diarrheal diseases represent a serious medical concern for developing nations (1, 2). Gastrointestinal infection with the genus Shigella alone is responsible for over 1.1 million deaths annually and disproportionately affects children under the age of five (3). Additionally, prior infection with Shigella has also been implicated in the etiology of persistent reactive arthritis and hemolytic-uremic syndrome (4). In the face of increasing antimicrobial resistance (5), a deeper understanding of host-pathogen interactions is crucial in developing additional therapeutic avenues. Shigella infections spread via a fecal-oral route and invade enterocytes through a complex multistep process that includes the paracellular invasion of intestinal epithelium and endocytic uptake of the bacterium through the basal membrane of the intestinal cells (6, 7). Once inside the host cell, the bacterium escapes the endocytic vacuole, gains motility through actin polymerization in the cytosol, and moves between cells via paracytosis (8–12). Each of these steps, as well as successful evasion of the host immune response, relies on the activity of over 2 dozen effector proteins secreted from the bacterium through the syringe-like type III secretion apparatus (13–15). Almost half of these secreted effectors are members of the IpaH family of enzymes, found either on the large virulence plasmid of Shigella or on the bacterial chromosome (16). The nearly complete structure of IpaH3 reveals the conserved architecture for the 10-member IpaH family, which consists of two domains: a highly conserved (>95%) C-terminal catalytic domain that assembles the polyubiquitin degradation signal on the protein target and a more variable N-terminal leucine-rich repeat domain that is responsible for recruiting target proteins (17).
The immunomodulatory effects of IpaH9.8 have been appreciated since 2003, when Haraga and Miller (18) demonstrated that expression of Shigella IpaH9.8 decreased NF-κB-mediated transcription of downstream genes, including IL-8, RANTES, GM-CSF, and IL-1β, subsequently shown to result from binding of IpaH9.8 to the U2AF35 splicing factor (19). The exact biochemical mechanism for inhibition of NF-κB signaling remained unclear until Rhode et al. (20) showed that IpaH9.8 (and presumably all IpaH paralogs) possesses intrinsic ubiquitin ligase activity localized to the conserved C-terminal domain. Because these enzymes contain an active site cysteine residue essential for E3 ligase function, the IpaH family has been termed HECT3-like, referring to the conserved HECT family of eukaryotic enzymes that also possess an active site cysteine (21). However, the primary sequences of the IpaH catalytic domains are unrelated to those of the eukaryotic HECT ligase superfamily or to the larger class of RING ligases (17, 20, 22). The dissimilarity to HECT domain ligases was confirmed with publication of structures for IpaH3 (PDB code 3CVR) and the catalytic domain of IpaH1.4 (PDB code 3CKD), revealing a unique fold for this new class of E3 ligase that does not resemble the multilobed subdomain architecture of the HECT superfamily (22). Thus, although IpaH9.8 possesses an essential active site cysteine to which a ubiquitin thioester intermediate forms (22), presumably by thiol exchange from its cognate E2∼ubiquitin co-substrate, other similarities between the prokaryotic IpaH and eukaryotic HECT ligases remain speculative. Further characterization of IpaH9.8 specificity identified NF-κB essential modulator (NEMO) as a target substrate for IpaH9.8-mediated polyubiquitination and subsequent proteasomal degradation, thereby providing a likely mechanism for Shigella to block the host cell from mounting an effective innate immune response to infection (23).
IpaH enzymes are one of several examples of pathogens co-opting the otherwise exclusively eukaryotic ubiquitin-proteasome system as a virulence strategy. The E6 protein of human papillomaviruses redirects the specificity of the archetype HECT ligase E6AP/UBE3A to target the tumor suppressor p53 during host cell transformation (24). Kaposi's sarcoma-associated herpesvirus possesses two ubiquitin ligases, MIR-1 and -2, that belong to the RING-like family of E3 ligases and function to down-regulate several cell membrane proteins, including MHC-1, required for antigen presentation (25). Enterohemmorrhagic Escherichia coli contains a family of NleG effectors that possess conserved U-Box ligase domain architecture (26). The Shigella-like Pseudomonas syringiae plant pathogen also introduces effectors through a type III secretion apparatus, one of which (AvrPtoB) possesses a plant homeodomain related to eukaryotic RING domain ligases and inhibits apoptosis of infected cells (27). Likewise, the Salmonella effector SopA and the E. coli effector NleL are ubiquitin ligases whose sequences are clearly related to those of the HECT domain superfamily (28, 29). Except for IpaH, this cohort of ubiquitin ligases are all related in sequence to known eukaryotic ubiquitin ligases, suggesting that they arose through lateral gene transfer of eukaryotic genes (17, 27, 30). Therefore, it is less likely that they would provide a target for mechanism-based therapeutic intervention, given their similarity to host ligases and the high potential for off-path effects. Alternatively, the IpaH enzymes presumably have arisen through convergent evolution to interact with the host ubiquitin system, making them inherently better potential therapeutic targets possessing less risk for host cell toxicity. Furthermore, marked conservation in the core catalytic domain of the IpaH enzymes predicts that a single inhibitor targeting this domain could be effective against all isoforms. This is especially important because the apparent redundancy of at least the chromosomal genes suggests that only quantitative ablation of IpaH activity will result in observable changes in the host immune response (16). Consistent with this conclusion, two identified targets for IpaH ligases, NEMO for IpaH9.8 and NF-κB p65 for IpaH4.5, participate in the same host innate immune response pathway (23, 31). With variable ubiquitination targets, inhibiting IpaH target interactions would be inadequate to inhibit the downstream consequences of IpaH ligase activity.
To this end, understanding the mechanism of IpaH-mediated ubiquitin conjugation and how it specifically interacts with the eukaryotic ubiquitination machinery is essential to understanding the virulence strategy used by Shigella. It also allows comparison of the IpaH mechanism with features of eukaryotic HECT domain ligases potentially to discover functional aspects of the enzymes by analogy. Recent findings from our group have revealed previously unsuspected complexity in the mechanism of the HECT ligase-catalyzed polyubiquitin chain formation (32, 33). Kinetic studies show that E6AP possesses two E2∼ubiquitin thioester binding sites that function in concert to assemble the polyubiquitin degradation signal, which presumably remains tethered to the active site cysteine of the HECT domain as a thioester until en bloc transfer to the targeted substrate (32, 34). More recent kinetic evidence identifies a radial symmetric E6AP trimer as the catalytically competent form of the enzyme (33).
In the present study, we have exploited the ability of ubiquitin ligases to synthesize polyubiquitin chains in the absence of target protein as a functional readout of ligase activity in order to probe mechanistic features of the IpaH catalytic cycle. These techniques allow us to establish unambiguously the cognate E2 supporting IpaH9.8-catalyzed chain formation and to confirm that the ligase synthesizes unanchored polyubiquitin chains. Furthermore, we show that IpaH9.8 exists as an obligate dimer that cooperatively binds its E2∼ubiquitin thioester substrate. Finally, in accordance with the newly proposed model for E6AP (32), IpaH9.8 displays a second functionally distinct substrate binding site for the E2∼ubiquitin thioester, suggesting that the IpaH ligase arose by convergent evolution. Together, these studies provide the first quantitative insights into the IpaH family of bacterial ligases and, by comparison, into the eukaryotic ubiquitin conjugation machinery.
MATERIALS AND METHODS
Creatine phosphokinase and bovine ubiquitin were purchased from Sigma. Ubiquitin was further purified to apparent homogeneity by FPLC and quantitated spectrophotometrically at 280 nm (35). Ubiquitin was radioiodinated using the chloramine-T method to yield specific activities of ∼15,000 cpm/pmol using carrier-free Na125I from GE Healthcare or PerkinElmer Life Sciences (36). Ubiquitin-activating enzyme (Uba1) was purified from outdated human erythrocytes (36) and was quantitated using the stoichiometric formation of Uba1∼125I-ubiquitin thioester (36–38).
Generation and Purification of Recombinant E2 Paralogs
Human recombinant E2 proteins Ubc2b (UBE2B), Ubc5A (UBE2D1), Ubc5B (UBE2D2), Ubc5C (UBE2D3), UbcH6 (UBE2E1), UbE2E2 (UBE2E2), UbcM2 (UBE2E3), and UbcH7 (UBE2L3) were those described previously (39). The Ubc5BC85S and Ubc5BC85A mutants were generated from pGEX4T1-HsUbc5B using the QuikChange site-directed mutagenesis kit (Agilent) and were individually sequenced to confirm the mutation and the absence of cloning artifacts. Recombinant wild-type and mutant Ubc5B enzymes were expressed in BL21 (DE3) E. coli harboring the specific wild-type or mutant pGEX-Ubc5B plasmid by growing four 1-liter cultures at 20 °C to A600 = 0.6. Expression of the GST fusion protein was induced with the addition of isopropyl-1-thio-β-d-galactopyranoside to a final concentration of 0.5 mm. After 15 h, the cells were harvested by centrifugation at 6,000 × g for 15 min; resuspended in ice-cold 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, and 1 mm DTT; and lysed by Emulsiflex (Avestin). The resulting lysate was centrifuged at 100,000 × g for 45 min, and GST-Ubc5 protein was purified from the 100,000 × g supernatant by glutathione-Sepharose (GE Life Sciences) affinity chromatography using a 10-ml bed volume gravity flow column (40). The GST-E2 fusion proteins were processed with 50 units/ml thrombin (GE Life Sciences) to cleave the GST tag, and the resulting Ubc5 polypeptide was collected in the unadsorbed fraction of a subsequent glutathione-Sepharose affinity purification step designed to remove free GST. Total protein concentration was determined spectrophotometrically using the calculated 280 nm molar extinction coefficient for each protein. Typical yields were 5–6 mg of apparently homogeneous protein/liter of culture. Total protein was compared with the active fraction of protein as determined by end point thioester formation with 125I-ubiquitin (38, 41). The Ubc5 isoforms were typically ∼15% active by total protein with a further ∼40% loss with each freeze/thaw cycle. Because Ubc5BC85A is unable to form a thioester bond with 125I-ubiquitin, the native fraction of this E2 mutant was approximated by that of a parallel wild-type Ubc5B preparation (41).
Generation and Purification of IpaH9.8, IpaH9.8(244–545), IpaH9.8L435R, IpaH9.8F393A, and SspH2
The wild-type IpaH9.8 coding sequence from Shigella flexneri (a generous gift of Dr. Malabi Venkatesan, Walter Reed Army Institute of Research) was subcloned into the BamHI/NotI sites of pGEX4T-1 to yield pGEX4T1-IpaH9.8. IpaH9.8(244–545) was subcloned from pGEX4T1-IpaH9.8 by PCR using appropriately designed primers and also inserted into the BamHI/NotI sites of pGEX4T-1 to generate pGEX4T-1-IpaH9.8(244–545). Mutant pGEX4T1-IpaH9.8L435R and pGEX4T1-IpaH9.8F393A were generated from pGEX4T1-IpaH9.8 using the QuikChange site-directed mutagenesis kit (Agilent). The SspH2 gene was amplified from Salmonella enterica genomic DNA (ATCC strain 13076, a generous gift of Dr. Jeffery Hobden, Louisiana State University Medical School) and subcloned into the BamHI/NotI sites of pGEX4T-1 to generate pGEX4T1-SspH2. E. coli BL21 DE3 cells harboring either wild-type or mutant pGEX4T1-IpaH9.8 were grown at 37 °C to an A600 of 0.8 and then induced with 0.5 mm isopropyl-1-thio-β-d-galactopyranoside. After an additional 4.5 h, the cells were harvested by centrifugation at 6,000 × g for 15 min; resuspended in ice-cold buffer containing 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, and 1 mm DTT; and then lysed by Emulsiflex (Avestin). The resulting extract was centrifuged at 100,000 × g for 45 min. The GST-IpaH9.8 fusion protein was purified from the 100,000 × g supernatant by glutathione-Sepharose affinity chromatography (39). The isolated fusion protein was processed with 50 units/ml thrombin (GE Life Sciences) to remove the GST moiety. The IpaH9.8 was collected in the unadsorbed fraction of an additional glutathione-Sepharose affinity purification step to remove free GST and any contaminating GST-IpaH9.8 remaining after thrombin treatment. The resulting IpaH9.8 was further purified by resolution on a Superdex 200 prep-grade column followed by dialysis against 50 mm Tris-HCl (pH 7.5) containing 1 mm DTT. Small aliquots were flash-frozen in liquid nitrogen and stored at −80 °C. Recombinant SspH2 was purified exactly as described for IpaH9.8, except that it was expressed at 20 °C for 17 h to enhance yield. Analysis by both reducing and non-reducing SDS-PAGE and Coomassie Brilliant Blue staining showed a single band of >95% purity migrating at the expected relative molecular mass of 62 kDa. Concentrations were determined spectrophotometrically at 280 nm using a theoretical molar extinction coefficient of 75,180 m−1 cm−1 for IpaH9.8 and its point mutants, 52,040 m−1 cm−1 for IpaH9.8(244–545), or 112,840 m−1 cm−1 for SspH2. Typical yields were ∼10 mg of purified protein/liter of culture. The enzymes were stable for at least 1 year when stored at −80 °C.
Size Exclusion Chromatography
The relative solution molecular weights of selected proteins were determined by FPLC using a Superose 12 (10/30) gel filtration column (Amersham Biosciences) equilibrated at 4 °C in 50 mm Tris-HCl (pH 7.5), 50 mm NaCl, and 1 mm DTT. One hundred microliters of each protein solution were resolved at 0.5 ml/min and monitored at 280 nm. The void volume of the column was determined using 100 μl of 2 mg/ml blue dextran 200. The column was calibrated using a set of standards of known molecular weight resolved under identical conditions.
IpaH9.8-catalyzed 125I-Ubiquitin Conjugation Assay
The E3 ligase activity of IpaH9.8 was quantitated in kinetic assays under initial velocity conditions (42). Rates of IpaH9.8-catalyzed 125I-polyubiquitin chain formation were determined at 37 °C in 25-μl reactions containing 50 mm Tris-HCl (pH 7.5), 2 mm ATP, 10 mm MgCl2, 1 mm DTT, 10 mm creatine phosphate, 1 IU of creatine phosphokinase, 5 μm 125I-ubiquitin, 50 nm human Uba1, and the indicated concentrations of E2 and IpaH9.8. Reactions were started with the addition of 125I-ubiquitin to a final concentration of 5 μm and then quenched after 10 min by the addition of 2× SDS sample buffer containing 4% (v/v) β-mercaptoethanol. After incubating samples at 100 °C for 2 min, conjugated 125I-ubiquitin was resolved from free 125I-ubiquitin by 12% (w/v) SDS-PAGE at 4 °C, after which the gels were dried and visualized by autoradiography (42). Unless otherwise indicated, unanchored polyubiquitin chain formation was determined by excising lanes and measuring associated radioactivity present at the top of the stacker gel, representing free 125I-polyubiquitin chains, by γ-counting and then calculating absolute rates using the corrected specific radioactivity of the radioiodinated ubiquitin (42). Kinetic data were calculated by non-linear regression analysis using Grafit version 5.0 (42).
Static Light Scattering
Static light scattering data were collected at 37 °C using a DynaPro NanoStar instrument (Wyatt Technologies) with two independent measurements of 10 5-s acquisitions. Each enzyme was present at 75 μm total protein concentration in 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, and 1 mm DTT (33).
GST-IpaH9.8 Subunit Exchange Assay
Non-chimeric IpaH9.8 and IpaH9.8L435R proteins were prepared from their corresponding GST fusions by processing with 50 units/ml thrombin and then passed through a glutathione-Sepharose affinity column to remove unprocessed GST fusion. The unabsorbed fraction was further purified on a 1 × 30-cm Sephadex 200 FPLC column to resolve thrombin from the processed IpaH9.8 proteins.
For the subunit exchange assays, 400-μl reactions containing 4.3 μm IpaH9.8 or IpaH9.8L435R in the absence or presence of 4.3 μm GST-IpaH9.8 or GST-IpaH9.8L435R in 50 mm Tris-Cl (pH 7.5), 150 mm NaCl, 1 mm DTT, and 50% (v/v) glutathione-Sepharose were incubated at 37 °C. At the indicated times, 100-μl aliquots were removed, and the glutathione-Sepharose matrix was collected by centrifugation for 1 min at 10,000 × g and then washed three times with 200 μl of 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, and 1 mm DTT. Bound proteins were eluted directly into 100 μl of 2× SDS sample buffer. Samples were then resolved by 10% (w/v) SDS-PAGE under reducing conditions and visualized by Coomassie Brilliant Blue staining. Quantitative densitometry was used to determine the amount of IpaH9.8 or IpaH9.8L435R co-purifying with the respective GST-IpaH9.8 protein. The amount purified in the absence of the GST-IpaH9.8 was subtracted as background due to nonspecific binding. Half-lives of subunit exchange were determined by comparing the relative amounts of GST-tagged and untagged IpaH9.8 or IpaH9.8L435R purified at the 24 h time point. Serial dilutions of each sample were analyzed to determine the appropriate range over which staining intensity was linear with respect to sample dilution. Intensity values for the GST-tagged enzymes were corrected for the additional binding of dye due to the GST moiety under the assumption that GST and IpaH9.8 exhibit similar color yields for Coomassie dye binding.
RESULTS
IpaH9.8 Functions with the Ubc5 Family of E2 Paralogs to Synthesize Unanchored Lysine48-linked Polyubiquitin Chains
All E3 ubiquitin ligases tested to date are capable of assembling polyubiquitin chains in the absence of their cognate target proteins (32, 39, 42). The initial rates of polyubiquitin chain formation can thus be exploited as a facile reporter function for detailed kinetic analysis of the E3 ligase mechanism even without knowledge of the cognate target protein(s) or post-translational modification(s) required for target protein recognition (32, 39, 42). Previous reports suggest that IpaH9.8 functions solely with the Ubc5 family of E2-conjugating enzymes (17, 20, 22); however, differences in the stability of various E2 families, ambiguity associated with establishing the rate-limiting step in such screens, and errors in estimating active E2 concentration based on total protein complicate such an assignment, as discussed previously (42). The E2 screen shown in Fig. 1A was conducted under E3-limiting initial velocity conditions using representative paralogs from the major E2 families. Each E2 species was present at a final concentration of 100 nm active protein, as determined in parallel by stoichiometric E2∼125I-ubiquitin thioester formation (42). Because most E2S∼Ub-E3 interactions identified to date exhibit affinities corresponding to Km values of ∼100 nm, performing the screen at a similar concentration provides maximum sensitivity to potential differences in Km, kcat, or both and reduces the likelihood of false positive results arising from favoring functionally irrelevant interactions due to excessive substrate concentrations, as discussed previously (42). Maintaining E3-limiting conditions, confirmed by the independence of rate on [Uba1]o, guarantees that all active E2 is present as its corresponding ubiquitin thioester, the actual substrate for the ligase (42). Only members of the Ubc5 family supported robust 125I-polyubiquitin chain formation, with the resulting conjugates accumulating at the top of the resolving and stacker gels (Fig. 1A, lanes 3–5). Because the assays are conducted under initial velocity conditions, the autoradiographic intensity is proportional to the initial rate; thus, the increased intensity of the signal with Ubc5B and Ubc5C compared with Ubc5A indicates that IpaH9.8 can distinguish among these otherwise highly conserved Ubc5 isozymes. The signal that accumulates in the resolving gel in the presence of UbcH6, UbE2E2, UbcM2, or E2epf (Fig. 1A, lanes 6–8 and 10) results from IpaH9.8-independent autoconjugation of the E2 paralogs (32, 39). These results confirm that members of the Ubc5 family of E2 paralogs are the cognate carrier proteins supporting IpaH9.8-catalyzed 125I-polyubiquitin chain formation.
FIGURE 1.

The Ubc5 family of E2 enzymes supports IpaH9.8-catalyzed assembly of unanchored lysine 48-linked polyubiquitin chains. A, 125I-ubiquitin conjugation assays containing 5 nm IpaH9.8 were incubated for 10 min in the absence (lane 1) or presence (lanes 2–10) of a 100 nm concentration of the indicated E2 paralog and then quenched with sample buffer and resolved by 12% (w/v) SDS-PAGE under reducing conditions as described under “Materials and Methods.” The resulting gel was dried and visualized by autoradiography. B, an incubation identical to that of lane 4 of A was quenched by the addition of 8 IU of apyrase, and DTT was added to a final concentration of 10 mm to reduce all thioester bonds (0 min). Isopeptidase T was added to a final concentration of 3.8 μm by total protein, and aliquots were removed at the indicated times and quenched with sample buffer. At 50 min, additional isopeptidase T was added to a final concentration of 7.6 μm by total protein, and the reaction was allowed to continue for an additional 20 min (lane 8). The samples were resolved and visualized as in A. C, after autoradiography of the gel in B, 125I-ubiquitin conjugates larger than 25 kDa were excised and quantified by γ-counting. The percentage of 125I radioactivity remaining compared with zero time was analyzed by a semilog plot. The dashed line represents extrapolation of the limiting linear rate to t0 in order graphically to determine the fraction of IsoT refractory (anchored) versus IsoT sensitive (unanchored) chains. D, 125I-ubiquitin conjugation assays containing 5 nm IpaH9.8 were incubated for 10 min with 5 μm 125I-ubiquitin (lane 3) or 1 μm 125I-ubiquitin and a 4 μm concentration of the indicated unlabeled wild type or single lysine-to-arginine ubiquitin point mutant (lanes 4–11). Samples were resolved and visualized as in A. Mobility of relative molecular weight markers and position of the stacker gel are shown to the left. Position of free 125I-ubiquitin is shown to the right.
The ubiquitin-specific protease IsoT is an isopeptidase that disassembles free unanchored polyubiquitin chains (43) and can be used to distinguish free from anchored forms of the degradation signal (32, 39, 42). When the products of an IpaH9.8-catalyzed 125I-ubiquitin conjugation reaction are incubated in the presence of IsoT, the high molecular weight conjugates are lost (Fig. 1B). Conjugates present in the stacker are completely disassembled during the course of the incubation, indicating that they represent unanchored 125I-polyubiquitin chains (42). Polyubiquitin chains present at the top of the resolving gel are much less sensitive to the action of IsoT, with the remaining signal present at 50 min being refractory to the addition of a second aliquot of the enzyme and incubation for an additional 20 min (Fig. 1B, lanes 7 and 8), indicating that this associated signal represents anchored chains. Conjugates of greater than 25 kDa in the lanes of Fig. 1B were quantitated by excising the lanes and determining associated 125I radioactivity. The time dependence for loss of conjugates in the presence of IsoT shows biphasic kinetics consistent with the qualitative data of Fig. 1B and indicates that 17% of the total conjugates, present at the top of the resolving gel, represent anchored chains, whereas 83% represent free chains (Fig. 1C) when the limiting rate for the slow phase is extrapolated to t0 (dashed line).
Proteomic studies demonstrate the existence of a variety of polyubiquitin chains distinguished by their linkages to the seven lysine residues of ubiquitin (44). Early reports indicated that IpaH9.8 synthesized Lys48-linked polyubiquitin chains (17), the canonical signal for targeting substrates to the 26 S proteasome (45). Later reports proposed that with ectopic expression of IpaH9.8, NEMO was modified with Lys27-linked polyubiquitin chains (23), whereas Seyedarabi et al. (46) suggested that the isolated catalytic domain synthesizes polyubiquitin chains linked through Lys6, Lys11, Lys29, and Lys33. To unambiguously define the linkage specificity of IpaH9.8, we exploited an isotope dilution/chain termination technique, which we term the extension termination (ExTerm) assay (Fig. 1D). The linkage assay is based on single mutants of ubiquitin serving as chain terminators if the position is required for chain formation. Conjugation assays are conducted in the presence of 5 μm 125I-ubiquitin (lane 3) or 1 μm 125I-ubiquitin diluted with 4 μm wild type (lane 4) or the indicated single lysine-to-arginine ubiquitin point mutants (lanes 5–11). The reduction in conjugate signal for lane 4 compared with lane 3 represents dilution of the 125I label by the addition of unlabeled wild-type ubiquitin. A similar reduction is expected for those point mutants for which the lysine is not required for chain formation. The nearly complete loss of signal for UbK48R (lane 10) indicates that IpaH9.8 principally forms Lys48-linked polyubiquitin chains. The ablation of signal for the UbK6R mutant suggests that the ligase is also able to catalyze a limited rate of Lys6 linkage formation. These results confirm that IpaH9.8 nearly exclusively forms free and anchored Lys48-linked polyubiquitin degradation signals.
IpaH9.8 Concentration Dependence for Polyubiquitin Chain Partitioning
In preliminary experiments establishing conditions for IpaH9.8-catalyzed 125I-ubiquitin conjugation, we observed that the partitioning of reaction products varied with IpaH9.8 concentration (Fig. 2A). At high concentrations, a ladder of polyubiquitin species emanates from a band corresponding to the expected size for monoubiquitinated IpaH9.8 (Fig. 2A, lane 2), whereas at lower concentrations, signal localized in the 5% stacker gel (Fig. 2A, lane 12). There was a progressive concentration-dependent shift in the distribution of conjugates with increasing [IpaH9.8]o that followed a hyperbolic dependence with a Kd = 75 nm (not shown). We also noted in a parallel non-reducing gel to that of Fig. 2A, performed to monitor steady state Uba1∼ and Ubc5A∼125I-ubiquitin thioester formation, that the overall rate of 125I-polyubiquitin chain formation became Uba1-limiting as the concentration of IpaH9.8 increased (not shown). Thus, at high [IpaH9.8]o, the rate of polyubiquitin chain formation exceeds the ability of Uba1 to maintain Ubc5A quantitatively in its 125I-ubiquitin-charged form, confirmed by an increase in rate when [Uba1]o was doubled in the assays (not shown). The presence of signal in the stacker gel does not preclude IpaH9.8 autoconjugates, but the IsoT digestion kinetics shown in Fig. 1, B and C, indicate that these species are almost exclusively free chains. We hypothesized that the partitioning of polyubiquitin chains into either anchored or free pools is dependent on IpaH9.8 concentration. We tested this by assessing the IsoT sensitivity of chains synthesized in reactions containing either 5 μm or 5 nm IpaH9.8 in reactions similar to those of Fig. 1, B and C (Fig. 2B). Based on IsoT sensitivity, we found that conjugates synthesized by 5 μm IpaH9.8 (closed circles) were composed of only 30% free chains, as indicated by the extrapolated Y intercept, whereas conjugates synthesized by 5 nm IpaH9.8 (open circles) comprised 82% free chains. The IpaH9.8 concentration dependence suggests that autoconjugation occurs in trans, requiring oligomerization of IpaH9.8 into a presumed Michaelis complex.
FIGURE 2.

IpaH9.8 concentration dependence for ubiquitin chain partitioning. A, autoradiogram of 125I-ubiquitin conjugation assays containing 15 nm Uba1 and 132 nm Ubc5A in the absence (lane 1) or presence (lanes 2–12) of varying amounts of IpaH9.8 (9.1 μm to 0.95 nm) were incubated for 10 min as described under “Materials and Methods.” Samples were resolved by 12% (w/v) SDS-PAGE and visualized by autoradiography. The mobility of relative molecular weight markers and position of the stacker gel are shown to the left. The position of free 125I-ubiquitin is shown to the right. B, 125I-ubiquitin conjugation reactions containing either 5 μm (closed circles) or 5 nm (open circles) IpaH9.8 were assayed for IsoT sensitivity as described in the legend to Fig. 1, B and C. The limiting rates for signal loss (dashed lines) were extrapolated to t0 in order graphically to determine the fraction of IsoT labile conjugates as the y intercept.
IpaH9.8 Exhibits Cooperative Kinetics
Previous studies from our laboratory indicate that initial rates of polyubiquitin chain formation can be used as a reporter function for kinetic study of E3 ubiquitin ligases (32, 39, 42). Using the same strategy, we investigated the dependence of [E2]o on the initial rate of IpaH9.8-catalyzed polyubiquitin chain formation. In the absence of E2, a conjugate band at ∼120 kDa represents E3-independent Uba1 autoubiquitination described previously (32, 39, 42) (Fig. 3A). For all three Ubc5 family members tested, 125I-polyubiquitin chain formation increased with [E2]o from 5 to 500 nm, as shown for Ubc5B (Fig. 3A). Rates of ubiquitin conjugation were calculated from associated radioactivity determined by γ-counting of the stacker region for each excised lane after the gel was dried and autoradiographed (32, 39, 42). The finding that the level of conjugated 125I-ubiquitin does not change when Uba1 concentration is doubled (lane 10) confirms that the assay remains under E3-limiting conditions at the highest Ubc5B concentration.
FIGURE 3.

IpaH9.8 exhibits ordered cooperative allosteric kinetics. A, autoradiogram of 10-min 125I-ubiquitin conjugation assays containing 2.5 nm IpaH9.8 in the absence (lane 1) or presence (lanes 2–9) of increasing concentrations of Ubc5B (5–500 nm) as described under “Materials and Methods.” The assay of lane 10 contained the highest concentration of Ubc5B and twice the concentration of Uba1 to verify IpaH9.8-limiting conditions. The assay of lane 11 was identical to that of lane 9 but in the absence of IpaH9.8. Gels were visualized by autoradiography and quantitated by γ-counting as described under “Materials and Methods.” B, concentration dependence of initial velocity versus [E2]o for Ubc5A (closed circles), Ubc5B (open circles), and Ubc5C (closed squares). Lines, theoretical nonlinear sigmoidal regression fits of the data for kinetic constants summarized in Table 1. C, concentration dependence of initial velocity on [Ubc5B]o. Line, nonlinear regression fit of the data to Equation 1 and the kinetic constants summarized under “Results,” where K′ = [S]0.5nH.
Unlike the hyperbolic kinetics for [E2]o dependence observed for the HECT ligase E6AP (32), IpaH9.8 displays sigmoidal kinetics with respect to [E2]o (Fig. 3B), confirmed by the linearity of the corresponding Hill plot for representative data (inset). Non-linear regression analysis of the data fit to the Hill equation indicates that IpaH9.8 displays similar apparent binding affinities, measured as [S]½, for each of the Ubc5 orthologs.4 The Ubc5B and Ubc5C orthologs are kinetically indistinguishable, with [S]½ ≃ 150 nm and kcat ≃ 0.07 s−1 (Fig. 3B and Table 1), where kcat = Vmax/[E3]o. The qualitatively lower activity exhibited by Ubc5A in Fig. 1A is reflected in a lower kcat (0.025 ± 0.001 s−1) compared with the other two orthologs, although Ubc5A exhibits a slightly higher apparent affinity ([S]½ = 115 ± 26 nm) (Fig. 3A and Table 1). The three Ubc5 orthologs additionally exhibit Hill coefficients (nH) of 1.7–2.0 (Table 1). Although IpaH9.8 has previously been assumed to exist as a monomer (17), observation of cooperativity requires the enzyme to exist functionally as an oligomer. Because the Hill coefficient represents the lower limit for the number of substrate binding sites that are conformationally linked during the catalytic cycle, the value of nH requires that IpaH9.8 exists minimally as a dimer, assuming a single binding site per subunit.
TABLE 1.
Summary of kinetic constants for IpaH9.8
| E2 enzyme | [S]½ | nH | kcat | kcat/[S]½ |
|---|---|---|---|---|
| nm | s−1 | m−1 s−1 | ||
| Ubc5A | 115 ± 26 | 1.7 ± 0.1 | 0.025 ± 0.001 | 2.2 × 105 |
| Ubc5B | 140 ± 32 | 1.8 ± 0.1 | 0.071 ± 0.004 | 5.1 × 105 |
| Ubc5C | 147 ± 53 | 2.0 ± 0.3 | 0.069 ± 0.004 | 4.7 × 105 |
IpaH9.8 Displays Substrate Inhibition
At E2 concentrations above 500 nm, substrate inhibition is observed with each of the three Ubc5 isozymes that trends to zero rate at infinite substrate concentration, representative data of which are shown for Ubc5B (Fig. 3C). Analogous substrate inhibition has been observed previously for the E6AP HECT ligase (32), the TRIM RING ligases (39), and the N-end rule RING ligase E3α/Ubr1 (41), consistent with ordered sequential binding at two functionally distinct E2∼ubiquitin thioester binding sites. Reasonably assuming cooperativity for both the “activation” (K′1 and nH1) and “inhibition” (K′2 and nH2) sites, the data for Ubc5B or Ubc5C provide excellent fits to a mathematical model for ordered cooperative binding shown in Equation 1,
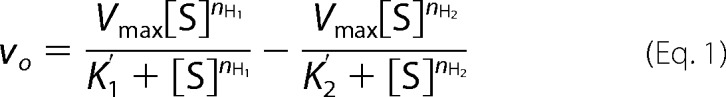 |
with Ubc5B (Fig. 3C), for which activation and inhibition sites were fit to [S]½ values of 150 ± 90 and 740 ± 240 nm, respectively, and nH values of 1.7 ± 0.2 and 1.8 ± 0.4, respectively. For Ubc5C, activation and inhibition sites were fit to [S]½ values of 220 ± 30 nm and 1,400 ± 170 nm, respectively, and nH values of 1.5 ± 0.01 and 1.1 ± 0.01, respectively. These results are consistent with IpaH9.8 possessing two kinetically distinguishable E2∼ubiquitin thioester binding sites having significant differences in apparent affinity.
The N-terminal Leucine-rich Repeat Domain Is Required for Cooperative Substrate Binding
Singer et al. (22) have previously estimated the binding of uncharged Ubc5B to the IpaH9.8 C-terminal domain by fluorescence polarization, the data of which displayed a hyperbolic binding dependence. To reconcile the earlier hyperbolic data with the cooperative kinetics observed here for the full-length enzyme, we also tested the E2 concentration dependence on the initial velocity for the truncated IpaH9.8 catalytic domain, IpaH9.8(244–545). Consistent with previous reports (17, 22, 47), IpaH9.8(244–545) is ∼50-fold more active than full-length IpaH9.8 when kcat values are compared (Tables 1 and 2). In contrast to full-length IpaH9.8, the IpaH9.8(244–545) truncation displays hyperbolic kinetics with respect to [E2]o for all three Ubc5 orthologs (Fig. 4), confirmed by the linearity of the corresponding double reciprocal plot for Ubc5B (inset). The same trends found for full-length IpaH9.8 are observed with IpaH9.8(244–545); the Ubc5B and Ubc5C orthologs are kinetically indistinguishable with Km = 180 nm and kcat = 4.8 s−1, whereas Ubc5A shows slightly higher affinity (Km = 147 ± 7 nm) but lower catalytic competence (kcat = 3.6 ± 0.4 s−1) (Fig. 4 and Table 2). Our kinetically determined Km value of 180 nm for Ubc5B is significantly lower than the Kd value of 300 μm reported by Singer et al. (22) for binding of uncharged Ubc5B to the isolated catalytic domain determined by fluorescence polarization. The marked apparent difference in binding affinity requires that the ubiquitin and E2 moieties of the E2∼ubiquitin thioester contribute approximately equally to overall binding to the catalytic domain or that there is a significant fraction of denatured Ubc5B in the earlier studies (22), as noted before for similar discrepancies in affinities for Ubc5 orthologs with E6AP (32). The loss of the sigmoidal dependence on [E2]o requires that the N-terminal leucine-rich repeat domain conformationally communicates substrate binding between subunits and is essential for the cooperative kinetics observed for full-length IpaH9.8.
TABLE 2.
Summary of kinetic constants for IpaH9.8(244–545)
| E2 enzyme | Km | kcat | kcat/Km |
|---|---|---|---|
| nm | s−1 | m−1 s−1 | |
| Ubc5A | 140 ± 7 | 3.6 ± 0.4 | 2.5 × 107 |
| Ubc5B | 180 ± 11 | 4.8 ± 0.3 | 2.7 × 107 |
| Ubc5C | 180 ± 20 | 4.8 ± 1.0 | 2.7 × 107 |
FIGURE 4.

IpaH9.8(244–545) exhibits hyperbolic kinetics. Shown is the concentration dependence of initial velocity versus [E2]o for Ubc5A (closed circles), Ubc5B (open circles), and Ubc5C (closed squares) in the presence of 25 pm IpaH9.8(244–545) in assays otherwise identical to those of Fig. 3. Lines, theoretical nonlinear hyperbolic regression fits of the data for kinetic constants summarized in Table 2. Inset, double reciprocal plot of the data of Ubc5B.
IpaH9.8 Possesses Two E2∼Ubiquitin Thioester Binding Sites
Because of the significant difference between values of [S]½ determined kinetically and Kd values reported previously from equilibrium fluorescence polarization studies (22), we asked whether [S]½ approximated the Kd for Ubc5B∼ubiquitin thioester binding to full-length IpaH9.8 by determining the [Ubc5B]o dependence on the initial velocity of polyubiquitin chain formation in the absence or presence of a 270 nm concentration of an isosteric nonreactive Ubc5BC85S-ubiquitin oxyester substrate analog (32, 41). The Ubc5BC85S-ubiquitin oxyester exhibited competitive inhibition with respect to wild-type Ubc5B∼ubiquitin thioester (Ki = 117 ± 29 nm), requiring that both compounds bind at the same or overlapping binding sites (Fig. 5A). Good agreement between the [S]½ observed with the Ubc5B∼ubiquitin thioester and the Ki of the substrate analog requires that [S]½ approximates the cooperative intrinsic dissociation constant describing IpaH9.8 binding of E2∼ubiquitin thioester. Similarly, the excellent agreement between the Ki for Ubc5B-ubiquitin oxyester binding to full-length IpaH9.8 (Fig. 5A) and the Km for Ubc5B∼ubiquitin thioester binding to the truncated catalytic domain (Fig. 4) is consistent with the full-length enzyme exhibiting the less frequently observed V-type cooperativity in which the kcat changes rather than affinity.
FIGURE 5.

IpaH9.8 possesses two distinct Ubc5B∼ubiquitin thioester binding sites. A, double reciprocal plots of initial rates of 125I-ubiquitin conjugation determined as in Fig. 3 under IpaH9.8-limiting conditions in assays containing 5 nm IpaH9.8 and the indicated concentrations of Ubc5B in the absence (closed circles) or presence (open circles) of 270 nm Ubc5BC85S-125I-ubiquitin oxyester. B, double reciprocal plot of initial rates of 125I-ubiquitin conjugation determined as in Fig. 3 under IpaH9.8-limiting conditions in assays containing 2.5 nm IpaH9.8 and the indicated concentrations of Ubc5B in the absence (closed circles) or presence (open circles) of 3.5 μm Ubc5BC85A product analog.
Conversely, a non-reactive E2 product analog was engineered by mutating the active site Cys85 to alanine (32, 41). The absence of a reactive nucleophile at the active site prevents this E2 species from becoming charged with ubiquitin by Uba1 and thus serves as a mimic of the uncharged product E2 following isopeptide bond formation of the ubiquitin moiety. The kinetics of IpaH9.8-catalyzed chain formation were assessed in the absence or presence of 3.5 μm Ubc5BC85A (Fig. 5B). The product analog was a non-competitive inhibitor with respect to Ubc5B∼125I-ubiquitin thioester and exhibited a Ki = 2.2 ± 0.5 μm, which is considerably lower than the Kd of 300 μm determined previously by fluorescence polarization (22). The marked differences in affinities determined here versus those reported by Singer et al. (22) probably results from determining Ubc5B concentration by total protein in the earlier study and the significant inherent instability of the protein discussed previously that results in overestimation of active protein concentration (32, 42). In contrast, as noted under “Materials and Methods,” Ubc5BC85A concentration is here estimated based on total protein concentration determined spectrophotometrically and corrected for active protein content of a parallel wild-type Ubc5B preparation determined by 125I-ubiquitin thioester formation (32). For the present kinetic results, non-competitive inhibition requires that Ubc5BC85A binds at a site distinct from the wild-type Ubc5B∼ubiquitin substrate, consistent with the two-site model indicated by observation of substrate inhibition (Fig. 3C).
IpaH3 Crystallizes as a Symmetric Dimer
The IpaH family enzymes are thought to exist as monomers (17); however, cooperative kinetics observed with the present data a priori require oligomerization of the enzyme. Therefore, we reexamined the extant crystal structures of the IpaH family of enzymes. The only structure for IpaH9.8 is of the isolated catalytic domain (46). Unfortunately, the covalently linked dimeric structure is probably an artifact because formation of a disulfide bond between active site Cys337 residues results in linearization of a helix-loop-helix fold that is conserved among all known paralogs into an extended helix by perturbation of the loop (Ala333–Arg340) within which resides Cys337 (46). In contrast, the sequence for IpaH3 is 82% identical to IpaH9.8, with the catalytic domain showing >98% identity and the leucine-rich repeat domain showing 60% identity (17). Given the high level of sequence conservation, key mechanistic features of polyubiquitin chain formation should also be conserved between the paralogs. The nearly full-length structure of IpaH3 consists of an N-terminal domain that is capped by two α-helices, followed by a series of leucine-rich repeats that are connected to the C-terminal catalytic domain by a short linker region (17).
Because the IpaH enzymes possess only a single active site defined by the catalytic cysteine, we asked whether IpaH3 forms a symmetrical dimer evident in the structural data that might have gone unnoticed. When the symmetry mates of IpaH3 are generated, a dimer emerges with an extensive interface that results in a buried surface area of 2,870 Å2, representing a solvent free binding energy of −20 kcal/mol (Kd = 8 × 10−15 m) (Fig. 6A). Remarks accompanying the coordinates deposited in the Protein Data Bank indicate that this dimer interface had previously been identified by the PISA analysis suite (European Bioinformatics Institute) as a significant interface with a high probability of representing the biologically relevant assembly of IpaH3, although this was not addressed at the time (17). The subunit interface encompasses two juxtaposed catalytic domains and involves a core of 16 hydrophobic residues (eight from each monomer) that are stabilized by interchain ionic and polar interactions (Fig. 6B). Additional buried surface area exists between the catalytic domain of one dimer mate and the convex face of the leucine-rich repeat domain of the opposite N-terminal domain, but no interactions are observed between the two N-terminal domains.
FIGURE 6.

Identification of a symmetric dimer in the crystal structure of the related IpaH3 ligase. A, surface rendering of the crystal structure for IpaH3 (PDB code 3CVR) in complex with its dimer mate identified by PISA analysis. Active site cysteine residues are colored yellow. B, ribbon diagram of the crystal structure for IpaH3 in complex with its dimer mate in the same orientation as in A. Residue side chains predicted by PISA analysis to be at the dimer interface are modeled as spheres. Active site cysteine residues are modeled as yellow spheres. C, gel filtration chromatography of IpaH9.8(244–545), as described under “Materials and Methods.” Inset, relative mobility of peaks 1 and 2 (closed circles) are shown on the calibration plot summarizing a series of standards (open circles). The predicted elution volume for monomeric IpaH9.8(244–545) is indicated (inset).
To test the oligomerization state of the ligase in solution, we first analyzed IpaH9.8 by gel filtration chromatography. When resolved by Superose 12 (10/30) chromatography, recombinant IpaH9.8 eluted as a symmetric peak with a relative molecular mass of 99 kDa, significantly larger than the predicted 62 kDa monomer molecular mass (data not shown). The crystal structure for IpaH3 predicts that monomeric IpaH9.8 has molecular dimensions of 113 × 47 Å, giving an axial ratio of 2.4 (17). Because of the anti-parallel arrangement of the dimer mates, the proposed dimer has molecular dimensions of 128 × 86 Å, corresponding to an axial ratio of 1.5. Therefore, the dimer is expected to elute at a smaller apparent relative molecular weight than predicted by its doubling in mass. We then asked whether multimerization was unique to the full-length enzyme because cooperative kinetics were not observed with the isolated catalytic domain (Fig. 4). The IpaH9.8(244–545) truncation migrated as two distinct populations when analyzed by gel filtration chromatography with relative molecular masses of 130 and 52 kDa (Fig. 6C). These are approximately 4- and 2-fold larger than the predicted relative molecular mass of 35 kDa for the truncated enzyme, indicating that the isolated catalytic domain forms multimers in solution, although cooperativity is lost with the truncation. When analyzed by either reducing or non-reducing SDS-PAGE, IpaH9.8(244–545) migrated as a single band of 35 kDa, indicating that these multimeric states are not the result of interchain disulfide formation (data not shown).
As a test of the structure, we next attempted to perturb the putative wild-type dimer by engineering a destabilizing residue into the subunit interface of IpaH9.8 predicted from the dimeric structure of IpaH3. The central residue of the IpaH3 hydrophobic core is Leu461, which corresponds to Leu435 in the IpaH9.8 sequence. This residue was mutated to an arginine to yield IpaH9.8L435R in order to introduce a charged residue into the otherwise apolar interface. In the native dimeric structure, both Leu435 residues are juxtaposed across the dimer interface, and their side chains are separated by 3.9 Å. The addition of two positively charged guanidinium groups within the space is expected to destabilize the predicted dimer interface by charge repulsion. The IpaH9.8L435R point mutant and wild-type IpaH9.8 were expressed and purified to apparent homogeneity in parallel, and then their kinetics were compared with respect to [Ubc5B]o as in Fig. 3. The initial velocity kinetics for IpaH9.8L435R with respect to [Ubc5B]o demonstrated that the point mutation had no effect on the affinity of Ubc5B∼ubiquitin thioester binding but decreased kcat 5-fold compared with wild-type IpaH9.8 (Fig. 7A). Parallel analysis of the two proteins by static light scattering indicates that wild-type IpaH9.8 has an apparent radius of 3.97 nm and a solution molecular mass of 85 kDa, whereas IpaH9.8L435R has a radius of 3.58 nm and a solution molecular mass of 65 kDa, consistent with the monomer molecular mass of 62 kDa. The difference in apparent radius is consistent with the predicted change in axial ratio between monomer and dimer. Wild-type IpaH9.8 exhibits a polydispersity of 6.4%, reflecting a relatively monodispersed structure, whereas the dimer-destabilized mutant has a 35.4% polydispersity, consistent with a shift in the monomer-dimer equilibrium to the monomeric state (Fig. 7B).
FIGURE 7.

IpaH9.8 dimer formation is necessary for activity. A, initial rates of 125I-ubiquitin conjugation were determined as in Fig. 3 under E3-limiting conditions in assays containing the indicated concentrations of Ubc5B and either 2.5 nm IpaH9.8 (closed circles) or 5 nm IpaH9.8L435R (open circles). B, static light scattering analysis of 75 μm of either IpaH9.8 (solid line) or IpaH9.8L435R (dashed line). C, SDS-PAGE resolution and Coomassie Brilliant Blue staining of glutathione-Sepharose precipitation assays of reactions containing an 8.5 μm concentration of either IpaH9.8 (left) or IpaH9.8L435R (right) in the absence or presence of 8.5 μm either GST-IpaH9.8 or GST-IpaH9.8L435R, respectively. D, quantitation of the amount of IpaH9.8 or IpaH9.8L435R co-purifying with the respective GST-IpaH9.8 or GST-IpaH9.8L435R compared with background binding in the absence of the GST-fused proteins at the indicated times as analyzed by quantitative densitometry described under “Materials and Methods.”
If IpaH9.8 normally exists as a stable dimer, then the subunits must exchange with a pool of free monomer at a finite rate, and the destabilized IpaH9.8L435R point mutant should exchange with an increased rate. To test this, we incubated IpaH9.8 in the absence or presence of GST-IpaH9.8 and then isolated the latter by glutathione-Sepharose precipitation to quantitate the amount of untagged IpaH9.8 that co-purified (Fig. 7C). Parallel reactions contained mutant IpaH9.8L435R and GST-IpaH9.8L435R. After 6 or 24 h, increasing amounts of IpaH9.8L435R co-purified with GST-IpaH9.8L435R, whereas no exchange was detected with wild-type IpaH9.8. That the increase in IpaH9.8L435R associated with the GST chimera results from subunit exchange and not further processing of the homodimeric fusion protein by contaminating thrombin can be seen by the absence of IpaH9.8 in the control lanes (Fig. 7C, left) and the absence of any loss in the corresponding GST-IpaH9.8L435R bands (Fig. 7C, right). By measuring the amount of IpaH9.8 as a percentage of total IpaH9.8 and GST-IpaH9.8 that was purified at each time point, we were able to estimate a lower limit for the half-life for subunit exchange of >140 h for wild-type IpaH9.8, which decreased to 15 h for the dimer-destabilized IpaH9.8L435R mutant, Fig. 7D.
Cooperativity Is Transduced through Phe395 of IpaH9.8
The loss of cooperativity (Fig. 4) and concomitant increase in kcat (Table 2) accompanying truncation of the leucine-rich repeat targeting domain of IpaH9.8 suggest that wild-type allostery functions to relieve catalytic repression imposed by the targeting domain. A fundamentally similar interpretation has been proposed by Chou et al. (48), who concluded that IpaH9.8 and its IpaH3 and SspH2 paralogs exist in “on” and “off” states, with truncation of the leucine-rich repeat domain creating a constitutively “on” state. Because two-state models are the basis of allosteric cooperativity (49, 50), we asked whether the cooperative kinetics observed here constituted the “on/off” states of the earlier study. Chou et al. (48) demonstrated for SspH2 that the increase in rate accompanying truncation of the leucine-rich repeat domain could be qualitatively replicated in part by mutation of Phe393 to alanine, a residue at the interface between the targeting and catalytic domains. The results of Fig. 8 demonstrate that for the paralogous mutation, the IpaH9.8F395A point mutant exhibits hyperbolic kinetics, as shown by the linearity of the double reciprocal plot (inset), yielding values of kcat and Km of 0.31 ± 0.04 s−1 and 290 ± 88 nm, respectively. The increase in kcat association with mutation of Phe395 does not quantitatively replicate the effect of truncation (Table 2), indicating that the presence of the N-terminal targeting domain continues to repress the observed kcat, although cooperativity is abrogated.
FIGURE 8.

Cooperativity is transduced through Phe393. Initial rates of 125I-polyubiquitin chain formation were determined as in Fig. 3 in the presence of 5 nm IpaH9.8F395A. Kinetic constants were determined by nonlinear regression analysis fit to hyperbolic kinetics. Inset, double reciprocal plot of the data.
The Distantly Related SspH2 Salmonella Paralog Also Exhibits Cooperative Kinetics
Finally, we asked whether the basic mechanistic features of IpaH9.8 were shared with a more distant homolog, the SspH2 protein of S. enterica. The individual domains of SspH2 share the same general structural features of IpaH9.8 and IpaH3, with an N-terminal leucine-rich repeat targeting domain and an α-helical catalytic domain, but also include an additional ∼150-residue region at the extreme N terminus, the structure of which has not been resolved (47). The SspH2 catalytic domain only shares 38% identity with the IpaH9.8 catalytic domain but retains a similar overall fold (47). However, the relative orientations of the domains differ in the extant crystal structures, with the leucine-rich repeat domain assuming an almost 180° rotation relative to the catalytic domain when compared with IpaH9.8 (47). Recombinant SspH2 displayed a specificity for Ubc5 isoforms, as found for IpaH9.8 (Fig. 1A), and a concentration dependence on partitioning of polyubiquitin chains between autoubiquitination at high concentration and formation of unanchored chains at low concentration similar to that observed for IpaH9.8 in Fig. 2 (data not shown). In a detailed kinetic study, recombinant SspH2 exhibited sigmoidal kinetics with respect to [Ubc5B]o corresponding to [S]½ = 660 ± 61 nm, nH = 1.6 ± 0.1, and kcat = 0.052 ± 0.003 s−1 by non-linear regression analysis (Fig. 9). Additionally, at Ubc5B∼ubiquitin concentrations above ∼2 μm, substrate inhibition was observed similar to that found for IpaH9.8 in Fig. 3C. Finally, full-length recombinant SspH2, with a predicted monomer molecular mass of 87 kDa migrated with a relative molecular mass of 184 kDa by Superose 12 (10/30) gel filtration chromatography consistent with a stable dimer (data not shown). These data indicate that SspH2 also utilizes a cooperative allosteric mechanism consistent with a functional dimer. Collectively, these observations require that oligomerization, cooperative kinetics, and allosteric regulation are conserved mechanistic features of this bacterial ligase superfamily.
FIGURE 9.

SspH2 exhibits cooperative kinetics. Concentration dependence of initial velocity versus [Ubc5B]o in the presence of 5 nm SspH2. Line, theoretical sigmoidal nonlinear regression fit of the data for kinetic constants under “Results.” Inset, Hill plot for the data.
DISCUSSION
Bacterial and viral pathogens have demonstrated remarkable adaptability in exploiting host pathways for ubiquitin and ubiquitin-like protein-dependent regulation (24–28). Many of these virulence strategies rely on molecular mimicry of eukaryotic ubiquitin ligases to remodel the host proteome by modifying the scope of targeted protein degradation (51). Among the latter cohort of E3-like virulence factors, the IpaH family of bacterial ligases is unique in sharing neither sequence nor structural homology with known eukaryotic ligase lineages (51), calling into question their evolutionary provenance. In the current work, we provide the first detailed quantitative kinetic analysis of the IpaH family using rates for assembly of 125I-polyubiquitin chains as a functional readout of ligase activity, the results of which reveal mechanistic details not previously suspected.
As with other ligases, IpaH9.8 catalyzes the assembly of polyubiquitin chains in the absence of substrate (42) and exhibits an absolute E2 specificity for the Ubc5 orthologs when compared under identical biochemically defined conditions (Fig. 1). Remarkably, IpaH9.8 is able kinetically to distinguish Ubc5A from Ubc5B/C under parallel conditions (Fig. 3 and Table 1). Although the sequences are highly conserved among the three isoforms, in a broader sequence analysis, we find that human Ubc5A is more closely related to S. cerevisiae Ubc4 than to either human Ubc5B or Ubc5C (not shown), suggesting that Ubc5A may actually be the human ortholog of S. cerevisiae Ubc4. Because essential functional differences have been reported between S. cerevisiae Ubc4 and Ubc5 (52), the present data may reflect related differences among the human orthologs.
Using the newly developed extension termination (ExTerm) assay, the polyubiquitin chains formed by IpaH9.8 are shown unambiguously to represent the canonical Lys48-linked degradation signal recognized by the 26 S proteasome (45) (Fig. 1D), in agreement with Rohde et al. (20) and Zhu et al. (17). By exploiting the specificity of IsoT to disassemble only free unanchored polyubiquitin chains (43), we demonstrate that the predominant products formed in the absence of substrate at low concentrations of IpaH9.8 (5 nm) are unanchored degradation signals (Figs. 1, B and C, and 2). The ability of IpaH9.8 to synthesize unanchored polyubiquitin chains in the absence of a target protein suggests that these chains may be preassembled as an E3-linked thioester before transfer en bloc to water in forming free chains or to target protein lysines in forming anchored chains, although we cannot rigorously preclude a model in which the degradation signal is sequentially elongated from the initially anchored ubiquitin molecule in the latter case (32). In contrast, at higher concentrations of the ligase (5 μm), anchored chains arising by autoubiquitination are the predominant products (Fig. 2). The latter observation suggests that autoubiquitination occurs in trans through formation of a Michaelis complex between IpaH9.8 molecules, supported by the hyperbolic IpaH9.8 dependence on partitioning of free versus anchored chains, for which the observed Kd of 75 nm probably represents the equilibrium constant for autoassociation. Under the latter conditions, the overall rate of chain formation becomes E1-limiting, as shown by loss of E1∼ and E2∼125I-ubiquitin thioesters in parallel reducing gels (see legend to Fig. 2). In reviewing earlier studies in the literature, nearly all have been conducted at Uba1 and E3 concentrations corresponding to the latter conditions for which the reactions are unlikely to be E3-limiting and, therefore, fail directly to report catalytic features of the bacterial ligase (17, 20, 22, 46–48). Under biochemically defined E3-limiting conditions, we demonstrate for the first time that IpaH9.8 displays cooperative kinetics in the assembly of 125I-polyubiquitin chains with respect to E2∼ubiquitin thioester concentration (Fig. 3, A and B). The empirically derived Hill coefficients (nH) for E2∼ubiquitin binding approach 2, indicating that IpaH9.8 functions minimally as a dimer. The only other published example of cooperativity among ubiquitin ligases is the TRIM superfamily of RING ligases (39), although non-cooperative oligomerization is also required for E6AP-catalyzed assembly of Lys48-linked polyubiquitin degradation signals (33). Cooperative substrate binding requires IpaH9.8 to function as an obligate oligomer. We examined in detail the extant crystal structures for IpaH1.4 (PDB entry 3CKD), IpaH3 (3CVR), and IpaH9.8 (3L3P) in an effort to identify cryptic oligomerization that might have gone unnoticed but found no such evidence in the form of duplicated domains, motifs, or catalytic cysteine residues. However, in IpaH3, the closest evolutionary neighbor of IpaH9.8 (82% identity) for which the catalytic domains are essentially identical, we noted that when the unit cell of the former was reconstructed, it became evident that the ligase crystallized as a symmetric dimer. Although not discussed in the literature (17), the 3CVR structure file contained an annotation indicating that the dimer had also been identified by the PISA (Proteins, Interfaces, Structures, and Assemblies) server as a likely assembly, independently corroborating our conclusion. Recognizing that biologically relevant higher order assemblies of multimeric proteins are often recapitulated in protein crystals, the structures of which can be used to identify important interfaces mediating oligomerization (53, 54), we hypothesized that this dimer is the functionally relevant biological assembly for IpaH3 and, by extension, the entire superfamily of IpaH bacterial ubiquitin ligases. To test this hypothesis, we engineered a destabilizing L435R mutation into the predicted apolar subunit interface of IpaH9.8 and demonstrated that this change markedly reduced the kcat for polyubiquitin chain formation but without consequence for the Km for E2∼ubiquitin binding (Fig. 7A), decreased the apparent solution molecular weight by static light scattering (Fig. 7B), and increased the rate of subunit exchange (Fig. 7, C and D). Given that the catalytic domains of IpaH enzymes are essentially identical, this implies that other members of the superfamily form homodimers as their catalytically relevant species.
Substrate inhibition with respect to E2∼ubiquitin thioester concentration has been observed with several RING and HECT ligases that have been examined in detail and arises from the presence of two independent and functionally distinct binding sites of different affinities (32, 39, 41). In the case of the E6AP/UBE3A HECT domain ligase, a cryptic higher affinity UbcH7∼ubiquitin binding site (site 1) is required for formation of the initial HECT∼ubiquitin thioester to the active site Cys820, whereas a lower affinity UbcH7∼ubiquitin binding site (site 2) is required for rate-limiting polyubiquitin chain elongation (32) and is equivalent to the “canonical” site identified in the original crystal structure of Huang et al. (55). In the present case, substrate inhibition with respect to Ubc5B/C (Fig. 3C) can be modeled by Equation 1, allowing graphical determination of the kinetic constants for both sites. The higher affinity site 1 ([S]½ = 150 ± 90 nm for Ubc5B) is identical to that for which Ubc5BC85S-ubiquitin oxyester is a competitive inhibitor (Ki = 129 ± 29 nm) (Fig. 5A), supported by the good correspondence of the two affinities. Cooperativity requires identical conformationally linked high affinity binding sites on each subunit of the dimer, corresponding to site 1 (Fig. 3B). Were there only a single Ubc5B∼ubiquitin binding site per subunit, the Ubc5BC85A product analog would be predicted to act as a competitive inhibitor of Ubc5B∼ubiquitin. Non-competitive inhibition observed with Ubc5BC85A at micromolar concentrations (Ki = 2.2 ± 0.5 μm; Fig. 5B) and substrate inhibition at higher concentrations requires a second lower affinity and functionally distinct binding site on each subunit that probably corresponds to the “inhibitory” site 2 (Fig. 3C). Such a mechanism is remarkably similar to that recently proposed for E6AP and the broader HECT ligase superfamily to account for their novel kinetic properties (32). Although we do not observe stable 125I-ubiquitin thioester formation to IpaH9.8 under the present assay conditions, presumably because the intermediate turns over rapidly by transfer to water, we propose by analogy to E6AP that site 1 is responsible for thioester formation to the catalytic Cys337 and that site 2 is required for chain elongation. The ordered addition mechanism required for substrate inhibition by Ubc5∼ubiquitin thioester is consistent with this functional site assignment. Interestingly, our observation that Ubc5BC85A is not a competitive inhibitor of Ubc5B∼125I-ubiquitin thioester requires that the E2 alone has a significantly diminished affinity for binding to the putative site 1 in the absence of the ubiquitin moiety, as also found for E6AP (32).
The present studies demonstrate for the first time significant quantitative similarities between the IpaH and HECT ligase superfamilies of ubiquitin ligases. Both have affinities for their cognate E2∼ubiquitin thioester co-substrates in the low nanomolar range (Table 2) (32) and kcat for polyubiquitin chain formation of 5–10 s−1 when corrected for negative cooperativity (Table 2) or degree of oligomerization (33). Both ligase families additionally exhibit empirically validated mechanisms requiring ordered binding of the E2∼ubiquitin thioester co-substrate to two functionally distinct sites, the requirement of oligomerization for polyubiquitin chain formation, and previously documented requirements for ubiquitin thioester formation to conserved cysteine catalytic residues (22, 24). Because the sequences and folds of the respective catalytic domains are distinct between these ligase superfamilies, the present observations represent a remarkable instance of convergent evolution in enzyme mechanism between prokaryotes and eukaryotes.
Acknowledgments
We are grateful to Dr. Malabi Venkatesan for the generous gift of the IpaH9.8 expression plasmid and to Dr. Jeffery Hobden for the S. enterica genomic DNA.
This work was supported, in whole or in part, by National Institutes of Health Grant GM034009 (to A. L. H.).
By convention, [S]½ is defined as the concentration of substrate yielding a rate that is half of Vmax. It is related to the apparent intrinsic dissociation constant K′ by [S]½ = n√K′, where n is the Hill coefficient.
- HECT
- homologous to E6AP C terminus
- E1
- ubiquitin-activating enzyme
- E2 or Ubc
- ubiquitin carrier protein/ubiquitin-conjugating enzyme
- E3
- ubiquitin-protein isopeptide ligase
- E6AP
- E6-associated protein
- IsoT
- isopeptidase T/Usp5
- NEMO
- NF-κB essential modifier
- RING
- really interesting new gene
- Uba1
- human ubiquitin-activating enzyme (UBE1)
- PDB
- Protein Data Bank.
REFERENCES
- 1. Kotloff K. L., Winickoff J. P., Ivanoff B., Clemens J. D., Swerdlow D. L., Sansonetti P. J., Adak G. K., Levine M. M. (1999) Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull. World Health Organ. 77, 651–666 [PMC free article] [PubMed] [Google Scholar]
- 2. Sur D., Ramamurthy T., Deen J., Bhattacharya S. K. (2004) Shigellosis: challenges and management issues. Indian J. Med. Res. 120, 454–462 [PubMed] [Google Scholar]
- 3. Lee H., Kotloff K., Chukaserm P., Samosornsuk S., Chompook P., Deen J. L., Von Seidlein L., Clemens J. D., Wanpen C. (2005) Shigellosis remains an important problem in children less than 5 years of age in Thailand. Epidemiol. Infect. 133, 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Townes J. M. (2010) Reactive arthritis after enteric infections in the United States: the problem of definition. Clin. Infect. Dis. 50, 247–254 [DOI] [PubMed] [Google Scholar]
- 5. Sack R. B., Rahman M., Yunus M., Khan E. H. (1997) Antimicrobial resistance in organisms causing diarrheal disease. Clin. Infect. Dis. 24, S102–S105 [DOI] [PubMed] [Google Scholar]
- 6. Perdomo J. J., Gounon P., Sansonetti P. J. (1994) Polymorphonuclear leukocyte transmigration promotes invasion of colonic epithelial monolayer by Shigella flexneri. J. Clin. Invest. 93, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wassef J. S., Keren D. F., Mailloux J. L. (1989) Role of M cells in initial antigen uptake and in ulcer formation in the rabbit intestinal loop model of shigellosis. Infect. Immun. 57, 858–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. High N., Mounier J., Prévost M. C., Sansonetti P. J. (1992) IpaB of Shigella flexneri causes entry into epithelial cells and escape from the phagocytic vacuole. EMBO J. 11, 1991–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bernardini M. L., Mounier J., d'Hauteville H., Coquis-Rondon M., Sansonetti P. J. (1989) Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc. Natl. Acad. Sci. U.S.A. 86, 3867–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Makino S., Sasakawa C., Kamata K., Kurata T., Yoshikawa M. (1986) A genetic determinant required for continuous reinfection of adjacent cells on large plasmid in S. flexneri 2a. Cell 46, 551–555 [DOI] [PubMed] [Google Scholar]
- 11. Goldberg M. B., Theriot J. A. (1995) Shigella flexneri surface protein IcsA is sufficient to direct actin-based motility. Proc. Natl. Acad. Sci. U.S.A. 92, 6572–6576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sansonetti P. J. (2001) Rupture, invasion and inflammatory destruction of the intestinal barrier by Shigella, making sense of prokaryote-eukaryote cross-talks. FEMS Microbiol. Rev. 25, 3–14 [DOI] [PubMed] [Google Scholar]
- 13. Schroeder G. N., Hilbi H. (2008) Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin. Microbiol. Rev. 21, 134–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parsot C. (2009) Shigella type III secretion effectors: how, where, when, for what purposes? Curr. Opin. Microbiol. 12, 110–116 [DOI] [PubMed] [Google Scholar]
- 15. Cornelis G. R. (2006) The type III secretion injectisome. Nat. Rev. Microbiol. 4, 811–825 [DOI] [PubMed] [Google Scholar]
- 16. Ashida H., Toyotome T., Nagai T., Sasakawa C. (2007) Shigella chromosomal IpaH proteins are secreted via the type III secretion system and act as effectors. Mol. Microbiol. 63, 680–693 [DOI] [PubMed] [Google Scholar]
- 17. Zhu Y., Li H., Hu L., Wang J., Zhou Y., Pang Z., Liu L., Shao F. (2008) Structure of a Shigella effector reveals a new class of ubiquitin ligases. Nat. Struct. Mol. Biol. 15, 1302–1308 [DOI] [PubMed] [Google Scholar]
- 18. Haraga A., Miller S. I. (2003) A Salmonella enterica serovar typhimurium translocated leucine-rich repeat effector protein inhibits NF-κB-dependent gene expression. Infect. Immun. 71, 4052–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Okuda J., Toyotome T., Kataoka N., Ohno M., Abe H., Shimura Y., Seyedarabi A., Pickersgill R., Sasakawa C. (2005) Shigella effector IpaH9.8 binds to a splicing factor U2AF35 to modulate host immune responses. Biochem. Biophys. Res. Commun. 333, 531–539 [DOI] [PubMed] [Google Scholar]
- 20. Rohde J. R., Breitkreutz A., Chenal A., Sansonetti P. J., Parsot C. (2007) Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host. Microbe 1, 77–83 [DOI] [PubMed] [Google Scholar]
- 21. Grau-Bovë X., Sebë-Pedrös A., Ruiz-Trillo I. (2013) A genomic survey of HECT ubiquitin ligases in eukaryotes reveals independent expansions of the HECT system in several lineages. Genome Biol. Evol. 5, 833–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singer A. U., Rohde J. R., Lam R., Skarina T., Kagan O., Dileo R., Chirgadze N. Y., Cuff M. E., Joachimiak A., Tyers M., Sansonetti P. J., Parsot C., Savchenko A. (2008) Structure of the Shigella T3SS effector IpaH defines a new class of E3 ubiquitin ligases. Nat. Struct. Mol. Biol. 15, 1293–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ashida H., Kim M., Schmidt-Supprian M., Ma A., Ogawa M., Sasakawa C. (2010) A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKγ to dampen the host NF-κB-mediated inflammatory response. Nat. Cell Biol. 12, 66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scheffner M., Huibregtse J. M., Vierstra R. D., Howley P. M. (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505 [DOI] [PubMed] [Google Scholar]
- 25. Coscoy L., Sanchez D. J., Ganem D. (2001) A novel class of herpesvirus-encoded membrane-bound E3 ubiquitin ligases regulates endocytosis of proteins involved in immune recognition. J. Cell Biol. 155, 1265–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu B., Skarina T., Yee A., Jobin M. C., Dileo R., Semesi A., Fares C., Lemak A., Coombes B. K., Arrowsmith C. H., Singer A. U., Savchenko A. (2010) NleG Type 3 effectors from enterohaemorrhagic Escherichia coli are U-Box E3 ubiquitin ligases. PLoS. Pathog. 6, e1000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Janjusevic R., Abramovitch R. B., Martin G. B., Stebbins C. E. (2006) A bacterial inhibitor of host programmed cell death defenses is an E3 ubiquitin ligase. Science. 311, 222–226 [DOI] [PubMed] [Google Scholar]
- 28. Diao J., Zhang Y., Huibregtse J. M., Zhou D., Chen J. (2008) Crystal structure of SopA, a Salmonella effector protein mimicking a eukaryotic ubiquitin ligase. Nat. Struct. Mol. Biol. 15, 65–70 [DOI] [PubMed] [Google Scholar]
- 29. Lin D. Y., Diao J., Zhou D., Chen J. (2011) Biochemical and structural studies of a HECT-like ubiquitin ligase from Escherichia coli O157:H7. J. Biol. Chem. 286, 441–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Y., Higashide W. M., McCormick B. A., Chen J., Zhou D. (2006) The inflammation-associated Salmonella SopA is a HECT-like E3 ubiquitin ligase. Mol. Microbiol. 62, 786–793 [DOI] [PubMed] [Google Scholar]
- 31. Wang F., Jiang Z., Li Y., He X., Zhao J., Yang X., Zhu L., Yin Z., Li X., Wang X., Liu W., Shang W., Yang Z., Wang S., Zhen Q., Zhang Z., Yu Y., Zhong H., Ye Q., Huang L., Yuan J. (2013) Shigella flexneri T3SS effector IpaH4.5 modulates the host inflammatory response via interaction with NF-κB p65 protein. Cell Microbiol. 15, 474–485 [DOI] [PubMed] [Google Scholar]
- 32. Ronchi V. P., Klein J. M., Haas A. L. (2013) E6AP/UBE3A ubiquitin ligase harbors two E2∼ubiquitin binding sites. J. Biol. Chem. 288, 10349–10360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ronchi V. P., Klein J. M., Edwards D. J., Haas A. L. (2014) The active form of E6-associated protein (E6AP)/UBE3A ubiquitin ligase is an oligomer. J. Biol. Chem. 289, 1033–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang M., Pickart C. M. (2005) Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. EMBO J. 24, 4324–4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baboshina O. V., Haas A. L. (1996) Novel multiubiquitin chain linkages catalyzed by the conjugating enzymes E2epf and Rad6 are recognized by the 26 S proteasome subunit 5. J. Biol. Chem. 271, 2823–2831 [DOI] [PubMed] [Google Scholar]
- 36. Haas A. L. (2005) Purification of E1 and E1-like enzymes. Methods Mol. Biol. 301, 23–35 [DOI] [PubMed] [Google Scholar]
- 37. Haas A. L., Rose I. A. (1982) The mechanism of ubiquitin activating enzyme: a kinetic and equilibrium analysis. J. Biol. Chem. 257, 10329–10337 [PubMed] [Google Scholar]
- 38. Haas A. L., Bright P. M. (1988) The resolution and characterization of putative ubiquitin carrier protein isozymes from rabbit reticulocytes. J. Biol. Chem. 263, 13258–13267 [PubMed] [Google Scholar]
- 39. Streich F. C., Jr., Ronchi V. P., Connick J. P., Haas A. L. (2013) Tripartite motif ligases catalyze polyubiquitin chain formation through a cooperative allosteric mechanism. J. Biol. Chem. 288, 8209–8221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tokgöz Z., Siepmann T. J., Streich F., Jr., Kumar B., Klein J. M., Haas A. L. (2012) E1-E2 interactions in ubiquitin and Nedd8 ligation pathways. J. Biol. Chem. 287, 311–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Siepmann T. J., Bohnsack R. N., Tokgöz Z., Baboshina O. V., Haas A. L. (2003) Protein interactions within the N-end rule ubiquitin ligation pathway. J. Biol. Chem. 278, 9448–9457 [DOI] [PubMed] [Google Scholar]
- 42. Ronchi V. P., Haas A. L. (2012) Measuring rates of ubiquitin chain formation as a functional readout of ligase activity. Methods Mol. Biol. 832, 197–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wilkinson K. D., Tashayev V. L., O'Connor L. B., Larsen C. N., Kasperek E., Pickart C. M. (1995) Metabolism of the polyubiquitin degradation signal- Structure, mechanism, and role of isopeptidase T. Biochemistry 34, 14535–14546 [DOI] [PubMed] [Google Scholar]
- 44. Ikeda F., Dikic I. (2008) Atypical ubiquitin chains: new molecular signals. EMBO Rep. 9, 536–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chau V., Tobias J. W., Bachmair A., Marriott D., Ecker D. J., Gonda D. K., Varshavsky A. (1989) A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 243, 1576–1583 [DOI] [PubMed] [Google Scholar]
- 46. Seyedarabi A., Sullivan J. A., Sasakawa C., Pickersgill R. W. (2010) A disulfide driven domain swap switches off the activity of Shigella IpaH9.8 E3 ligase. FEBS Lett. 584, 4163–4168 [DOI] [PubMed] [Google Scholar]
- 47. Quezada C. M., Hicks S. W., Galán J. E., Stebbins C. E. (2009) A family of Salmonella virulence factors functions as a distinct class of autoregulated E3 ubiquitin ligases. Proc. Natl. Acad. Sci. U.S.A. 106, 4864–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chou Y. C., Keszei A. F., Rohde J. R., Tyers M., Sicheri F. (2012) Conserved structural mechanisms for autoinhibition in IpaH ubiquitin ligases. J. Biol. Chem. 287, 268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Monod J., Wyman J., Changeux J. P. (1965) On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 12, 88–118 [DOI] [PubMed] [Google Scholar]
- 50. Koshland D. E., Jr., Némethy G., Filmer D. (1966) Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5, 365–385 [DOI] [PubMed] [Google Scholar]
- 51. Stebbins C. E., Galán J. E. (2001) Structural mimicry in bacterial virulence. Nature 412, 701–705 [DOI] [PubMed] [Google Scholar]
- 52. Stoll K. E., Brzovic P. S., Davis T. N., Klevit R. E. (2011) The essential Ubc4/Ubc5 function in yeast is HECT-E3 dependent and RING-E3 dependent pathways require only mono-Ub transfer by Ubc4. J. Biol. Chem. 286, 15165–15170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Krissinel E. (2010) Crystal contacts as nature's docking solutions. J. Comput. Chem. 31, 133–143 [DOI] [PubMed] [Google Scholar]
- 54. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 55. Huang L., Kinnucan E., Wang G., Beaudenon S., Howley P. M., Huibregtse J. M., Pavletich N. P. (1999) Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science 286, 1321–1326 [DOI] [PubMed] [Google Scholar]