

Background: E2F1 and FOXO3 are two transcription factors participating in cellular senescence.
Results: E2F1 represses FOXO3 transactivity through interaction.
Conclusion: E2F1-FOXO3 regulatory mechanism is conserved and participates in regulating cellular senescence.
Significance: Drugs and/or therapies that inhibit the physical interaction might be candidates for reducing cellular senescence and increasing longevity.
Keywords: E2F Transcription Factor, FOXO, Oxidative Stress, Senescence, Transcription Regulation
Abstract
E2F1 and FOXO3 are two transcription factors that have been shown to participate in cellular senescence. Previous report reveals that E2F1 enhanced cellular senescence in human fibroblast cells, while FOXO transcription factors play against senescence by regulation reactive oxygen species scavenging proteins. However, their functional interplay has been unclear. Here we use E2F1 knock-out murine Embryonic fibroblasts (MEFs), knockdown RNAi constructs, and ectopic expression of E2F1 to show that it functions by negatively regulating FOXO3. E2F1 attenuates FOXO3-mediated expression of MnSOD and Catalase without affecting FOXO3 protein stability, subcellular localization, or phosphorylation by Akt. We mapped the interaction between E2F1 and FOXO3 to a region including the DNA binding domain of E2F1 and the C-terminal transcription-activation domain of FOXO3. We propose that E2F1 inhibits FOXO3-dependent transcription by directly binding FOXO3 in the nucleus and preventing activation of its target genes. Moreover, knockdown of the Caenorhabditis elegans E2F1 ortholog efl-1 significantly extends lifespan in a manner that requires the activity of the C. elegans FOXO gene daf-16. We conclude that there is an evolutionarily conserved signaling connection between E2F1 and FOXO3, which regulates cellular senescence and aging by regulating the activity of FOXO3. We speculate that drugs and/or therapies that inhibit this physical interaction might be good candidates for reducing cellular senescence and increasing longevity.
Introduction
E2F transcription factor 1 (E2F1)3 plays a central role in cell proliferation, development, apoptosis, and senescence (1–3). While two genes encoding E2F family members have been identified in Caenorhabditis elegans, efl-1 and efl-2, eight E2F members have been identified in mammals with E2F1 being the best characterized. E2F1 protein consists of several highly conserved domains including a nuclear localization signal, DNA binding domain, a protein-binding site for its heterodimeric partner DP, a transcriptional activation domain, and a retinoblastoma (Rb) protein-binding domain, among others (1). Rb contacts and inhibits E2F1 through two different regions, including the E2F1 trans-activation domain and the E2F1 coiled-coil marked box domain, which also participates in DP binding. When a cell prepares to enter the S phase of the cell cycle, the Rb-E2F1 complex is disrupted, allowing E2F1 to activate transcription of downstream target genes responsible for normal entry into S phase (4). A seeming contradiction was noted when abnormal replication resulting in tumor formation was observed in E2F1 knock-out mice (5, 6). Further complication of our understanding of E2F1 function came from reports that DNA damage resulting from certain anti-cancer drugs elicited the ectopic activation of E2F1 and its subsequent induction of the pro-apoptotic genes BH3-only protein (Bim) and p73 (7, 8). Interestingly, E2F1 and its target gene p14 (ARF) tumor suppressor have been shown to induce senescence in normal human fibroblasts (3).
The Forkhead box O (FOXO) family of transcription factors also plays a central role in many biological processes including cell proliferation, differentiation, and apoptosis (9, 10). While one member of this family has been identified in C. elegans, DAF-16 (11, 12), four mammalian family members are known. This protein family is highly conserved and consists of a DNA binding domain, nuclear localization signal, nuclear export signal, transcriptional activation domain, and numerous sites for various post-translational modifications, which play critical roles in regulating FOXO activity (9, 10). For example, Akt-mediated phosphorylation of FOXO at three conserved sites results in FOXO cytoplasm localization. DAF-16 has been shown to affect the lifespan of C. elegans (13). Mammalian FOXO proteins also regulate aging and cellular senescence through the transcriptional activation of genes encoding reactive oxygen species (ROS) scavenging proteins such as MnSOD and catalase (14).
We report here a mechanism by which E2F1 regulates cellular and organismal senescence. E2F1 KO mouse embryonic fibroblasts (MEFs) show attenuated senescence and ROS levels through the elevated transcription of genes encoding detoxifying enzymes such as MnSOD and Catalase. This reduction in senescence requires the activity of FOXO3. We demonstrate that E2F1 and FOXO3 physically interact, and that this results in the inhibition of FOXO3. This requirement is conserved across species as we show that reducing levels of efl-1 in C. elegans increase their longevity but only in the presence of functional DAF-16. We conclude that one mechanism through which E2F1 regulates the cell cycle and senescence is by inhibiting the activity of FOXO3.
EXPERIMENTAL PROCEDURES
Cell Culture
Primary MEFs from wild-type and E2F1 KO mice were harvested from E12.5 embryos. HEK293T and H1299 cells were maintained in MEM medium supplemented with 10% fetal bovine serum (FBS) at 5% CO2 concentration culture.
Plasmid Constructs
The lentivirus vector plko.1-puro was used to express shRNA; either this vector or control empty vector was co-transfected with the lentiviral packaging plasmids VSVG and pCMV-deltaR8.2 into HEK293T cells for virus production. 48 h after transfection, supernatant was filtered through a 0.45-μm filter, and used to infect cells. 72 h after infection, cells were selected with 1.5 μg/ml puromycin in culture medium.
Visualization and Quantitation of Protein and RNA
Western blots were performed as described (15). Total RNA was isolated from cells using Trizol reagent (Invitrogen). The products from reverse transcription were performed to real-time PCR with specific primers. Primer sequences used are available on request.
Chromatin Immunoprecipitation Assay
At passage 5, wide-type or E2F1 KO MEFs was performed with the ChIP assay kit (Millipore, 17–295) following manufacturer's instructions. Briefly, MEFs were cross-linked with 1% formaldehyde for 10 min at room temperature. DNA-protein immunocomplexes were immunoprecipitated by FOXO3 antibody (Abcam, ab-12162) or normal rabbit IgG. Cross-linking was reversed by heating over night at 65 °C. The purified DNA was subjected to quantitative real-time PCR. Primers were as follows.
mMnSOD-ChIP-F: 5-GGCTTAATGGGTCATCCTAG-3, mMnSOD-ChIP-R: 5-GTCTTTATGCCACATACTGA-3; mCatalase-ChIP-F: 5-TGACATTTGGCACCATTAGG-3, mCatalase-ChIP-R: 5-CGCCAGAATCACATAAACAG-3; hMnSOD-ChIP-F: 5-GCCCTAGTTACATTCTTCTG-3, hMnSOD-ChIP-R: 5-ACAGTCAGGCGAAGAGGAA-3; hCatalase-ChIP-F: 5-AATTGACTTCAGAGAACAGC-3, hCatalase-ChIP-R: 5-ACCAAACCAATACAATTACC-3.
Luciferase Reporter Assay
pGL2-promoter-FKRE has been described (16). HEK293T cells were plated onto 24-well culture dishes and transfected with indicated plasmids. 24 h after transfection, cells were harvested. Luciferase assay was performed with Promega dual-luciferase reporter assay system following the manufacturer's instructions.
DNA Pull-down Assay
1 pm biotin-labeled MnSOD FKRE DNA was incubated with streptavidin-coated beads for 15 min with shaking in a DNA binding solution (5 mm Tris-HCl, pH 7.0, 0.5 mm EDTA and 1 m NaCl). Then, the beads were mixed with 100 μl of the cell lysates combined with 400 μl of incubation buffer (50 mm Tris-HCl, pH 7.0, 100 mm KCl, 1 mm EDTA, 0.1% Triton X-100, 5% glycerol) for 3 h at 4 °C. Beads were washed three times with incubation buffer and boiled with 1× SDS loading buffer. Samples were subjected to Western blots to detect proteins. DNA sequences corresponding to MNSOD FKRE were listed as follows. MnSOD-FKRE-biotin-F: 5-CCTAGTTACATTCTTCTGACGTCTGTAAACAAGCCCAGCCCTTCCTGTTG-3, MnSOD-FKRE-biotin-R: 5-CAACAGGAAGGGCTGGGCTTGTTTACAGACGTCAGAAGAATGTAACTAGG-3.
Measurement of Senescence Markers
SA-β-gal was detected using the Cellular Senescence Assay kit (Millipore, KAA002) following the manufacturer's protocol. Cells were labeled with 10 mm BrdU (Sigma-Aldrich) in growth medium for 24 h at 37 °C. BrdU-labeled DNA was detected with mouse monoclonal anti-BrdU (GE Healthcare, RPN202) according to the manufacturer's protocol. Intracellular ROS generation was assessed using 20,70-dichlorofluorescein diacetate (DCF) (Molecular Probes) following to the manufacturer's protocol.
C. elegans Knock-down Experiments
Worm RNAi was carried out at 20 °C using the feeding method as previously described (17). The efl-1 RNAi #1 construct was generated by inserting 200bp efl-1 cDNA into pAD12, an empty RNAi vector (18). The efl-2 RNAi #2 construct was obtained from the Ahringer RNAi library. Escherichia coli HT115 (DE3) transformed with pAD12 was used as control.
RESULTS
E2F1 Regulates Cellular Senescence in MEFs
It has been reported that E2F1 regulates intracellular ROS generation through various mechanisms (17, 19, 20). Multiple experiments have shown that intracellular ROS play a critical role in cellular senescence of mammalian cells such as MEFs (18, 21). We compared wild-type (WT) and E2F1 knock-out (E2F1 KO) MEFs to determine the role of E2F1 in the regulation of cellular senescence. Bromodeoxyuridine (BrdU) incorporation, marking proliferating cells undergoing DNA synthesis, and β-galactosidase hydrolysis by the enzyme, senescence-associated β-galactosidase (SA-β-gal), found only in senescing cells, served as readouts for cellular senescence. The ability of MEFs to proliferate decreases with cell passage; knocking out E2F1 attenuates this effect. The proliferation of passage 5 WT MEFs was greatly reduced compared with E2F1 KO MEFs, with all cells visualized by Hoechst stained DNA but DNA synthesis was greatly reduced only in WT cells (Fig. 1A, left panel). While the levels of BrdU incorporation of passage 2 and 5 E2F1 KO MEFs were not significantly different, passage 2 WT MEFs showed a significant reduction in BrdU incorporation compared with E2F1 KO cells, and this decrease was much greater at passage 5 (Fig. 1A, right panel). We did not see any difference in the percent of senescing passage 2 E2F1 KO and WT MEFs; however, by passage 5 a significantly greater proportion of WT MEFs were testing positive for senescence compared with E2F1 KO MEFs, indicated by SA-β-gal activity (Fig. 1B).
FIGURE 1.

E2F1 deficiency attenuates MEF senescence. A, proliferation rate of WT and E2F1 KO MEFs at passage 2 and passage 5 as measured by BrdU labeling for 24 h prior to fixation and staining. The left panel shows a representative picture of passage 5 cells visualized by the DNA stain Hoechst and dividing cells by BrdU incorporation. The right panel shows the percent of BrdU-positive cells with the bars representing the mean ± S.E. (t test, **, p < 0.01, n = 3). E2F1 KO cells have been set to 100% to show the percent reduction of division of WT cells. B, WT and E2F1 KO MEFs were stained for senescence-associated β-galactosidase (SA-β-Gal) activity at passage 2 and passage 5. The percentage of SA-β-gal-positive cells was calculated from five randomly chosen fields. At least 150 cells were analyzed per experiment. Data represent the mean ± S.E. (t test, **, p < 0.01, n = 3). C, WT passage 2 MEFs were more sensitive to hydrogen peroxide treatment than E2F1 KO MEFs. Cells were stained for SA-β-Gal activity 48 h after 100 μm H2O2 treatment. Data represent the mean ± S.E. (t test, **, p < 0.01; n = 3). D, ROS levels in WT and E2F1 KO MEFs, treated with hydrogen peroxide or not, were measured by staining with dichlorofluorescein (DCF). Non-treated WT cells have been set to 100%. ROS levels of WT cells are higher than E2F1 KO cells (t test, *, p < 0.05; **, p < 0.01, n = 3). Data represent the mean ± S.E.
It is well established that ROS increase robustly with continued cell division of MEFs (22). While passage 2 WT and E2F1 KO MEFs showed similar levels of SA-β-gal-positive cells (Fig. 1B), the addition of oxidative stress by the treatment of these cells with hydrogen peroxide greatly increased the percent WT MEFs with SA-β-gal activity but did so only modestly in E2F1 KO MEFs (Fig. 1C). Moreover, the levels of intracellular ROS, as measured by dichlorofluorescein (DCF) levels, whether in the steady state or under oxidative stress, were significantly reduced in E2F1 KO MEFs compared with WT cells (Fig. 1D). Together, these results suggest that in WT MEFs, E2F1 promotes cellular senescence and ROS production, or inhibits ROS clearance.
E2F1 Regulates Cellular Senescence through FOXO
It is well known that FOXO transcription factors have a conserved role in ROS metabolism and senescence control via the regulation of detoxifying enzymes (23). We next asked whether the regulation of cellular senescence by E2F1 is coordinated with the activity of FOXO. Wild-type and E2F1 KO MEFs infected with a lentivirus vector carrying the U6 promoter but not expressing an interference RNA show the expected reduction in SA-β-gal staining in the latter cells; however, U6 directed expression of an interference shRNA for FOXO3 abrogates this reduction (Fig. 2A). Knockdown efficiency of FOXO3 shRNA was confirmed (Fig. 2B). This increase in senescence is correlated with a decrease in the E2F1 KO caused elevated levels of MnSOD and Catalase mRNA (Fig. 2, C and D). In contrast, mRNA levels of the stress induced protein Sesn3 are unaffected by the loss of E2F1 or by the loss of E2F1 and reduction of FOXO3 (Fig. 2E).
FIGURE 2.

E2F1 regulation of senescence requires FOXO3. A, WT and E2F1 KO passage 3 MEFs were infected with lentivirus constitutively expressing FOXO3 (U6/FOXO) shRNA or a control (U6) vector. Senescent cells were examined by SA-β-gal staining and the percent positive cells determined, shown as the mean ± S.E. (t test, **, p < 0.01; ns, not significant, n = 3). B, WT and E2F1 KO MEFs were infected with U6/FOXO or U6 empty lentivirus. Proteins were extracted and detected by immunoblotting with the anti-FOXO3 and E2F1 antibodies. GAPDH serves as the loading control. C-E, WT and E2F1 KO passage 2 MEFs were infected as in A. The mRNA levels of MnSOD (C), catalase (D), and Sesn3 (E) were measured compared relative to WT MEFs infected with control vector. Data are shown as the mean ± S.E. (t test, **, p < 0.01; *, p < 0.05; ns, not significant, n = 3). F, WT passage 3 MEFs were transfected with constitutively expressing GFP-E2F1 (E2F1) or control (GFP) vector. Proteins were extracted and detected by immunoblotting with the indicated antibodies. G, WT passage 3 MEFs were transfected as in F, and senescent cells were visualized by SA-β-gal staining. A representative example is shown in the left panel, and the percent positive cells shown in the right panel as the mean ± S.E. (t test, **, p < 0.01; n = 3).
Since reducing the levels of E2F1 reduces senescence and increases the levels of MnSOD and Catalase, we wanted to see what the effect of overexpression of E2F1 would be. Passage 3 WT MEFs were transfected with vectors expressing the control GFP or with GFP-E2F1. Protein was isolated from these transfected cells, size separated on acrylamide gels, blotted and probed with antibodies on Westerns. The expression of GFP-E2F1 is clearly visible; concomitant with this, the levels of MnSOD and catalase proteins are decreased compared with GFP control, with the control GAPDH protein unchanged (Fig. 2F). Staining these cells for SA-β-gal shows a marked increase in senescence as visualized by SA-β-gal staining (Fig. 2G). Based on these results, we hypothesize that E2F1 controls senescence by regulating the levels of MnSOD and Catalase through FOXO3 activity.
E2F1 Interacts with FOXO3
To elucidate the relationship between E2F1 and FOXO3, we investigated whether the two proteins interact with each other. When expressed in HEK293T cells, GFP-E2F1 and Flag-FOXO3 formed a complex as shown by anti-Flag immunoprecipitation (IP) (Fig. 3A). More importantly, endogenous E2F1 was found associated with endogenous FOXO3 in primary MEFs. This was observed by immunoprecipitating with either IgG, as control, or a FOXO3 antibody and detecting proteins with anti-E2F1 and anti-FOXO3 antibodies (Fig. 3B).
FIGURE 3.

E2F1 and FOXO3 physically interact in vivo. A, immunoprecipitation of FOXO3 precipitates E2F1. Lysates of HEK293T cells transfected with plasmids encoding GFP-E2F1 and Flag-FOXO3 or control vector were immunoprecipitated with anti-Flag antibody. Western blot were followed with anti-GFP or -Flag antibody. B, immunoprecipitation of endogenous FOXO3 from primary MEFs precipitates E2F1. Anti-FOXO3 or IgG immunoprecipitates from primary MEFs were immunoblotted with FOXO3 or E2F1 antibody. Two bands of endogenous E2F1 were recognized by E2F1 antibody (E2F1 C-20, Santa Cruz Biotechnology). C, The P5 fragment of FOXO3 binds to E2F1 in vitro. GST was fused to 5 different fragments of FOXO3 (P1-P5) and used in a pull-down assay of Myc-E2F1. Precipitated proteins were blotted and detected with the Myc antibody in the upper panel. The lower panel is Coomassie Blue (CB) staining of GST-FOXO3 peptides. D, P5 fragment of FOXO3 is required to bind E2F1. Lysates of HEK293T cells transfected with plasmids encoding GFP-E2F1 and control vector (−), full-length Flag-FOXO3 (WT) or Flag-FOXO3ΔP5 were immunoprecipitated with anti-Flag antibody. Western blot of the immunoprecipitated (top panel) or input (bottom panel) protein were detected with anti-GFP or -Flag antibody. E, P5 fragment of FOXO3 binds a region of E2F1 including its DNA binding domain. GST pull-down assay of His-fusion E2F1 fragments (NT, DBD, CT) with recombinant GST or GST fused with the fragment of FOXO3-P5. F, upper panel: HEK293T was transfected as in (A). 24 h after transfection, 100 μm H2O2 was added into medium. Co-IP assay was performed as in A to show the interaction between FOXO3 and E2F1. Bottom panel: statistical results from three independent experiments. Relative interaction was determined by quantification of GFP-E2F1/Flag-FOXO3 (t test, *, p < 0.05; **, p < 0.01, n = 3).
Next, using recombinant GST fusion proteins containing five non-overlapping regions of FOXO3 (P1–P5), we delineated the region within FOXO3 that mediated the interaction with E2F1. Only P5 (amino acids 542–672), which includes the transcription-activation domain of FOXO3, pulled down E2F1 (Fig. 3C). Further supporting this result, immunoprecipitation of the Flag-tagged version of FOXO3-ΔP5, which lacks amino acids 542–672, failed to co-immunoprecipitate the GFP-tagged E2F1 (Fig. 3D). To identify the region of E2F1 that interacts with FOXO3, we recombined His fusion proteins with three non-overlapping regions of E2F1 (NT, DBD, and CT). We found that the central region of E2F1, including its DNA binding domain (DBD), is responsible for binding to the P5 domain of FOXO3 (Fig. 3E). Taken together, we conclude that E2F1 and FOXO3 form a physical complex, which negatively regulates FOXO3 activity.
We next wanted to determine the effects of oxidative stress on the E2F1/FOXO3 interaction. HEK293T cells were transfected with GFP-E2F1 and Flag-FOXO3 expressing plasmids. Cells were then treated with H2O2 and lysates collected after various times and immunoprecipitated with Flag antibody. Increased exposure time to H2O2 reduced the interaction between E2F1 and FOXO3 (Fig. 3F). This result suggests that cells may respond to oxidative stress by weakening the interaction between E2F1 and FOXO3, thus allowing FOXO3 to activate ROS genes and combat the stress.
E2F1 Suppresses the Transcriptional Activity of FOXO3
To test more directly if E2F1 represses FOXO3 activity by preventing the activation of target genes, we used the Forkhead Response Element (FKRE) fused to a firefly luciferase reporter gene. The transcription-activation domain of FOXO proteins is essential for their activation of target genes (24). A dual luciferase assay was used to assess the transcriptional activity of FOXO3. HEK293T cells were transfected with FKRE-firefly and tk-Renilla luciferase reporters, with or without FOXO1/3 and/or E2F1 expressing plasmids. The reporters alone show some activation of the FKRE-luciferase gene, presumably from endogenous FOXO1/3 proteins. This activation is significantly repressed when the E2F1 plasmid is co-transfected (Fig. 4A). Transfection of the FOXO1/3 plasmids strongly stimulated expression from the FKRE-luciferase reporter. This activation is largely repressed by co-transfection with E2F1 plasmid (Fig. 4A). To rule out squelching effect of E2F1, we used an unrelated transcription factor SP1 as a control. Unlike E2F1, SP1 has no significant effect on FOXO1/3 transcriptional activity (Fig. 4A). The protein levels of FOXO1 and FOXO3 are shown in bottom panel. This repression is dose-dependent as increasing the amount of E2F1 plasmid inversely correlates with FOXO3 activation of FKRE-luciferase (Fig. 4B). Our previous results suggested the hypothesis that E2F1 inhibits FOXO3 in the nucleus through direct binding and blocking the activity of FOXO3. We utilized the dual luciferase assay to test this hypothesis. HEK293T cells transfected with FOXO3 expressing plasmid showed elevated levels of FKRE-luciferase activity (Fig. 4C). Co-transfection with a wild-type E2F1 expressing construct abrogated most of this activation. However, co-transfection with a truncated E2F1 that lacks amino acids 1–125, which includes its nuclear localization sequence (E2F1Δ125), failed to repress the transcription activity of FOXO3 (Fig. 4C). Additionally, deletion of the FOXO3 binding region of E2F1, E2F1ΔDBD, also blocked its ability to inhibit FOXO3 (Fig. 4D). Furthermore, we constructed a transcription activity domain deficiency E2F1 (E2F1ΔTA), and performed FKRE-luciferase reporter assay. Accordingly, E2F1ΔTA significantly inhibits FOXO3 activity (Fig. 4E). These results suggest E2F1 represses FOXO3 activity specifically, but largely independent of E2F1 transcription activity.
FIGURE 4.

E2F1 inhibits FOXO3 activity. A, upper panel: FOXO3 and FOXO1 activity is repressed by E2F1. HEK293T cells were transfected with the FOXO3-responsive element, FKRE-luciferase reporter gene, the constitutively active tk-Renilla reporter as a control, and plasmids encoding E2F1 and/or FOXO3/FOXO1,SP1. Data are shown as the mean ± S.E. firefly/Renilla luciferase activity (t test, ***, p < 0.001; n = 3). Bottom Panel: FOXO1 and FOXO3 protein levels were detected by anti-Flag antibody. B, FOXO3 activity is measured as in A. E2F1 expression significantly repressed FOXO3 activity in a dose dependent manner (dose from 0.02–0.1 μg). Data are shown as the mean ± S.E. firefly/Renilla luciferase activity (t test, **, p < 0.01; ***, p < 0.001; n = 3). C, constitutively cytoplasmic E2F1 cannot repress FOXO3 activity. Deletion of the nuclear localization signal in the N-terminal 125 amino acids of E2F1 abolishes the inhibition of FOXO3 by E2F1. HEK293T cells were transfected with FKRE-luciferase and tk-Renilla reporter genes as described in A, together with plasmids encoding full-length (WT) or truncated (Δ125) E2F1 and the FOXO3 expressing plasmid. The data are shown as the mean ± S.E. firefly/Renilla luciferase activity (t test, **, p < 0.01; ns, not significant, n = 3). D, deletion of the FOXO3-binding domain of E2F1 (DBD) abrogates repression. HEK293T cells were transfected as in A with the indicated plasmids. The data are shown as the mean ± S.E. firefly/Renilla luciferase activity (t test, *, p < 0.05; **, p < 0.01; n = 3). E, deletion of the transcription activity domain of E2F1 (E2F1 ΔTA) significantly represses FOXO3a activity. HEK293T cells were transfected as in A with the indicated plasmids. The data are shown as the mean ± S.E. firefly/Renilla luciferase activity (t test, *, p < 0.05; **, p < 0.01; ***, p < 0.001; n = 3). F, E2F1 repression of FOXO3 is independent of Akt phosphorylation. HEK293T cells were transfected as in A except that the Akt independent, activated form of FOXO3, FOXOTM, was used. Data are shown as the mean ± S.E. firefly/Renilla luciferase activity (t test, **, p < 0.01; n = 3). G, E2F1 repression of FOXO3 is independent of p53. The p53 defective cell line H1299 was transfected as in A with indicated plasmids and relative luciferase activity was measured. Data are shown as the mean ± S.E. firefly/Renilla luciferase activity (t test, **, p < 0.01; ***, p < 0.001; n = 3). H, E2F1 KO MEFs have elevated FOXO3 activity. WT and E2F1KO MEFs are transfected with the FKRE-luciferase reporter gene and the tk-Renilla control. Data are shown as the mean ± S.E. firefly/Renilla luciferase activity (t test, **, p < 0.01; n = 3). I–J, FOXO3 enrichment on MnSOD or catalase promoter is elevated in E2F1 KO MEFs. WT and E2F1KO MEFs at passage 5 were cross-linked and subjected to ChIP assay with anti-FOXO3 antibody. Primers corresponding to FKRE of MnSOD or catalase promoter are used to detecting FOXO3 binding. FOXO3 occupancy on MnSOD or catalase increased in E2F1KO MEFs (t test, ***, p < 0.001 on MnSOD promoter, *, p < 0.05 on catalase promoter; n = 3). K–L, FOXO3 enrichment on MnSOD or Catalase promoter is attenuated when E2F1 ectopically expression in HEK293T cells. HEK293T cells were transient transfected with Flag-FOXO3a and/or GFP-E2F1 plasmids as indicated. 24h after transfection, cells were fixed and subjected to ChIP assay with anti-Flag antibody. Primers corresponding to human FKRE of MnSOD or catalase promoter are used to detecting FOXO3 binding. FOXO3a occupancy was decreased on MnSOD and catalase promoter when GFP-E2F1 overexpressed (t test, *, p < 0.05 on MnSOD and catalase promoter, n = 3). M–N, E2F1 enrichment on MnSOD or catalase promoter is not affected by FOXO3a. Cells were transfected as in K and subjected to ChIP assay with anti-GFP antibody. E2F1 binding on FKRE of MnSOD or Catalase promoter were detected qRT-PCR. No significant differences were detected among groups (t test, ns: p > 0.05 on MnSOD and Catalase promoter, n = 3). O, HEK293T cells were transfected with FOXO3 together with GFP-E2F1 WT or ΔDBD. Whole cell extracts were pulled down by biotin-labeled MnSOD FKRE DNA. GFP-E2F1 blocked FOXO3-DNA binding. E2F1 ΔDBD had no significant effect on FOXO3-DNA binding. P, ChIPed DNA from Fig. 4K was analyzed with primers corresponding to E2F1 binding site of Bim promoter. FOXO3 did not affect E2F1 occupancy on Bim promoter.
Akt phosphorylation of FOXO proteins facilitates their nuclear export, blocking the transcriptional activation of target genes (24). We asked whether a constitutively nuclear FOXO3 protein, mutant at all three Akt phosphorylation sites, could be affected by E2F1. A plasmid expressing the Akt-independent protein, FOXO3TM, strongly activated transcription from the FKRE-luciferase reporter. This activation is repressed by co-transfection with the E2F1-expressing plasmid (Fig. 4F).
The p53 transcription factor has been shown to interact and cooperate with both E2F1 and FOXO in multiple cellular processes (25, 26). To find out if the repression of FOXO3 activity by E2F1 requires p53, we repeated the transfections shown in Fig. 4A but instead used the p53-null cell line H1299. The results are shown (Fig. 4G). The relative activation by transfection of the FOXO3-expressing plasmid in H1299 cells is less than in HEK293T cells. This could result from a higher level of activation of the endogenous FOXO3 protein or a lesser ability of transfected FOXO3 to activate transcription. Nevertheless, co-transfection with the E2F1-expressing plasmid abrogated this activation. Thus, repression of FOXO3 by E2F1 does not require p53 protein.
Then a comparison of WT and E2F1 KO MEFs showed a significantly higher level of FKRE-luciferase activity in the knock-out cells compare with WT MEFs (Fig. 4H). We also performed a chromatin immunoprecipitation (ChIP) assay in WT and E2F1 KO MEFs to investigate the FOXO3 enrichment on MnSOD and Catalase promoter (Fig. 4, I–J). The elevated levels of FOXO occupancy on the promoter of both MnSOD and Catalase in E2F1 KO MEFs compared with WT MEFs. To further elucidate the relationship between E2F1 and FOXO3, we overexpress E2F1 and FOXO3 in HEK293T cell line ectopically. ChIP assay revealed that E2F1 disrupted the binding between FOXO3 and MnSOD/Catalase promoter (Fig. 4, K–L). On another aspect, E2F1 does not bind to the FKRE of MnSOD/Catalase promoter, indicating that this blocking effect of E2F1 is not caused by direct binding of E2F1 with the gene promoter (Fig. 4, M–N). In addition, we performed a DNA pulldown assay to confirm this conclusion. As expected, FOXO3 was pulled down by FKRE of MnSOD promoter. E2F1 WT, but not the ΔDBD mutation blocked FOXO3-FKRE binding in vitro (Fig. 4O). We also found that FOXO3 overexpression did not affect the binding affinity of E2F1 to its putative target, Bim, through ChIP assay (Fig. 4P). Therefore, we conclude that E2F1 binds to FOXO3 and represses FOXO3-DNA binding, therefore blocking its ability to activate FOXO3 target genes.
E2F1 Does Not Affect the Stability and Subcellular Localization of FOXO3
A number of proteins including Murine Double Minute 2 (MDM2) and the major catalytic subunit of IκB kinase, IKKβ, can regulate FOXO3 by post-translational modification, resulting in its degradation (27, 28). Additionally, FOXO3 phosphorylation by Akt at Ser-252, among others, results in exportation of FOXO3 to the cytoplasm where it cannot activate target genes (29). We wanted to examine whether E2F1 might work by a similar mechanisms. HEK293T cells were transfected with a plasmid expressing FOXO3 alone or with increasing amounts of a plasmid expressing Myc-E2F1. A Western blot of protein isolated from these cells showed that the level of FOXO3 protein was unchanged with increasing amounts E2F1 (Fig. 5A). Wild-type and E2F1 KO MEFs were next compared. Protein isolated from these cells was examined by Western blot for the indicated proteins (Fig. 5B). None of the levels of the indicated proteins, FOXO3, pS252-FOXO3, Akt, nor pS473-Akt were changed relative to the control GAPDH; E2F1 is absent in E2F1 KO MEFs, as expected. We conclude that E2F1 does not affect the protein abundance of FOXO3 nor proteins known to modify its abundance.
FIGURE 5.
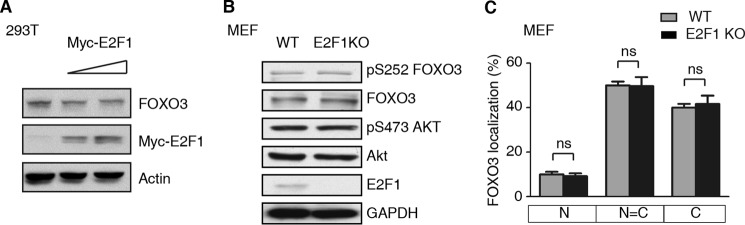
E2F1 inhibits FOXO activation independent of Akt-mediated phosphorylation and without changing its subcellular localization. A, increasing levels of E2F1 do not affect FOXO3 protein levels. Lysates of HEK293T cells transfected with a constant level of a FOXO3-expressing plasmid and increasing doses Myc-E2F1 were immunoblotted with the indicated antibodies. FOXO3 levels were unchanged relative to an actin control. B, WT and E2F1 KO MEFs have comparable levels of the indicated proteins. Lysates were immunoblotted with the indicated antibodies. C, subcellular localization of GFP-FOXO3 transfected WT or E2F1 KO MEFs is similar. The data are shown as the mean ± S.E. (t test, ns, not significant; n = 3).
We next wanted to examine whether E2F1 might negatively regulate FOXO3 by changing its subcellular localization. Wild-type or E2F1 KO MEFs transfected with GFP-FOXO3 were compared. The subcellular localization of GFP-FOXO3 was unchanged (Fig. 5C). Taken together, these results suggest that E2F1 does not regulate FOXO3 by altering its abundance, subcellular localization, or indirectly by affecting Akt signaling.
The E2F1-FOXO Signaling Pathway Regulates Longevity in C. elegans
To find out whether the regulation of FOXO3 by E2F1 is conserved through evolution, we turned to the C. elegans genetic system. In C. elegans, the FOXO ortholog DAF-16 occupies a critical point in the regulatory network of aging (13). There are two orthologs of E2F1, efl-1 and efl-2, encoded in the C. elegans genome (30). Interestingly, two different RNAi constructs of efl-1 (Fig. 6A, left) but not efl-2 (Fig. 6A, right) significantly extended the lifespan of wild-type C. elegans. This increased longevity requires the FOXO ortholog DAF-16 as the effect of efl-1 RNAi was completely abolished in a daf-16 loss of function mutation (mu86) mutant (Fig. 6B).
FIGURE 6.

E2F (EFL-1) regulation of C. elegans longevity requires FOXO (DAF-16). A and B, reducing expression of EFL-1 by feeding wild-type worms bacteria expressing either of two different RNAi efl-1 clones, compared with a control clone, extended lifespan (A, left), (**, p < 0.001 for efl-1#1 or efl-1#2 RNAi versus control RNAi), but not efl-2 RNAi (A, right) (not significant for efl-2#1 or efl-2#2 RNAi versus control RNAi). Daf-16 (mu86), a putative null mutation of FOXO, doesn't extend lifespan, when knockdown efl-1 (B) (not significant for efl-1#1 or efl-1#2 RNAi versus control RNAi). C–D, increased longevity of efl-1 knockdown worms correlates with increased levels of stress response genes sod3, mtl-1, and sip1. Relative mRNA levels of the indicated genes were determined by quantitative real-time qPCR in wild type (C) (t test, **, p < 0.01; n = 3) and daf-16 (mu86) worms (D) (t test, *, p < 0.05; n = 3), fed bacteria as in A and B. E, GFP-DAF-16 transgenic worms were used for subcellular localization analysis of DAF-16. EFL-1 did not affect the subcellular localization of DAF-16 in worms (not significant for efl-2#1 or efl-2#2 RNAi versus control RNAi).
Finally, we examined the effect on DAF-16 target genes of reducing efl-1 expression. Wild-type worms fed efl-1 RNAi constructs showed elevated levels of the mRNAs of the DAF-16 target genes sod-3, mtl-1 and sip-1, compared with worms feed vector alone (Fig. 6C). When daf-16 worms were fed the same RNAi constructs we still saw elevated levels of these mRNAs but to significantly lower levels (Fig. 6D). Consistent with our finding in Fig. 5C that E2F1 does not affect the subcellular localization of FOXO3, we also observed that there is no significant difference of DAF-16 subcellular localization between WT and EFL-1 knockdown in worms (Fig. 6E). We conclude that EFL-1 acts to limit C. elegans lifespan by negatively regulating DAF-16. Taken together, the our results show that E2F1 and EFL-1 bind to and inhibit the transcriptional activity of FOXO3 and DAF-16, respectively, to regulate cellular senescence and lifespan and that this function is conserved from nematodes to mammals.
DISCUSSION
We report here an analysis of the functional relationship between certain E2F1 and FOXO transcription factor family members in MEFs, human tissue culture cells and the worm C. elegans. Both E2F1 and FOXO had previously been implicated in the regulation of cellular and organismal senescence (1, 2, 9, 10, 13); however, their relationship to each other was unclear. Here we demonstrate that E2F1 physically interacts with FOXO resulting in the inhibition of certain FOXO transcription target genes encoding the antioxidant enzymes MnSOD and catalase. This most likely directly results in an increase in ROS levels and senescence.
Cultured WT MEFs can undergo only a limited number of passages before entering senescence; however, here we demonstrate that E2F1 KO MEFs have reduced senescence with comparable passage numbers. This can be seen as a relative increase in DNA replication, a decrease in the senescence markers SA-β-gal and ROS and an increase in MnSOD and Catalase. These E2F1-dependent changes in senescence are largely abrogated by the knock down FOXO3 in mouse and human cells and the FOXO ortholog DAF-16 in nematodes. This is also measurable at the organismal level as the increased longevity seen in efl-1 knock down worms is abrogated in a daf16 mutant background.
We are able to see a physical interaction between E2F1 and FOXO3, using co-immunoprecipitation techniques, both from ectopically expressed proteins in human tissue culture cells and from the endogenous proteins in MEFs. We identified the C-terminal 125 amino acids of FOXO3, including the transcriptional activation domain, and the middle third of E2F1, including the DNA binding domain, as necessary and sufficient for their binding to each other. Removal of either of these interaction domains blocks the ability of E2F1 to negatively regulate FOXO3-induced transcription from a reporter construct.
Previous studies have shown that the FOXO proteins are exquisitely regulated by a variety of post-translational modifications including phosphorylation, acetylation, ubiquitination, and arginine and lysine methylation (15). Additionally, the activity of FOXO can be modulated through direct binding to other proteins including PGC-1α and β-catenin (31, 32). Our discovery of E2F1 as a novel negative regulator of FOXO3 adds to this regulatory complexity. We did not detect any effect of E2F1 on FOXO3 protein stability, subcellular localization, or phosphorylation by Akt and similarly, we also observed EFL-1 did not affect the subcellular localization of DAF-16 in worms. As our data show that E2F1 directly binds to the C-terminal transcription-activation domain of FOXO3, we postulate that it acts by blocking its ability to transcriptionally activate certain FOXO3 target genes such as MnSOD and catalase. This mode of regulation by E2F1 is reminiscent to HCF-1, the C. elegans homolog of mammalian host cell factor 1, which directly binds to DAF-16 in the nucleus and negatively regulates DAF-16, possibly by preventing DAF-16 from binding to the promoters of its target genes (33).
A recent report by another group looking at E2F1 transcriptional specificity has identified FOXO transcription factors as essential cofactors in regulating apoptosis (34). They describe a positive regulatory loop mediated by the physical interaction of E2F1 and FOXO transcription factors to up regulate apoptotic genes and speculate that induction of E2F1, which is deregulated in many cancer cells, might be an effective cancer therapy. However, E2F1 has also been implicated in cell proliferation and senescence. Here we have shown that the physical interaction of E2F1 and FOXO transcription factors can also negatively regulate FOXO transcription factors blocking the induction of the antioxidant genes MnSOD and Catalase. Therapies aimed at blocking this interaction might allow FOXO proteins to attenuate senescence and increase longevity. This dichotomy of E2F1 activation having an anti-cancer but a pro-senescence function serves as a note of caution for potential therapies affecting E2F1. Identifying therapies that can affect one or the other set of pathways will be key to their development.
Acknowledgments
We thank members of the Yuan laboratory and Dr. Duan Enkui for critical reading of the manuscript and helpful discussions. We also thank Dr. Mark Mortin for critical reading and editorial assistance.
This work was supported by the Ministry of Science and Technology of China (973-2012CB910701 and 2013DFA31990) (to Z. Y.), (973-2010CB835203) (to M. D.), and the National Science Foundation of China (81125010 and 81030025) (to Z. Y.).
- E2F1
- E2F transcription factor 1
- DCF
- 20,70-dichlorofluorescein diacetate
- FOXO3
- Forkhead box O3
- FKRE
- Forkhead response element
- MDM2
- Murine Double Minute 2
- MEF
- murine embryonic fibroblast
- RB
- retinoblastoma
- ROS
- reactivated oxygen species
- H2O2
- hydrogen peroxide.
REFERENCES
- 1. Wu Z., Zheng S., Yu Q. (2009) The E2F family and the role of E2F1 in apoptosis. Int. J. Biochem. Cell Biol. 41, 2389–2397 [DOI] [PubMed] [Google Scholar]
- 2. Matsumura I., Tanaka H., Kanakura Y. (2003) E2F1 and c-Myc in cell growth and death. Cell Cycle 2, 333–338 [PubMed] [Google Scholar]
- 3. Dimri G. P., Itahana K., Acosta M., Campisi J. (2000) Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol. Cell. Biol. 20, 273–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeGregori J., Kowalik T., Nevins J. R. (1995) Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 15, 4215–4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Field S. J., Tsai F. Y., Kuo F., Zubiaga A. M., Kaelin W. G., Jr., Livingston D. M., Orkin S. H., Greenberg M. E. (1996) E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell 85, 549–561 [DOI] [PubMed] [Google Scholar]
- 6. Yamasaki L., Jacks T., Bronson R., Goillot E., Harlow E., Dyson N. J. (1996) Tumor induction and tissue atrophy in mice lacking E2F-1. Cell 85, 537–548 [DOI] [PubMed] [Google Scholar]
- 7. Rödicker F., Stiewe T., Zimmermann S., Pützer B. M. (2001) Therapeutic efficacy of E2F1 in pancreatic cancer correlates with TP73 induction. Cancer Res. 61, 7052–7055 [PubMed] [Google Scholar]
- 8. Zhao Y., Tan J., Zhuang L., Jiang X., Liu E. T., Yu Q. (2005) Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc. Natl. Acad. Sci. U.S.A. 102, 16090–16095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Calnan D. R., Brunet A. (2008) The FoxO code. Oncogene 27, 2276–2288 [DOI] [PubMed] [Google Scholar]
- 10. van der Horst A., Burgering B. M. (2007) Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 8, 440–450 [DOI] [PubMed] [Google Scholar]
- 11. Lin K., Dorman J. B., Rodan A., Kenyon C. (1997) daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278, 1319–1322 [DOI] [PubMed] [Google Scholar]
- 12. Ogg S., Paradis S., Gottlieb S., Patterson G. I., Lee L., Tissenbaum H. A., Ruvkun G. (1997) The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389, 994–999 [DOI] [PubMed] [Google Scholar]
- 13. Murphy C. T., McCarroll S. A., Bargmann C. I., Fraser A., Kamath R. S., Ahringer J., Li H., Kenyon C. (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–283 [DOI] [PubMed] [Google Scholar]
- 14. Nogueira V., Park Y., Chen C. C., Xu P. Z., Chen M. L., Tonic I., Unterman T., Hay N. (2008) Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 14, 458–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xie Q., Chen J., Yuan Z. (2012) Post-translational regulation of FOXO. Acta Biochim. Biophys. Sin 44, 897–901 [DOI] [PubMed] [Google Scholar]
- 16. Tang E. D., Nuñez G., Barr F. G., Guan K. L. (1999) Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 274, 16741–16746 [DOI] [PubMed] [Google Scholar]
- 17. Song Y. S., Lee B. Y., Hwang E. S. (2005) Dinstinct ROS and biochemical profiles in cells undergoing DNA damage-induced senescence and apoptosis. Mech Ageing Dev 126, 580–590 [DOI] [PubMed] [Google Scholar]
- 18. Kodama R., Kato M., Furuta S., Ueno S., Zhang Y., Matsuno K., Yabe-Nishimura C., Tanaka E., Kamata T. (2013) ROS-generating oxidases Nox1 and Nox4 contribute to oncogenic Ras-induced premature senescence. Genes Cells 18, 32–41 [DOI] [PubMed] [Google Scholar]
- 19. Espada L., Meo-Evoli N., Sancho P., Real S., Fabregat I., Ambrosio S., Tauler A. (2012) ROS production is essential for the apoptotic function of E2F1 in pheochromocytoma and neuroblastoma cell lines. PLoS One 0051544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Raimundo N., Song L., Shutt T. E., McKay S. E., Cotney J., Guan M. X., Gilliland T. C., Hohuan D., Santos-Sacchi J., Shadel G. S. (2012) Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell 148, 716–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weyemi U., Lagente-Chevallier O., Boufraqech M., Prenois F., Courtin F., Caillou B., Talbot M., Dardalhon M., Al Ghuzlan A., Bidart J. M., Schlumberger M., Dupuy C. (2012) ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 31, 1117–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Han Y. H., Kwon J. H., Yu D. Y., Moon E. Y. (2006) Inhibitory effect of peroxiredoxin II (Prx II) on Ras-ERK-NFκB pathway in mouse embryonic fibroblast (MEF) senescence. Free Radic. Res. 40, 1182–1189 [DOI] [PubMed] [Google Scholar]
- 23. Kops G. J., Dansen T. B., Polderman P. E., Saarloos I., Wirtz K. W., Coffer P. J., Huang T. T., Bos J. L., Medema R. H., Burgering B. M. (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419, 316–321 [DOI] [PubMed] [Google Scholar]
- 24. Perrot V., Rechler M. M. (2003) Characterization of insulin inhibition of transactivation by a C-terminal fragment of the forkhead transcription factor Foxo1 in rat hepatoma cells. J. Biol. Chem. 278, 26111–26119 [DOI] [PubMed] [Google Scholar]
- 25. You H., Mak T. W. (2005) Crosstalk between p53 and FOXO transcription factors. Cell Cycle 4, 37–38 [DOI] [PubMed] [Google Scholar]
- 26. Pan H., Yin C., Dyson N. J., Harlow E., Yamasaki L., Van Dyke T. (1998) Key roles for E2F1 in signaling p53-dependent apoptosis and in cell division within developing tumors. Mol. Cell 2, 283–292 [DOI] [PubMed] [Google Scholar]
- 27. Yang J. Y., Zong C. S., Xia W., Yamaguchi H., Ding Q., Xie X., Lang J. Y., Lai C. C., Chang C. J., Huang W. C., Huang H., Kuo H. P., Lee D. F., Li L. Y., Lien H. C., Cheng X., Chang K. J., Hsiao C. D., Tsai F. J., Tsai C. H., Sahin A. A., Muller W. J., Mills G. B., Yu D., Hortobagyi G. N., Hung M. C. (2008) ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 10, 138–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu M. C., Lee D. F., Xia W., Golfman L. S., Ou-Yang F., Yang J. Y., Zou Y., Bao S., Hanada N., Saso H., Kobayashi R., Hung M. C. (2004) IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 117, 225–237 [DOI] [PubMed] [Google Scholar]
- 29. Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., Greenberg M. E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 [DOI] [PubMed] [Google Scholar]
- 30. Ceol C. J., Horvitz H. R. (2001) dpl-1 DP and efl-1 E2F act with lin-35 Rb to antagonize Ras signaling in C. elegans vulval development. Mol. Cell 7, 461–473 [DOI] [PubMed] [Google Scholar]
- 31. Puigserver P., Rhee J., Donovan J., Walkey C. J., Yoon J. C., Oriente F., Kitamura Y., Altomonte J., Dong H., Accili D., Spiegelman B. M. (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 423, 550–555 [DOI] [PubMed] [Google Scholar]
- 32. Essers M. A., de Vries-Smits L. M., Barker N., Polderman P. E., Burgering B. M., Korswagen H. C. (2005) Functional interaction between β-catenin and FOXO in oxidative stress signaling. Science 308, 1181–1184 [DOI] [PubMed] [Google Scholar]
- 33. Li J., Ebata A., Dong Y., Rizki G., Iwata T., Lee S. S. (2008) Caenorhabditis elegans HCF-1 functions in longevity maintenance as a DAF-16 regulator. PLoS Biol. 0060233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shats I., Gatza M. L., Liu B., Angus S. P., You L., Nevins J. R. (2013) FOXO transcription factors control E2F1 transcriptional specificity and apoptotic function. Cancer Res. 73, 6056–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]