

Background: Bacterial homologues of neurotransporters served as structural paradigms for interpretation of the functional data available for their eukaryotic counterparts.
Results: We identified and characterized a close bacterial homologue of the rat vesicular monoamine transporter rVMAT2.
Conclusion: BbMAT is a multidrug antiporter. Conserved membrane-embedded carboxyls play a role in substrate and proton transport.
Significance: Understanding of the bacterial homologue should provide insights into rVMAT2.
Keywords: Antibiotic Resistance, Membrane Transport, Multidrug Transporter, Neurotransmitter, Neurotransmitter Transport, Proton Transport
Abstract
Transporters essential for neurotransmission in mammalian organisms and bacterial multidrug transporters involved in antibiotic resistance are evolutionarily related. To understand in more detail the evolutionary aspects of the transformation of a bacterial multidrug transporter to a mammalian neurotransporter and to learn about mechanisms in a milieu amenable for structural and biochemical studies, we identified, cloned, and partially characterized bacterial homologues of the rat vesicular monoamine transporter (rVMAT2). We performed preliminary biochemical characterization of one of them, Brevibacillus brevis monoamine transporter (BbMAT), from the bacterium B. brevis. BbMAT shares substrates with rVMAT2 and transports them in exchange with >1H+, like the mammalian transporter. Here we present a homology model of BbMAT that has the standard major facilitator superfamily fold; that is, with two domains of six transmembrane helices each, related by 2-fold pseudosymmetry whose axis runs normal to the membrane and between the two halves. The model predicts that four carboxyl residues, a histidine, and an arginine are located in the transmembrane segments. We show here that two of the carboxyls are conserved, equivalent to the corresponding ones in rVMAT2, and are essential for H+-coupled transport. We conclude that BbMAT provides an excellent experimental paradigm for the study of its mammalian counterparts and bacterial multidrug transporters.
Introduction
Vesicular neurotransmitter transporters mediate the uptake and storage of neurotransmitter molecules in synaptic vesicles and are essential for regulated synaptic release (1–4). The transport of monoamines (serotonin, dopamine, histamine, adrenaline, and noradrenaline) is carried out by the vesicular monoamine transporter (VMAT)3 family, which includes two isoforms: VMAT1 and VMAT2. The influx of charged substrates is coupled to the opposite movement of two protons (1, 2). In addition to their native substrates, VMATs interact with many clinically relevant drugs, including the psychostimulant 3,4-methylene-dioxymethamphetamine and the parkinsonian toxin 1-methyl-4-phenylpyridinium (MPP+) (2–6). Heterologous expression of VMATs protects mammalian and yeast cells against MPP+ toxicity by sequestering the toxin in vesicles, away from its primary site of action in mitochondria (7, 8).
Phylogenetic analysis reveals that VMAT proteins belong to the drug/H+ antiporters (DHA12) subfamily of the major facilitator superfamily (MFS), i.e. they are evolutionarily related to bacterial drug transporters and multidrug transporters (2, 7, 9, 10). Moreover, using the power of yeast genetics, we were able to demonstrate that three mutations are sufficient to transform rVMAT2 back into an MDT. These mutations cause rVMAT2 to lose the ability to transport neurotransmitters while still conferring resistance against the toxic substrates MPP+, ethidium, and acriflavine (8).
The above findings led us to study bacterial homologues to try to understand in greater detail the evolutionary aspects of the transformation of a bacterial MDT to a mammalian vesicular neurotransmitter transporter and to unveil mechanisms in a milieu more amenable to structural, biochemical, and biophysical studies. Crystal structures and subsequent mechanistic studies of prokaryotic homologues of mammalian neurotransporters have had a major impact on the field (11, 12). Notably, in the case of the plasma membrane transporters, they have served as structural paradigms for the interpretation of a wealth of biochemical and electrophysiological data available on their eukaryotic counterparts (12–14).
We identified over a dozen candidate transporters and chose to biochemically characterize one from Brevibacillus brevis (B. brevis monoamine transporter, BbMAT). It displays high sequence homology (25% identity and 52% similarity) to and shares some of the substrates (MPP+ and acriflavine) with rVMAT2, using exchange for multiple protons as the driving force. Because these bacterial multidrug transporters translocate toxic compounds and antibiotics away from the cytoplasm, they confer resistance against these toxins, and they have been associated with the phenomenon of resistance to multiple antibiotics. Antimicrobial resistance has been identified recently as one of the most serious health threats in a 2013 Centers for Disease Control and Prevention report (15). Therefore, study of these homologues should also aid in understanding basic mechanisms of antibiotic resistance.
Despite exciting recent progress, three-dimensional structures are available for only 13 MFS proteins, none of which have been shown to function as an H+-coupled antiporter (16). Here we present a homology model of BbMAT on the basis of the crystal structure of YajR, a putative proton-driven MFS transporter from Escherichia coli (17). As expected, the model contains 12 transmembrane (TM) helices, arranged in two domains of six TMs each, which are related by 2-fold pseudosymmetry with an axis that runs normal to the membrane and between the two halves. According to this model, there are four membrane-embedded carboxylic amino acids, but we show that only two, Asp-25 (TM1) and Glu-229 (TM7), are essential for transport. Given that the equivalent residues in rVMAT2 are crucial for transport activity, we suggest analogous roles for these residues in the two proteins.
EXPERIMENTAL PROCEDURES
Bioinformatics
A BLAST search using the rVMAT2 sequence as a query against the non-redundant database of the available microbial genomes was performed using the NCBI server. The rVMAT2 and BbMAT sequences were aligned using Clustal Omega (18). Conservation analysis was carried out using 500 homologues of BbMAT obtained using three iterations of PSI-BLAST with BbMAT as a query and aligned using Muscle (19).
Molecular Modeling of BbMAT
To build a molecular model of BbMAT, we searched for a template structure using the following search tools: PSI-BLAST v2.2.29 (20) on the non-redundant PDB database, I-Tasser v4.0 (21) on the basis of multiple-threading alignments obtained from the LOMETS (22) metaserver, and HHblits (from HH-suite v2.0) (23) using a non-redundant PDB sequence database clustered at 70% sequence identity. Databases were dated July 12, 2014. The crystal structure of a putative proton-driven E. coli transporter, YajR, in an outward-facing conformation (PDB code 3WDO) (17) exhibited the highest sequence identity (∼20%) and excellent coverage (∼90%), with E-values from PSI-BLAST and HHblits of 6 × 10−38 and 8.6 × 10−34, respectively. The structure of YajR was therefore selected as the most suitable template. In preparation for modeling, a preliminary pair-wise sequence alignment of YajR and BbMAT was constructed using the AlignMe PST mode on the AlignMe server v1.1 (24). Conserved MFS and DHA12 sequence motifs were aligned correctly (10, 17, 25). Alternative sequence matches were identified on the basis of a multiple sequence alignment obtained using the PRALINETM server (26) and on a multiple structure alignment generated using MAMMOTH-mult (27) of 36 MFS proteins. These proteins were identified with the search tools used to find the template. Adjustments to the AlignMe alignment were made in the following residues of the BbMAT sequence: 42–43, 113–116, 133–134, 199–206, 241–301, and 324–334. The adjustments were selected to optimize the agreement with PSIPRED v3.2 (28) secondary structure and TOPCONS (29) transmembrane predictions and to reduce the number of residues in generously allowed and disallowed regions of the Ramachandran plot as evaluated by PROCHECK v3.5.4 (30). For each adjustment, the effect on the per-residue and total ProQM (31) scores was tracked. In the final alignment, ∼17% of the residues are identical, and the similarity reaches ∼44%, as estimated over the whole sequence but excluding residues 1–6 in the N terminus and residues 389–405 in the C terminus. The four helices that contain the pore-lining residues investigated in this study, namely TM1, TM4, TM7, and TM11, exhibit a similarity with the template of 45, 54, 43, and 63%, respectively. The final alignment (Fig. 3B) was used to build a total of 300 homology models of BbMAT on the basis of YajR using MODELLER (version 9.13) (32). α Helix constraints were applied for residues 113–119 and 311–315 of TM4 and TM10, respectively. The best BbMAT model (Fig. 3A) was selected as that with the lowest Molpdf energy value of MODELLER (32) and the highest PROCHECK (30) and global ProQM (31) scores. The final model is of excellent quality according to PROCHECK (30), with zero residues in the generously allowed and disallowed regions of the Ramachandran plot. The ProQM score was 0.707, which compares well with that of the template structure (0.741). The model of BbMAT is available upon request.
FIGURE 3.

Structural model of BbMAT on the basis of the YajR transporter structure. A, the BbMAT model in an outward-facing conformation is represented as helical cartoons, viewed from the plane of the membrane with the periplasm at the top (left, with TM4 and part of TM11 omitted for clarity) and from the periplasm (right). The model predicts four negatively charged and two positively charged residues exposed to the central cavity, which could be involved in the transport activity of BbMAT. Side chains of these residues are shown as sticks. The figures were generated using PyMOL v1.7.05 (Schrödinger). B, sequence alignment between YajR and BbMAT used for modeling. The alignment was colored according to the chemical nature of the residues as described in Fig. 1. The helices assigned for the YajR structure using DSSP (67) and the PSIPRED prediction for helices in BbMAT are shown as blue bars above and below the sequences, respectively. Red rectangles mark the MFS and DHA12 motifs (10, 25), and black triangles indicate the residues that have been investigated in this study. C, sequence logo illustrating conservation of Asp-25, Asp-128, Glu-222, and Glu-229 among 500 homologues belonging to the MFS family. Multiple sequence alignment was performed using Muscle, and the logo was generated using Weblogo 3.3 (68). The residue colors are as follows: polar (Gly, Ser, Thr, Tyr, and Cys) in green, neutral (Gln and Asn) in purple, basic (Lys, Arg, and His) in blue, acidic (Asp and Glu) in red, and hydrophobic (Ala, Val, Leu, Ile, Pro, Trp, Phe, and Met) in black. Numbering is according to the sequence of BbMAT.
Plasmids and Strains
E. coli JM109 (33), BW25113 ΔemrE ΔmdfA (34), and C41 (35) were used throughout this work. The desired gene was cloned into pT7-7-Myc-His6 plasmid by PCR using genomic DNA from B. brevis as the template (DSM30, ATCC, catalog no. 8246). Primers were designed to overlap the ends of the gene and included restriction sites for NdeI and EcoRI. The plasmid was named pT7-7 BbMAT. For simplicity, the BbMAT-Myc-His6 protein is termed BbMAT. Site-directed mutagenesis was accomplished with the QuikChange® II site-directed mutagenesis kit (Stratagene), and the sequences of all constructs were verified by DNA sequencing. The sequence of BbMAT cloned and sequenced from the above strain differs from Uniprot entry C0ZB03 as follows: K42M, I59V, M75V, G102A, T137A, K194R, V196E, M260L, G270S, Y275F, K276Q, M283I, W294L, I308V, G321A, T394A, S395R, S397C, and I398T (the first letter denotes the amino acid in the Uniprot entry C0ZB03 sequence, the number is the position in the sequence, and the second letter is the amino acid as sequenced in our laboratory). The new, experimentally determined sequence was used in all our studies.
Drug Resistance Assays
Resistance to toxic compounds was assessed essentially as described in Ref. 36, 37, using E. coli JM109 cells transformed with the indicated plasmids. For experiments on solid medium, cells were diluted to an A600 of 0.2, and 4.5 μl of a series of 10-fold dilutions were spotted on LB plates containing 30 mm BisTris propane (pH 7) and 100 μg/ml ampicillin with or without the addition of the indicated concentration of the toxic compound. Growth was analyzed after overnight incubation at 37 °C. For experiments in liquid medium, cells were diluted to an A600 of 0.01 and grown for 12 h in LB medium containing 30 mm BisTris propane (pH 7) and 100 μg/ml ampicillin with or without the addition of an increasing concentration of the toxic compound. The cells were grown in a Synergy 2 Alpha microplate reader (BioTek) at 37 °C, and A600 was measured automatically by the device. IC50 was calculated from growth observed after 6 h.
Membrane Preparation, Solubilization, Purification, and Reconstitution
Overexpression, purification, and reconstitution of BbMAT were performed essentially as described for EmrE, with the following modifications (38). E. coli C41 cells bearing the pT7-7 plasmid with either wild-type BbMAT or different mutants were grown in LB medium to an A600 of 0.5. Protein expression was induced by adding isopropyl 1-thio-β-d-galactopyranoside to a final concentration of 300 μm, followed by a 10- to 12-h incubation at 20 °C. Cells were harvested by centrifugation and washed twice with lysis buffer (150 mm NaCl, 15 mm Tris (pH 7.5), and 250 mm sucrose) before further handling or storage at −70 °C. Cells were then resuspended in sodium-sucrose buffer containing 2.5 mm MgSO4, 15 μg/liter culture DNase, and 1 mm PMSF and lysed with a LV1 microfluidizer (Microfluidics) at 16,000 psi. Unbroken cells were sedimented by centrifugation, and the membrane fraction was collected by ultracentrifugation at 245,000 × g for 2 h at 4 °C and resuspended in sodium-sucrose buffer. The membranes were rapidly frozen in liquid nitrogen and stored at −70 °C.
Reconstitution was performed by solubilizing membranes containing ∼10 μg of His-tagged protein in sodium buffer (150 mm NaCl, 15 mm Tris (pH 7.5)) containing 0.5% lauryl maltose neopentyl glycol (Anatrace) and 0.5 mm PMSF. After 60 min at 4 °C, unsolubilized material was pelleted by centrifugation (208,000 × g for 15 min), imidazole was added to 10 mm, and the His-tagged protein was incubated with 10 μl Ni2+-nitrilotriacetic acid beads (Qiagen) for 1 h at 4 °C. Beads were then washed once with 10 volumes of sodium buffer with 0.005% lauryl maltose neopentyl glycol and 20 mm imidazole and washed three times with 10 volumes of the same solution but with 1% octyl-glucoside (Anatrace) instead of lauryl maltose neopentyl glycol. The protein was eluted with 500 μl of the octyl-glucoside buffer containing 450 mm imidazole and mixed with 5 mg of E. coli polar lipid extract (Avanti) suspended in 500 μl of sodium buffer and 1.2% octyl-glucoside. Eluted protein and phospholipids were sonicated together in a bath-type sonicator to clarity. To remove detergent, the mixture was dialyzed (dialysis membrane molecular weight cutoff 12–14 kDa) overnight at 4 °C against 300 volumes of the appropriate buffer (140 mm (NH4)2-SO4 with 15 mm Tris-SO4 (pH 7.4)). Dialysis buffer was exchanged with fresh buffer for an additional 2 h, and then the mixture was ultracentrifuged for 70 min at 200,000 × g. The liposome pellet was resuspended in 150 μl of the above buffer, divided into 25-μl aliquots, and stored at −70 °C. To quantify the amount of protein, liposomes were solubilized in a denaturing buffer (15 mm Tris-Cl (pH 7.5), 150 mm NaCl, 2% SDS, and 6 m urea) at room temperature for 20 min. The solubilized liposomes were then centrifuged (208,000 × g, 20 min) to remove the insoluble fraction. The supernatant was incubated at 4 °C with Ni2+-nitrilotriacetic acid-agarose beads for 1 h, and the beads were subsequently washed with denaturing buffer. Protein was eluted with sample buffer containing 450 mm imidazole, samples were separated by SDS-PAGE on 15% Laemmli gels (39), stained with Coomassie Brilliant Blue, and digitally analyzed with Image Gauge 3.46 Fujifilm software. To evaluate expression levels, membranes were solubilized, separated via SDS-PAGE, and analyzed as described above.
Uptake of [3H]MPP+
Liposomes were thawed and sonicated to clarity in a bath-type sonicator. Uptake was measured in reaction buffer containing 140 mm K2SO4, 15 mm Tricine, 15 mm glycine, and 5 mm MgCl2 (pH 9). Liposomes (1.5 μl) were diluted into 200 μl of reaction buffer with 50 nm valinomycin (unless indicated otherwise) and the indicated concentrations of the radiolabeled MPP+, usually 1 μm. Nonspecific accumulation of [3H]MPP+ was measured in the presence of the ionophore nigericin (15 μm) and subtracted from total transport. The reaction was terminated at the indicated time points by diluting the mixture in 2 ml of ice-cold buffer, filtering through 0.2-μm Supor®-200 filters (PALL), and washing with an additional 2 ml of ice-cold buffer. Radioactivity in the liposomes was measured via liquid scintillation. To assess inhibition by different compounds, liposomes were incubated in the presence of increasing concentrations of the indicated compound together with [3H]MPP+ (1 μm). Data were analyzed with Origin 8.6 (OriginLab). All experiments were performed in duplicate and repeated at least twice (the lowest R-squared value is 0.95).
Transport in Whole Cells
Transport of ethidium in whole cells can be followed conveniently because of the intense increase in ethidium fluorescence after binding with DNA. In this assay, the endogenous energy sources and the proton electrochemical gradient are depleted by treatment with an uncoupler. Under these conditions, ethidium transport is driven only by its chemical gradient. The proton electrochemical gradient can be restored quickly, after removal of the uncoupler, by the addition of glucose. The protocol used for this assay was essentially as described previously (40), with the following modifications. EmrE and MdfA are the primary endogenous ethidium transporters in E. coli, so we used the knockout strain BW251113 ΔemrEΔmdfA, which has very low background activity (34). Cells transformed with “empty” pT7-7 plasmid (negative control) or one coding for the either the wild-type BbMAT or the indicated mutants were grown at 37 °C in minimal medium A supplemented with 100 μg/ml ampicillin and 0.36% glucose. When the cultures reached an A600 of 0.5, cells were harvested by centrifugation, washed twice with minimal media A without glucose and ampicillin, and resuspended in the same medium to an A600 of 0.5. After adding 40 μm of the protonophoric uncoupler carbonyl cyanide m-chlorophenylhydrazone (Sigma-Aldrich, St. Louis, MO), the cultures were incubated for 1 h at 37 °C. Cells were harvested by centrifugation, quickly washed twice, and resuspended in carbonyl cyanide m-chlorophenylhydrazone-free medium without glucose. Transport was initiated by adding 5 μm ethidium and monitoring fluorescence for 60 min. At this point, 0.36% glucose was added, and fluorescence was monitored for an additional 15 min. Fluorescence was measured at 25 °C using a Synergy 2 Alpha microplate reader (BioTek) with excitation and emission wavelengths of 535 and 610 nm, respectively.
RESULTS
BbMAT Is a Bacterial Homologue of rVMAT2 That Confers Resistance to Aromatic Cations
To identify prokaryotic homologues of the mammalian VMAT2, we performed a BLAST search and found several significant hits, all of which were cloned from genomic DNA. After cursory assessment of the drug resistance phenotype via a disc-based diffusion assay and appraisal of expression levels, we chose to characterize in depth the protein cloned from B. brevis (BbMAT). Sequence analysis reveals that BbMAT contains all the MFS as well as the DHA12 motifs (Fig. 1A) and is one of the closest bacterial homologues of rVMAT2 with 25% identity and 52% similarity. To assess the function of BbMAT in vivo, JM109 E. coli cells were transformed with a pT7-7 plasmid bearing the gene encoding BbMAT. 4.5 μl of serial 10-fold dilutions of BbMAT-expressing E. coli cells were spotted on LB plates containing various drugs together with mock (pT7-7 vector)-transformed cells as the negative control (Fig. 1B). The drugs included aromatic cations that are substrates of VMAT and/or other multidrug transporters, namely ethidium, acriflavine, and MPP+. After incubating overnight, cells expressing BbMAT exhibited very robust growth on the three compounds, whereas the mock transformed cells did not. These results suggest that BbMAT acts in vivo to confer resistance against the abovementioned toxic compounds.
FIGURE 1.
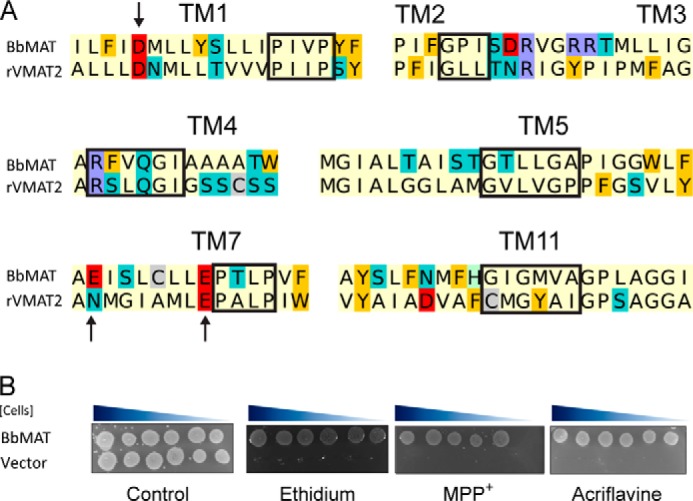
BbMAT is a multidrug resistance transporter homologous to rVMAT2. A, sequence alignment of key regions demonstrating the conservation between rVMAT2 and BbMAT. The alignment is colored according to the chemical properties of the residues: aliphatic (Ala, Gly, Ile, Leu, Met, Pro, and Val) in pale yellow, polar uncharged (Asn, Gln, Ser, and Thr) in teal, aromatic (Phe, Trp, and Tyr) in yellow/orange, acidic (Asp and Glu) in red, basic (Lys and Arg) in blue, His in pale green, and Cys in gray. Red rectangles mark the MFS and DHA12 motifs (10, 25). Arrows indicate residues studied in this work. B, BbMAT confers resistance against ethidium, MPP+, and acriflavine. E. coli JM109 cells harboring BbMAT or empty vector were assayed for drug resistance. 4.5 μl of serial 10-fold dilutions of cells were spotted on plates supplemented with ethidium (100 μg/ml), MPP+ (3 mm), or acriflavine (100 μg/ml). The plates are representative of at least three independent experiments.
BbMAT Is an H+-coupled Antiporter
To investigate its function in more detail, BbMAT was purified, reconstituted into proteoliposomes, and evaluated for its ability to transport [3H]MPP+(Fig. 2). For this purpose, we overexpressed His6-C terminus tagged BbMAT in E. coli C41 cells, purified with immobilized metal affinity chromatography (Fig. 2B, inset), and reconstituted into proteoliposomes. Because BbMAT displays significant similarity to VMAT2 and other members of the DHA12 family, we also tested whether the activity of BbMAT is coupled to a proton gradient. To generate a pH gradient, the proteoliposomes were loaded with (NH4)2SO4 and diluted into an ammonium-free buffer. A proton gradient (acidic inside) was generated upon dilution that drove [3H]MPP+ accumulation in liposomes, which increased over time (Fig. 2A). Accumulation was inhibited by nigericin, a K+/H+ ionophore that collapses the pH gradient (Fig. 2C). The transport process was also highly dependent on the addition of the K+ ionophore valinomycin (Fig. 2A), implying that the reaction catalyzed by BbMAT is electrogenic. The addition of valinomycin prevented development of the membrane potential generated by BbMAT and had, therefore, a strong stimulatory effect on the rate of the transport reaction. On the basis of the finding that the transport process is electrogenic, we tentatively propose that BbMAT exchanges substrates and protons with a stoichiometry greater than 1 (H+ > substrate).
FIGURE 2.

BbMAT catalyzes exchange of cationic substrates with protons. Proteoliposomes (1.5 μl) reconstituted with 9 ng of purified BbMAT were assayed for [3H]MPP+ uptake. A, transport time course with (■) and without (●) the K+ ionophore valinomycin. Also shown for reference (★) are liposomes reconstituted with no protein (mock). B, rates of transport in the presence of increasing concentrations of MPP+. The calculated Km value is 25 ± 2 μm, and Vmax is 5 ± 0.1 pmol/min/ng BbMAT protein. Inset, purified BbMAT analyzed by SDS-PAGE and visualized with Coomassie. C, inhibition of MPP+ transport by nigericin. D, inhibition of MPP+ transport by 10 μm of known substrates (ethidium, acriflavine, methyl viologen) or inhibitors (tetrabenazine, reserpine) of multidrug resistance transporters. Results presented are from duplicate experiments. Error bars indicate mean ± S.E. Experiments were repeated at least twice.
We next characterized the kinetic constants of BbMAT by measuring rates of [3H]MPP+ transport at various concentrations (Fig. 2B). We calculated a Michaelis-Menten constant (Km) value of 25 ± 2 μm and a maximum velocity (Vmax) of 5 ± 0.1 pmol/min/ng protein, the latter of which corresponds to an approximate turnover rate of 230 min−1. We then defined the substrate specificity of BbMAT by screening the ability of several putative substrates to inhibit [3H]MPP+ uptake. As predicted from the phenotype assay, MPP+ uptake into the liposomes was inhibited by acriflavine and ethidium (Fig. 2D), with IC50 values of 2.5 ± 0.1 μm and 3.0 ± 0.4 μm, respectively. Tetrabenazine and reserpine, two known potent inhibitors of VMAT2 (41, 42), as well as the divalent drug methyl viologen, all failed to inhibit BbMAT transport activity (Fig. 2D).
BbMAT Has Four Membrane-embedded Carboxylic Amino Acids, but Only Two Are Responsible for Drug Resistance Activity
As an initial step in discerning the three-dimensional architecture of the protein and in pinpointing functionally critical residues, we generated a homology model of BbMATon the basis of the crystallographic structure of YajR (PDB code 3WDO) in an outward-facing conformation (Fig. 3A). The sequence of YajR, a putative proton-driven MFS transporter from E. coli (17), exhibited the highest sequence identity (∼20%) and coverage (∼90%) among all MFS transporters with known structure, with PSI-BLAST and HHblits E-values of ∼10−36, and was therefore selected as the most suitable template (see “Experimental Procedures”). In both the initial AlignMe PST alignment and refined alignments, 17–18% of the residues are identical, and the conserved motifs of the DHA12 subfamily are positioned correctly (Fig. 3B). As expected, the model (Fig. 3A) corresponds to the familiar MFS fold (43), with two bundles of six TMs each (TM1–6 and TM7–12), related by 2-fold pseudosymmetry. The model predicts that six charged residues are located within the membrane-spanning helices: four carboxyl residues, Asp-25 (TM1), Asp-128 (TM4), Glu-222, and Glu-229 (TM7), one Arg (Arg-108), and one His (His-346), all of which are exposed to the aqueous cavity (Fig. 3A).
Residues similar to Arg-108 are also placed near the periplasmic ends of TM4 in many other MFS multidrug transporters, including MdfA, QacA/B, CaMDR1, TetA(B), rVMAT, Bcr, and Bmr (see Ref. 44 for a review). In many cases, the corresponding residue is essential, and mutating it is proposed to affect the conformational preference of the transporter. Unfortunately, all non-conservative mutants of this residue tested in several transporters were inactive, so it is difficult to elucidate its precise impact on the transport cycle. Nevertheless, its conservation suggests that Arg-108 plays an important role, at least in some MFS multidrug transporters (44). H346 is not conserved significantly, and its mutation to Ala did not affect BbMAT activity, as judged by the ability of the H346A mutant to confer resistance to acriflavine or ethidium (Fig. 4).
FIGURE 4.

Two carboxyl groups are important for conferring resistance by BbMAT. A, drug resistance of E. coli cells harboring the indicated BbMAT mutant. Serial dilutions of cells were spotted on plates supplemented with ethidium (100 μg/ml) or acriflavine (100 μg/ml). The plates are representative of at least three independent experiments. B, IC50 values for acriflavine were assessed by calculating the amount of drug required to inhibit cell growth by 50%, as described under “Experimental Procedures.” The experiments were repeated at least twice. C, expression of the different mutants assessed by Coomassie staining. ND, not determined.
In a sequence conservation analysis, we found that Asp-25 and Glu-229 are completely conserved among the 500 closest homologues from the MFS family (Fig. 3C). Glu-222 is frequently substituted with Asn, and Asp-128 is occasionally replaced with another polar or charged residue, either Ser or Glu. Strikingly, Asp-25 and Glu-229 are also the only two membrane-embedded carboxyls that are conserved between BbMAT and rVMAT2 (Fig. 1A).
Given the conservation of Asp-25 and Glu-229, we sought to discern their possible roles in the transport cycle. The abovementioned membrane-embedded carboxyls were mutated either alone or in combinations as described below. The mutant proteins were then tested for their ability to confer resistance to ethidium and acriflavine (Fig. 4). Resistance was measured in E. coli on plates containing concentrations of ethidium and acriflavine that do not support growth of the mock-transformed cells (Fig. 4A). IC50 values were estimated from the ability of cells to grow in liquid medium in the presence of a range of acriflavine concentrations (Fig. 4B). The expression of wild-type and mutants was assayed as shown in Fig. 4C. All of the replacements at positions 128 (D128E, D128N, and D128A) and 222 (E222D, E222Q, and E222A) were fully active and conferred drug resistance at levels similar to those of wild-type, indicating that neither of these residues is crucial for the functions tested. Furthermore, the combination D128N-E222Q, which results in a protein with only two carboxyls within the transmembrane domains, is also fully active, as judged by the phenotype assay (Fig. 4). Replacements at position 25, by contrast, had a drastic effect on protein activity. The mutant proteins D25N and D25A lost the ability to confer drug resistance to the bacteria, whereas a mutant bearing the conservative mutation D25E conferred resistance similar to that of wild-type protein. These results indicate that a carboxylic (and presumably negatively charged) residue at position 25 is essential for proper in vivo activity. In TM7, there are two carboxyls facing the aqueous cavity in the model: Glu-222 and Glu-229 (Fig. 3A). Although Glu-222 is not conserved and was not essential for activity, Glu-229 is conserved in the MFS family, and the equivalent residue in rVMAT2 is also essential for activity (25). Accordingly, we predicted that an ionizable residue at this position would be essential and that BbMAT would not be functional with only a polar residue at this position. Unexpectedly, E229Q supported weak but detectable drug resistance ability. For the E229Q mutant, the IC50 for acriflavine was 48 ± 4 μm, well below that of the wild-type protein (200 ± 9 μm) but significantly higher than that of the negative control (22 ± 1 μm). These results led us to postulate that the presence of the other carboxyl in TM7 (Glu-222) might partially compensate for the loss of a basic side chain at position 229. To test this hypothesis, we created a double mutant, E222Q/E229Q, and tested its ability to confer drug resistance. In support of our hypothesis, cells bearing the double replacement did not grow in the presence of ethidium or acriflavine (Fig. 4), and the IC50 value calculated for cells bearing E222Q/E229Q was 20 ± 4 μm, essentially the same as that of mock-transformed cells (22 ± 1 μm).
Glu-222 Can Partially Compensate for Glu-229
The growth phenotype described above suggests that Glu-222 can partially compensate for Glu-229. To examine this phenomenon in a more direct assay, we reconstituted the mutated proteins into proteoliposomes. As shown in Fig. 5, proteoliposomes reconstituted with either the D25N or E222Q/E229Q mutants catalyze weak, time-dependent uptake of [3H]MPP+, only marginally higher than liposomes reconstituted with no protein (Fig. 5, inset, mock). On the other hand, liposomes reconstituted with the E229Q mutation support substantial uptake, albeit at slower rates and to lower levels relative to the wild-type protein. These results validate the notion that Asp-25 and Glu-229 are key positions necessary for proper uptake activity but that Glu-222 can partially compensate for the requirement of a carboxylic amino acid in TM7 when Glu-229 is mutated.
FIGURE 5.

[3H]MPP+ uptake into proteoliposomes. [3H]MPP+ uptake was assayed as described in the legend for Fig. 2, with liposomes prepared from wild-type (■) and D25N-expressing (♦), E229Q-expressing (●), and E222Q/E229Q-expressing (▴) cells. Inset, for comparison, the results not corrected for protein content are shown for the wild type (■), E222Q/E229Q (▴), and mock liposomes (★).
Ethidium Transport in Whole Cells Provides an Assay Independent of the Proton Motive Force
Identification of the inactive (D25N, E222Q/E229Q), or partially active (E229Q) mutants emphasizes the role of the corresponding carboxyls in the overall transport process. Identification of the steps in which these residues are necessary requires an assay independent of the proton motive force. We were not able to reliably assay downhill transport of MPP+ in proteoliposomes because of the relatively high passive permeability of the substrate. We therefore adapted a whole-cell ethidium uptake system used previously with other transporters (40, 45). This assay enabled us to monitor both downhill passive transport driven by the ethidium gradient as well as uphill active efflux driven by the proton gradient. In this system, we used the proton uncoupler carbonyl cyanide m-chlorophenylhydrazone to collapse the proton electrochemical gradient and deplete the cells of endogenous energy sources. Monitoring downhill movement of ethidium into the cells is possible by measuring the increase in fluorescence resulting from association of intracellular ethidium with DNA (Fig. 6A). Because BbMAT conferred resistance to ethidium, it was expected that it would also transport ethidium. Ethidium uptake catalyzed by wild-type BbMAT in de-energized cells remained linear for about 15 min, slowed down, and reached a steady state after about 40 min (Fig. 6B, wt), whereas uptake by mock-transformed cells was much slower and reached 40% steady state levels only after 60 min (Fig. 6B, mock). The mutant D128N/E222Q has only two membrane-embedded carboxyls because the non-essential ones are replaced with non-ionizable polar residues. The rate of ethidium uptake catalyzed by D128N/E222Q is essentially the same as that of wild-type BbMAT (Fig. 6B). The mutants D25N and E222Q/E229Q were considered to be inactive (see above) because they did not confer resistance to the tested compounds and did not display detectable proton-driven [3H]MPP+ uptake. However, both mutants could still catalyze downhill transport of ethidium. Although the transport rate is slower than that of the wild-type protein, the value reached after 60 min is about the same (Fig. 6B). Remarkably, replacing only Glu-229 (E229Q), which had a significant but partial effect on the ability of BbMAT to confer resistance (Fig. 4) and to transport [3H]MPP+ (Fig. 5), instead had only a very minor effect on the ability of the protein to transport ethidium downhill (Fig. 6B). The lower rates observed with the mutant proteins suggest that the mutations may affect affinity for the substrate or a rate-limiting step in the catalytic cycle.
FIGURE 6.
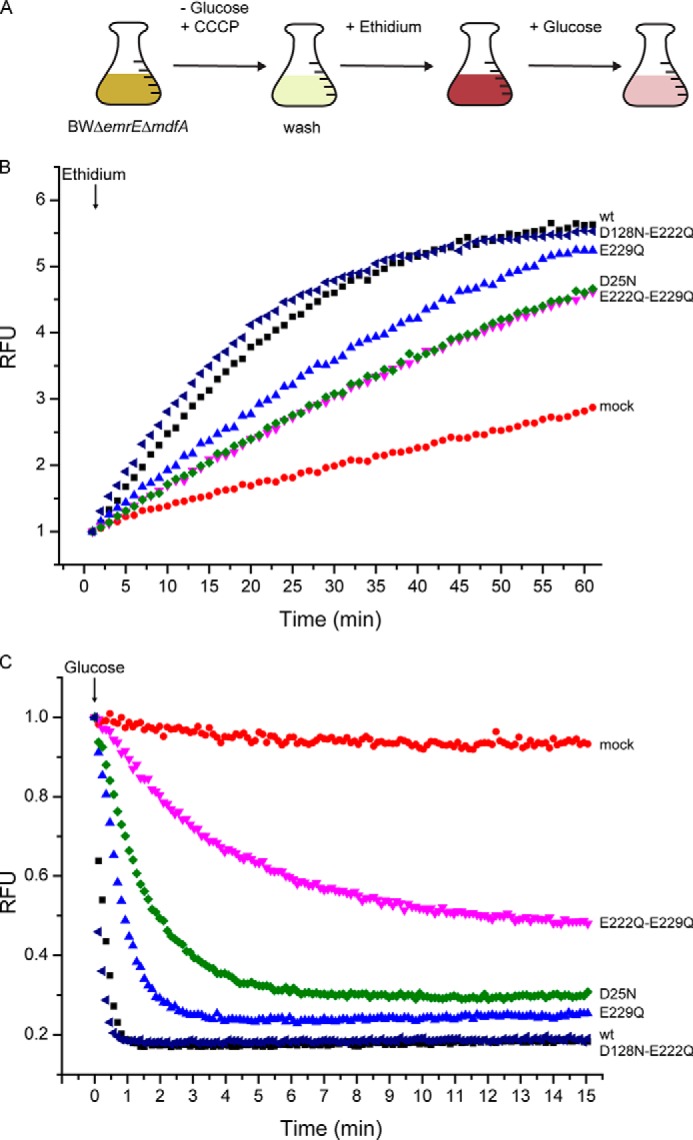
Asp-25 and Glu-229 are important for proton and substrate binding. A, schematic illustrating the protocol of the experiment. CCCP, carbonyl cyanide m-chlorophenylhydrazone. B and C, ethidium transport activity of BbMAT mutants measured using ethidium fluorescence and shown in relative fluorescence units (RFU). E. coli BWΔemrEΔmdfA cells expressing the wild type (black ■), D128N/E222Q (dark blue ◀), E229Q (light blue ▴), E222Q/E229Q (pink ▾), and D25N (green ▴) BbMAT or mock-transformed (red ●) were assayed for downhill passive ethidium uptake (B) and uphill efflux initiated by the addition of glucose (C). In C, the initial value of fluorescence for each strain was taken as 1.
When glucose was added to regenerate the proton gradient in wild-type BbMAT-expressing cells, we observed a rapid decrease in fluorescence as the ethidium was actively removed from the cells, coupled to the downhill movement of protons (Fig. 6, A and C). As judged by the drop in fluorescence intensity, more than 80% of the ethidium was removed from the cell in less than a minute. Furthermore, as expected from the experiments described above, the behavior of the double mutant D128N/E222Q was practically indistinguishable from that of the wild type (Fig. 6C). Remarkably, mock-transformed cells showed only a minor decrease in fluorescence, providing an extremely wide range for assessing proton-coupled ethidium efflux in the various mutants. In cells bearing the D25N or E222Q/E229Q mutants, the rate of decrease in efflux was substantially slower than that observed with wild-type protein, and the E222Q/E229Q mutant did not reach a new equilibrium, even 15 min after addition of glucose. The equilibrium level reflects the ability of the transporter to remove ethidium in an energy-dependent manner and, thus, generate a gradient of ethidium. The maximal gradient that a transporter is capable of generating is essentially determined by the H+/substrate stoichiometry. We speculate that the lower level of ethidium efflux achieved by D25N and E222Q/E229Q is the result of a different H+/substrate stoichiometry, implying that these mutants only exchange one proton per substrate molecule. The lower rates, more significant in the E222Q/E229Q mutant, could also result from effects on ethidium affinity alone, as suggested from the downhill experiments and/or from a lower effective driving force if the stoichiometry is only 1H+/substrate. Taken together, these data implicate Asp-25 and Glu-229 in proton transport and, possibly, also in substrate binding.
The single mutant E229Q displayed a marginal resistance phenotype (Fig. 4) with impaired but detectable [3H]MPP+ uptake (Fig. 5). In the efflux assay, the behavior of E229Q was also intermediate (Fig. 6C). Specifically, the active removal of ethidium was slower than that of the wild type but faster than that of the E222Q/E229Q mutant. Moreover, the steady state level was intermediate between those of the wild type and E222Q/E229Q. These results add further support to the proposal that Glu-222 can partially compensate for Glu-229.
DISCUSSION
The extraordinary advance in our understanding of the mechanism of ion-coupled transporters is largely due to the increasing availability of high-resolution structures. One reason behind this progress is the use of bacterial homologues as research paradigms because they are generally more stable and, therefore, more amenable to structural and biochemical analysis. For example, the use of the bacterial homologues GltPh and LeuT has propelled studies of the plasma membrane neurotransmitter transporter families SLC1 and SLC6, respectively (11–14).
In the MFS family, the bacterial transporters XylE, GlcPSe, and PepTst/so have been used to obtain structural and functional data for their mammalian counterparts, the glucose (GLUT) and oligopeptide transporters (PepT1 and 2), respectively (46–50). In the case of the vesicular monoamine transporter 2, the use of a bacterial homologue is particularly attractive because VMAT2 is evolutionarily and functionally related to bacterial multidrug transporters (8, 10). Moreover, the current surge in available sequences has allowed the identification of a number of closely related homologues. After an initial screening of more than a dozen homologues on the basis of phenotype and expression, we chose to study in detail a multidrug transporter from the bacterium B. brevis (BbMAT). BbMAT shares 25% identity and 52% similarity with rVMAT2 and contains all of the conserved MFS and DHA12 motifs (Fig. 1A). Furthermore, BbMAT shares some of the substrates (MPP+ and acriflavine) of rVMAT2 and appears to catalyze their transport in exchange for at least two H+, making BbMAT a reasonable investigative model.
The majority of structural and functional data available for MFS transporters comes from symporters (16, 51–53). At present, there are only two structures available of putative MFS H+-coupled antiporters, YajR and EmrD, but neither has been functionally characterized (17, 54, 55). Nevertheless, in many cases, analysis of the available MFS structures has revealed a significant role for residues in TM1 and/or TM7 in substrate binding and ion coupling (16). For MFS symporters, there is ample evidence that carboxyl groups participate in proton binding and that protons must be bound prior to binding of a substrate (e.g., see Refs. 48, 49, 56). For antiporters, a competition mechanism is suggested in which the carboxylate alternately binds the proton or substrate (57).
Because neither the transport mechanism nor the substrate and proton binding sites have been fully characterized for VMAT2, we decided to study the proton and binding sites in BbMAT with the goal of increasing our mechanistic understanding of VMAT2. Among the four carboxyls identified in the transmembrane helices of BbMAT, two (Asp-25 and Glu-229) are absolutely conserved in rVMAT2 (Figs. 1 and 7). Interestingly, these residues are also highly conserved among other MFS transporters and are the only two carboxylates found to be essential for drug resistance. A mutant protein in which the two other membrane-embedded carboxyls (Asp-128 and Glu-222) are replaced is fully active. By contrast, replacing either Asp-25 or Glu-229 has a substantial effect on the ability of the mutant transporters to confer resistance (Fig. 4) and to transport both MPP+ (Fig. 5) and ethidium (Fig. 6).
FIGURE 7.
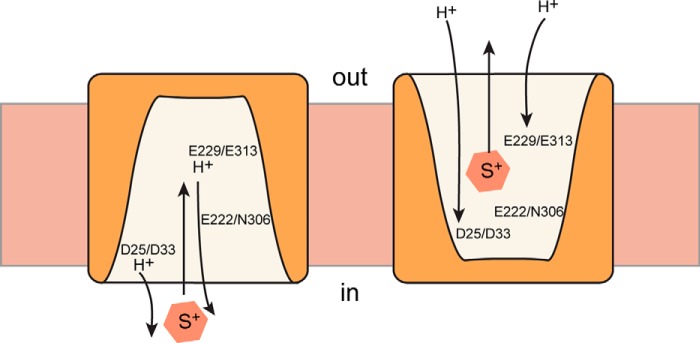
A simplified view of the catalytic cycle of BbMAT and rVMAT2. For the sake of simplicity, the schematic includes only the conformations where the protein faces the inside (in) or the outside (out), with two protons moving to the inside of the cell in the case of BbMAT or to the vesicular lumen in the case of rVMAT2. In the schematic, the conserved amino acids in BbMAT and the corresponding ones in rVM AT2 are shown, and their hypothetical role in substrate and proton binding is presented.
The corresponding positions in rVMAT2 (Asp-33 and Glu-313) are also important for transport activity, although their precise role is unclear. Specifically, when Asp-33 is mutated to Asn, practically no transport activity could be detected, but ΔμH+-dependent reserpine binding was unaffected (58). This indicates that the mutant protein is still coupled to at least one proton (59). Moreover, serotonin failed to inhibit reserpine binding to the mutated transporter, suggesting a role for Asp-33 in substrate binding as well (58).
Compared with Glu-229 in BbMAT, mutating Glu-313 in TM7 had a more drastic effect on rVMAT2 function because the conservative replacement E313D led to a dramatic decrease in transport ability, whereas a less conservative mutation (E313Q) abolished it completely (25). It is therefore surprising to observe that the corresponding residue in BbMAT (Glu-229) can be mutated to Gln without the same catastrophic effect. Given that VMAT2, unlike BbMAT, lacks a second Glu in TM7, the difference leads us conclude that Glu-222 can partially compensate for Glu-229. Experiments in whole cells and proteoliposomes clearly support the hypothesis that one carboxyl in TM7 is sufficient for partial activity (Figs. 4–6).
Our experiments demonstrate that Asp-25 and Glu-229 are both essential for proton-driven transport (Fig. 5) but that BbMAT can still catalyze downhill ethidium flux in their absence (Fig. 6). As discussed above, the lower rates of ethidium uptake catalyzed by mutants D25N and E222Q/E229Q (Fig. 6) might reflect modified affinities for the substrate, a lower driving force as a result of the implied change in stoichiometry, or an effect of the mutation on one or more of the conformational changes necessary for the transport cycle.
When assayed for their ability to remove ethidium coupled to ΔμH+, both D25N and E222Q/E229Q exhibited a much slower rate of ethidium removal and an inability to extrude ethidium to the same extent as the wild-type protein, as reflected by the fact that they reach very different steady-state levels. The effects on rates, as mentioned before, and the steady state shown both here and in the [3H]MPP+ transport experiments, can be explained by a change in stoichiometry and, therefore, a diminished driving force acting on the mutants. The [3H]MPP+ transport experiments are more quantitative because they directly measure the amount of substrate in the liposome. However, they are more limited in their dynamic range because the 90% decrease in MPP+ uptake expected from a change in stoichiometry of 2H+/drug to 1H+/drug results in transport levels that are only marginally higher than the background level (transport/absorption in the presence of nigericin). The glucose-induced efflux of ethidium, by contrast, provides an excellent dynamic range, but the results are mainly qualitative because the ethidium levels are measured indirectly. On the basis of these measurements, we suggest that both Asp-25 and Glu-229 are involved in proton binding and that, when one is neutralized, BbMAT is coupled to one proton only.
Interestingly, for MdfA, a proton-coupled MDT from the MFS family, a phenomenon similar to that of Glu-222 and Glu-229 was reported for two carboxyls (Glu-26 and Asp-34) located two helical turns apart in TM1, the symmetry-equivalent helix to TM7. That is, each charged residue can compensate for the other (60). It has been suggested that Glu-26 in MdfA is important for substrate but not proton binding (61). MdfA has a 1:1 H+ to substrate stoichiometry, and D34 is suggested to be the proton-binding site, indicating an indirect competition mechanism in which protonation of Glu-6 affects the protonation state of Asp-34. The situation in TM7 of BbMAT may be similar, but, because an additional proton is required, carboxyls may be needed in both TM1 and TM7.
In two other well characterized but more distant MFS multidrug antiporters from Gram-positive bacteria, LmrP and QacA, membrane-embedded carboxylic acids in various TMs have been shown to play a role in substrate binding and proton translocation, although the location of the carboxylic amino acids is not conserved (62–66). Among the well characterized MFS antiporters, the highest similarity and, presumably, mechanism is indeed observed between BbMAT and rVMAT2 (Fig. 7). The tentatively proposed stoichiometry of two protons per substrate, the common location of essential residues, and the overlap in some substrates together support the conclusion that BbMAT is an excellent experimental paradigm for the study of its VMAT mammalian counterparts as well as the bacterial multidrug transporters.
This work was supported, in whole or in part, by National Institutes of Health Grants NS16708 (to S. S.) and R21MH098180 (to S. K. S.) and by the Division of Intramural Research of the NINDS, National Institutes of Health (to L. R. F. and A. V. J.). This work was also supported by the Israel-USA Binational Science Foundation (to S. S. and S. K. S.).
- VMAT
- vesicular monoamine transporter
- MPP+
- 1-methyl-4-phenylpyridinium
- MFS
- major facilitator superfamily
- BbMAT
- B. brevis monoamine transporter
- MDT
- multidrug transporter
- TM
- transmembrane
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
REFERENCES
- 1. Eiden L. E. (2000) The vesicular neurotransmitter transporters: current perspectives and future prospects. FASEB J. 14, 2396–2400 [DOI] [PubMed] [Google Scholar]
- 2. Schuldiner S., Shirvan A., Linial M. (1995) Vesicular neurotransmitter transporters: from bacteria to humans. Physiol. Rev. 75, 369–392 [DOI] [PubMed] [Google Scholar]
- 3. Chaudhry F. A., Edwards R. H., Fonnum F. (2008) Vesicular neurotransmitter transporters as targets for endogenous and exogenous toxic substances. Annu. Rev. Pharmacol. Toxicol. 48, 277–301 [DOI] [PubMed] [Google Scholar]
- 4. Edwards R. (1992) The transport of neurotransmitters into synaptic vesicles. Curr. Opin. Neurobiol. 2, 586–594 [DOI] [PubMed] [Google Scholar]
- 5. Yelin R., Schuldiner S. (1995) The pharmacological profile of the vesicular monoamine transporter resembles that of multidrug transporters. FEBS Lett. 377, 201–207 [DOI] [PubMed] [Google Scholar]
- 6. Darchen F., Scherman D., Desnos C., Henry J.-P. (1988) Characteristics of the transport of the quaternary ammonium 1-methyl-4-phenylpyridinium by chromaffin granules. Biochem. Pharmacol. 37, 4381–4387 [DOI] [PubMed] [Google Scholar]
- 7. Liu Y., Peter D., Roghani A., Schuldiner S., Privé G. G., Eisenberg D., Brecha N., Edwards R. H. (1992) A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell 70, 539–551 [DOI] [PubMed] [Google Scholar]
- 8. Gros Y., Schuldiner S. (2010) Directed evolution reveals hidden properties of VMAT, a neurotransmitter transporter. J. Biol. Chem. 285, 5076–5084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vardy E., Arkin I. T., Gottschalk K. E., Kaback H. R., Schuldiner S. (2004) Structural conservation in the major facilitator superfamily as revealed by comparative modeling. Protein Sci. 13, 1832–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vardy E., Steiner-Mordoch S., Schuldiner S. (2005) Characterization of bacterial drug antiporters homologous to mammalian neurotransmitter transporters. J. Bacteriol. 187, 7518–7525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh S.K. (2008) LeuT: a prokaryotic stepping stone on the way to a eukaryotic neurotransmitter transporter structure. Channels 2, 380–389 [DOI] [PubMed] [Google Scholar]
- 12. Gouaux E. (2009) Review: the molecular logic of sodium-coupled neurotransmitter transporters. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Forrest L. R., Rudnick G. (2009) The rocking bundle: a mechanism for ion-coupled solute flux by symmetrical transporters. Physiology 24, 377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boudker O., Verdon G. (2010) Structural perspectives on secondary active transporters. Trends Pharmacol Sci. 31, 418–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Center for Disease Control (2013) Annual Report of the Centers for Disease Control. USA Center for Disease Control and Prevention, Atlanta, GA [Google Scholar]
- 16. Yan N. (2013) Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 38, 151–159 [DOI] [PubMed] [Google Scholar]
- 17. Jiang D., Zhao Y., Wang X., Fan J., Heng J., Liu X., Feng W., Kang X., Huang B., Liu J., Zhang X. C. (2013) Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc. Natl. Acad. Sci. U.S.A. 110, 14664–14669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goujon M., McWilliam H., Li W., Valentin F., Squizzato S., Paern J., Lopez R. (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu S., Zhang Y. (2007) LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375–3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Remmert M., Biegert A., Hauser A., Söding J. (2012) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9, 173–175 [DOI] [PubMed] [Google Scholar]
- 24. Stamm M., Staritzbichler R., Khafizov K., Forrest L. R. (2014) AlignMe: a membrane protein sequence alignment web server. Nucleic Acids Res. 42, W246–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yaffe D., Radestock S., Shuster Y., Forrest L. R., Schuldiner S. (2013) Identification of molecular hinge points mediating alternating access in the vesicular monoamine transporter VMAT2. Proc. Natl. Acad. Sci. U.S.A. 110, E1332–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Simossis V. A., Heringa J. (2005) PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 33, W289–W294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lupyan D., Leo-Macias A., Ortiz A. R. (2005) A new progressive-iterative algorithm for multiple structure alignment. Bioinformatics 21, 3255–3263 [DOI] [PubMed] [Google Scholar]
- 28. Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 29. Bernsel A., Viklund H., Hennerdal A., Elofsson A. (2009) TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res. 37, W465–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laskowski R. A., Macarthur M. W., Moss D. S., Thornton J. M. (1993) Procheck: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 31. Ray A., Lindahl E., Wallner B. (2010) Model quality assessment for membrane proteins. Bioinformatics 26, 3067–3074 [DOI] [PubMed] [Google Scholar]
- 32. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2007) Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 50, 2.9.1–2.9.31. [DOI] [PubMed] [Google Scholar]
- 33. Yanisch-Perron C., Vieira J., Messing J. (1985) Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33, 103–119 [DOI] [PubMed] [Google Scholar]
- 34. Tal N., Schuldiner S. (2009) A coordinated network of transporters with overlapping specificities provides a robust survival strategy. Proc. Natl. Acad. Sci. U.S.A. 106, 9051–9056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miroux B., Walker J. E. (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260, 289–298 [DOI] [PubMed] [Google Scholar]
- 36. Rotem D., Schuldiner S. (2004) EmrE, a multidrug transporter from Escherichia coli, transports monovalent and divalent substrates with the same stoichiometry. J. Biol. Chem. 279, 48787–48793 [DOI] [PubMed] [Google Scholar]
- 37. Elbaz Y., Salomon T., Schuldiner S. (2008) Identification of a glycine motif required for packing in EmrE, a multidrug transporter from Escherichia coli. J. Biol. Chem. 283, 12276–12283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Soskine M., Adam Y., Schuldiner S. (2004) Direct evidence for substrate-induced proton release in detergent-solubilized EmrE, a multidrug transporter. J. Biol. Chem. 279, 9951–9955 [DOI] [PubMed] [Google Scholar]
- 39. Laemmli U. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 40. Yerushalmi H., Lebendiker M., Schuldiner S. (1995) EmrE, an Escherichia coli 12-kDa multidrug transporter, exchanges toxic cations and H+ and is soluble in organic solvents. J. Biol. Chem. 270, 6856–6863 [DOI] [PubMed] [Google Scholar]
- 41. Scherman D., Jaudon P., Henry J. (1983) Characterization of the monoamine carrier of chromaffin granule membrane by binding of [2–3H]dihydrotetrabenazine. Proc. Natl. Acad. Sci. U.S.A. 80, 584–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scherman D., Henry J. (1984) Reserpine binding to bovine chromaffin granule membranes: characterization and comparison with dihydrotetrabenazine binding. Mol. Pharmacol. 25, 113–122 [PubMed] [Google Scholar]
- 43. Radestock S., Forrest L. R. (2011) The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J. Mol. Biol. 407, 698–715 [DOI] [PubMed] [Google Scholar]
- 44. Fluman N., Bibi E. (2009) Bacterial multidrug transport through the lens of the major facilitator superfamily. Biochim. Biophys. Acta 1794, 738–747 [DOI] [PubMed] [Google Scholar]
- 45. Lambert B., Le Pecq J. B. (1984) Effect of mutation, electric membrane potential, and metabolic inhibitors on the accessibility of nucleic acids to ethidium bromide in Escherichia coli cells. Biochemistry 23, 166–176 [DOI] [PubMed] [Google Scholar]
- 46. Sun L., Zeng X., Yan C., Sun X., Gong X., Rao Y., Yan N. (2012) Crystal structure of a bacterial homologue of glucose transporters GLUT1–4. Nature 490, 361–366 [DOI] [PubMed] [Google Scholar]
- 47. Newstead S., Drew D., Cameron A. D., Postis V. L., Xia X., Fowler P. W., Ingram J. C., Carpenter E. P., Sansom M. S., McPherson M. J., Baldwin S. A., Iwata S. (2011) Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J. 30, 417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Solcan N., Kwok J., Fowler P. W., Cameron A. D., Drew D., Iwata S., Newstead S. (2012) Alternating access mechanism in the POT family of oligopeptide transporters. EMBO J. 31, 3411–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Doki S., Kato H. E., Solcan N., Iwaki M., Koyama M., Hattori M., Iwase N., Tsukazaki T., Sugita Y., Kandori H., Newstead S., Ishitani R., Nureki O. (2013) Structural basis for dynamic mechanism of proton-coupled symport by the peptide transporter POT. Proc. Natl. Acad. Sci. U.S.A. 110, 11343–11348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Iancu C. V., Zamoon J., Woo S. B., Aleshin A., Choe J. Y. (2013) Crystal structure of a glucose/H+ symporter and its mechanism of action. Proc. Natl. Acad. Sci. U.S.A. 110, 17862–17867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kumar H., Kasho V., Smirnova I., Finer-Moore J. S., Kaback H. R., Stroud R. M. (2014) Structure of sugar-bound LacY. Proc. Natl. Acad. Sci. U.S.A. 111, 1784–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kaback H. R., Smirnova I., Kasho V., Nie Y., Zhou Y. (2011) The alternating access transport mechanism in LacY. J. Membr. Biol. 239, 85–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Forrest L. R., Krämer R., Ziegler C. (2011) The structural basis of secondary active transport mechanisms. Biochim. Biophys. Acta 1807, 167–188 [DOI] [PubMed] [Google Scholar]
- 54. Yin Y., He X., Szewczyk P., Nguyen T., Chang G. (2006) Structure of the multidrug transporter EmrD from Escherichia coli. Science 312, 741–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Steed P. R., Zou P., Trone K. E., Mchaourab H. S. (2013) Structure and pH-induced structural rearrangements of the putative multidrug efflux pump EmrD in liposomes probed by site-directed spin labeling. Biochemistry 52, 7964–7974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smirnova I., Kasho V., Sugihara J., Vázquez-Ibar J. L., Kaback H. R. (2012) Role of protons in sugar binding to LacY. Proc. Natl. Acad. Sci. U.S.A. 109, 16835–16840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schuldiner S. (2014) Competition as a way of life for H+-coupled antiporters. J. Mol. Biol. 426, 2539–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Merickel A., Rosandich P., Peter D., Edwards R. (1995) Identification of residues involved in substrate recognition by a vesicular monoamine transporter. J. Biol. Chem. 270, 25798–25804 [DOI] [PubMed] [Google Scholar]
- 59. Rudnick G., Steiner-Mordoch S. S., Fishkes H., Stern-Bach Y., Schuldiner S. (1990) Energetics of reserpine binding and occlusion by the chromaffin granule biogenic amine transporter. Biochemistry 29, 603–608 [DOI] [PubMed] [Google Scholar]
- 60. Sigal N., Fluman N., Siemion S., Bibi E. (2009) The secondary multidrug/proton antiporter MdfA tolerates displacements of an essential negatively charged side chain. J. Biol. Chem. 284, 6966–6971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fluman N., Ryan C. M., Whitelegge J. P., Bibi E. (2012) Dissection of mechanistic principles of a secondary multidrug efflux protein. Mol. Cell 47, 777–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Masureel M., Martens C., Stein R. A., Mishra S., Ruysschaert J. M., Mchaourab H. S., Govaerts C. (2014) Protonation drives the conformational switch in the multidrug transporter LmrP. Nat. Chem. Biol. 10, 149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mazurkiewicz P., Konings W. N., Poelarends G. J. (2002) Acidic residues in the lactococcal multidrug efflux pump LmrP play critical roles in transport of lipophilic cationic compounds. J. Biol. Chem. 277, 26081–26088 [DOI] [PubMed] [Google Scholar]
- 64. Mitchell B. A., Paulsen I. T., Brown M. H., Skurray R. A. (1999) Bioenergetics of the staphylococcal multidrug export protein QacA: identification of distinct binding sites for monovalent and divalent cations. J. Biol. Chem. 274, 3541–3548 [DOI] [PubMed] [Google Scholar]
- 65. Xu Z., O'Rourke B. A., Skurray R. A., Brown M. H. (2006) Role of transmembrane segment 10 in efflux mediated by the staphylococcal multidrug transport protein QacA. J. Biol. Chem. 281, 792–799 [DOI] [PubMed] [Google Scholar]
- 66. Schaedler T. A., van Veen H. W. (2010) A flexible cation binding site in the multidrug major facilitator superfamily transporter LmrP is associated with variable proton coupling. FASEB J. 24, 3653–3661 [DOI] [PubMed] [Google Scholar]
- 67. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 68. Crooks G. E., Hon G., Chandonia J. M., Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]