

Background: Extracellular loop 2 of GAT-1 contains two conserved cysteines.
Results: Transport by an extracellular loop 4 cysteine mutant is inhibited under oxidizing conditions.
Conclusion: Transport is accompanied by proximity changes between extracellular loops 2 and 4.
Significance: This work provides new insights into the molecular mechanism of neurotransmitter transport.
Keywords: Conformational Change, Cysteine-mediated Cross-linking, Disulfide, Neurotransmitter Transport, Sulfhydryl, GABA
Abstract
The sodium- and chloride-coupled GABA transporter GAT-1 is a member of the neurotransmitter:sodium:symporters, which are crucial for synaptic transmission. Structural work on the bacterial homologue LeuT suggests that extracellular loop 4 closes the extracellular solvent pathway when the transporter becomes inward-facing. To test whether this model can be extrapolated to GAT-1, cysteine residues were introduced at positions 359 and 448 of extracellular loop 4 and transmembrane helix 10, respectively. Treatment of HeLa cells, expressing the double cysteine mutant S359C/K448C with the oxidizing reagent copper(II)(1,10-phenantroline)3, resulted in a significant inhibition of [3H]GABA transport. However, transport by the single cysteine mutant S359C was also inhibited by the oxidant, whereas its activity was almost 4-fold stimulated by dithiothreitol. Both effects were attenuated when the conserved cysteine residues, Cys-164 and/or Cys-173, were replaced by serine. These cysteines are located in extracellular loop 2, the role of which in the structure and function of the eukaryotic neurotransmitter:sodium:symporters remains unknown. The inhibition of transport of S359C by the oxidant was markedly reduced under conditions expected to increase the proportion of inward-facing transporters, whereas the reactivity of the mutants to a membrane-impermeant sulfhydryl reagent was not conformationally sensitive. Our data suggest that extracellular loops 2 and 4 come into close proximity to each other in the outward-facing conformation of GAT-1.
Introduction
Sodium-coupled neurotransmitter transporters remove their substrates from the synaptic cleft and thereby enable efficient neurotransmission. With the exception of the transporters for glutamate, the transporters for other neurotransmitters, such as GABA, serotonin, dopamine, norepinephrine, and glycine, are sodium- and chloride-dependent and belong to the neurotransmitter:sodium:symporter (NSS)2 family (reviewed in Refs. 1 and 2). These eukaryotic NSS transporters mediate electrogenic sodium- and chloride-coupled neurotransmitter transport. In the case of the GABA transporter GAT-1, the stoichiometry of the process is 2:1:1 (Na+/Cl−/GABA) (3–6).
High-resolution structures of the bacterial NSS homologue LeuT (7, 8) have been described. LeuT consists of 12 transmembrane domains (TMs) with TM1 to -5 related to TM6 to -10 by a pseudo-2-fold axis in the membrane plane, and its binding pocket is located at the interface of these two domains (7). In the outward-occluded conformation, the binding pocket is separated from the cytoplasm by ∼20 Å of ordered protein. Although LeuT is an excellent model for the NSS neurotransmitter transporters (9–12), extracellular loop 2 (EL2) of the homologue is considerably shorter than that of their eukaryotic counterparts (7). Very recently, a crystal structure of the Drosophila melanogaster dopamine transporter has been reported (13). Because it was necessary to delete 43 residues from EL2 (13), the role of this loop in the structure and function of the eukaryotic NSS transporters remains unknown. EL2 of the eukaryotic transporters, but not of LeuT, contains two conserved cysteine residues capable of forming a disulfide bond (14, 15). Such a disulfide bond was also seen in the D. melanogaster dopamine transporter (13).
Comparison of the different LeuT structures suggest that EL4, which connects TM7 to TM8, may play a key role in the occlusion of the binding pocket from the extracellular medium during transition of the outward- to inward-facing conformation. Upon this transition, one of the EL4 residues, Gly-318, comes close to Asp-401 located in the extracellular part of TM10 (Fig. 1). To test if this prediction can be extrapolated to GAT-1, cysteine residues were introduced into GAT-1 at the corresponding positions, namely 359 from EL4 and 448 from TM10. Treatment of the double cysteine mutant with copper(II)(1,10-phenantroline)3 (CuPh) resulted in inhibition of GABA transport. However, inhibition of transport was also seen without a cysteine at position 448. In this study, we have addressed the question of which of the endogenous cysteine residues of GAT-1 can potentially form a disulfide bond with the cysteine introduced at position 359 under oxidizing conditions. Our results indicate that under conditions favoring outward-facing conformations of GAT-1, each of the two cysteine residues from EL2 is in proximity to the cysteine introduced at position 359.
FIGURE 1.
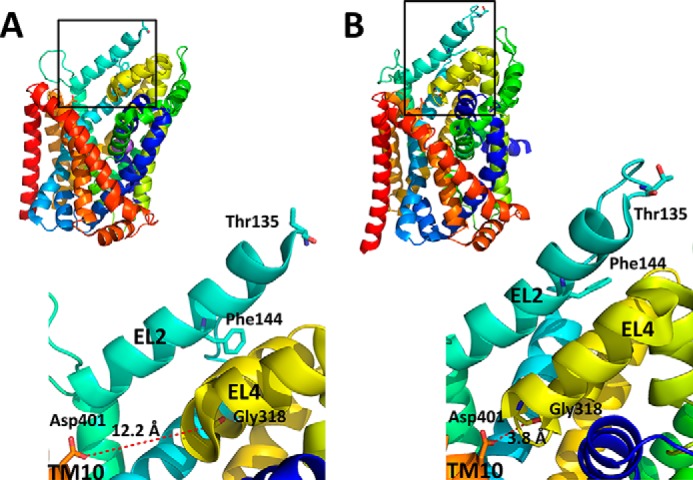
Distances between Gly-318 and Asp-401 in outward- and inward-open LeuT structures. LeuT structures in the outward-open (A) and inward-open LeuT (B) conformations (Protein Data Bank entries 3TT1 and 3TT3, respectively), show the distances (Å) between Gly-318 and Asp-401 in the enlarged black boxes. Gly-318 from EL4 and Asp-401 from TM10 correspond to Ser-359 and Lys-448 of GAT-1, respectively. The LeuT EL2 counterparts (Thr-135 and Phe-144) of Cys-164 and Cys-173 of GAT-1, derived from the alignments shown in supplemental Fig. 8 from Ref. 13, are also indicated in the enlargements. The figure was prepared using the PyMOL Molecular Graphics System, version 1.5.0.4 (Schrödinger, LLC, New York).
EXPERIMENTAL PROCEDURES
Generation and Subcloning of Mutants
Mutations were made by site-directed mutagenesis of the wild type (WT) GAT-1 in the vector pBluescript SK(−) (Stratagene) using single-stranded uracil-containing DNA as described previously (16, 17). Briefly, the GAT-1-containing plasmid was used to transform Escherichia coli CJ236 (dut−, ung−). From one of the transformants, single-stranded uracil-containing DNA was isolated upon growth in uridine-containing medium, according to the standard protocol from Stratagene, using helper phage R408. This yields the sense strand, and consequently mutagenic primers were designed to be antisense. The mutants were subcloned into C74A-GAT-1 in the vector pBluescript SK(−) and in the oocyte expression vector pOG1, using unique restriction enzymes. The mutations were verified by sequencing the entire coding region of the cDNA.
Cell Growth and Expression
HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 200 units/ml penicillin, 200 μg/ml streptomycin, and 2 mm glutamine. Infection with recombinant vaccinia/T7 virus vTF7-3 (18) and subsequent transfection with plasmid DNA, as well as GABA transport, were done as described previously (19). In all of the experiments with the HeLa cells, the expression vector was pBluescript SK(−), whereas it was pOG1 for the oocyte expression.
Inhibition Studies with the Impermeant Sulfhydryl Reagent MTSET
Before the transport measurements, the cells adhering to 24-well plates were washed with 1 ml of the transport medium containing 150 mm choline chloride instead of NaCl. Each well was then incubated at room temperature with 200 μl of the preincubation medium (the different compositions are indicated in the figure legends) supplemented with 1 mm MTSET. After 5 min, the medium was aspirated, and the cells were washed twice with 1 ml of the transport solution, followed by [3H]GABA transport. MTSET was purchased from Anathrace, Inc.
Inhibition by Copper(II)(1,10-Phenanthroline)3
This was basically done as described (20, 21). HeLa cells transfected with the indicated construct were washed once with choline solution (150 mm choline chloride, 5 mm KPi, pH 7.4, and 0.5 mm MgSO4) and preincubated for 5 min with the solutions of different compositions containing the indicated concentrations of CuPh. The CuPh stock solution was prepared for each experiment by mixing 0.4 ml of 1.25 m 1,10-phenanthroline in water/ethanol (1:1) and 0.6 ml of 250 mm CuSO4.
Cell Surface Biotinylation
Labeling of wild type and mutant transporters at the cell surface, using Sulfo-NHS-SS-Biotin (Pierce), quenching of the reaction, cell lysis, and isolation of the biotinylated proteins by streptavidin-agarose beads (Pierce) were done as described (22). After SDS-PAGE (10% gel) and transfer to nitrocellulose, the GAT-1 protein was detected with an affinity-purified antibody, directed against an epitope from the cytoplasmic C-terminal tail of GAT-1, at a 1:500 dilution, with horseradish peroxidase-conjugated secondary antibody at a 1:40,000 dilution, and with ECL. 1% goat serum was present in all antibody, blocking, and washing solutions to minimize the appearance of nonspecific bands. The films were scanned using a standard scanner, and quantitative densitometry was done using ImageJ version 1.43u; statistical analysis was done with Origin version 6.1 software (OriginLab Corp.).
Expression in Oocytes and Electrophysiology
cRNA was transcribed using mMESSAGE-mMACHINE (Ambion) and injected into Xenopus laevis oocytes, as described (23). Oocytes were placed in the recording chamber, penetrated with two agarose-cushioned micropipettes (1%/2 m KCl; resistance varied between 0.5 and 3 megaohms), voltage-clamped using GeneClamp 500 (Axon Instruments), and digitized using Digidata 1322 (Axon Instruments), both controlled by the pClamp9.0 suite (Axon Instruments). Voltage jumping was performed using a conventional two-electrode voltage clamp as described previously (24). The standard buffer, termed ND96, was composed of 96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm Na-HEPES, pH 7.5. The transient currents are defined as the currents in ND96 minus those in ND96 plus 10 μm tiagabine and Qmax as the magnitude of the charge moved between −140 and +60 mV, respectively. The GABA-induced currents are defined as the currents in ND96 subtracted from those in the same medium supplemented with 1 mm GABA at −140 mV. Treatment of oocytes, expressing GAT-1 WT or S359C/K448A, with CuPh was done exactly as described (25, 26).
Statistical Evaluations
The significance of differences between mutants and WT in transport, sodium-dependent transient currents, GABA-induced currents, inhibition by CuPh, and the stimulation by DTT was determined using a one-way analysis of variance with a post hoc Dunnett multiple-comparison test, where p < 0.05 or smaller was taken as significant (*). Results on the inhibition by CuPh were plotted using normalized data for each mutant, where the untreated activity levels were normalized to 100%.
RESULTS
Effect of CuPh on Transport by Cysteine Mutants
Cysteine residues were introduced, both individually and simultaneously, in positions 359 (EL4) and 448 (TM10) of a GAT-1 construct where Cys-74 was replaced by Ala (C74A). We shall refer to this C74A construct as WT. The S359C/K448C double mutant exhibited low but significant transport of [3H]GABA (Fig. 2A). Preincubation of HeLa cells expressing the double mutant with 30 μm CuPh resulted in inhibition of transport by more than 60% (Fig. 2B). Very little inhibition was seen with K448C and S359A/K448C (Fig. 2B), similar to results with WT (data not shown). However, in contrast to our expectations, also the transport activity of S359C was inhibited by CuPh, albeit less than S359C/K448C (Fig. 2B). Moreover, also the activity of S359C/K448A was inhibited by the oxidant to a similar extent as the double cysteine mutant (Fig. 2B). It is noteworthy that all of the mutants with a cysteine at position 359 have a reduced transport activity, whereas with a serine or alanine, the transport activity was similar to the WT construct (Fig. 2A).
FIGURE 2.
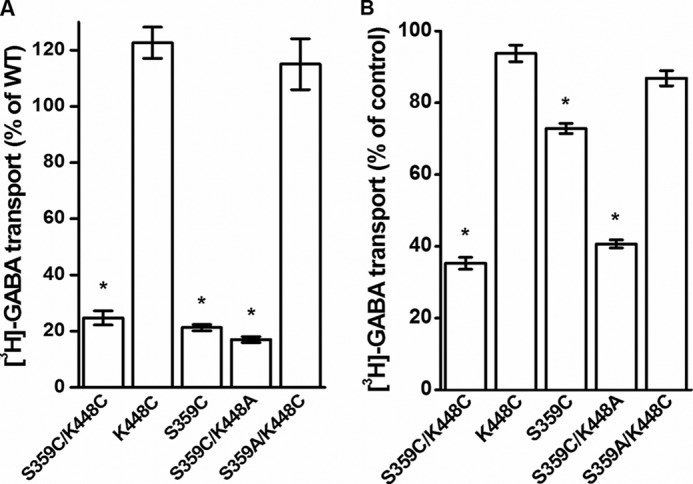
Transport activity and the effect of CuPh on inactivation. WT and the indicated mutants were transiently expressed in HeLa cells as described under “Experimental Procedures.” A, sodium-dependent [3H]GABA transport was measured at room temperature for 10 min. B, cells were incubated in the NaCl-containing medium with or without 30 μm CuPh for 5 min at room temperature, prior to the transport measurement. For A, the data are given as percentage of the activity of WT. Results represent mean ± S.E. (error bars) of at least three different experiments performed in quadruplicate. For B, the data are expressed as a percentage of untreated control and represent mean ± S.E. *, p < 0.05.
The decreased transport activity by S359C and other mutants analyzed in this study could be due to decreased levels of the mutants at the plasma membrane or an intrinsic defect of the mutations on transport activity. To address this, the surface expression levels of several of the mutants (Fig. 3) were compared with their transport activity (Table 1). All of the observed bands are specific for GAT-1 because they were absent from the biotinylated fraction of cells transfected with the vector alone (Fig. 3, SK). The fast running diffuse band, observed only with WT and S359C, and the fastest running species represent the monomeric forms of the fully glycosylated and deglycosylated transporter, respectively (27), and the two slower bands probably represent deglycosylated or glycosylated dimeric forms of the transporter. As can be seen in Table 1, the decreased transport activity of S359C is due to an intrinsic effect of the mutation, because its surface expression was similar to that of WT. With S359C/K448A, the surface expression was decreased, and there was a marked decrease in the proportion of the fully glycosylated monomeric species (Table 1). The transport activity and the surface expression decreased to the same extent, so that the intrinsic transport activity of this mutant was similar to that of WT.
FIGURE 3.
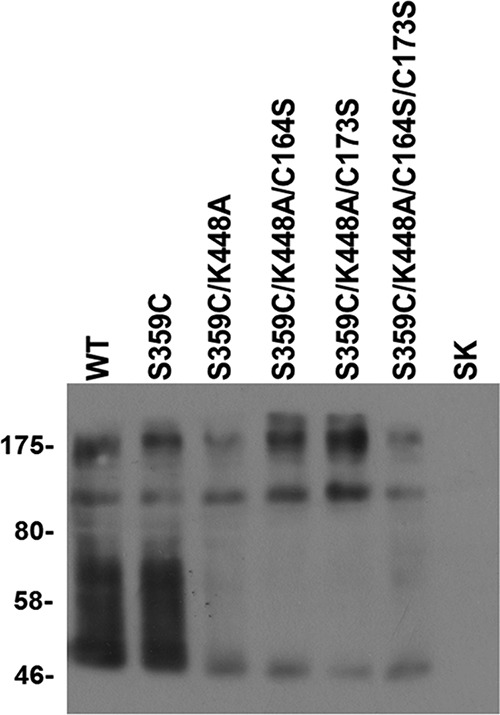
Cell surface biotinylation of S359C mutants. HeLa cells expressing WT and the indicated mutants, as well as HeLa cells transfected with the vector alone (SK), were biotinylated and processed as explained under “Experimental Procedures.” The results shown are typical of three independent determinations. The markers shown were run in the lane to the left of WT and contain “Prestained Protein Marker, Broad Range” (catalog no. P7708S, New England Biolabs).
TABLE 1.
Surface biotinylation and GABA transport by the S359C mutants
All values are given as a percentage of WT. The intensity in the linear range (10 s of exposure) of all of the transporter-specific bands was summed. Transport activity was determined in parallel with the surface expression (n = 3). For normalized activity, the values of the transport activity were divided by those of the corresponding intensity. All relative transport and intensity results were significant (p < 0.05) except for the relative intensity of S359C. For these series of experiments, the transport rates of WT and the mutants as well as the percentage of transport of the mutants relative to WT were higher than those given in Figs. 2 and 5, although the trend was similar. This is possibly due to the fact that the experiments documented in this table were performed at the stage of the revision of this paper, and passage numbers of the HeLa cells were lower than those for the earlier experiments.
| [3H]GABA transport | Intensity | Normalized | |
|---|---|---|---|
| % | % | % | |
| S359C | 27.9 ± 4.3 | 90 ± 3 | 31 |
| S359C/K448A | 36.6 ± 0.9 | 40 ± 9 | 107 |
| S359C/K448A/C164S | 9.2 ± 0.9 | 31 ± 6 | 30 |
| S359C/K448A/C173S | 12.8 ± 0.6 | 40 ± 16 | 32 |
| S359C/K448A/C164S/C173S | 25.6 ± 1.9 | 22 ± 4 | 130 |
In agreement with the results shown in Fig. 2B, the maximal inhibition by CuPh was lower for S359C than that for S359C/K448C or S359C/K448A (Fig. 4A), and the activity of the latter two mutants was much more potently inhibited by low concentrations of CuPh than S359C (Fig. 4A, inset). To test the idea that this maximal inhibition is determined by the nature of the amino acid residue at position 448, other substitutions were introduced. With a Glu or a Gly at this position, the inhibition of S359C was similar to that with Cys or Ala (Fig. 4B). On the other hand, the lower inhibition seen with a Lys at position 448 was also seen when it was replaced by Gln (Fig. 4B).
FIGURE 4.

Sensitivity of S359C mutants to CuPh. A, HeLa cells expressing the indicated mutants were preincubated in NaCl-containing medium supplemented with the indicated concentrations of CuPh for 5 min at room temperature. Subsequently, the cells were washed and assayed for [3H]GABA transport as described under “Experimental Procedures.” Inset, the abscissa is expanded, to show the effects of CuPh on S359C/K448A and S359C/K448C at submicromolar concentrations of the oxidant. B, HeLa cells expressing the indicated mutants were incubated with or without 30 μm CuPh prior to the transport measurement. Values are given as a percentage of activity in the absence of CuPh. The data are expressed as a percentage of untreated control and represent mean ± S.E. (error bars). The transport activity of the non-treated mutants (percentage of WT) was 21.3 ± 1.1% (n = 22) for S359C and 17 ± 4, 14 ± 4, and 58 ± 7% (n = 3) for S359C/K448G, S359C/K448E, and S359C/K448Q, respectively (*, p < 0.05).
The data presented in Fig. 2B indicate that the inhibition of S359C mutants by CuPh is not due to oxidative cross-linking of this cysteine with the one introduced at position 448. It is, however, possible that this inhibition could be due to the formation of a disulfide bond between an endogenous cysteine residue and Cys-359. Because position 359 is on an external loop, it was reasonable to assume that the potential partner of Cys-359 would be one of the three externally accessible cysteine residues. These cysteine residues, located at positions 74, 164, and 173, are exposed to the extracellular medium. Because Cys-74 was already replaced by Ala, we mutated the two other cysteines, which are located on EL2, to serine. The replacement of these two cysteine residues by serine in the background of either S359C or S359C/K448A resulted in reduced transport activity (see legend to Fig. 5) (p < 0.05 in all cases), consistent with results obtained with SERT and DAT (14, 15). Nevertheless, the sensitivity of our assay permits detection of transport activity as low as 0.5% of that of WT, so that there was no problem in obtaining reliable and significant data on the mutant transporters. It is noteworthy that the deletion of only one of the EL2 cysteines resulted in a shift of the distribution of the transporter-specific bands toward oligomeric forms (Table 1). Interestingly, when both mutations were introduced, there was no decrease in the intrinsic transport activity (Table 1).
FIGURE 5.
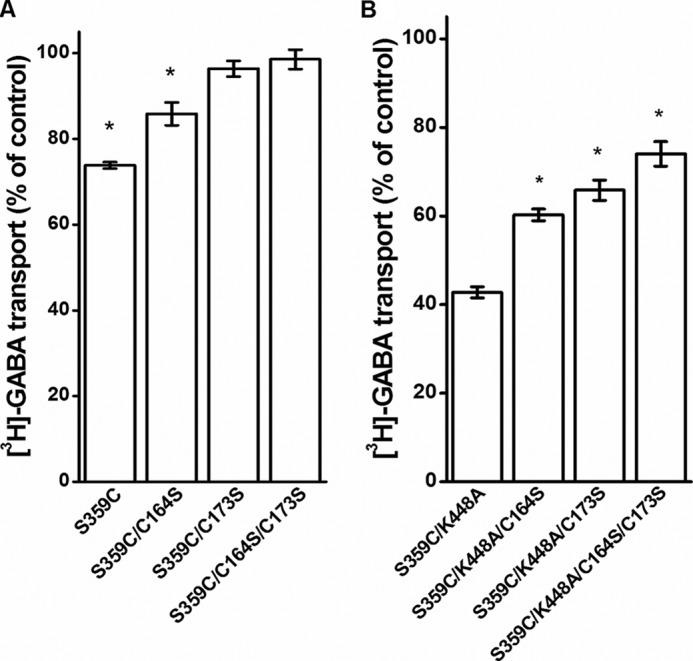
Effect of EL2 mutations on the sensitivity of S359C mutants to CuPh. HeLa cells expressing the indicated S359C (A) or S359C/K448A (B) mutants were preincubated in NaCl-containing medium in the presence or absence of 30 μm CuPh for 5 min at room temperature. Subsequently, the cells were washed and assayed for [3H]GABA transport as described under “Experimental Procedures.” The data are given as a percentage of control. The transport activity of the non-treated mutants (percentage of WT) was 3.8 ± 1.1% (n = 8), 5.5 ± 1.6% (n = 9), 9.3 ± 1.7% (n = 10), 9.6 ± 1.6% (n = 8), 7.3 ± 2.0% (n = 9), and 12.2 ± 2.2% (n = 9) for S359C/C164S, S359C/C173S, S359C/C164S/C173S, S359C/K448A/C164S, S359C/K448A/C173S, and S359C/K448A/C164S/C173S, respectively (*, p < 0.05). Error bars, S.E.
The extent of inhibition of the activity of S359C by CuPh was significantly reduced when Cys-164 was replaced with serine and even more with C173S and C164S/C173S (Fig. 5A). Because the inhibition of the activity of S359C by CuPh was larger in the presence of the K448A mutation, the cysteine residues at positions 164 and 173 were also replaced by serine in the S359C/K448A background. The findings were basically the same, except that now it was possible to see that the largest reduction of the inhibition was obtained when both cysteine residues were replaced (Fig. 5B). Nevertheless, there was still some inhibition of transport by CuPh in the C164S/C173S background, which was more than the small nonspecific inhibition of 10–15% by CuPh, seen for instance in Fig. 2B (columns without S359C). Therefore, we additionally replaced other endogenous cysteine residues with the aim of reducing the inhibition by CuPh even further. However, the transport activity of these mutants was too low for further analysis (data not shown).
Effects of Dithiothreitol on Transport by Cysteine Mutants
The mutants with a cysteine at position 359 exhibit a much lower transport activity than those with a serine or alanine (Fig. 2A). Therefore, we considered the possibility that, even without preincubation with CuPh, the cysteine residue at position 359 is already cross-linked to endogenous cysteine residues, at least in a considerable fraction of the S359C transporters. To address this possibility, HeLa cells expressing S359C were incubated with DTT, prior to the transport assay. Indeed, this maneuver resulted in an almost 4-fold higher transport activity (Fig. 6). The stimulation was only seen with cysteine at position 359. Treatment of WT or S359A with DTT had very little effect on transport (Fig. 6).
FIGURE 6.
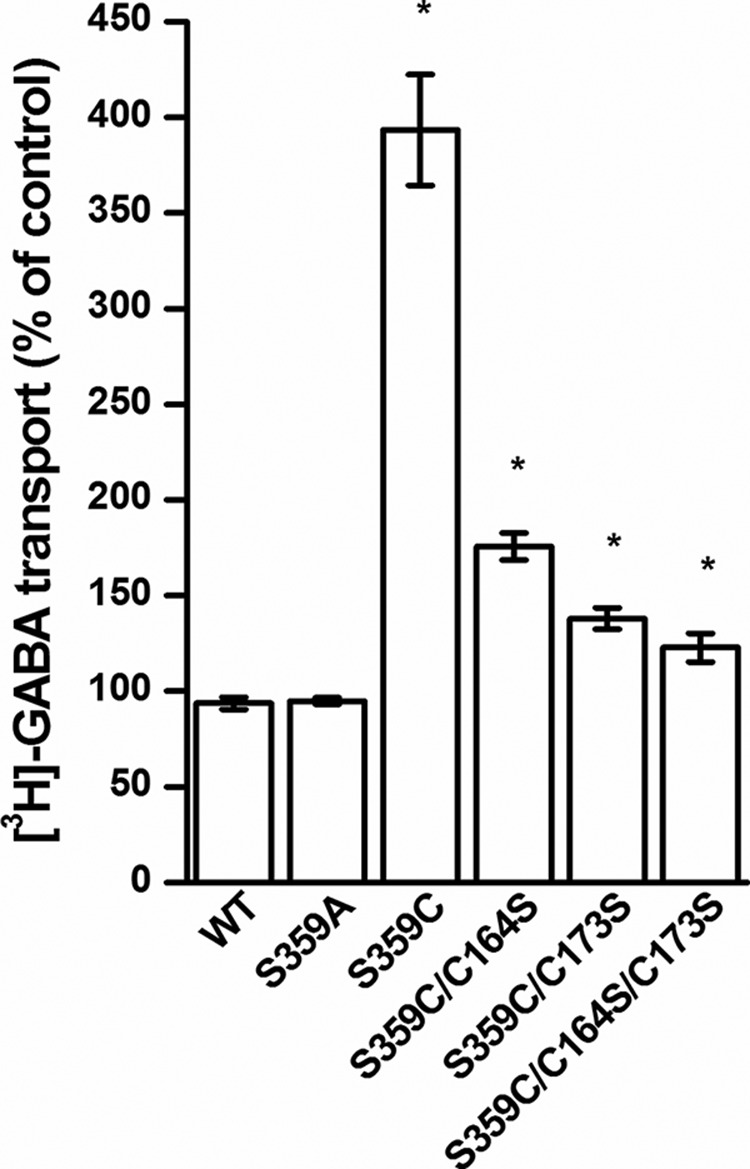
Effect of DTT on transport. HeLa cells, expressing the indicated mutants, were preincubated in the presence or absence of 12 mm DTT in a NaCl-containing medium for 5 min at room temperature. Subsequently, the cells were washed and assayed for [3H]GABA transport as described under “Experimental Procedures.” The data are given as a percentage of the untreated control.
The stimulation of the activity of S359C by DTT was dramatically diminished when the EL2 cysteines were replaced by serine. Again, the effect seen with S359C/C173S was more pronounced than with S359C/C164S (Fig. 6). Very little stimulation by DTT was seen when both EL2 cysteine residues were replaced (Fig. 6).
Effect of GABA and Coupling Ions on the Inhibition by CuPh and MTSET
Our observations suggest that the cysteine residue placed at position 359 is sufficiently close to the endogenous Cys-164 and Cys-173 for disulfide formation (Figs. 5 and 6), at least in one of the conformations of the transporter. The inhibition of the transport activity of S359C by CuPh was highest when the cells expressing this transporter were exposed to the oxidant in the presence of sodium (Fig. 7A). Under these conditions, a large proportion of the transporters reside in outward-facing conformations (28). The inhibition by CuPh was diminished when sodium was replaced by choline, and the same was true when the NaCl-containing medium was supplemented with GABA (Fig. 7A). A significantly smaller effect was observed in the absence of chloride (NaCl replaced by sodium gluconate). Because the inhibition of transport of S359C by CuPh was modest, the experiments were also done with the S359C/K448A and S359C/K448C mutants, which exhibited a more robust inhibition (Fig. 4A). Also with both double mutants, the effects of the medium composition on the inhibition by CuPh were similar to those with S359C alone (Fig. 7, B and C).
FIGURE 7.
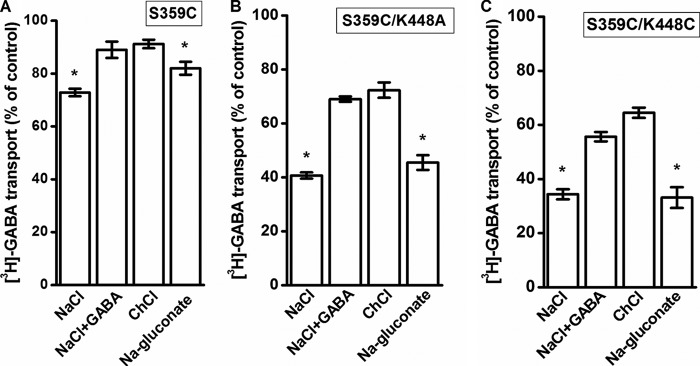
Effect of the composition of the external medium on the inhibition of S359C mutants by CuPh. HeLa cells expressing S359C (A), S359C/K448A (B), or S359C/K448C (C) were preincubated for 5 min in the presence or absence of 30 μm CuPh in media containing NaCl, NaCl + 1 mm GABA, choline chloride (ChCl), or sodium gluconate, as indicated. Values are given as a percentage of control (preincubation without CuPh in each condition) and represent the mean ± S.E. (error bars) of at least three different experiments done in quadruplicate. The mean values with the different conditions were compared with those from choline chloride-containing medium using one-way analysis of variance with a post hoc Dunnett multiple-comparison test: *, p < 0.05.
The effects of CuPh are dependent not only on the distance of the cysteine pairs but also on their aqueous accessibility. On the other hand, modification of a cysteine residue by the membrane-impermeant sulfhydryl reagent MTSET depends on, besides its reactivity, its aqueous accessibility only. Preincubation of cells expressing S359C with 1 mm MTSET for 5 min resulted in an inhibition of transport of around 50% (Fig. 8A). However, in contrast to the inhibition by CuPh (Fig. 7A), the extent of inhibition of transport by MTSET was not dependent on the composition of the preincubation medium (Fig. 8A). In the case of S359C/K448A, the inhibition by the sulfhydryl reagent was more potent than with S359C, but also here a similar inhibition of transport was observed under each of the preincubation conditions (Fig. 8B). Very little inhibition by the sulfhydryl reagent, if any, was seen with C164S, C173S, and C164S/C173S under any of the indicated conditions (Fig. 9, A–C).
FIGURE 8.
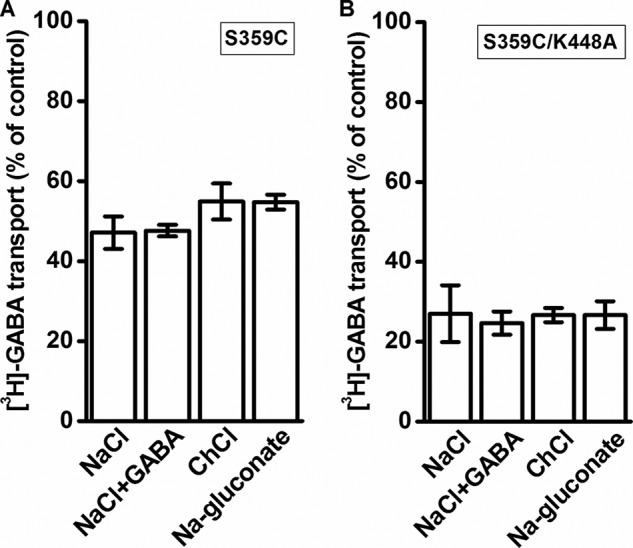
Effect of the composition of the external medium on the inhibition of S359C mutants by MTSET. HeLa cells expressing S359C (A) or S359C/K448A (B) were preincubated in the presence or absence of 1 mm MTSET in media containing NaCl, NaCl + 1 mm GABA, choline chloride (ChCl), or sodium gluconate, as indicated. Values are given as a percentage of control (preincubation without MTSET in each condition) and represent the mean ± S.E. (error bars) of at least three different experiments done in quadruplicate.
FIGURE 9.
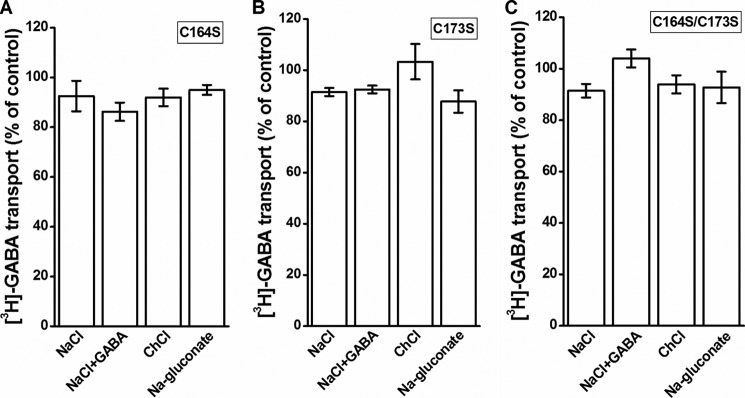
Effect of the composition of the external medium on the inhibition of EL2 mutants by MTSET. HeLa cells expressing C164S (A), C173S (B), or C164S/C173S (C) were preincubated in the presence or absence of 1 mm MTSET in media containing NaCl, NaCl + 1 mm GABA, choline chloride (ChCl), or sodium gluconate, as indicated. Values are given as a percentage of control (preincubation without MTSET in each condition) and represent the mean ± S.E. (error bars) of at least three different experiments done in quadruplicate. The transport activity of the non-treated mutants (percentage of WT) was 19.1 ± 2.9, 18.9 ± 3.4, and 31.8 ± 4.4% (n = 3) for C164S, C173S, and C164S/C173S, respectively, p < 0.05.
The radioactive transport by S359C/K448A, expressed in HeLa cells, was markedly inhibited by CuPh (Figs. 2B, 4A, and 5B). We used GABA-induced steady state currents as an independent assay to measure the impact of CuPh of S359C/K448A, expressed in Xenopus laevis oocytes. The GABA-induced currents by this mutant were also inhibited by 20 μm CuPh (Fig. 10, A and B), although the extent of the inhibition was lower than that observed in the HeLa cells (Fig. 4A). The remaining activity was 61 ± 5% as compared with 97 ± 3% in WT (Fig. 10A). The sodium-dependent transient currents are a read-out of a partial reaction of the transport cycle, namely the transition between the inward- and outward-facing conformation of the empty transporter (28). The impact of CuPh on the magnitude of the charge moved between −140 and +60 mV was less than that on the transport currents, with 85 ± 3% remaining after the treatment with the oxidant as compared with 100 ± 3% in the WT (Fig. 10A). The voltage dependence of the transient currents by the mutant was “right-shifted” by around 30 mV as compared with WT (Fig. 10C), indicating a higher apparent affinity for sodium. However, this voltage dependence was hardly changed by the treatment with the oxidant (Fig. 10C).
FIGURE 10.
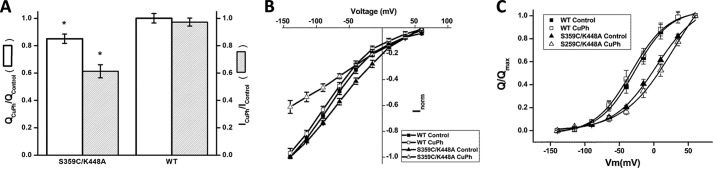
Effects of CuPh on the transient currents and GABA-induced steady state currents by WT and S359C/K448A. The membrane voltage of oocytes expressing WT and S359C/K448A was stepped from a holding potential of −25 mV to voltages between −140 and +60 mV in 25-mV increments. Each potential was held clamped for 500 ms, followed by 500 ms of a potential clamped at −25 mV. A, charge movements (open bars, Qmax) and GABA-induced currents (gray bars) for WT and S359C/K448A following treatment with 20 μm CuPh were normalized to those before the exposure to the oxidant, QCuPh/QControl and ICuPh/IControl, respectively. The data are given as mean ± S.E. (error bars) of at least three oocytes. *, p < 0.05. B, voltage dependence of the GABA-induced currents by WT and S359C/K448A before and after application of 20 μm CuPh. For WT and S359C/K448A, the GABA-induced currents at each potential from 420 to 480 ms were averaged and normalized to the GABA-induced current at −140 mV. These currents were then plotted against the corresponding potential (mV). The data are the means ± S.E. (error bars) of at least three repeats. Wherever error bars are not visible, the error was smaller than the size of the symbols. The currents at −140 mV induced by 1 mm GABA ranged from −177 to −331 nA in WT and from 147 to −186 nA in S359C/K448A. C, the charge movements of oocytes expressing WT and S359C/K448A in 100 mm sodium, before and after treatment with 20 μm CuPh, were plotted as a function of the voltage. Charge movements were normalized to Qmax and were fit, using the Boltzmann distribution non-linear curve fit function in Origin version 6.1 (OriginLab Corp.). Wherever error bars are not visible, the error was smaller than the size of the symbols. The Qmax values before and after application of CuPh were 16 ± 2.5 and 16.1 ± 2.6 nanocoulombs for WT and 13.5 ± 0.6 and 11.5 ± 0.7 nanocoulombs for S359C/K448A, respectively. Data points are averaged from at least three oocytes for each transporter.
DISCUSSION
The data reported here indicate that, under oxidative conditions, a cysteine introduced at position 359 of EL4 of GAT-1 can form a disulfide bond with the endogenous cysteines at positions 164 and 173 of EL2 (Figs. 5 and 6). We arrived at these inferences by serendipity. Namely, our original aim was to provide support for the idea that, not only in LeuT but also in GAT-1, EL4 moves to occlude the binding pocket from the extracellular side during the transition from the outward- to inward-open transition (8). However the data depicted in Fig. 2B indicate that the inhibition of transport activity of S359C/K448C by CuPh is not due to the formation of a disulfide bond between the cysteine residues at positions 359 and 448. On the other hand, the inhibition of transport by S359C under oxidative conditions was diminished when Cys-164 and Cys-173 were replaced. The impact of the substitution of Cys-173 was more pronounced than that of Cys-164, but the strongest attenuation was seen when both were substituted (Fig. 5). This indicates that either cysteine residue can react with the cysteine introduced at position 359 (Fig. 11, top), but Cys-173 is probably in closer proximity to S359C than Cys-164. The mirror image of these results was obtained on the stimulation of transport of S359C by DTT (Fig. 6). The almost 4-fold stimulation of the transport activity by the reductant shows that in cells expressing the single mutant, a large proportion of the cysteine residues at position 359 is already cross-linked. The reduction of this stimulation upon replacing Cys-164 and Cys-173 also indicates that either of these residues can be close to Cys-359, presumably because of flexibility of EL2. Consistently, the activity of S359C is around 4-fold lower than that of S359A (Fig. 2A), apparently because with the latter mutant, the cross-linking with the cysteine residues from EL2 is impossible.
FIGURE 11.
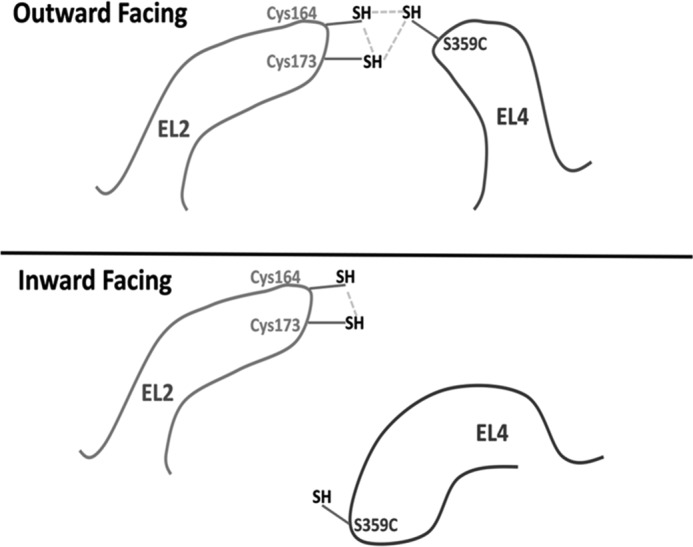
Proposed disulfide formation between cysteine residues for EL2 and EL4. In the outward-facing form of S359C, disulfide formation is possible between each of the two endogenous EL2 cysteines and the cysteine introduced in position 359 of EL4. As described under “Discussion,” we propose that the distance between EL2 and EL4 increases when the transporter becomes inward-facing and, as a result, the oxidative cross-linking of Cys-359 and either Cys-164 or Cys-173 is diminished.
Our results do not contradict the conclusions that the dopamine transporter DAT and the serotonin transporter SERT equivalents of Cys-164 and Cys-173 can form an intramolecular disulfide bridge (14, 15). Further support for disulfide formation between the EL2 cysteines is provided by the fact that in the recently obtained structure of the dopamine transporter from D. melanogaster, the equivalent cysteine residues are indeed cross-linked. However, this result has to be interpreted with caution, because 43 residues from EL2 were removed in the Drosophila construct, used to determine the structure (13). Transport by the S359C mutants is partly inhibited by low concentrations of CuPh, but the extent of this inhibition is not much increased at high concentrations of the oxidant (Fig. 4A). A likely explanation is that only in part of the S359C transporters are Cys-164 and Cys-173 free to form a disulfide bound with the cysteine introduced at position 359, whereas in the others, the two EL2 cysteine residues form a disulfide with each other or are already cross-linked to the Cys at position 359. It is not clear if also in WT the Cys-164/Cys-173 disulfide bond is present in only a fraction of the transporters or if this is the result of the mutation at position 359. Because the S359C transporters have considerable activity that is stimulated around 4-fold by DTT to very similar levels as WT (Figs. 2A and 6), we believe that EL2 and EL4 are also in close proximity in WT. The extent of inhibition by CuPh depends on the nature of the amino acid residue at position 448 (Fig. 4, A and B). Clearly, a much more pronounced inhibition is seen with a cysteine or an alanine at position 448 (Fig. 4A). The results shown in Fig. 4B indicate that this is not merely due the smaller size of the side chain, which could potentially enable better accessibility to the oxidant. Nevertheless, the inhibition of transport activity of S359C/K448A by MTSET is larger than that of S359C (Fig. 8). This suggests that the nature of the residue at position 448 may influence the accessibility of Cys-359, at least in some of the substitution mutants. Alternatively or additionally, it is possible that the degree of reduction of the disulfide bridges is dependent on the nature of the side chain at position 448 by an allosteric mechanism.
The higher intrinsic transport activity of S359C/K448A than that of S359C (Table 1) is compatible with the idea that the K448A mutation results in an increased reduction of the disulfide bridges between the cysteines at positions 164, 173, and 359. On the other hand, it is not clear why this is accompanied by low levels of the fully glycosylated transporter. It is possible that there is less glycosylation of the transporter when the number of disulfide bridges is reduced. As a consequence, there is an increased proportion of transporters in the plasma that are not fully processed but are nevertheless active. The data in Table 1 also indicate that the number of transporters at the plasma membrane is related to the very presence of Cys-164 and Cys-173. Elimination of both EL2 cysteines has less of an impact on the transport activity than the elimination of only one of them (Table 1). It is not clear why replacing either Cys-164 or Cys-173 by Ser has such a serious impact on the intrinsic transport activity. The increased proportion of oligomeric forms suggests that the oligomeric forms of the transporter, observed with these mutants, may be inactive (Table 1). Indeed, when both EL2 cysteines are replaced, the distribution of the bands is very similar to that of S359C/K448A, consistent with the idea that the deglycosylated monomeric form of the transporter is active.
For all of the S359C single and double mutants tested, the most significant inhibition of transport by CuPh was observed in the presence of sodium (Fig. 7, A–C). Under these conditions, a large proportion of the transporters is outward-facing (28). This inhibition is attenuated in the presence of GABA or the absence of sodium, both conditions favoring the inward-facing conformation of the transporter. The cysteine introduced at position 359 was accessible to MTSET under all conditions tested (Fig. 8). Therefore, it is most likely that the attenuation of the inhibition by CuPh is not due to a decreased accessibility but rather to an increased distance between position 359 and positions 164 and 173 in the inward-facing conformation of the transporter (Fig. 11).
The inhibition of GABA-induced transport currents of S359C/K448A by CuPh (Fig. 10, A and B) was less than when radioactive transport was used as an assay (Figs. 2, 4, and 5). A possible reason could be that when S359C/K448A is expressed in oocytes, a larger proportion of Cys-359 is already cross-linked to the cysteines from EL2 than when the HeLa cell expression system is used. The lower inhibition of the sodium-dependent transient currents by CuPh than that of the GABA-induced steady-state currents (Fig. 10A) suggests that the ability of EL4 to “close in” on the binding pocket is more important for the translocation of the substrate-loaded than that of the substrate-free transporter. The “right-shifted” voltage dependence of the transient currents by S359C (Fig. 10C) indicates an increased apparent affinity for sodium. This is probably due to an increased proportion of outward-facing transporters resulting from cross-linking between EL2 and EL4.
It is important to point out that although there is structural information on EL2 of LeuT, this loop is much longer in the NSS neurotransmitter transporters. Thus, our results, indicating proximity of EL2 and EL4 in the outward-facing conformation of the transporter, could not have been predicted by the comparison of the various LeuT structures. Interestingly, our results are in harmony with earlier observations on the inhibition of the dopamine transporter DAT by Zn2+ (29, 30). The target of the divalent cation is an endogenous zinc binding site formed by four amino acid residues, two from EL2 and two from EL4 (31). Because the inhibition by zinc is not due to the formation of a covalent bound, it is impossible to monitor inhibition following preincubation under conditions favoring outward- or inward-facing conformations. Nevertheless, it appears that the binding of the divalent cation increases the proportion of outward-facing transporters (32). Probably, our observations that EL2 and EL4 of GAT-1 approach each other in an outward-facing conformation can also be extended to DAT. Thus, the Zn2+ site could only be formed when these loops come together. We anticipate that our functional results will be reinforced by future studies of active neurotransmitter transporters, crystallized in multiple conformations.
Acknowledgment
We thank Assaf Ben-Yona for the preparation of Fig. 1 and for help with the surface biotinylation.
This work was supported by Israel Science Foundation Grant 804/11 and by a grant from the Rosetrees Trust.
- NSS
- neurotransmitter:sodium:symporter
- TM
- transmembrane domain
- EL
- extracellular loop
- CuPh
- copper(II)(1,10-Phenanthroline)3
- MTSET
- (2-trimethylammonium)methanethiosulfonate.
REFERENCES
- 1. Kanner B. I. (1994) Sodium-coupled neurotransmitter transport: structure, function and regulation. J. Exp. Biol. 196, 237–249 [DOI] [PubMed] [Google Scholar]
- 2. Nelson N. (1998) The family of Na+/Cl− neurotransmitter transporters. J. Neurochem. 71, 1785–1803 [DOI] [PubMed] [Google Scholar]
- 3. Keynan S., Kanner B. I. (1988) γ-Aminobutyric acid transport in reconstituted preparations from rat brain: coupled sodium and chloride fluxes. Biochemistry 27, 12–17 [DOI] [PubMed] [Google Scholar]
- 4. Kavanaugh M. P., Arriza J. L., North R. A., Amara S. G. (1992) Electrogenic uptake of γ-aminobutyric acid by a cloned transporter expressed in Xenopus oocytes. J. Biol. Chem. 267, 22007–22009 [PubMed] [Google Scholar]
- 5. Mager S., Naeve J., Quick M., Labarca C., Davidson N., Lester H. A. (1993) Steady states, charge movements, and rates for a cloned GABA transporter expressed in Xenopus oocytes. Neuron 10, 177–188 [DOI] [PubMed] [Google Scholar]
- 6. Lu C. C., Hilgemann D. W. (1999) GAT1 (GABA:Na+:Cl−) cotransport function. Steady state studies in giant Xenopus oocyte membrane patches. J. Gen. Physiol. 114, 429–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 8. Krishnamurthy H., Gouaux E. (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou Y., Zomot E., Kanner B. I. (2006) Identification of a lithium interaction site in the γ-aminobutyric acid (GABA) transporter GAT-1. J. Biol. Chem. 281, 22092–22099 [DOI] [PubMed] [Google Scholar]
- 10. Zhang Y. W., Rudnick G. (2006) The cytoplasmic substrate permeation pathway of serotonin transporter. J. Biol. Chem. 281, 36213–36220 [DOI] [PubMed] [Google Scholar]
- 11. Vandenberg R. J., Shaddick K., Ju P. (2007) Molecular basis for substrate discrimination by glycine transporters. J. Biol. Chem. 282, 14447–14453 [DOI] [PubMed] [Google Scholar]
- 12. Dodd J. R., Christie D. L. (2007) Selective amino acid substitutions convert the creatine transporter to a γ-aminobutyric acid transporter. J. Biol. Chem. 282, 15528–15533 [DOI] [PubMed] [Google Scholar]
- 13. Penmatsa A., Wang K. H., Gouaux E. (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen J. G., Liu-Chen S., Rudnick G. (1997) External cysteine residues in the serotonin transporter. Biochemistry 36, 1479–1486 [DOI] [PubMed] [Google Scholar]
- 15. Chen R., Wei H., Hill E. R., Chen L., Jiang L., Han D. D., Gu H. H. (2007) Direct evidence that two cysteines in the dopamine transporter form a disulfide bond. Mol. Cell. Biochem. 298, 41–48 [DOI] [PubMed] [Google Scholar]
- 16. Kunkel T. A., Roberts J. D., Zakour R. A. (1987) Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 154, 367–382 [DOI] [PubMed] [Google Scholar]
- 17. Kleinberger-Doron N., Kanner B. I. (1994) Identification of tryptophan residues critical for the function and targeting of the γ-aminobutyric acid transporter (subtype A). J. Biol. Chem. 269, 3063–3067 [PubMed] [Google Scholar]
- 18. Fuerst T. R., Niles E. G., Studier F. W., Moss B. (1986) Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 83, 8122–8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Keynan S., Suh Y. J., Kanner B. I., Rudnick G. (1992) Expression of a cloned γ-aminobutyric acid transporter in mammalian cells. Biochemistry 31, 1974–1979 [DOI] [PubMed] [Google Scholar]
- 20. Brocke L., Bendahan A., Grunewald M., Kanner B. I. (2002) Proximity of two oppositely oriented reentrant loops in the glutamate transporter GLT-1 identified by paired cysteine mutagenesis. J. Biol. Chem. 277, 3985–3992 [DOI] [PubMed] [Google Scholar]
- 21. Zomot E., Zhou Y., Kanner B. I. (2005) Proximity of transmembrane domains 1 and 3 of the γ-aminobutyric acid transporter GAT-1 inferred from paired cysteine mutagenesis. J. Biol. Chem. 280, 25512–25516 [DOI] [PubMed] [Google Scholar]
- 22. Ben-Yona A., Bendahan A., Kanner B. I. (2011) A glutamine residue conserved in the neurotransmitter:sodium:symporters is essential for the interaction of chloride with the GABA transporter GAT-1. J. Biol. Chem. 286, 2826–2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kanner B. I. (2003) Transmembrane domain I of the γ-aminobutyric acid transporter GAT-1 plays a crucial role in the transition between cation leak and transport modes. J. Biol. Chem. 278, 3705–3712 [DOI] [PubMed] [Google Scholar]
- 24. Borre L., Kanner B. I. (2004) Arginine 445 controls the coupling between glutamate and cations in the neuronal transporter EAAC-1. J. Biol. Chem. 279, 2513–2519 [DOI] [PubMed] [Google Scholar]
- 25. Borre L., Kavanaugh M. P., Kanner B. I. (2002) Dynamic equilibrium between coupled and uncoupled modes of a neuronal glutamate transporter. J. Biol. Chem. 277, 13501–13507 [DOI] [PubMed] [Google Scholar]
- 26. Shabaneh M., Rosental N., Kanner B. I. (2014) Disulfide cross-linking of transport and trimerization domains of a neuronal glutamate transporter restricts the role of the substrate to the gating of the anion conductance. J. Biol. Chem. 289, 11175–11182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bennett E. R., Su H., Kanner B. I. (2000) Mutation of arginine 44 of GAT-1, a (Na(+) + Cl(−))-coupled γ-aminobutyric acid transporter from rat brain, impairs net flux but not exchange. J. Biol. Chem. 275, 34106–34113 [DOI] [PubMed] [Google Scholar]
- 28. Ben-Yona A., Kanner B. I. (2012) An acidic amino acid transmembrane helix 10 residue conserved in the neurotransmitter:sodium:symporters is essential for the formation of the extracellular gate of the γ-aminobutyric acid (GABA) transporter GAT-1. J. Biol. Chem. 287, 7159–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Norregaard L., Frederiksen D., Nielsen E. O., Gether U. (1998) Delineation of an endogenous zinc-binding site in the human dopamine transporter. EMBO J. 17, 4266–4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loland C. J., Norregaard L., Gether U. (1999) Defining proximity relationships in the tertiary structure of the dopamine transporter. Identification of a conserved glutamic acid as a third coordinate in the endogenous Zn2+-binding site. J. Biol. Chem. 274, 36928–36934 [DOI] [PubMed] [Google Scholar]
- 31. Stockner T., Montgomery T. R., Kudlacek O., Weissensteiner R., Ecker G. F., Freissmuth M., Sitte H. H. (2013) Mutational analysis of the high-affinity zinc binding site validates a refined human dopamine transporter homology model. PLoS Comput. Biol. 9, e1002909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Loland C. J., Norregaard L., Litman T., Gether U. (2002) Generation of an activating Zn2+ switch in the dopamine transporter: mutation of an intracellular tyrosine constitutively alters the conformational equilibrium of the transport cycle. Proc. Natl. Acad. Sci. U.S.A. 99, 1683–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]