

Background: Constitutive activation of STAT3 is associated with the progression of hepatocellular carcinoma (HCC), and abrogation of STAT3 signaling is a potential target for HCC treatment.
Results: A novel azaspirane modulates the JAK-STAT pathway in HCC.
Conclusion: The lead compound induces apoptosis by down-regulating STAT3 signaling.
Significance: This investigation reports a novel inhibitor of the JAK-STAT pathway with the potential to target various cancers.
Keywords: Bioinformatics, Cell Migration, Hepatocellular Carcinoma, Janus Kinase (JAK), STAT3
Abstract
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that regulates genes involved in cell growth, proliferation, and survival, and given its association with many types of cancers, it has recently emerged as a promising target for therapy. In this work, we present the synthesis of N-substituted azaspirane derivatives and their biological evaluation against hepatocellular carcinoma (HCC) cells (IC50 = 7.3 μm), thereby identifying 2-(1-(4-(2-cyanophenyl)1-benzyl-1H-indol-3-yl)-5-(4-methoxy-phenyl)-1-oxa-3-azaspiro(5,5) undecane (CIMO) as a potent inhibitor of the JAK-STAT pathway with selectivity over normal LO2 cells (IC50 > 100 μm). The lead compound, CIMO, suppresses proliferation of HCC cells and achieves this effect by reducing both constitutive and inducible phosphorylation of JAK1, JAK2, and STAT3. Interestingly, CIMO displayed inhibition of Tyr-705 phosphorylation, which is required for nuclear translocation of STAT3, but it has no effect on Ser-727 phosphorylation. CIMO accumulates cancer cells in the sub-G1 phase and decreases STAT3 in the nucleus and thereby causes down-regulation of genes regulated via STAT3. Suppression of STAT3 phosphorylation by CIMO and knockdown of STAT3 mRNA using siRNA transfection displayed a similar effect on the viability of HCC cells. Furthermore, CIMO significantly decreased the tumor development in an orthotopic HCC mouse model through the modulation of phospho-STAT3, Ki-67, and cleaved caspase-3 in tumor tissues. Thus, CIMO represents a chemically novel and biologically in vitro and in vivo validated compound, which targets the JAK-STAT pathway as a potential cancer treatment.
Introduction
Hepatocellular carcinoma (HCC)7 is a fatal liver cancer affecting 600,000 people worldwide annually (1), and it ranks third in terms of global cancer mortality (2). The development and progression of HCC is largely associated with endemic hepatitis B or hepatitis C virus infection, alcoholic hepatitis, non-alcoholic steatohepatitis, hemochromatosis, obesity, and consumption of aflatoxin B1 (3–5). Surgical therapies, including liver resection and liver transplantation, as well as non-surgical therapies, such as embolization, systemic chemotherapy, and radiation therapy, have exhibited the highest efficacies in the treatment of the neoplasm (6), and as with many cancers, the detection and treatment of HCC in early stages can enhance the prognosis.
Signal transducer and activator of transcription 3 (STAT3) is an inducible transcription factor present in the cytoplasm of most cell types, and it is involved in extracellular signal transduction to the nucleus by cytokines of the IL-6 family and epidermal and platelet-derived growth factors and hence governs cell differentiation, proliferation, and survival and, in a tumor, its proliferation, development, survival, angiogenesis, metastasis, and evasion (7, 8). Activation of STAT3 is also known to transmit various survival signals by promoting the expression of genes involved in cell cycle progression (cyclin D1), angiogenesis (VEGF and HIF-1α), cell migration (MMP-2/9), and immune evasion (RANTES (regulated on activation normal T cell expressed and secreted)) and antiapoptotic genes (Bcl2, Bcl-xL, and survivin) (9, 10). JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2) are the upstream kinases that phosphorylate different STAT proteins and are involved in different functions. Structurally, the activation of Janus kinase (JAK) and c-Src kinase leads to the phosphorylation of tyrosine 705 and homodimerization of STAT3, followed by its nuclear translocation to transcribe the target genes. Constitutive activation of STAT3 is observed in more than 15 types of solid and hematological tumors, including hepatocellular carcinoma, leukemia, lymphoma, prostate cancer, breast cancer, ovarian cancer, and multiple myeloma (11). To summarize, the critical role of STAT3 in progression of in particular hepatocellular carcinoma renders it a unique target for cancer treatment.
Azaspirane-based compounds have been studied extensively and have been shown to have very good antioncogenic activity in various tumor models (12). We previously reported sugar mimetic (2-(2,6-difluorophenyl)-5-(4-methoxyphenyl)-1-oxa-3-azaspiro(5,5) undecane) as an antitumor agent, which possesses binding affinity to cytokines and various growth factors (13). Many discoveries have revealed that azaspiranes are good inhibitors of various classes of tyrosine kinases. Specifically, small molecules, such as atiprimod, azaspirane, staurosporines, and lestaurtinib, are some of the known azaspirane-based tyrosine kinase inhibitors (14–16). Atiprimod and azaspirane are structurally related compounds that were shown to inhibit JAK2/JAK3 in preclinical studies, thereby suppressing cell proliferation, along with angiogenesis-activating caspases, to drive cell death (17, 18). Furthermore, it has been reported that staurosporines show strong JAK3 inhibition (19). Lestaurtinib is an orally bioavailable JAK2 inhibitor that is in phase II clinical trials for acute myeloid leukemia (20, 21). However, the development of atiprimod has been stalled due to commercial reasons. The synthesis of staurosporines or lestaurtinib is a complex phenomenon because they are isolated from Streptomyces. Hence, we evolved the new scaffold shown in Fig. 1A.
FIGURE 1.
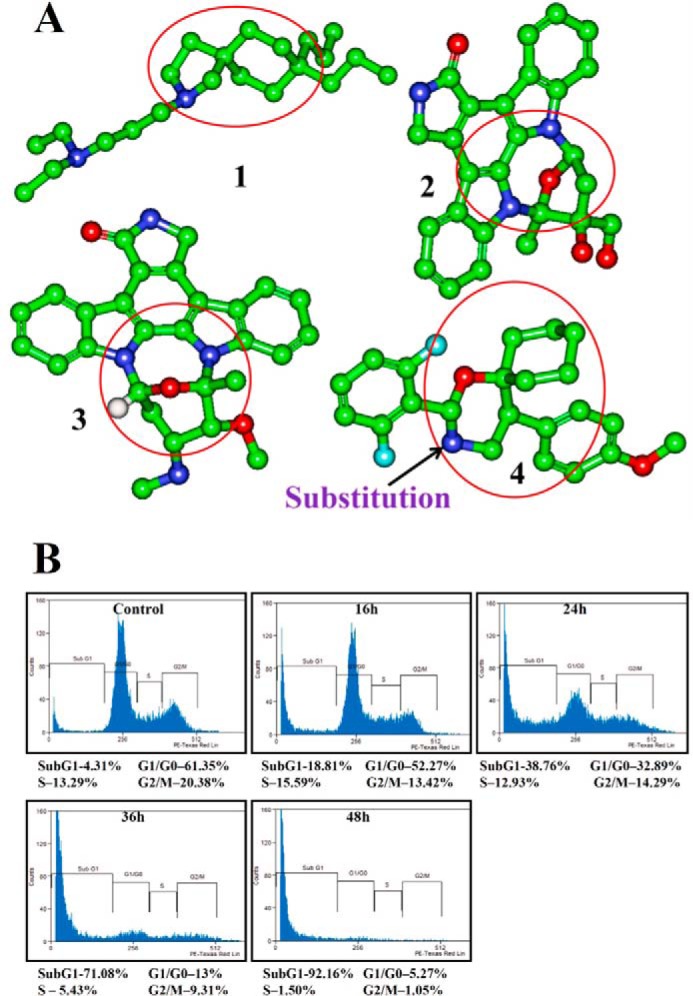
A, azaspirane scaffold (highlighted with red circle) evolution. 1, atiprimod; 2, staurosporine; 3, lestaurtinib; 4, (2-(2,6-difluorophenyl)-5-(4-methoxyphenyl)-1-oxa-3-azaspiro[5.5]undecane) (DMBO). B, CIMO causes the accumulation of HepG2 cells in the sub-G1 phase. HepG2 cells (5 × 105/ml) were treated with 10 μmol/liter CIMO for the indicated times, after which the cells were washed, fixed, stained with propidium iodide, and analyzed for DNA content by flow cytometry.
Inhibition of upstream tyrosine kinases, such as JAK2/3, by a small molecule now results in a decline of STAT3-targeted gene expression, thereby inhibiting various biological processes crucial for cancer cell survival and invasion. In the current work, we synthesized and evaluated the effect of N-substituted azaspirane derivatives on the JAK-STAT3 pathway in hepatocellular carcinoma cells and found 2-(1-(4-(2-cyanophenyl)1-benzyl-1H-indol-3-yl)-5-(4-methoxy-phenyl)-1-oxa-3-azaspiro(5,5) undecane (CIMO) as a potent inhibitor of this pathway. CIMO enhances cytotoxicity, depletes the nuclear pool of STAT3, and down-regulates constitutively active and inducible upstream kinases (STAT3) and expression of target genes in vitro and in vivo, as will be described in more detail below.
EXPERIMENTAL PROCEDURES
Reagents
Hoechst 33342, MTT, Tris, glycine, NaCl, SDS, and BSA were purchased from Sigma-Aldrich. Dulbecco's modified Eagle's medium (DMEM), FBS, and antibiotic/antimycotic mixture were obtained from Invitrogen. Rabbit polyclonal antibodies to STAT3 and mouse monoclonal antibodies against phospho-STAT3 (Tyr-705) and Bcl-2, Bcl-xL, cyclin D1, survivin, Bak, Bid, PTP1B, SHPTP1, SHPTP2, pro-caspase-3, and PARP were obtained from Santa Cruz Biotechnology, Inc. Antibodies to phospho-specific Src (Tyr-416), Src, phospho-specific JAK1 (Tyr-1022/1023), JAK1, phosphospecific JAK2 (Tyr-1007/1008), and JAK2 were purchased from Cell Signaling Technology (Beverly, MA). Goat anti-rabbit-HRP conjugate and goat anti-mouse HRP were purchased from Sigma-Aldrich. Nuclear extraction and DNA binding kits were obtained from Active Motif (Carlsbad, CA). Bacteria-derived recombinant human IL-6 was purchased from ProSpec-TanyTechnoGene Ltd. (Rehovot, Israel).
Chemistry; Synthesis of 1-(2-Amino)-1-(4-methoxyphenylethyl) Cyclohexanol
We initially prepared the compound 1-(2-amino-1-(4-methoxyphenyl)-ethyl)-cyclohexanolmonoacetate as described earlier (22).
Synthesis of 5-(4-Methoxyphenyl)-1-oxa-3-aza-spiro-(5,5) Undecane
To a stirred solution of 1-(2-amino)-1-(4-methoxyphenylethyl)-cyclohexanol 3 (1 eq) in methanol (10 ml), we added various aldehydes (1.2 eq) and anhydrous potassium carbonate (2.5 eq), and the reaction mixture was stirred at room temperature for 4–5 h. After the completion of the reaction, water was added and extracted with ethyl acetate (15 ml). The combined organic layer was dried over anhydrous sodium sulfate. The crude solid was obtained upon evaporation of the solvent under reduced pressure and recrystallized from hexane and ethyl acetate to furnish crystalline solid.
Cell Lines
HCC cell lines HepG2 and PLC/PRF5 cells were obtained from the American Type Culture Collection (Manassas, VA). Huh7-Luc, Hep3B, and LO2 cells were kindly provided by Prof. Kam Man Hui (National Cancer Centre, Singapore). All of the cells were cultured in DMEM containing 1× antibiotic-antimycotic solution with 10% FBS.
Western Blotting
For detection of phosphoproteins, CIMO-treated whole-cell extracts were lysed in lysis buffer (20 mm Tris (pH 7.4), 250 mm NaCl, 2 mm EDTA (pH 8.0), 0.1% Triton X-100, 0.01 mg/ml aprotinin, 0.005 mg/ml leupeptin, 0.4 mm PMSF, and 4 mm NaVO4). Lysates were then spun at 14,000 rpm for 10 min to remove insoluble material and resolved on SDS gel. After electrophoresis, the proteins were electrotransferred to a nitrocellulose membrane, blocked with 5% nonfat milk, and probed with various antibodies (1:1000) overnight at 4 °C. The blot was washed, exposed to HRP-conjugated secondary antibodies for 1 h, and finally examined by chemiluminescence (ECL; GE Healthcare).
To detect STAT3-regulated proteins and PARP, cells (2 × 106/ml) were treated with CIMO for the indicated times in the respective figures. The cells were then washed, and protein was extracted by incubation for 30 min on ice in 0.05 ml of buffer containing 20 mm HEPES, pH 7.4, 2 mm EDTA, 250 mm NaCl, 0.1% Nonidet P-40, 2 μg/ml leupeptin, 2 μg/ml aprotinin, 1 mm PMSF, 0.5 μg/ml benzamidine, 1 mm DTT, and 1 mm sodium vanadate. The lysate was centrifuged, and the supernatant was collected. Whole-cell extract protein (30 μg) was resolved on SDS-PAGE; electrotransferred onto a nitrocellulose membrane; blotted with antibodies against survivin, Bcl-2, Bcl-xL, cyclin D1, Bak, Bid, ICAM-1, procaspase-3, and PARP; and then detected by chemiluminescence (ECL; GE Healthcare).
Immunocytochemistry for STAT3 Localization
HepG2 cells were plated in chamber slides in DMEM containing 10% FBS and allowed to adhere for 24 h. Following treatment with CIMO for 6 h, the cells were fixed with cold acetone for 10 min, washed with PBS, and blocked with 5% normal goat serum for 1 h. The cells were then incubated with rabbit polyclonal anti-human STAT3 antibody (dilution, 1:100). After overnight incubation, the cells were washed and then incubated with goat anti-rabbit IgG-Alexa 594 (1:100) for 1 h and counterstained for nuclei with Hoechst (50 ng/ml) for 5 min. Stained cells were mounted with mounting medium (Sigma-Aldrich) and analyzed under a fluorescence microscope (DP 70, Olympus, Tokyo, Japan).
DNA Binding Assay
DNA binding was performed using a STAT3 DNA binding TransAMTM ELISA kit (Active Motif, Carlsbad, CA). Briefly, nuclear extracts (5 μg) from CIMO-treated cells were incubated in a 96-well plate coated with oligonucleotide containing the STAT3-specific DNA probe. Bound STAT3 was then detected by a specific primary antibody. An HRP-conjugated secondary antibody was subsequently applied to detect the bound primary antibody and provided the basis for colorimetric quantification. The enzymatic product was measured at 450 nm with a microplate reader (Tecan Systems, San Jose, CA). The specificity of this assay was tested by the addition of wild-type or mutated STAT3 consensus oligonucleotide in the competitive or mutated competitive control wells before the addition of the nuclear extracts.
Transfection with STAT3 siRNA and STAT3-mediated Transcription Activity
Human STAT3, small interfering RNA (siRNA), and the α2-macroglobulin luciferase reporter constructs have been described previously (23). Briefly, 60–70% confluent cells in a 6-well plate were transfected using FuGENE6 (Roche Applied Science) transfection reagent. A luciferase assay was performed using the Dual-Luciferase assay kit (Promega Corp., Singapore), Transfections were carried out in triplicate using 1 μg of the appropriate α2-macroglobulin luciferase reporter plasmid and empty vector per transfection along with 0.2 μg of Renilla expression plasmid as a control for transfection efficiency. Luciferase activities were assayed 48 h after transfection using the Dual-Luciferase assay system (Promega) as described previously (23).
MTT Assay
The antiproliferative effect of CIMO against HepG2 cells was determined by the MTT dye uptake method as described previously (24, 25). Briefly, the cells (2.5 × 104/ml) were incubated in triplicate in a 96-well plate in the presence or absence of different concentrations of CIMO in a final volume of 0.2 ml for up to 72 h at 37 °C. Thereafter, 20 μl of MTT solution (5 mg/ml in PBS) was added to each well. After 2 h of incubation at 37 °C, 0.1 ml of lysis buffer (20% SDS, 50% dimethylformamide) was added; incubation was continued overnight at 37 °C; and the optical density at 570 nm was measured by a Tecan plate reader.
Flow Cytometric Analysis
To determine the effect of CIMO on the cell cycle, cells were treated with CIMO at the indicated time points (Fig. 1B) up to 48 h. Thereafter, cells were washed, fixed with 70% ethanol, and incubated for 30 min at 37 °C with 0.1% RNase A in PBS. Cells were then washed again, resuspended, and stained in PBS containing 25 μg/ml propidium iodide for 30 min at room temperature. Cell distribution across the cell cycle was analyzed with a Beckman Coulter flow cytometer.
Migration Assay
An IBIDI culture insert (IBIDI GmbH) with two reservoirs separated by a 500-μm-thick wall created by a culture insert in a 35-mm Petri dish was used. 70 μl of HepG2 cells (5 × 105 cells/ml) were added into the two reservoirs of the same insert and incubated at 37 °C. After 12 h, the insert was gently removed, creating a gap of ∼500 μm. The cells were treated with 5 μm CIMO for 8 h before being exposed to 100 ng/ml CXCL12 for 24 h. The width of the wound was measured at time zero and after 24 h of incubation with and without CIMO in the absence or presence of CXCL12. Graphs were plotted against the percentage of migration distance the cells moved before and after treatment, normalized to control, as described previously (26).
Invasion Assay
The invasion assay was performed with slight modifications in a method described previously (27). A BD BiocoatTM MatrigelTM invasion chamber with 8-μm pores in the light-tight polyethylene terephthalate membrane and was coated with a reconstituted basement membrane gel (BD Biosciences). 2 × 105 cells were suspended in serum-free DMEM and seeded into the Matrigel transwell chambers. The cells were incubated with CIMO for 8 h. After incubation, the outer surfaces of the transwell chambers were wiped with cotton swabs, and the invading cells were fixed and stained with crystal violet solution. The invading cells were then counted in five randomly selected areas under microscopic observation.
Orthotopic Implantation of HCC in Nude Mice
100 μl containing 3 × 106 Huh 7-Luc cells were injected subcutaneously in the right flank of nude mice. When the tumor volume reached ∼1 cm3, the tumor was harvested, cut into 2-mm3 pieces, and then implanted orthotopically into the liver of nude mice. A midline abdominal incision (3–5 cm) was made to expose the whole liver, and the liver capsule was mechanically injured with a needle. A single piece of human HCC tissue (∼2 mm3) was filled into the liver tissue (which was visible as a white spot) with forceps, and the abdominal wall was closed. The skin incisions were closed with wound clips. The development of tumors was monitored by imaging and quantification of the bioluminescence signals using the Xenogen IVIS system (Caliper Life Sciences).
In Vivo Experiments
All animal experiments were performed according to protocols approved by the SingHealth Institutional Animal Use and Care Committee. For drug efficacy study, 8-week-old athymic nu/nu female mice (Biolasco, Taiwan) were implanted with the Huh 7-Luc cells orthotopically as described above. When the bioluminescence signal reached 106, mice were treated either with vehicle (1% DMSO), 2 mg/kg CIMO, or 10 mg/kg CIMO 5 days a week intraperitoneally. Tumor development was monitored twice a week by measuring the bioluminescence signals. Mice were euthanized when the humane end-point criterion is met by CO2 inhalation. Primary liver tumor and lung tissues were excised, snap-frozen, and stored at −80 °C until further analysis.
Immunohistochemical Analysis of Tumor Samples
Solid tumors from control and drug-treated groups were fixed with 10% phosphate-buffered formalin, processed, and embedded in paraffin. The sections were cut to 5-μm size and deparaffinized in xylene, dehydrated in graded alcohol, and finally hydrated in water. Antigen retrieval was conducted by boiling the slide in 10 mm sodium citrate (pH 6.0) for 30 min. Immunohistochemistry was conducted following the manufacturer's instructions (Dako LSAB kit). Briefly, endogenous peroxidases were quenched with 3% hydrogen peroxide. Sections were incubated with primary antibodies for 2 h as follows: anti-phospho-STAT3, anti-Ki-67 and anti-caspase-3 (each at a 1:100 dilution). The slides were subsequently washed several times in TBS with 0.1% Tween 20 and were incubated with biotinylated linker for 45 min, provided in the LSAB kit, according to the manufacturer's instructions. Immunoreactive species were detected using 3,3′-diaminobenzidine tetrahydrochloride as a substrate. The sections were counterstained with Gill's hematoxylin and mounted under glass coverslips. Images were taken using an Olympus BX51 microscope (magnification, 40×). Quantitative analyses of immunohistochemistry images were performed by visual scores between the control and treated images. In this expression quantitation technique, each image is divided into four parts, and each part is individually quantitated for the biomarker expression. A cell scored as positive refers to the presence of brown staining (peroxidase) in any part of the studied tissue. A negative score refers to no staining or weak staining.
Statistical Analysis
An unpaired t test with Welch's correction was used for statistical comparisons between groups; p < 0.05 was considered statistically significant (GraphPad Prism version 5.0, GraphPad Software).
RESULTS
Chemistry
Synthesis and Characterization of Novel Azaspiranes
Multicomponent reactions are a powerful tool to generate the libraries of bioactive compounds. Herein, we synthesized a new set of azaspiranes by utilizing the multicomponent reaction involving 1-[2-amino-1-(4-methoxyphenyl)-ethyl]-cyclohexanolmonoacetate, aryl/benzyl/hetaryl halides, and various aldehydes via single step condensation and nucleophilic substitution reactions in one step. The title compounds were prepared and recrystallized from hexane and ethyl acetate to furnish crystalline solids. The structures of new azaspiranes were deduced based on IR, 1H NMR, 13C NMR, and LCMS spectroscopic analysis.
Pharmacology
CIMO Suppresses Proliferation of HCC Cells in a Dose- and Time-dependent Manner
We first investigated the antiproliferative activity of the novel azaspiranes on HepG2 cells using an MTT assay. Among the tested compounds, CIMO was found to be the most effective with an IC50 of 7.3 μm, compared with other structurally related azaspiranes, with an IC50 ranging from 9.8 to >50 μm. Additionally, CIMO was tested on a panel of six cell lines, including Hep3B, PLC/PRF5, AGS, DU145, MDA MB231, and CAL27 cells. CIMO exhibited a substantial decrease of viable cells in all six tested cell lines. However, CIMO did not show a high cytotoxic effect on LO2 cells up to 72 h at 100 μm, thereby indicating that the CIMO does not have a cytotoxic effect on this non-diseased cell line.
CIMO Causes Accumulation of HepG2 Cells in Sub-G1 Phase
In late apoptosis, activation of endonucleases leads to fragmentation of genomic DNA into oligomers, thereby contributing to a decrease in DNA content, which in turn leads to the buildup of cells in sub-G1 phase. In order to evaluate the effect of CIMO on cell cycle distribution of HepG2 cells, we performed flow cytometric analysis. HepG2 cells were treated with CIMO at different time intervals up to 48 h and analyzed cell cycle distribution after propidium iodide staining. Interestingly, CIMO increased the accumulation of the sub-G1 cell population to 18.8, 38.7, 71, and 92.1% at 16, 24, 36, and 48 h, respectively (Fig. 1B).
CIMO Potently Inhibits Constitutive STAT3 Phosphorylation in HCC Cells
Azaspiranes are well known for their inhibitory action against JAK-STAT pathway. Therefore, we further tested the library of new azaspiranes toward the inhibition of constitutive activation of STAT3 in HepG2 cells by Western blotting via antibodies recognizing phospho-STAT3 (Tyr-705). It was found that CIMO potently inhibited the phosphorylation of STAT3 compared with other structural analogues. As shown in Fig. 2, A and C, levels of phospho-STAT3 were found to be substantially down-regulated in a dose- and time-dependent manner, with maximum inhibition identified at 20 μm and 6 h. At the same time, STAT3 protein expression remained unchanged (Fig. 2, A and C, bottom). We observed that exposure to AG490, a well known inhibitor of JAK2, decreased the phosphorylation of STAT3 in a dose-dependent manner in HepG2 cells with the maximum inhibition at 200 μm for 6 h (Fig. 2B) (28). However, CIMO showed a comparable effect already at a 10-fold lower concentration and hence much higher potency.
FIGURE 2.

A, CIMO suppresses phospho-STAT3 in a dose-dependent manner. HepG2 cells (5 × 105/ml) were treated with the indicated concentrations of CIMO for 6 h, after which whole-cell extract was prepared and resolved on SDS-polyacrylamide gel, electrotransferred onto nitrocellulose membrane, and probed for phospho-STAT3, and the same blot was stripped and reprobed with STAT3 antibody to verify equal protein loading. B, AG490 suppresses phospho-STAT3 in a dose-dependent manner. HepG2 cells (5 × 105/ml) were treated with the indicated concentrations of AG490 for 6 h, after which Western blotting was done as described for A. C, CIMO suppresses phospho-STAT3 levels in a time-dependent manner. HepG2 cells (5 × 105/ml) were treated with 10 μmol/liter CIMO for the indicated times, after which Western blotting was done as described for A. D, CIMO had no effect on phospho-STAT3(Ser-727) and STAT3 protein expression. HepG2 cells (5 × 105/ml) were treated with 10 μmol/liter CIMO for the indicated times, after which Western blotting was done as described for A, and the membrane was probed using antibodies against phospho-STAT3(Ser-727) and STAT3. E, CIMO inhibits the translocation of STAT3 to the nucleus. HepG2 cells (1 × 105/ml) were incubated with or without 10 μmol/liter CIMO for 6 h and then analyzed for the intracellular distribution of STAT3 by immunocytochemistry. The same slides were counterstained for nuclei with Hoechst (50 ng/ml) for 5 min and analyzed under a fluorescence microscope. F, CIMO suppresses phospho-Src, phospho-JAK1, and phospho-JAK2 levels in a time-dependent manner. HepG2 cells (5 × 105/ml) were treated with 10 μmol/liter CIMO, after which whole-cell extracts were prepared, resolved in SDS-PAGE, electrotransferred onto nitrocellulose membranes, and probed with phospho-Src, phospho-JAK1, and phospho-JAK2 antibodies. The same blots were stripped and reprobed with Src, JAK1, and JAK2 antibodies to verify equal protein loading. G, CIMO suppresses STAT3 DNA binding ability in HepG2 cells. HepG2 cells were treated with 10 μmol/liter CIMO for the indicated time, nuclear extracts were prepared, and 5 μg of the nuclear extract protein was used for the ELISA-based DNA-binding assay. *, p < 0.05.
Effect of CIMO on STAT3 Phosphorylation Is Specific for Tyr-705
Given that STAT3 can undergo phosphorylation at Tyr-705 or Ser-727, which is mediated by Janus kinase and Akt, respectively (29), and that phospho-STAT3(Ser-727) has been reported to regulate transcription activation in MAPK pathway as well (30), the effect of CIMO on phosphorylation of serine 727 in STAT3 was investigated next. Here it was found that CIMO completely inhibited the phosphorylation of Tyr-705; however, it had no effect on phosphorylation of Ser-727 (Fig. 2, C and D), indicating that CIMO inhibits upstream tyrosine kinases.
CIMO Depletes Nuclear Localization of STAT3 in HCC Cells
Inhibition of phosphorylation of STAT3 at Tyr-705 suppresses nuclear translocation and down-regulates the expression of target genes (31). We hence evaluated whether CIMO can inhibit nuclear translocation of STAT3. Fig. 2E clearly demonstrates that CIMO causes a significant decrease of STAT3 in the nucleus of HepG2 cells. This overall represents conclusive evidence that CIMO inhibits phosphorylation of STAT3 and accumulates in the cytoplasm.
CIMO Suppresses Constitutive Activation of c-Src, JAK1, and JAK2 in HCC Cells
Given that the activation of STAT3 is regulated by soluble tyrosine kinases of c-Src and JAK family proteins (32, 33). CIMO treatment presented significant inhibition of phosphorylation of c-Src kinase, JAK1, and JAK2 (Fig. 2F), without affecting the levels of c-Src, JAK1, and JAK2 proteins. Therefore, the results obtained confirmed that inhibition of STAT3 is due to the inhibition of c-Src and JAK family proteins.
CIMO Inhibits STAT3 DNA Binding Activity in HCC Cells
We further investigated whether CIMO modulates STAT3 DNA binding activity in HepG2 cells because STAT3 dimer translocates into the nucleus and binds to specific DNA nucleotide sequence to regulate gene expression (34, 35). CIMO suppressed the binding of STAT3 to the DNA in a time-dependent manner in HepG2 cells (Fig. 2G).
CIMO Inhibits STAT3-mediated Transcription Activity in HepG2 Cells
Increased STAT3 activity has been previously reported to stimulate oncogenicity of hepatocellular carcinoma (14, 36). Therefore, we first evaluated the level of phosphorylated STAT3(Tyr-705) in HepG2 cells with siRNA-mediated deletion of STAT3 transcripts and/or exposure to CIMO, using Western blot analysis. Transient transfection of STAT3-directed siRNA in HepG2 cells resulted in decreased levels of phosho-STAT3 and total STAT3 protein compared with their vector control cells, demonstrated using Western blot. On the other hand, application of CIMO to HepG2 cells resulted in decreased phospho-STAT3 levels compared with their control cells exposed with DMSO (Fig. 3A). In contrast, the protein levels of total STAT3 were not significantly altered in HepG2 cells upon exposure to CIMO, when compared with their DMSO-exposed control.
FIGURE 3.

A, comparative study of cell viability between STAT3-siRNA-transfected and CIMO-treated HepG2 cells. Correspondingly, Western blot analysis was used to assess the levels of phospho-STAT3 and STAT3 in HepG2 cells with siRNA-mediated depletion of STAT3 expression and upon exposure to 4 μm CIMO. Whole-cell extract was prepared and resolved on SDS-polyacrylamide gel, electrotransferred onto nitrocellulose membrane, and probed for phospho-STAT3, and the same blot was stripped and reprobed with STAT3 antibody and β-actin to verify equal protein loading. B, CIMO modulates STAT3-mediated transcription and α2-M promoter activity in HepG2 cells. C, CIMO inhibits IL-6-induced phosphorylation of STAT3, JAK1, and JAK2. Hep3B cells (5 × 105/ml) were treated with 10 μmol/liter CIMO for the indicated times and then stimulated with IL-6 (10 ng/ml) for 15 min. Whole-cell extracts were then prepared, resolved on an SDS-polyacrylamide gel, electrotransferred onto nitrocellulose membrane, and probed with phospho-STAT3, phospho-JAK1, and phospho-JAK2 antibodies. The same blot was stripped and reprobed with STAT3, JAK1, and JAK2 antibody to verify equal protein loading. D and E, CIMO suppresses STAT3-regulated gene products involved in cell proliferation and survival. HepG2 cells (5 × 105/ml) were treated with the 10 μmol/liter CIMO for the indicated time intervals, after which whole-cell extract was prepared, resolved on an SDS-polyacrylamide gel, and electrotransferred onto nitrocellulose membrane, and the membrane was sliced according to molecular weight and probed against Bcl-2, cyclin D1, Survivin, Bak, ICAM-1, Bcl-xL, and Bid. The same blot was stripped and reprobed with β-actin antibody to verify equal protein loading. Error bars, S.E.
In addition, we subsequently assessed STAT3-mediated transcriptional activity using an α2-macroglobulin (α2-M) promoter in HepG2 cells with either siRNA-mediated depletion of STAT3 expression or by exposure to CIMO (Fig. 3B). The α2-M reporter construct contains a fragment of the α2-M gene promoter (−215 to +8 bp) to which STAT3 binds and induces transcription of this gene. siRNA-mediated depletion of STAT3 expression in HepG2 cells exhibited decreased α2-M promoter activity when compared with their vector control cells. Similarly, upon exposure to the CIMO compound, HepG2 cells exhibited decreased α2-M promoter activity when compared with their control cells exposed with DMSO.
CIMO Down-regulates IL-6-induced JAK1, JAK2, and STAT3 Phosphorylation in HCC Cells
Elevated levels of serum IL-6 have been reported in various types of cancers, leading to the overactivation of STAT3 (37, 38). Hep3B are HCC cells that lack constitutively active JAK and STAT3 proteins. CIMO substantially down-regulated the IL-6-induced phosphorylation of JAK1, JAK2, and STAT3 in Hep3B cells (Fig. 3C). These results clearly demonstrate that CIMO modulates both constitutive and inducible activation of proteins of the JAK-STAT pathway.
CIMO Regulates the Expression of STAT3-targeted Genes Involved in Cell Proliferation and Survival
STAT3 activation has been reported to regulate the expression of proapoptotic and antiapoptotic proteins (39, 40). Therefore, we investigated whether CIMO modulates the expression of various STAT3-regulated proapoptotic and antiapoptotic genes. We found the down-regulation of antiapoptotic proteins, including Bcl-2, Bcl-xL, Survivin, ICAM-1, Bid, and cell cycle regulator cyclin D1. We also found the up-regulation of proapoptotic protein Bak in a time-dependent manner with maximum activity at 36 h (Fig. 3, D and E). This finding provides evidence that CIMO inhibits survival signaling on multiple levels, hence rendering the cell more prone to apoptosis induction.
CIMO Activates Procaspase-3 and Induces Cleavage of PARP
Activated caspase-3 cleaves the full-length PARP (116 kDa) into 85- and 24-kDa fragments, and PARP is involved in the DNA repair mechanism and drives the cell to apoptosis (41). We investigated whether suppression of constitutively active STAT3 by CIMO leads to apoptosis. Fig. 4A demonstrates the activation of procaspase-3 and subsequent decline of full-length PARP with an increase in the cleaved 85-kDa fragment in a time-dependent manner. These results clearly indicate that CIMO induces caspase-3-mediated apoptosis in HepG2 cells.
FIGURE 4.
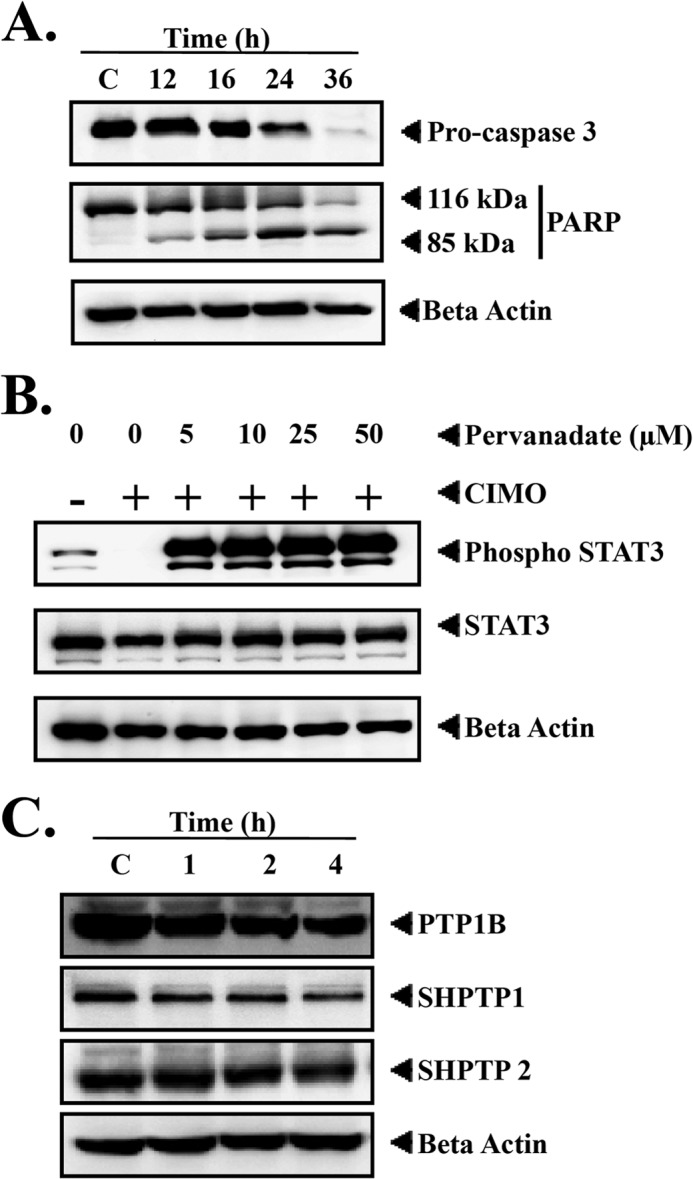
A, CIMO activates caspase-3 and induces apoptosis. HepG2 cells (5 × 105/ml) were treated with 10 μmol/liter CIMO for the indicated times, and whole-cell extracts were prepared, separated on SDS-PAGE, and subjected to Western blotting against caspase-3 and PARP antibody. The same blot was stripped and reprobed with β-actin antibody to show equal protein loading. B, pervanadate reversed the inhibitory effect of CIMO on phospho-STAT3. HepG2 cells (5 × 105/ml) were treated with the indicated concentrations of pervanadate and 10 μmol/liter CIMO for 4 h, after which whole-cell extracts were prepared, resolved on an SDS-polyacrylamide gel, electrotransferred onto nitrocellulose membrane, and probed for phospho-STAT3 and STAT3. C, inhibitory activity of CIMO on phospho-STAT3 is mediated by protein-tyrosine phosphatase. HepG2 cells (5 × 105/ml) were treated with 10 μmol/liter CIMO for the indicated times; whole-cell extracts were prepared, separated on SDS-PAGE, and subjected to Western blotting against PTP1B, SHPTP1, and SHPTP2 antibody; and the same blot was stripped and reprobed with β-actin antibody to show equal protein loading.
Tyrosine Phosphatases Are Involved in CIMO-induced Inhibition of STAT3 Activation
Protein-tyrosine phosphatases have been implicated in STAT3 activation (42). We determined whether CIMO-induced inhibition of STAT3 tyrosine phosphorylation could be due to activation of a protein-tyrosine phosphatase. Treatment of HepG2 cells with the broad spectrum tyrosine phosphatase inhibitor sodium pervanadate prevented CIMO-induced inhibition of STAT3 activation (Fig. 4B). This suggests that tyrosine phosphatases are involved in CIMO-induced inhibition of STAT3 activation. Based on this, we also analyzed the expression of various tyrosine phosphatases, including SHP-1, SHPTP-2, and PTP1B, upon treatment with CIMO for up to 4 h and found no change in the levels of these phosphatases (Fig. 4C). Based on these results, we are hypothesizing the involvement of some other phosphatases in the reversal of the effect of CIMO on STAT3 phosphorylation.
CIMO Suppresses CXCL12-induced HepG2 Cell Migration and Invasion
STAT3-targeted gene products are known to be involved in cancer cell migration (10). Fig. 5A interprets the movement of the cells in the presence and absence of CIMO and CXCL12. CIMO limits the HepG2 cell migration by nearly 50%, both in the presence and absence of CXCL12, compared with the respective controls. In the case of the invasion assay, more than 50% of HepG2 cell motility was inhibited both in the presence and absence of CXCL12 across the polyethylene terephthalate membrane, suggesting that CIMO interferes with cell invasion (Fig. 5B).
FIGURE 5.
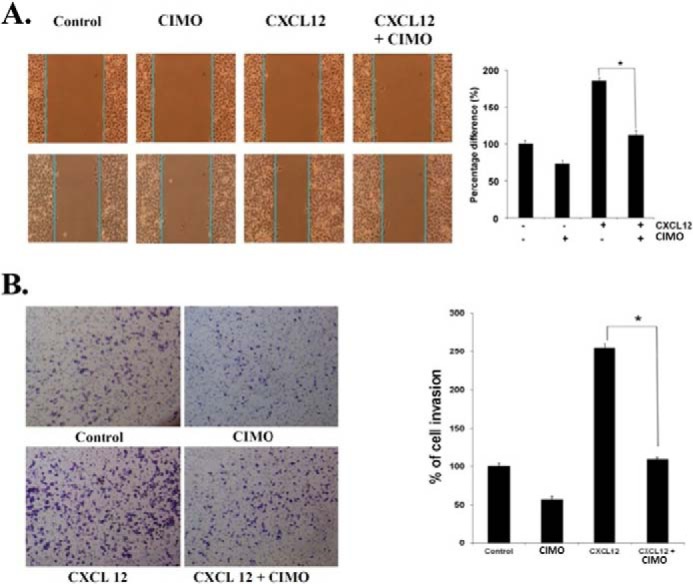
A, CIMO inhibits cell migration. 70 μl of HepG2 cells (5 × 105/ml) were seeded into each compartment of the culture insert, and the insert was removed after 12 h. The width of the wound was measured initially, and cells were incubated with and without CIMO (5 μm, 8 h) and CXCL12 (100 ng/ml, 24 h). B, CIMO inhibits the cell invasion. HepG2 (2 × 105) cells were seeded in the top chamber of BD BioCoatTM MatrigelTM. After preincubation with or without 5 μmol/liter CIMO for 8 h, transwell chambers were placed into the wells of a 24-well plate that contained either only basal medium or basal medium with CXCL12 (100 ng/ml) for 24 h. After incubation, the chamber was assessed for cell invasion by staining with crystal violet. Error bars, S.E.
CIMO Suppresses the Growth of Human HCC in Vivo and STAT3 Activation in Tumor Tissues
We also tested the antitumor potential of CIMO in vivo via intraperitoneal administration in an orthotopic model of human HCC using Huh 7-Luc-transfected cells. It was found that CIMO at a concentration of 10 mg/kg induced significant inhibition of tumor growth compared with the DMSO-treated controls (Fig. 6A). The unpaired t test showed a statistically significant difference in tumor growth between the CIMO-treated and control groups (p value = 0.0385 as compared with the DMSO-treated control group). We further analyzed the effect of CIMO on constitutive phospho-STAT3 levels in HCC tumor tissues by immunohistochemical analysis and found that CIMO significantly inhibited constitutive STAT3 activation in the treated versus control group (Fig. 6B). The effect of CIMO was also analyzed upon the expression of Ki-67 (marker of proliferation) and cleaved caspase-3 (marker of apoptosis). As shown in Fig. 7, expression of Ki-67 was down-regulated, and that of cleaved caspase-3 was significantly increased in the CIMO-treated group, compared with control.
FIGURE 6.
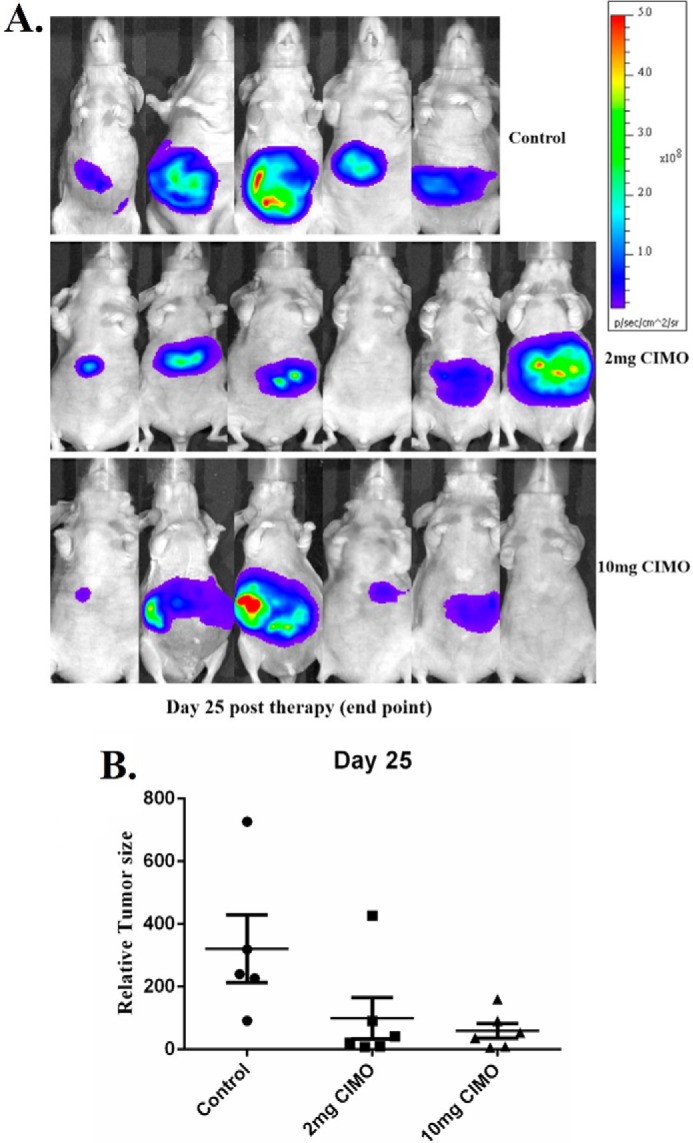
CIMO inhibits the growth of human HCC in vivo. A, representative images of mice from bioluminescent imaging. B, relative tumor burden in athymic mice bearing orthotopically implanted Huh 7-Luc2 tumors treated with vehicles alone (n = 5) or 2 mg/kg (n = 6) or 10 mg/kg (n = 6) CIMO. Points, mean; bars, S.E. *, p < 0.05 (unpaired Student's t test).
FIGURE 7.
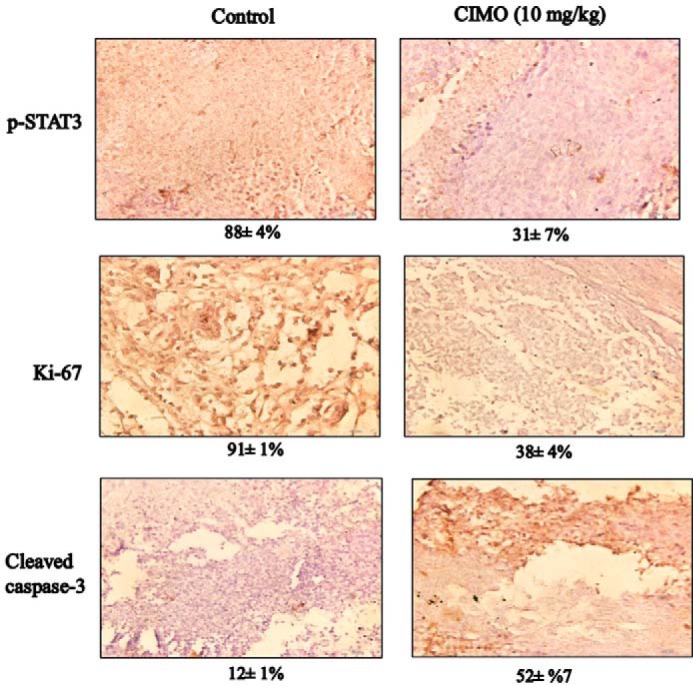
Immunohistochemical analysis of phospho-STAT3 (p-STAT3), Ki-67, and caspase-3 showed the inhibition in expression of phospho-STAT3, and Ki-67 and increased levels of cleaved caspase-3 expression in CIMO-treated samples as compared with the control group. The percentage indicates positive staining for the given biomarker. The photographs were taken at a magnification of ×40.
In Silico Interaction of CIMO with the Kinase Domain of JAK2
In order to better understand compound action on a mechanistic level, we also performed computational studies in the next step. In the current study, CIMO showed potent anti-cancer activity via the JAK2-STAT3 pathway; hence, we considered the possibility that CIMO interacts with the kinase domain of JAK2 directly. Thus, the JAK2 inhibitor 1-methyl-1H-imidazole, which modulated the JAK/STAT pathway, was considered for our studies (20). A molecular docking study was carried out to examine the possibility of CIMO binding to the kinase domain of JAK2. The docking scores of the biologically active ligands with the kinase domain of JAK2 (Protein Data Bank entry 4C61) are summarized (Fig. 8A) (20). Based on Ligand Fit docking score calculations, CIMO shows a docking score of 95.07 kcal/mol, which is higher when compared with other structurally related azaspiranes. The known STAT3 inhibitors, such as Stattic and staurosporine, bound to the kinase domain of JAK2 with predicted binding energies of 37.2 and 83.2 kcal/mol, respectively, which was comparable with CIMO. Fig. 8 shows the best docked pose of CIMO as defined by the highest DOCK score. The protein-ligand interactions as shown in Fig. 8C have been classified into four clusters in order to enable identification and comparison of interaction patterns of CIMO moieties with specific JAK2 residues across other known JAK2 inhibitors. In cluster 1, the cyano-biphenyl moiety of CIMO bound to the hydrophobic pocket comprising Leu-855, Gly-856, Ala-880, Met-929, Val-863, Leu-932, and Gly-935. In cluster II, the indole moiety interacted with Gly-993, Asp-994, Gly-882, and Ser-862. In cluster III, the cyclohexyl-attached azaspirane moiety of CIMO bound to Gly-996, Leu-997, Glu-898, Phe-895, Gly-861, and Leu-884. In addition, the methoxy phenyl moiety of CIMO interacted with Asp-894, His-891, and Glu-890. These results indicate that CIMO could bind strongly to the kinase domain of JAK2.
FIGURE 8.

Bioinformatics approach of CIMO interaction toward the kinase domain of JAK2. A, ribbon diagram of the monomer of the JAK2 and its ligand binding site (LBS) of kinase domain (green). B, surface view of JAK2 and the bound CIMO in the ligand binding site region. C, interaction map of ligand binding site domain of JAK2 that interacted with CIMO. The labeled key amino acids are represented as a line model with the carbon atom in black, and other atoms in their parent colors. Shown is the binding of CIMO, whose carbon atom is green, and other atoms with their parent colors.
Cheminformatics-based Mode-of-action Rationalization for CIMO Predicts the JAK-STAT Pathway as a Possible Target
Rationalizing mode-of-action hypotheses utilizing the wealth of experimental data available in the public domain is now possible with chemogenomics and cheminformatics applications (43). In this regard, we applied the well established Laplacian-modified naive Bayesian classifier as implemented by Koutsoukas et al. (44) and predicted potential targets of CIMO. It was found that membrane-associated phospholipase A2, histamine H2 receptor, proteinase-activated receptor 1, steroid hormone receptor ERR1, gonadotrophin-releasing hormone receptor, and prostaglandin E synthase had a normalized likelihood of 10.41, 4.98, 4.96, 4.43, 4.04, and 4.04 for CIMO, respectively. These predicted targets for CIMO are known to cross-talk with the JAK-STAT pathway (45). Therefore, the in silico rationalization of the mode-of-action analysis for CIMO suggested the involvement of its anti-cancer effect through the JAK-STAT pathway.
DISCUSSION
STAT3 is an inducible monomeric transcription factor that dimerizes upon phosphorylation at Tyr-705 and translocates to the nucleus. It relays the oncogenic signals by permitting the expression of the target genes involved in uncontrolled cell proliferation, angiogenesis, apoptotic resistance, and tumor evasion. Therefore, inhibition of STAT3 signaling is an attractive approach in order to inhibit cell proliferation. The aim of this study was to synthesize new azaspirane-based small molecules and to evaluate whether new compounds can disrupt STAT3 signaling. This work reported the synthesis of 13 new azaspirane derivatives, which resulted in the discovery of CIMO as a lead molecule that exhibited good cytotoxic effect on HCC cells. CIMO inhibits constitutive and IL-6-induced activation of STAT3, and its inhibitory effect is specific to Tyr-705 in HCC cells. The role of STAT3 phosphorylation at Tyr-705 in tumorigenesis is well established; on the other hand, STAT3 can undergo phosphorylation at Ser-727, and its role in cancer progression remains controversial (46, 47). Moreover, CIMO proved to be a more effective inhibitor of the JAK-STAT pathway than AG490. Our results indicate that CIMO specifically inhibits Tyr-705 phosphorylation while showing no effect on Ser-727 phosphorylation in HCC cells. An inhibitory effect of CIMO on STAT3 phosphorylation was evident with the down-regulation of JAK1, JAK2, and c-Src proteins. JAK and c-Src proteins are the foremost tyrosine kinases with a critical role in STAT3 phosphorylation (48). This result confirms the CIMO-mediated blockade of upstream protein-tyrosine kinases in regulating aberrant behavior of STAT3 in the cancer cells.
Restriction of nuclear translocation and accumulation of STAT3 in the cytoplasm is a hallmark of abrogation of the JAK-STAT pathway. After understanding the critical role of phospho-STAT3 as a latent transcription factor, we analyzed the distribution of STAT3 in CIMO-treated HepG2 cells. The results obtained provide strong evidence demonstrating the link between phosphorylation and shuttling of STAT3 into the nucleus. A reduction in nuclear localization of STAT3 directly correlated with the decreased phosphorylation of STAT3 in the previous experiment. Gritsko et al. (49) have reported that STAT3 induces survivin gene expression and leads to apoptotic resistance. We also found that down-regulation of STAT3 regulated tumorigenic proteins, including Bcl-2, Bcl-xL, cyclin D1, survivin, ICAM-1, and Bid. This suggests the role of CIMO in limiting the expression of IAP (inhibitor of apoptosis) and cell cycle-regulating proteins. In addition, PARP and CAD (caspase-activated DNase) are two well known substrates of caspase-3. Activated caspase-3 catalyzes activation of CAD and cleavage of PARP, which results in formation of DNA oligomers. Our results on the cleavage of procaspase-3 and PARP and the deposition of hypodiploid cells in sub-G1 phase markedly indicated the apoptosis-inducing effect of CIMO in HCC cells.
SHP1, SH-PTP2, PTP-1B, and PTEN are some of the important protein-tyrosine phosphatases connected with STAT3 signaling. We further investigated the involvement of protein-tyrosine phosphatases in the suppression of STAT3 activation. Treatment of sodium pervanadate reversed the effect of CIMO on inhibition of STAT3 activation. This observation suggests the involvement of phosphatases in CIMO-induced STAT3 inhibition. Our experiments suggested that SHP1, SHPTP2, and PTP1B are not involved in the reversal of CIMO-induced STAT3 inhibition, but it is not clear which specific phosphatase is involved in this process, and it requires further investigation. We also demonstrated the substantial decline in HCC development in an orthotopic mouse model. This observation was supported by immunohistochemistry analysis data in which Ki-67 (biomarker of proliferation) and phospho-STAT3 levels were significantly down-regulated with a simultaneous increase in cleaved caspase-3 (biomarker of apoptosis) in tumor tissues treated with CIMO. Our results overall demonstrate the CIMO is a potent agent with an antiproliferative effect in vitro and in vivo, which shows its effect on HCC via abrogation of the JAK-STAT signaling cascade.
This work was supported by University Grants Commission Grant 41-257-2012-SR and Vision Group Science and Technology, Department of Science and Technology, Grant SR/FT/LS-142/2012 (to B.). This work was also supported by a grant from the National Medical Research Council of Singapore, Academic Research Fund Tier 1, and National University Health System Bench-to-Bedside and Bench-to-Bedside-to-Product Grant (to G. S.).
- HCC
- hepatocellular carcinoma
- CIMO
- 2-(1-(4-(2-cyanophenyl)1-benzyl-1H-indol-3-yl)-5-(4-methoxyphenyl)-1-oxa-3-aza spiro(5,5) undecane
- PARP
- poly(ADP-ribose polymerase)
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
REFERENCES
- 1. Whang-Peng J., Cheng A.-L., Hsu C., Chen C.-M. (2010) Clinical development and future direction for the treatment of hepatocellular carcinoma. J. Exp. Clin. Med. 2, 93–103 [Google Scholar]
- 2. Rajendran P., Ong T. H., Chen L., Li F., Shanmugam M. K., Vali S., Abbasi T., Kapoor S., Sharma A., Kumar A. P., Hui K. M., Sethi G. (2011) Suppression of signal transducer and activator of transcription 3 activation by butein inhibits growth of human hepatocellular carcinoma in vivo. Clin. Cancer Res. 17, 1425–1439 [DOI] [PubMed] [Google Scholar]
- 3. Sun B., Karin M. (2012) Obesity, inflammation, and liver cancer. J. Hepatol. 56, 704–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wörns M. A., Galle P. R. (2010) Future perspectives in hepatocellular carcinoma. Dig. Liver Dis. 42, S302–S309 [DOI] [PubMed] [Google Scholar]
- 5. Nakagawa H., Maeda S. (2012) Molecular mechanisms of liver injury and hepatocarcinogenesis: focusing on the role of stress-activated MAPK. Pathol. Res. Int. 2012, 172894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gish R. G. (2006) Hepatocellular carcinoma: overcoming challenges in disease management. Clin. Gastroenterol. Hepatol. 4, 252–261 [DOI] [PubMed] [Google Scholar]
- 7. McMurray J. S. (2006) A new small-molecule Stat3 inhibitor. Chem. Biol. 13, 1123–1124 [DOI] [PubMed] [Google Scholar]
- 8. Turkson J., Kim J. S., Zhang S., Yuan J., Huang M., Glenn M., Haura E., Sebti S., Hamilton A. D., Jove R. (2004) Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol. Cancer Ther. 3, 261–269 [PubMed] [Google Scholar]
- 9. Frank D. A. (2007) STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 251, 199–210 [DOI] [PubMed] [Google Scholar]
- 10. Huang S. (2007) Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin. Cancer Res. 13, 1362–1366 [DOI] [PubMed] [Google Scholar]
- 11. Bromberg J. (2002) Stat proteins and oncogenesis. J. Clin. Invest. 109, 1139–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Temple C., Jr., Wheeler G. P., Comber R. N., Elliott R. D., Montgomery J. A. (1983) Synthesis of potential anticancer agents: pyrido[4,3-b][1,4]oxazines and pyrido[4,3-b][1,4]thiazines. J. Med. Chem. 26, 1614–1619 [DOI] [PubMed] [Google Scholar]
- 13. Basappa, Murugan S., Kavitha C. V., Purushothaman A., Nevin K. G., Sugahara K., Rangappa K. S. (2010) A small oxazine compound as an anti-tumor agent: a novel pyranoside mimetic that binds to VEGF, HB-EGF, and TNF-α. Cancer Lett. 297, 231–243 [DOI] [PubMed] [Google Scholar]
- 14. Quintás-Cardama A., Manshouri T., Estrov Z., Harris D., Zhang Y., Gaikwad A., Kantarjian H. M., Verstovsek S. (2011) Preclinical characterization of atiprimod, a novel JAK2 and JAK3 inhibitor. Invest. New Drugs 29, 818–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang S. M., Malaviya R., Wilson L. J., Argentieri R., Chen X., Yang C., Wang B., Cavender D., Murray W. V. (2007) Simplified staurosporine analogs as potent JAK3 inhibitors. Bioorg. Med. Chem. Lett. 17, 326–331 [DOI] [PubMed] [Google Scholar]
- 16. Tam C. S., Verstovsek S. (2013) Investigational Janus kinase inhibitors. Expert Opin. Investig. Drugs 22, 687–699 [DOI] [PubMed] [Google Scholar]
- 17. Amit-Vazina M., Shishodia S., Harris D., Van Q., Wang M., Weber D., Alexanian R., Talpaz M., Aggarwal B. B., Estrov Z. (2005) Atiprimod blocks STAT3 phosphorylation and induces apoptosis in multiple myeloma cells. Br. J. Cancer 93, 70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hamasaki M., Hideshima T., Tassone P., Neri P., Ishitsuka K., Yasui H., Shiraishi N., Raje N., Kumar S., Picker D. H., Jacob G. S., Richardson P. G., Munshi N. C., Anderson K. C. (2005) Azaspirane (N-N-diethyl-8,8-dipropyl-2-azaspiro [4.5] decane-2-propanamine) inhibits human multiple myeloma cell growth in the bone marrow milieu in vitro and in vivo. Blood 105, 4470–4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilson L. J., Malaviya R., Yang C., Argentieri R., Wang B., Chen X., Murray W. V., Cavender D. (2009) Synthetic staurosporines via a ring closing metathesis strategy as potent JAK3 inhibitors and modulators of allergic responses. Bioorg. Med. Chem. Lett. 19, 3333–3338 [DOI] [PubMed] [Google Scholar]
- 20. Su Q., Ioannidis S., Chuaqui C., Almeida L., Alimzhanov M., Bebernitz G., Bell K., Block M., Howard T., Huang S., Huszar D., Read J. A., Rivard Costa C., Shi J., Su M., Ye M., Zinda M. (2014) Discovery of 1-methyl-1H-imidazole derivatives as potent Jak2 inhibitors. J. Med. Chem. 57, 144–158 [DOI] [PubMed] [Google Scholar]
- 21. Furqan M., Mukhi N., Lee B., Liu D. (2013) Dysregulation of JAK-STAT pathway in hematological malignancies and JAK inhibitors for clinical application. Biomark. Res. 1, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Basappa, Kavitha C. V., Rangappa K. S. (2004) Simple and an efficient method for the synthesis of 1-[2-dimethylamino-1-(4-methoxy-phenyl)-ethyl]-cyclohexanol hydrochloride: (+/-) venlafaxine racemic mixtures. Bioorg. Med. Chem. Lett. 14, 3279–3281 [DOI] [PubMed] [Google Scholar]
- 23. Pandey V., Jung Y., Kang J., Steiner M., Qian P. X., Banerjee A., Mitchell M. D., Wu Z. S., Zhu T., Liu D. X., Lobie P. E. (2010) Artemin reduces sensitivity to doxorubicin and paclitaxel in endometrial carcinoma cells through specific regulation of CD24. Transl. Oncol. 3, 218–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keerthy H. K., Mohan C. D., Siveen K. S., Fuchs J. E., Rangappa S., Sundaram M. S., Li F., Girish K. S., Sethi G., Basappa, Bender A., Rangappa K. S. (2014) Novel synthetic biscoumarins target tumor necrosis factor-α in hepatocellular carcinoma in vitro and in vivo. J. Biol. Chem. 289, 31879–31890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keerthy H. K., Garg M., Mohan C. D., Madan V., Kanojia D., Shobith R., Nanjundaswamy S., Mason D. J., Bender A., Basappa, Rangappa K. S., Koeffler H. P. (2014) Synthesis and characterization of novel 2-amino-chromene-nitriles that target Bcl-2 in acute myeloid leukemia cell lines. PloS One 9, e107118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manu K. A., Shanmugam M. K., Ong T. H., Subramaniam A., Siveen K. S., Perumal E., Samy R. P., Bist P., Lim L. H., Kumar A. P., Hui K. M., Sethi G. (2013) Emodin suppresses migration and invasion through the modulation of CXCR4 expression in an orthotopic model of human hepatocellular carcinoma. PLoS One 8, e57015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar B., Paricharak S., Dinesh K., Siveen K. S., Fuchs J., Rangappa S., Mohan C. D., Mohandas N., Kumar A. P., Sethi G. (2014) Synthesis, biological evaluation and in silico and in vitro mode-of-action analysis of novel dihydropyrimidones targeting PPAR-γ. RSC Adv. 4, 45143–45146 [Google Scholar]
- 28. Meydan N., Grunberger T., Dadi H., Shahar M., Arpaia E., Lapidot Z., Leeder J. S., Freedman M., Cohen A., Gazit A., Levitzki A., Roifman C. M. (1996) Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature 379, 645–648 [DOI] [PubMed] [Google Scholar]
- 29. Murase S., McKay R. D. (2014) Neuronal activity-dependent STAT3 localization to nucleus is dependent on Tyr-705 and Ser-727 phosphorylation in rat hippocampal neurons. Eur. J. Neurosci. 39, 557–565 [DOI] [PubMed] [Google Scholar]
- 30. Decker T., Kovarik P. (2000) Serine phosphorylation of STATs. Oncogene 19, 2628–2637 [DOI] [PubMed] [Google Scholar]
- 31. Li F., Shanmugam M. K., Chen L., Chatterjee S., Basha J., Kumar A. P., Kundu T. K., Sethi G. (2013) Garcinol, a polyisoprenylated benzophenone modulates multiple proinflammatory signaling cascades leading to the suppression of growth and survival of head and neck carcinoma. Cancer Prev. Res. 6, 843–854 [DOI] [PubMed] [Google Scholar]
- 32. Ihle J. N. (1996) STATs: signal transducers and activators of transcription. Cell 84, 331–334 [DOI] [PubMed] [Google Scholar]
- 33. Schreiner S. J., Schiavone A. P., Smithgall T. E. (2002) Activation of STAT3 by the Src family kinase Hck requires a functional SH3 domain. J. Biol. Chem. 277, 45680–45687 [DOI] [PubMed] [Google Scholar]
- 34. Rajendran P., Li F., Shanmugam M. K., Kannaiyan R., Goh J. N., Wong K. F., Wang W., Khin E., Tergaonkar V., Kumar A. P., Luk J. M., Sethi G. (2012) Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of STAT3/JAK2 signaling cascade in vitro and in vivo. Cancer Prev. Res. 5, 631–643 [DOI] [PubMed] [Google Scholar]
- 35. Yu C. L., Meyer D. J., Campbell G. S., Larner A. C., Carter-Su C., Schwartz J., Jove R. (1995) Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 269, 81–83 [DOI] [PubMed] [Google Scholar]
- 36. Rosmorduc O., Desbois-Mouthon C. (2011) Targeting STAT3 in hepatocellular carcinoma: sorafenib again. J. Hepatol. 55, 957–959 [DOI] [PubMed] [Google Scholar]
- 37. Moran D. M., Mattocks M. A., Cahill P. A., Koniaris L. G., McKillop I. H. (2008) Interleukin-6 mediates G0/G1 growth arrest in hepatocellular carcinoma through a STAT 3-dependent pathway. J. Surg. Res. 147, 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zauberman A., Zipori D., Krupsky M., Ben-Levy R. (1999) Stress activated protein kinase p38 is involved in IL-6 induced transcriptional activation of STAT3. Oncogene 18, 3886–3893 [DOI] [PubMed] [Google Scholar]
- 39. Aggarwal B. B., Vijayalekshmi R. V., Sung B. (2009) Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin. Cancer Res. 15, 425–430 [DOI] [PubMed] [Google Scholar]
- 40. Rajendran P., Li F., Manu K. A., Shanmugam M. K., Loo S. Y., Kumar A. P., Sethi G. (2011) γ-Tocotrienol is a novel inhibitor of constitutive and inducible STAT3 signalling pathway in human hepatocellular carcinoma: potential role as an antiproliferative, pro-apoptotic and chemosensitizing agent. Br. J. Pharmacol. 163, 283–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boulares A. H., Yakovlev A. G., Ivanova V., Stoica B. A., Wang G., Iyer S., Smulson M. (1999) Role of poly(ADP-ribose) polymerase (PARP) Cleavage in apoptosis: caspase 3-resistant parp mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 274, 22932–22940 [DOI] [PubMed] [Google Scholar]
- 42. Han Y., Amin H. M., Franko B., Frantz C., Shi X., Lai R. (2006) Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood 108, 2796–2803 [DOI] [PubMed] [Google Scholar]
- 43. Koutsoukas A., Simms B., Kirchmair J., Bond P. J., Whitmore A. V., Zimmer S., Young M. P., Jenkins J. L., Glick M., Glen R. C., Bender A. (2011) From in silico target prediction to multi-target drug design: current databases, methods and applications. J. Proteomics 74, 2554–2574 [DOI] [PubMed] [Google Scholar]
- 44. Koutsoukas A., Lowe R., Kalantarmotamedi Y., Mussa H. Y., Klaffke W., Mitchell J. B., Glen R. C., Bender A. (2013) In silico target predictions: defining a benchmarking data set and comparison of performance of the multiclass Naive Bayes and Parzen-Rosenblatt window. J. Chem. Inf. Model. 53, 1957–1966 [DOI] [PubMed] [Google Scholar]
- 45. Yellaturu C. R., Rao G. N. (2003) Cytosolic phospholipase A2 is an effector of Jak/STAT signaling and is involved in platelet-derived growth factor BB-induced growth in vascular smooth muscle cells. J. Biol. Chem. 278, 9986–9992 [DOI] [PubMed] [Google Scholar]
- 46. Hazan-Halevy I., Harris D., Liu Z., Liu J., Li P., Chen X., Shanker S., Ferrajoli A., Keating M. J., Estrov Z. (2010) STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 115, 2852–2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wen Z., Darnell J. E., Jr. (1997) Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 25, 2062–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nam S., Wen W., Schroeder A., Herrmann A., Yu H., Cheng X., Merz K. H., Eisenbrand G., Li H., Yuan Y. C., Jove R. (2013) Dual inhibition of Janus and Src family kinases by novel indirubin derivative blocks constitutively-activated Stat3 signaling associated with apoptosis of human pancreatic cancer cells. Mol. Oncol. 7, 369–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gritsko T., Williams A., Turkson J., Kaneko S., Bowman T., Huang M., Nam S., Eweis I., Diaz N., Sullivan D., Yoder S., Enkemann S., Eschrich S., Lee J. H., Beam C. A., Cheng J., Minton S., Muro-Cacho C. A., Jove R. (2006) Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 12, 11–19 [DOI] [PubMed] [Google Scholar]