

Background: Inflammatory bowel disease management lacks therapies that heal the epithelial barrier.
Results: PAR2 activation increases activities of MEK1/2 and PI3K in intestinal epithelial cells, which blocks apoptosis.
Conclusion: Cytokine-induced apoptosis in colonic epithelial cells is inhibited by PAR2 signaling.
Significance: PAR2 is important in maintaining intestinal epithelial homeostasis.
Keywords: Apoptosis, G Protein-coupled Receptor (GPCR), Intestinal Epithelium, Mitogen-activated Protein Kinase (MAPK), Protease
Abstract
Mucosal biopsies from inflamed colon of inflammatory bowel disease patients exhibit elevated epithelial apoptosis compared with those from healthy individuals, disrupting mucosal homeostasis and perpetuating disease. Therapies that decrease intestinal epithelial apoptosis may, therefore, ameliorate inflammatory bowel disease, but treatments that specifically target apoptotic pathways are lacking. Proteinase-activated receptor-2 (PAR2), a G protein-coupled receptor activated by trypsin-like serine proteinases, is expressed on intestinal epithelial cells and stimulates mitogenic pathways upon activation. We sought to determine whether PAR2 activation and signaling could rescue colonic epithelial (HT-29) cells from apoptosis induced by proapoptotic cytokines that are increased during inflammatory bowel disease. The PAR2 agonists 2-furoyl-LIGRLO (2f-LI), SLIGKV and trypsin all significantly reduced cleavage of caspase-3, -8, and -9, poly(ADP-ribose) polymerase, and the externalization of phosphatidylserine after treatment of cells with IFN-γ and TNF-α. Knockdown of PAR2 with siRNA eliminated the anti-apoptotic effect of 2f-LI and increased the sensitivity of HT-29 cells to cytokine-induced apoptosis. Concurrent inhibition of both MEK1/2 and PI3K was necessary to inhibit PAR2-induced survival. 2f-LI was found to increase phosphorylation and inactivation of pro-apoptotic BAD at Ser112 and Ser136 by MEK1/2 and PI3K-dependent signaling, respectively. PAR2 activation also increased the expression of anti-apoptotic MCL-1. Simultaneous knockdown of both BAD and MCL-1 had minimal effects on PAR2-induced survival, whereas single knockdown had no effect. We conclude that PAR2 activation reduces cytokine-induced epithelial apoptosis via concurrent stimulation of MEK1/2 and PI3K but little involvement of MCL-1 and BAD. Our findings represent a novel mechanism whereby serine proteinases facilitate epithelial cell survival and may be important in the context of colonic healing.
Introduction
The inflammatory bowel diseases (IBD)2, Crohn disease, and ulcerative colitis are chronic, debilitating conditions of the intestinal tract. IBD prevalence is highest in developed nations, although the incidence of IBD has been steadily increasing in all regions of the world (1). These diseases are difficult to manage medically and, other than colonic resection in the case of ulcerative colitis; there are no cures. A prevailing model for the pathogenesis of IBD implicates the development of an inappropriate immune response to intestinal microbiota that develops in genetically susceptible individuals with a defective epithelial barrier (2). Thus, understanding the pathways that regulate intestinal epithelial function and homeostasis may provide additional targets for therapy.
Epithelial homeostasis is maintained by a finely regulated balance between proliferation and apoptotic cell death. Intestinal epithelial cells arise from a stem cell region at the base of the mucosal crypts and migrate to the mucosal surface where they differentiate and perform functions associated with nutrient digestion, absorption, and host defense before proceeding through a physiological apoptotic program that results in shedding into the lumen (3). However, mucosal tissue from inflamed intestine displays significantly increased apoptosis of intestinal epithelial cells along the entire crypt-to-surface axis (4, 5). The inflammatory cytokine, TNF-α, is thought to be responsible for the increase in epithelial apoptosis observed in IBD. The resultant loss of epithelial homeostasis is associated with increased epithelial permeability to enhance the contact between luminal antigens and intestinal immune cells (6, 7). IFN-γ increases the expression of the TNF receptor and enhances TNF-α-induced apoptosis (8). The ongoing epithelial defect hinders the resolution of intestinal inflammation, which may be mitigated by growth factors that block epithelial apoptosis. For example, epidermal growth factor (EGF) can decrease the severity of dextran sodium sulfate-induced colitis and induce remission in patients with severe ulcerative colitis (9, 10). Mucosal healing has become an important end-point in the clinical management of IBD (11), and the reestablishment of normal intestinal mucosal structure is predictive of long term remission (12). Thus, the identification of new therapies that reestablish epithelial homeostasis to promote mucosal healing may provide new options for IBD patients.
A potential candidate for maintaining intestinal epithelial homeostasis is the G protein-coupled receptor, proteinase-activated receptor 2 (PAR2), which is expressed on intestinal epithelial cells (13, 14). PARs have a unique mechanism of activation, wherein proteolytic cleavage of the extracellular N terminus of the receptor by serine proteinases, which in the case of PAR2 includes trypsin, mast cell trypstase, coagulation factors VIIa/Xa, and kallikrein-related peptidase 14 (15), reveals a new N-terminal amino acid sequence that interacts with the second extracellular loop of the receptor to stimulate G protein- and β-arrestin-dependent signaling (16, 17). Synthetic peptides similar to the revealed “tethered ligand” sequence have been instrumental in identifying the specific functions of PAR2 while controlling for the receptor-independent actions of its activating proteinases. PAR2 activation increases cell proliferation in colonic epithelial cells in an ERK1/2-dependent manner (18). In other cell models, PAR2 activation also stimulates PI3K signaling (19). In this study, we sought to determine whether PAR2 signaling could decrease cytokine-stimulated apoptosis of colonic epithelial cells, based on the contention that even modest changes in the proliferation-apoptosis balance could promote the reestablishment of epithelial homeostasis to drive the resolution of inflammation in IBD. Using the HT-29 colonic epithelial cell line, we show that activation of PAR2 decreases TNF-α- and IFN-γ-induced apoptosis in a MEK1/2- and PI3K-dependent manner. Phosphorylation and inactivation of the proapoptotic protein, Bcl-2-associated death promoter (BAD), as well as up-regulation of anti-apoptotic myeloid cell leukemia 1 (MCL-1), are not fully responsible for the prosurvival effects of PAR2 activation on intestinal epithelial cells.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
The PAR2-activating peptides, SLIGRL, SLIGKV, and 2-furoyl-LIGRLO (2f-LI), as well as the inactive reverse peptide, 2-furoyl-OLRGIL-NH2, were synthesized by Dr. Denis McMaster (Peptide Core Facility, University of Calgary, Calgary, Alberta, Canada). Porcine pancreatic trypsin was purchased from Sigma-Aldrich. Recombinant human interferon γ (IFN-γ) and tumor necrosis factor α (TNF-α) were purchased from R&D Systems (Minneapolis, MN). The PI3K inhibitor, LY294002, was purchased from Cayman Chemical (Ann Arbor, MI). The MEK1/2 inhibitors, PD98059 and U0126, were purchased from Promega (Nepean, ON). The p90RSK inhibitor, SL0101, was purchased from Tocris Bioscience (Minneapolis, MN).
Cell Culture
The human colonic adenocarcinoma cell line HT-29 (ATCC, Manassas, VA) was grown in Dulbecco's modified Eagle's medium/nutrient mixture F-12 (DMEM/F12, Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS, Invitrogen), 100 units/ml penicillin, 100 μg/ml streptomycin, and 5 μg/ml plasmocin (InvivoGen, San Diego, CA). Trypsin was used to detach cells when passaging and plating for experiments. Although trypsin does activate PAR2, the receptor has been shown to recycle to the cell surface after 2 h (20). In all of our experiments, cells were used at least 2 days after lifting with trypsin. Cells were serum-starved for 1 h prior to stimulation with PAR2-activating peptides or trypsin. Apoptosis was then induced through the addition of 40 ng/ml IFN-γ for 5 min followed by 10 ng/ml of TNF-α for various time points as described previously (8). Both floating and attached cells were collected for analysis of apoptosis.
Immunoblot
Whole cell lysates were prepared as described previously (21). Whole cell lysates were prepared using a lysis buffer containing 40 mm Tris-HCl, 8 mm EDTA, 1% Triton X-100, and protease inhibitor mixture (Sigma-Aldrich). The phosphatase inhibitors NaF and Na3VO4 were added to the lysis buffer when determining levels of phosphorylated proteins. The protein concentrations of whole cell lysates were quantified using the DC Protein Assay Kit by following the manufacturer's instructions (Bio-Rad). A maximum of 45 μg of protein from each sample was separated by electrophoresis in a Criterion XT Bis-Tris Gel (Bio-Rad), followed by electrical transfer to a nitrocellulose membrane (0.2 μm pore size, Bio-Rad). Membranes were blocked for 1 h at room temperature with either 5% milk or 5% BSA in Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBST). Primary antibodies were diluted in 5% milk or 5% BSA TBST and incubated with the membranes overnight at 4 °C, followed by three 5-min washes in TBST and a 1-h incubation at room temperature with 5% milk TBST containing the appropriate secondary antibody conjugated to HRP. Unbound secondary antibody was then removed with three additional 5-min washes in TBST. Immunoblots were developed by enhanced chemiluminescence (ECL, GE Healthcare) using the ChemiDoc gel imaging system (Bio-Rad). Exposure times were varied to eliminate signal saturation. All the membranes were blotted for actin or for total kinases, which were used as loading controls. Background subtraction was applied to all blots using ImageJ (version 1.47v) with the sliding paraboloid algorithm (22). Band intensity was also calculated with ImageJ, which were then normalized by the sum of the replicates as described by Degasperi et al. (23). Antibodies, sources and catalogue numbers are as follows: cleaved caspase-3 (Cell Signaling Technology, Danvers, MA, catalogue no. 9661), cleaved caspase-9 (Cell Signaling, catalogue no. 9501), cleaved caspase-8 (Cell Signaling, catalogue no. 9496), cleaved poly(ADP-ribose) polymerase (PARP, Cell Signaling, catalogue no. 9542), p44/42 MAPK (Cell Signaling, phospho no. 9106, total no. 9102), Akt (Cell Signaling, phospho no. 9271, total no. 9272), p90RSK (Cell Signaling, phospho no. 9335, total no. 9355), BAD (Cell Signaling, phospho-Ser112 no. 9291, phospho-Ser136 no. 4366, total no. 9292), actin clone AC-40 (Sigma-Aldrich, no. A4700). Anti-mouse and anti-rabbit IgGs conjugated to HRP (The Jackson Laboratory, Bar Harbor, ME) were used as secondary antibodies.
Flow Cytometry
Floating cells were collected and pooled with attached cells, which were detached using a non-trypsin-containing proprietary solution to prevent activation of PAR2 (HyQtase, Thermo Fisher, Waltham, MA). Harvested cells were washed twice in calcium buffer supplied by the BD Pharmigen FITC annexin V apoptosis detection kit (San Diego, CA) to remove any remaining EDTA. Single cell suspensions were stained using the kit in accordance with manufacturer's instructions. Autofluorescence of cells was examined from the sample expected to have the greatest level of apoptosis. Care was taken to exclude cell debris and doublets. Compensation-adjusted florescence from at least 30,000 events was recorded using the Attune flow cytometer (Applied Biosystems). The percentage of early apoptotic cells (annexin V+/PI−) in untreated samples was subtracted from all other treatments to control for experimental error. Data were analyzed using Attune Cytometric Software (version v1.2.5, Applied Biosystems).
siRNA Knockdown
HT-29 cells were grown until confluent, trypsinized, and resuspended in antibiotic-free DMEM/F12 medium supplemented with 10% FBS. Cells were washed twice with medium and resuspended in antibiotic-free 10% FBS medium. Nonspecific siRNA (Qiagen, AllStars negative control), PAR2-specific siRNA (Qiagen, Hs_F2RL1_5; 5′-AGUCGUGAAUCUUGUUCAATT-3′ (sense) and 5′-UUGAACAAGAUUCACGACUAT-3′) (antisense)); BAD siRNA (Qiagen, Hs_BAD_4, 5′-ACUACCAAAUGUUAAUAAATT-3′ (sense) and 5′-UUUAUUAACAUUUGGUAGUGA-3′ (antisense)), or MCL-1 siRNA (Ambion by Invitrogen, Silencer Validated siRNA no. 120642, 5′-GCUGGUUUGGCAUAUCUAATT-3′ (sense) and 5′-UUAGAUAUGCCAAACCAGCTC-3′ (antisense)) were added to 200 μl of Invitrogen Opti-Mem reduced serum medium (Invitrogen), gently mixed by pipetting, and transferred into a well of a 12-well plate. Ten μl of Lipofectomine RNAiMAX (Invitrogen) was added to each siRNA solution, mixed by gently shaking the plate, and left undisturbed for 15 min at room temperature. One million cells were added to each well in a volume of 800 μl and left to incubate overnight at 37 °C and 5% CO2. The medium was changed the next day with DMEM/F12 supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin.
Quantitative PCR
Total RNA was extracted using the RNeasy mini kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. The reverse transcription reaction was performed using 700 ng of total RNA that was reverse-transcribed into cDNA using a random hexamer primer and SuperScript II Reverse Transcriptase (Invitrogen). Quantitative PCR was carried out using Qiagen's QuantiTect SYBR® Green PCR Kit in accordance with the manufacturer's instructions and performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems, Streetsville, ON). The annealing temperatures for both PAR2 and β-actin primers was 54 °C. Human PAR2 primers were as follows: 5′-AGTGGCACCATCCAAGGAAC-3′ (forward) and 5′-TGGAAGGAAGACAGTGGTCA-3′ (reverse). Human β-actin primers were as follows: 5′-AGAGGGAAATCGTGCGTGAC-3′ (forward) and 5′-CAATAGTGATGACCTGGCCGT-3′ (reverse). Relative quantities of mRNA were calculated using the ΔΔCT method using β-actin as the housekeeping gene (24).
Calcium Signaling
Functional knockdown of PAR2 by siRNA was verified by monitoring mobilization of intracellular calcium in response to 2f-LI as described previously (17) as an index of cell activation. Cells were detached after siRNA transfection with sterile 0.2 mm EDTA/PBS and incubated with 5 μm of Fluo4-acetoxymethyl ester no-wash (Invitrogen) for 30 min at room temperature in the presence of sulfinpyrazone (0.25 mm). Immediately before the experiment, Fluo4-loaded cells were diluted in calcium buffer (150 mm NaCl, 2 mm KCl, 1.5 mm CaCl2, 20 mm glucose, and 20 mm HEPES in deionized H2O, pH 7.4). Agonist-stimulated calcium mobilization was monitored with the Victor X4 fluorescent plate reader (PerkinElmer). Fluorescence data are expressed as the change in Fluo-4 fluorescence (excitation, 485 nm; emission, 535 nm) between baseline and maximum emission. The continued presence of intracellular Fluo4 in each sample was verified by the addition calcium ionophore, A23187 (Sigma-Aldrich), at the end of the experiment.
Statistical Analysis
Comparisons between two groups were made using the Student's t test. Comparisons between three or more groups were made using a one-way ANOVA, followed by a post hoc Bonferroni's test for significance. All statistical analyses were made using GraphPad Prism (version 6.0e, GraphPad Software, La Jolla, CA). A p value of < 0.05 was considered significant.
RESULTS
Activation of PAR2 in Colonic Epithelial Cells Decreases IFN-γ and TNF-α-induced Apoptosis
We chose to use the human colonic epithelial cell line HT-29 because they are known to express PAR2 and are sensitive to apoptosis stimulated by IFN-γ and TNF-α (8). This cell line resembles differentiated intestinal epithelia, but not crypt stem cells, due to the absence of an important anti-apoptotic protein, BCL-2, which makes them an appropriate model to investigate the cause of increased cell death of differentiated epithelia in IBD (25, 26). Serum-starved HT-29 cells were incubated with 40 ng/ml IFN-γ and 10 ng/ml TNF-α to model an inflammatory environment and investigate the effects of PAR2 activation on epithelial apoptosis. Increased levels of cleaved caspase-8, -9, and -3, and PARP were detected in whole cell lysates after 6 h of incubation (Fig. 1A). Pretreating HT-29 cells with the PAR2 activating peptides 2f-LI or SLIGKV, or with trypsin, significantly decreased cleavage of all caspases studied, as well as PARP (Fig. 1, A–E). In experiments using trypsin, the trypsin was removed after 5 min of incubation and replaced with serum-free medium to avoid proteolysis of IFN-γ and TNF-α. A hallmark of apoptosis is the loss of phospholipid symmetry in the plasma membrane resulting in the externalization of phosphatidylserine (PS) from the inner to the outer leaflet (27). The externalization of PS occurs in a caspase-3-dependent manner (28). Thus, PS exposure occurs after caspase-3 and PARP cleavage. An 8-h incubation with IFN-γ and TNF-α was found to maximally increase the percentage of HT-29 cells with externalized PS (data not shown). Preactivation of PAR2 with 2f-LI significantly decreased the percentage of cells displaying this apoptotic marker (Fig. 1, F–G). The observed reduction in apoptosis was dependent on the sequence of 2f-LI because the reverse peptide, 2-furoyl-OLRGIL, did not alter levels of cytokine-induced cleaved PARP stimulated by IFN-γ and TNF-α (Fig. 1H). Activation of PAR2 prior to the addition of cytokines was not able to decrease levels of cleaved caspases and PARP indefinitely as the inhibitory effect was lost after 8 h (Fig. 2). Thus, activation of PAR2 on colonic epithelial cells with either synthetic activating peptides or trypsin is able to significantly delay the kinetics of proinflammatory cytokine-induced apoptosis.
FIGURE 1.

PAR2 agonists decrease cytokine-induced cleavage of caspases and PARP as well as phosphatidylserine externalization in colonic epithelial cells. HT-29 cells were serum-starved and then incubated with PAR2-activating peptides 2f-LI or SLIGKV for 1 h. Trypsin was removed after 5 min and replaced with serum-free medium for the remainder of the hour. Apoptosis was induced with the proinflammatory cytokines IFN-γ and TNF-α. A, whole cell lysates were blotted for cleaved caspases and PARP (representative blot of n = 4). Densitometry was performed on cleaved caspase-9 (B), cleaved caspase-8 (C), cleaved caspase-3 (D) and cleaved PARP (E). Statistical analysis was done by one-way ANOVA followed by Bonferroni's multiple comparisons test. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 compared with cells treated with IFN-γ and TNF-α alone. F, proportion of cells undergoing apoptosis was determined by annexin V and propidium iodide flow cytometry. Representative dot plots showing healthy (annexin V− PI−) and apoptotic (annexin V+/PI−) cell populations. G, percentage of cells undergoing apoptosis (annexin V+ PI−) (n = 4; ***, p < 0.001 compared with untreated cells; $, p < 0.01 compared with cells treated with IFN-γ and TNF-α. H, HT-29 cells were pretreated with 2f-LI or the reverse peptide, 2f-OLRGIL, prior to the addition of IFN-γ and TNF-α (n = 5; *, p < 0.05 compared with cells treated with IFN-γ and TNF-α alone). a.u., arbitrary units; U, units.
FIGURE 2.
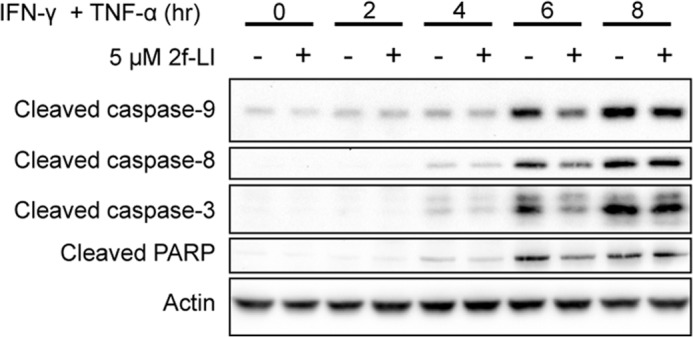
Cytokine-induced apoptosis is delayed by PAR2 stimulation. Serum-starved HT-29 cells were incubated with 5 μm 2f-LI for 1 h prior to the addition of IFN-γ and TNF-α for the time indicated. Whole cell lysates were blotted for cleaved caspases and PARP. Shown is a representative blot of n = 3.
Knockdown of PAR2 Sensitizes Colonic Epithelial Cells to Cytokine-induced Apoptosis
siRNA was used to decrease PAR2 expression to rule out any nonspecific effects of 2f-LI. PAR2 mRNA was significantly decreased after 24 h with 50 or 100 nm PAR2-specific siRNA compared with cells transfected with a scrambled siRNA (Fig. 3A). Due to the marked differences in the ability of commercially available antibodies to detect PAR2 (29), we recorded intracellular calcium release in response to 2f-LI as a measure of PAR2 function after knockdown. PAR2 siRNA significantly prevented the liberation of intracellular calcium compared with scrambled siRNA when cells were stimulated with 5 and 50 μm of 2f-LI (Fig. 3B). HT-29 cells transfected with siRNA were then incubated with cytokines to stimulate apoptosis, and the externalization of PS was measured by flow cytometry. 2f-LI decreased the proportion of cells with externalized PS in scrambled siRNA-transfected cells, but not in PAR2 siRNA-transfected cells (Fig. 3C). Furthermore, a greater proportion of PAR2 siRNA-transfected cells were observed to have externalized PS in response to cytokines compared with scrambled siRNA-transfected cells. These results indicate that 2f-LI acts specifically through PAR2 to decrease apoptosis. Additionally, these results show that HT-29 cells deficient in PAR2 are more susceptible to cytokine-induced apoptosis and that a baseline level of PAR2 activity may be occurring that prevents greater apoptosis when cells are stimulated with cytokines.
FIGURE 3.

Knockdown of PAR2 sensitizes colonic epithelial cells to cytokine-induced apoptosis. A, HT-29 cells were transfected with Lipofectamine alone (Lipo), nonspecific siRNA (Scr) or siRNA specific for PAR2. Total mRNA was isolated 24 h later, and quantitative PCR was used to detect relative levels of PAR2 mRNA (n = 3). *, p < 0.05 compared with scrambled (Scr) siRNA. B, a suspension of HT-29 cells was loaded with a calcium-sensitive dye 48 h after siRNA transfection. Intracellular calcium mobilization was recorded in response to increasing concentrations of 2f-LI to assess knockdown of PAR2 protein. Fluorescence data are expressed as the change in fluorescence between baseline and maximum emission (n = 4). ***, p < 0.0001 compared with scrambled siRNA. C, percentage of cells undergoing apoptosis (annexin V+ PI−) determined 48 h after siRNA transfection. Statistical analysis was done by one-way ANOVA followed by Bonferroni's multiple comparisons test (n = 4). *, p < 0.05.
PAR2 Anti-apoptotic Signaling Is Dependent on the Simultaneous Activities of MEK1/2 and PI3K
ERK1/2 and Akt are kinases that have well established roles in promoting cell survival and are activated by MEK1/2 and PI3K, respectively (30, 31). We tested whether PAR2 activates these kinases to block cytokine-induced apoptosis in intestinal epithelial cells. Blocking either the upstream kinase MEK1/2 with U0126 (Fig. 4, A–C) or PI3K with LY294002 (Fig. 4, D–F) did not block the ability of PAR2 to decrease the cleavage of PARP or caspase-3. Blocking either MEK1/2 or PI3K alone did, however, lower the ability of 2fLI to decrease detected cleaved PARP and cleaved caspase-3, providing evidence that these kinases were important in blocking apoptosis. When these inhibitors were applied concurrently, we observed complete blockage of the ability of PAR2 activation to inhibit caspase-3 and PARP cleavage (Fig. 4, G–I). Additionally, activation of PAR2 decreased PS externalization in the presence of a PI3K inhibitor but not an MEK1/2 inhibitor (Fig. 5). Again, both inhibitors together blocked the PAR2-mediated inhibition of PS externalization. Thus, the concurrent activation of both MEK1/2 and PI3K is required for the anti-apoptotic effect of PAR2 activation.
FIGURE 4.

Inhibition of both MEK1/2 and PI3K is necessary to block the actions of PAR2 activation on caspase-3 and PARP cleavage. HT-29 cells were serum-starved in the presence of either dimethyl sulfoxide (DMSO) or the inhibitors of MEK1/2 (U0126) (A–C), PI3K (LY294002) (D–F), or both (G–I) for 1 h prior to the addition of 2f-LI for one more hour. Apoptosis was stimulated by the addition of IFN-γ and TNF-α. Whole cell lysates were blotted for cleaved PARP and cleaved caspases-3. Blots are representative of n = 4. Statistical analysis was done by one-way ANOVA followed by Bonferroni's multiple comparisons test. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001. n.s., not significant.
FIGURE 5.
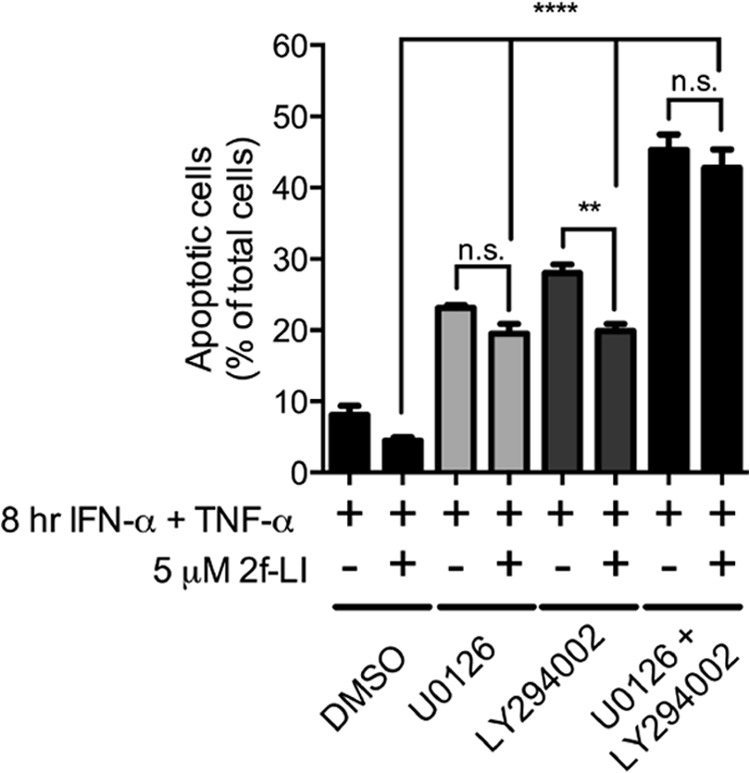
Inhibition of both MEK1/2 and PI3K are necessary to block the actions of PAR2 activation on phosphatidylserine externalization. HT-29 cells were serum-starved in the presence of either dimethyl sulfoxide (DMSO) or the inhibitors of MEK1/2 (U0126), PI3K (LY294002), or both for 1 h prior to the addition of 2f-LI for one more hour. Apoptosis was stimulated by the addition of IFN-γ and TNF-α for 8 h followed by determination of annexin V+/PI− cells by flow cytometry. Statistical analysis was done by one-way ANOVA followed by Bonferroni's multiple comparisons test (n = 3). **, p < 0.01; ****, p < 0.0001. n.s., not significant.
PAR2 Signaling Increases the Phosphorylation of BAD and Expression of MCL-1 During Cytokine-induced Apoptosis
Increased mitochondrial outer membrane permeability drives the intrinsic apoptotic pathway and is regulated by pro- and anti-apoptotic members of the B-cell lymphoma 2 (BCL-2) protein family (32). The expression of BAX, BAK, BIMEL, BIML+S, and BCL-XL were not altered by PAR2 activation during cytokine-induced apoptosis, whereas the expression of BCL-2 was not detected in HT-29 whole cell lysates (data not shown) and is in agreement with previous reports (33). Phosphorylation of the sensitizer BH3-only protein BAD and the subsequent release of anti-apoptotic BCL-XL represents a mechanism by which growth factors such as glucagon-like peptide-1 and EGF increase cell survival (34, 35). HT-29 cells were incubated with 2f-LI for up to 3 h to determine whether PAR2 activation resulted in the phosphorylation of BAD. 2f-LI increased the phosphorylation of ERK1/2, the downstream p90 ribosomal S6 kinase (p90RSK) and BAD at the S112 residue after 5 min, which returned to baseline phosphorylation levels after 2 h (Fig. 6A). Phosphorylation of BAD at Ser112 was markedly reduced by inhibition of MEK1/2 or p90RSK using U0126 or SL0101, respectively (Fig. 6B). The kinetics of PAR2-stimulated Akt phosphorylation was similar to ERK1/2 and was accompanied by phosphorylation of BAD at the Ser136 residue (Fig. 6C). The PI3K-selective inhibitor LY294002 blocked the phosphorylation of Akt as well as BAD at Ser136 (Fig. 6D). HT-29 cells undergoing apoptosis displayed increased BAD phosphorylation at Ser112 and Ser136 when pretreated with 2f-LI compared with cytokines alone at 2 and 4 h but not after 6 h (Fig. 6, F and G). These data show that PAR2 activation results in the phosphorylation of BAD at both the Ser112 and Ser136 positions and that this is driven by the activities of the MEK1/2-ERK1/2-p90RSK and the PI3K-Akt pathways, respectively. Additionally, PAR2 activation in cells undergoing apoptosis displayed increased levels of BAD phosphorylation.
FIGURE 6.

PAR2 signaling simultaneously activates MEK1/2-ERK1/2-p90RSK and PI3K-Akt pathways to phosphorylate BAD at Ser112 and Ser136 residues, respectively. Serum-starved HT-29 cells were treated with 2f-LI and harvested after the indicated times. A, activation of MAPK pathways was determined by blotting whole cell lysates for phosphorylated ERK1/2, p90RSK, and BAD at the Ser112 residue. B, cells were incubated with inhibitors to MEK1/2 (U0126) and p90RSK (SL0101) 1 h prior to the addition of 2f-LI for 5 min. Activation of the PI3K pathway was investigated by blotting for phosphorylated Akt and BAD at the Ser136 residue (C) and by using inhibitors to PI3K (LY294002) (D). All blots are representative of at least four independent experiments. E, serum-starved HT-29 cells were pretreated with 2f-LI for 1 h followed by incubation with IFN-γ and TNF-α for the indicated time. Whole cell lysates were blotted for BAD phosphorylation at Ser112 and Ser136. Representative blot of n = 6 is shown, and arrows indicate the predicted band for p-BAD(Ser112). Densitometry of phosphorylated BAD at Ser112 (F) and Ser136 (G) was acquired and expressed as a value relative to total BAD. Statistical analysis was done by unpaired t test; *, p < 0.05. DMSO, dimethyl sulfoxide; a.u., arbitrary units.
MCL-1 is an anti-apoptotic member of the BCL-2 family under the fine transcriptional control of PI3K and MEK1/2 (36, 37). HT-29 cells were found to express low basal levels of the MCL-1 protein (Fig. 7A). Activation of PAR2 with 2f-LI led to a significant increase in MCL-1 expression after 2 and 4 h (Fig. 7B). Pretreatment with 2f-LI led to a further increase in MCL-1 protein expression over cells treated with cytokines alone after 30 min or 1 h of incubation; however, this effect was not observed at later time points (Fig. 7, C and D). Thus, PAR2 activation led to an increase in the expression of anti-apoptotic MCL-1 in intestinal epithelial cells undergoing cytokine-induced apoptosis.
FIGURE 7.
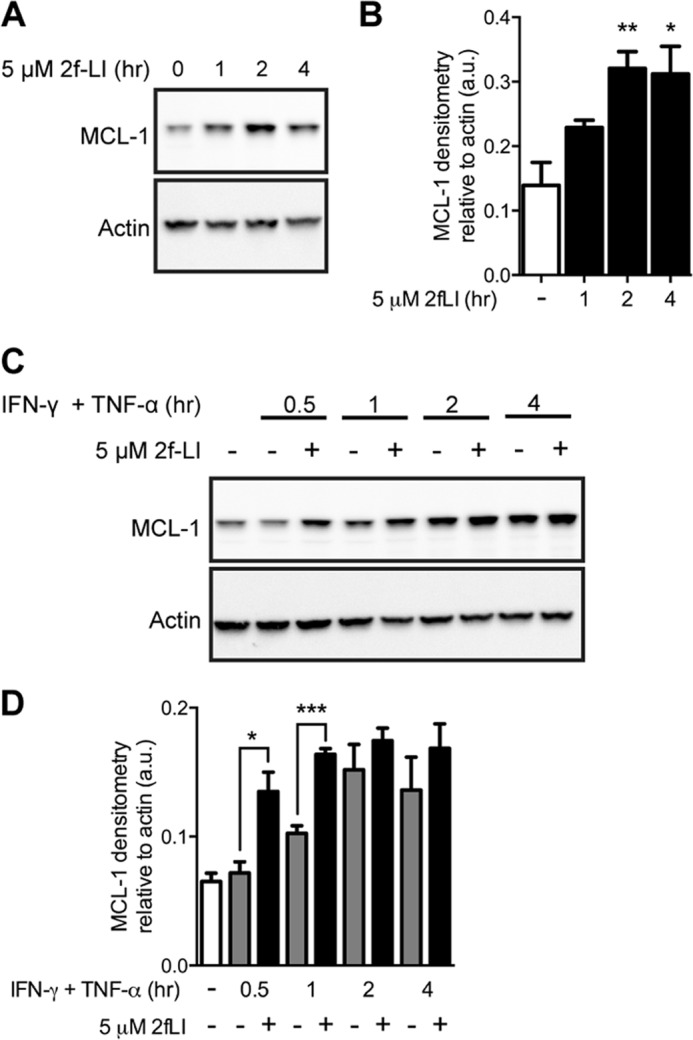
Activation of PAR2 increases MCL-1 expression. A, serum-starved HT-29 cells were stimulated with 2f-LI and harvested at the indicated times. MCL-1 protein expression was analyzed by blotting whole cell lysates. Representative blot of n = 3 is shown. B, MCL-1 protein densitometry relative to actin. Statistical analysis was done by one-way ANOVA followed by Bonferroni's multiple comparisons test (n = 3). *, p < 0.05; **, p < 0.01 compared with untreated cells. C, serum-starved HT-29 cells were pretreated with 2f-LI for 1 h followed by incubation with IFN-γ and TNF-α for the indicated time. Whole cell lysates were blotted for MCL-1. Representative blot of n = 4 is shown. D, densitometry of MCL-1 protein relative to actin. Statistical analysis was done by unpaired t test (n = 4). *, p < 0.05; ***, p < 0.001. a.u., arbitrary units.
PAR2 Signaling Requires Both BAD and MCL-1 Simultaneously to Inhibit Cytokine-induced Apoptosis in Intestinal Epithelial Cells
We next sought to determine whether the observed effects of PAR2 activation on BAD or MCL-1 were actually responsible for blocking apoptosis in HT-29 cells. We achieved the concurrent knockdown of BAD and MCL-1 expression by co-transfecting with specific siRNAs (Fig. 8). Knockdown of either BAD or MCL-1 alone did not alter the ability of PAR2 to decrease caspase-3 and PARP cleavage in HT-29 cells treated with cytokines (Fig. 8, A–F). Knockdown of MCL-1 on its own did, however, increase the amount of cleaved PARP detected when cells were pretreated with 2f-LI (Fig. 8, D and E). Concurrent knockdown of BAD and MCL-1 increased the detection of cleaved caspase-3 and cleaved PARP when cells were pretreated with 2f-LI (Fig. 8, G–I). Thus, the absence of both BAD and MCL-1 decreased the ability of PAR2 activation to decrease cleavage of these two apoptotic markers. Neither BAD nor MCL-1 were observed to contribute to decreased PS externalization triggered by PAR2 activation because 2f-LI could decrease detected PS in the absence of one or both of these proteins to levels detected in scrambled siRNA-treated cells (Fig. 9). Absence of BAD in HT-29 cells was observed to increase the effectiveness of 2f-LI in decreasing cytokine-stimulated PS externalization. Taken together, neither BAD nor MCL-1 participate significantly in PAR2-mediated inhibition of cytokine-induced apoptosis in HT-29 colonic epithelial cells.
FIGURE 8.

Knockdown of both BAD and MCL-1 inhibit the actions of PAR2 activation on caspase-3 and PARP cleavage. HT-29 cells were transfected with nonspecific siRNA (Scr) or siRNA specific for BAD (100 nm; A–C), MCL-1 (200 nm; D–F), or both (G–I). The concentration of scrambled siRNA added was equal to the total amount of specific siRNA added. After 48 h, cells were serum-starved, pretreated with 2f-LI for 1 h, and apoptosis was induced with IFN-γ and TNF-α for 6 h. Whole cell lysates were blotted for cleaved PARP and cleaved caspase-3. Blots are representative of at least n = 3. Statistical analysis was done by one-way ANOVA followed by Bonferroni's multiple comparisons test. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. n.s., not significant; a.u., arbitrary units.
FIGURE 9.
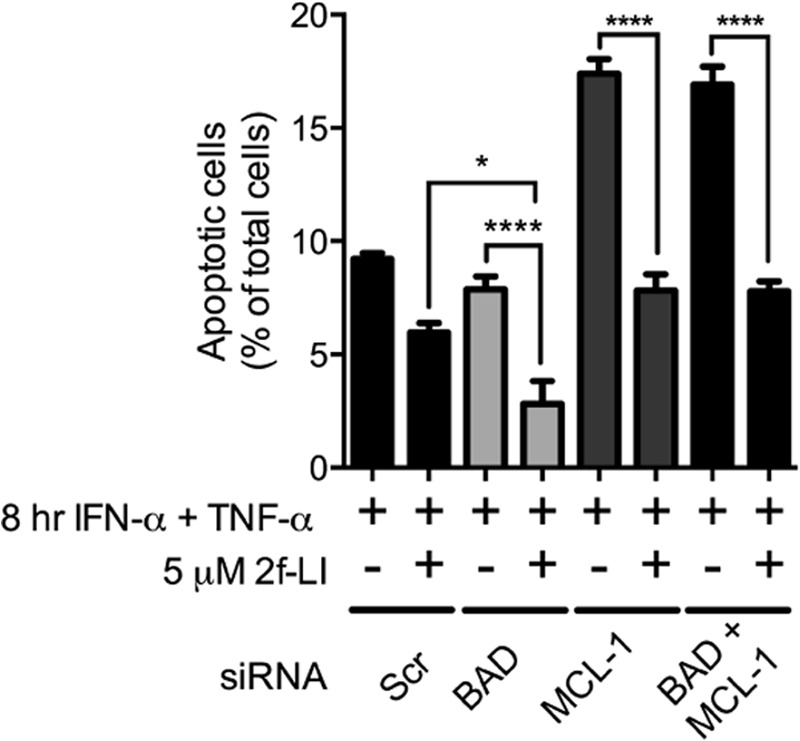
Knockdown of BAD and/or MCL-1 does not alter the actions of PAR2 activation on phosphatidylserine externalization. HT-29 cells were transfected with nonspecific siRNA (Scr) or siRNA specific for BAD (100 nm; A–C), MCL-1 (200 nm; D–F) or both (G–I). The concentration of scrambled siRNA added was equal to the total amount of specific siRNA added. After 48 h, cells were serum-starved and pretreated with 2f-LI for 1 h. Apoptosis was then induced with IFN-γ and TNF-α for 6 h followed by determination of annexin V+/PI− cell by flow cytometry. Statistical analysis was done by one-way ANOVA followed by Bonferroni's multiple comparisons test (n = 4). *, p < 0.05; ****, p < 0.0001.
DISCUSSION
Loss of epithelial homeostasis, through apoptosis induced by the cytokines IFN-γ and TNF-α, is a key feature of the pathophysiology that characterizes IBD. Here, we describe a new role for PAR2 in decreasing the kinetics of cytokine-induced apoptosis in colonic epithelial cells. The selective PAR2 agonist, 2f-LI, as well as the prototypical PAR2-activating serine protease, trypsin, were found to slow the cleavage of caspase-9, -8, and -3, and PARP when HT-29 cells were exposed to IFN-γ and TNF-α. PAR2 was found to be directly responsible because its knockdown eliminated the survival response induced by 2f-LI and increased the sensitivity of HT-29 cells to cytokine-stimulated apoptosis. The anti-apoptotic effect of PAR2 activation in HT-29 cells could be blocked when inhibitors of MEK1/2 and PI3K were applied concurrently, thus providing evidence for the requirement of the activity of both kinases downstream of PAR2. We next examined alterations in the BCL-2 protein family, which regulate mitochondrial outer membrane permeability to cytochrome c and activation of caspases. We observed that PAR2 signaling directly regulated the intrinsic apoptotic machinery by stimulating the phosphorylation of BAD and increasing MCL-1 expression at time points that preceded changes in caspase cleavage. Through double knockdown studies, however, we found that these BCL-2 family members played only a minimal role in the anti-apoptotic effect of PAR2 activation. A scheme proposing the mechanism whereby PAR2 inhibits cytokine-induced apoptosis is presented in Fig. 10.
FIGURE 10.

Activation of PAR2 inhibits cytokine-induced apoptosis. A, the cytokine TNF-α activates the extrinsic apoptotic pathway by binding the TNF receptor (TNFR), which then recruits and stimulates the cleavage of caspase-8. Activated caspase-8 in turn cleaves caspase-3 as well as BID into a truncated form (tBID) to stimulate the intrinsic apoptotic pathway. Truncated BID induces conformational changes in both BAX and BAK, which oligomerize and form channels on the mitochondria's outer membrane. Cytochrome c, normally found within the mitochondria, passes through these channels into the cytosol where it stimulates the activation of caspase-9. The intrinsic and extrinsic apoptotic pathways intersect by the activation of caspases-3 by cleaved caspase-9. Many proteins vital to the normal functioning of the cell, including PARP, are digested by activated casapse-3 leading to cellular apoptosis. In healthy cells, phosphatidylserine is found on the inner leaflet of the membrane but is externalized during apoptosis in a caspase-3-dependent manner. B, activation of PAR2 stimulates both PI3K and MEK1/2, leading to the phosphorylation of BAD through downstream kinases. Phosphorylated BAD binds 14-3-3 and releases anti-apoptotic BCL-XL into the cytosol. PAR2 also increases the expression of anti-apoptotic MCL-1. Both BCL-XL and MCL-1 inhibit the oligomerization of BAX and BAK stimulated by truncated BID resulting in less mitochondrial permeability. The intrinsic apoptotic pathway is inhibited as a result leading to decreased cleavage and activation of caspase-9 and caspase-3. Cleavage of caspase-8 and PARP is also reduced due to less activated casapse-3 being present. The externalization of PS is also decreased as a result of PAR2 activation and is dependent on the activities of both MEK1/2 and PI3K, but not the presence of MCL-1 and BAD. Thus, we hypothesize that PAR2 activation blocks apoptosis through other mechanisms that are dependent on ERK1/2 and PI3K activity.
Intestinal epithelial cells are uniquely situated to respond to PAR2-activating serine proteases. For example, during inflammation, microvascular leakage allows PAR2-activating coagulation cascade proteases to gain access to the basolateral aspect of the epithelium (38, 39). Indeed, the inhibition of apoptosis by factor VIIa and its co-factor, tissue factor, which can activate PAR2, has been described previously (40, 41). However, while levels of active caspase-3 and DNA fragmentation were all lowered by tissue factor in serum-starved BHK-21 cells expressing factor VIIa (40, 42), these studies did not examine the direct involvement of PAR2 in the anti-apoptotic mechanism, although it is known that BHK-21 cells express PAR2 and the factor VIIa·tissue factor complex activates PAR2 to stimulate intracellular Ca2+ release (39, 40). In addition to coagulation factors, mast cell-derived tryptase, which can also activate PAR2, has been shown to block Fas-induced apoptosis in rheumatoid synovial fibroblasts, but a role for PAR2 activation was not demonstrated in that study (43). Other proteases, such as the membrane tethered serine protease, matriptase, are expressed by intestinal epithelial cells and regulate barrier function (44), although an anti-apoptotic effect has not been ascribed to them at this point.
We found that colonic epithelial cells became sensitized to apoptosis in the presence of proinflammatory cytokines when PAR2 expression was knocked down with siRNA. These results suggest that HT-29 cells have a basal level of PAR2-mediated signaling activity in the absence of exogenous proteinases or activating peptides. HT-29 cells, along with other colon cancer cells, have been shown to produce active kallikrein-related peptidase 14 and matriptase, which are both capable of activating PAR2 (45–48). Normal human colonic epithelia also expresses trypsinogen IV that, after being cleaved by enteropeptidase to become enzymatically active trypsin, can stimulate PAR2-dependent calcium signaling (49). Apoptosis may be inhibited physiologically through this basal autocrine signaling and is abrogated when PAR2 is absent, leading to greater cell death in the presence of IFN-γ and TNF-α.
It was necessary to block both PI3K and MEK1/2 activities simultaneously in our model to completely reverse the ability of PAR2 activation to reduce levels of cleaved caspase-3, cleaved PARP, and the ratio of cells with externalized phosphatidylserine. This finding indicates that the activity of one pathway could compensate for the loss of the other, which may occur at the level of BAD phosphorylation and the subsequent release of BCL-XL. BAD is known to have three serine residues that are phosphorylated, which prevents BAD from dimerizing with BCL-XL and increases its binding to 14-3-3 (50–52). The resulting free BCL-XL inhibits apoptosis by preventing truncated BID from activating BAX and BAK, which stimulate the release of cytochrome c from the mitochondria (Fig. 10) (53). The phosphorylation of either Ser112 or Ser136 on BAD by MEK1/2- and PI3K-dependent pathways, respectively, are sufficient to stimulate dimerization with 14-3-3 and inhibit apoptosis (54, 55). In our study, PAR2 activation stimulated BAD phosphorylation at both of these residues, which could explain why the inhibition of both PI3K and MEK1/2 was required to reverse the effect of PAR2 activation. Another possible reason for the observed compensatory activities between MEK1/2 and PI3K is that both of these kinases are also involved in regulating MCL-1 expression, a BCL-2 family member with the same anti-apoptotic function as BCL-XL (56, 57). Furthermore, MCL-1 has been reported to be stabilized by ERK1/2-dependent phosphorylation (58).
Knockdown of BAD on its own had no effect on the ability of PAR2 activation to decrease apoptosis as measured by cleaved caspase-3, cleaved PARP, and the ratio of annexin V+/PI− HT-29 cells. Similarly, we did not see a loss of the PAR2-mediated survival response when MCL-1 expression was decreased. We hypothesized that PAR2 signaling might be acting on both BAD and MCL-1 simultaneously to decrease apoptosis. However, when expression of both proteins was decreased, we observed a minimal effect on the ability of PAR2 activation to inhibit the cleavage of capase-3 and PARP. Similarly, simultaneous knockdown of BAD and MCL-1 did not prevent PAR2 activation from reducing the degree of apoptosis as measured by PS externalization in cytokine-stimulated cells. Taken together, the data suggest concurrent activation of the MEK1/2 and PI3K pathways but little involvement of the mitochondrion-associated apoptotic and anti-apoptotic proteins investigated. This was interesting given the role of MAPK and PI3K pathways in regulating proteins such as BAD. For example, PI3K and its downstream messengers have been shown to inhibit the proteolytic activity of caspase-9 as well as preventing BAX translocation to the mitochondria via phosphorylation (59–61). Activation of ERK1/2 also promotes survival by increasing the degradation of proapoptotic BIMEL (62, 63); however, we did not observe any alterations in the expression of BIMEL in our experiments (data not shown). It may be that other proteins known to regulate mitochondrial outer membrane permeability, but not investigated here, may be involved in the anti-apoptotic effect of PAR2 activation. In addition, other targets of MAPK and PI3K activities could play a role, but to elucidate these will require further investigation.
In conclusion, we describe a mechanism by which PAR2 activation can inhibit cytokine-induced apoptosis in colonic epithelial cells. Although the effect was transient, we contend that even a subtle change in the level of apoptosis can have profound effects on reestablishing epithelial homeostasis to drive the resolution of mucosal structure and function in IBD. Activators of PAR2 now comprise a novel group of intestinal growth factors capable of stimulating epithelial survival, in addition to proliferation (18). PAR2 agonists should thus be investigated for their efficacy in ameliorating intestinal diseases such as IBD in which epithelial apoptosis contributes to pathology.
Acknowledgment
We thank Laurie Kennedy from the University of Calgary Flow Cytometry Facility for help with flow cytometry experimental design and data analysis.
This work was supported by a grant-in-aid from Crohn's and Colitis Canada (to W. K. M.).
- IBD
- inflammatory bowel disease(s)
- 2f-LI
- 2-furoyl-LIGRLO
- BAD
- Bcl-2-associated death promoter
- BCL-2
- B-cell lymphoma 2
- KLK
- kallikrein-related peptidase
- MCL-1
- myeloid cell leukemia 1
- p90RSK
- p90 ribosomal S6 kinase
- PAR
- proteinase-activated receptor
- PARP
- poly(ADP-ribose) polymerase
- PS
- phosphatidylserine
- ANOVA
- analysis of variance.
REFERENCES
- 1. Molodecky N. A., Soon I. S., Rabi D. M., Ghali W. A., Ferris M., Chernoff G., Benchimol E. I., Panaccione R., Ghosh S., Barkema H. W., Kaplan G. G. (2012) Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142, 46–54.e42; quiz e30 [DOI] [PubMed] [Google Scholar]
- 2. Kaser A., Zeissig S., Blumberg R. S. (2010) Inflammatory bowel disease. Annu. Rev. Immunol. 28, 573–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grossmann J., Walther K., Artinger M., Rümmele P., Woenckhaus M., Schölmerich J. (2002) Induction of apoptosis before shedding of human intestinal epithelial cells. Am. J. Gastroenterol. 97, 1421–1428 [DOI] [PubMed] [Google Scholar]
- 4. Zeissig S., Bojarski C., Buergel N., Mankertz J., Zeitz M., Fromm M., Schulzke J. D. (2004) Downregulation of epithelial apoptosis and barrier repair in active Crohn's disease by tumour necrosis factor α antibody treatment. Gut 53, 1295–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dirisina R., Katzman R. B., Goretsky T., Managlia E., Mittal N., Williams D. B., Qiu W., Yu J., Chandel N. S., Zhang L., Barrett T. A. (2011) p53 and PUMA independently regulate apoptosis of intestinal epithelial cells in patients and mice with colitis. Gastroenterology 141, 1036–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gitter A. H., Wullstein F., Fromm M., Schulzke J. D. (2001) Epithelial barrier defects in ulcerative colitis: characterization and quantification by electrophysiological imaging. Gastroenterology 121, 1320–1328 [DOI] [PubMed] [Google Scholar]
- 7. Kiesslich R., Duckworth C. A., Moussata D., Gloeckner A., Lim L. G., Goetz M., Pritchard D. M., Galle P. R., Neurath M. F., Watson A. J. (2012) Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut 61, 1146–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ossina N. K., Cannas A., Powers V. C., Fitzpatrick P. A., Knight J. D., Gilbert J. R., Shekhtman E. M., Tomei L. D., Umansky S. R., Kiefer M. C. (1997) Interferon-gamma modulates a p53-independent apoptotic pathway and apoptosis-related gene expression. J. Biol. Chem. 272, 16351–16357 [DOI] [PubMed] [Google Scholar]
- 9. Procaccino F., Reinshagen M., Hoffmann P., Zeeh J. M., Lakshmanan J., McRoberts J. A., Patel A., French S., Eysselein V. E. (1994) Protective effect of epidermal growth factor in an experimental model of colitis in rats. Gastroenterology 107, 12–17 [DOI] [PubMed] [Google Scholar]
- 10. Sinha A., Nightingale J., West K. P., Berlanga-Acosta J., Playford R. J. (2003) Epidermal growth factor enemas with oral mesalamine for mild-to-moderate left-sided ulcerative colitis or proctitis. N. Engl. J. Med. 349, 350–357 [DOI] [PubMed] [Google Scholar]
- 11. Neurath M. F., Travis S. P. (2012) Mucosal healing in inflammatory bowel diseases: a systematic review. Gut 61, 1619–1635 [DOI] [PubMed] [Google Scholar]
- 12. Baert F., Moortgat L., Van Assche G., Caenepeel P., Vergauwe P., De Vos M., Stokkers P., Hommes D., Rutgeerts P., Vermeire S., D'Haens G., Belgian Inflammatory Bowel Disease Research Group, and North-Holland Gut Club (2010) Mucosal healing predicts sustained clinical remission in patients with early-stage Crohn's disease. Gastroenterology 138, 463–468; quiz e10-1 [DOI] [PubMed] [Google Scholar]
- 13. Kong W., McConalogue K., Khitin L. M., Hollenberg M. D., Payan D. G., Böhm S. K., Bunnett N. W. (1997) Luminal trypsin may regulate enterocytes through proteinase-activated receptor 2. Proc. Natl. Acad. Sci. U.S.A. 94, 8884–8889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. D'Andrea M. R., Derian C. K., Leturcq D., Baker S. M., Brunmark A., Ling P., Darrow A. L., Santulli R. J., Brass L. F., Andrade-Gordon P. (1998) Characterization of protease-activated receptor-2 immunoreactivity in normal human tissues. J. Histochem. Cytochem. 46, 157–164 [DOI] [PubMed] [Google Scholar]
- 15. Adams M. N., Ramachandran R., Yau M. K., Suen J. Y., Fairlie D. P., Hollenberg M. D., Hooper J. D. (2011) Structure, function and pathophysiology of protease activated receptors. Pharmacol. Ther. 130, 248–282 [DOI] [PubMed] [Google Scholar]
- 16. DeFea K. A., Zalevsky J., Thoma M. S., Déry O., Mullins R. D., Bunnett N. W. (2000) β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 148, 1267–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramachandran R., Mihara K., Mathur M., Rochdi M. D., Bouvier M., Defea K., Hollenberg M. D. (2009) Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol. Pharmacol. 76, 791–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Darmoul D., Gratio V., Devaud H., Laburthe M. (2004) Protease-activated receptor 2 in colon cancer: trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J. Biol. Chem. 279, 20927–20934 [DOI] [PubMed] [Google Scholar]
- 19. Wang P., Kumar P., Wang C., Defea K. A. (2007) Differential regulation of class IA phosphoinositide 3-kinase catalytic subunits p110 α and β by protease-activated receptor 2 and β-arrestins. Biochem. J. 408, 221–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Déry O., Thoma M. S., Wong H., Grady E. F., Bunnett N. W. (1999) Trafficking of proteinase-activated receptor-2 and β-arrestin-1 tagged with green fluorescent protein. β-Arrestin-dependent endocytosis of a proteinase receptor. J. Biol. Chem. 274, 18524–18535 [DOI] [PubMed] [Google Scholar]
- 21. Hirota C. L., Moreau F., Iablokov V., Dicay M., Renaux B., Hollenberg M. D., MacNaughton W. K. (2012) Epidermal growth factor receptor transactivation is required for proteinase-activated receptor-2-induced COX-2 expression in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G111–G119 [DOI] [PubMed] [Google Scholar]
- 22. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Degasperi A., Birtwistle M. R., Volinsky N., Rauch J. (2014) Evaluating strategies to normalise biological replicates of Western blot data. PLoS One 10.1371/journal.pone.0087293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schmittgen T. D., Livak K. J. (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 25. Hockenbery D. M., Zutter M., Hickey W., Nahm M., Korsmeyer S. J. (1991) BCL2 protein is topographically restricted in tissues characterized by apoptotic cell death. Proc. Natl. Acad. Sci. U.S.A. 88, 6961–6965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iwamoto M., Koji T., Makiyama K., Kobayashi N., Nakane P. K. (1996) Apoptosis of crypt epithelial cells in ulcerative colitis. J. Pathol. 180, 152–159 [DOI] [PubMed] [Google Scholar]
- 27. Fadok V. A., Voelker D. R., Campbell P. A., Cohen J. J., Bratton D. L., Henson P. M. (1992) Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207–2216 [PubMed] [Google Scholar]
- 28. Frasch S. C., Henson P. M., Kailey J. M., Richter D. A., Janes M. S., Fadok V. A., Bratton D. L. (2000) Regulation of phospholipid scramblase activity during apoptosis and cell activation by protein kinase Cδ. J. Biol. Chem. 275, 23065–23073 [DOI] [PubMed] [Google Scholar]
- 29. Adams M. N., Pagel C. N., Mackie E. J., Hooper J. D. (2012) Evaluation of antibodies directed against human protease-activated receptor-2. Naunyn Schmiedebergs Arch. Pharmacol. 385, 861–873 [DOI] [PubMed] [Google Scholar]
- 30. Duronio V. (2008) The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem. J. 415, 333–344 [DOI] [PubMed] [Google Scholar]
- 31. Anjum R., Blenis J. (2008) The RSK family of kinases: emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 9, 747–758 [DOI] [PubMed] [Google Scholar]
- 32. Tait S. W., Green D. R. (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632 [DOI] [PubMed] [Google Scholar]
- 33. O'Connell J., Bennett M. W., Nally K., O'Sullivan G. C., Collins J. K., Shanahan F. (2000) Interferon-γ sensitizes colonic epithelial cell lines to physiological and therapeutic inducers of colonocyte apoptosis. J. Cell. Physiol. 185, 331–338 [DOI] [PubMed] [Google Scholar]
- 34. Gilmore A. P., Valentijn A. J., Wang P., Ranger A. M., Bundred N., O'Hare M. J., Wakeling A., Korsmeyer S. J., Streuli C. H. (2002) Activation of BAD by therapeutic inhibition of epidermal growth factor receptor and transactivation by insulin-like growth factor receptor. J. Biol. Chem. 277, 27643–27650 [DOI] [PubMed] [Google Scholar]
- 35. Quoyer J., Longuet C., Broca C., Linck N., Costes S., Varin E., Bockaert J., Bertrand G., Dalle S. (2010) GLP-1 mediates antiapoptotic effect by phosphorylating Bad through a β-arrestin 1-mediated ERK1/2 activation in pancreatic β-cells. J. Biol. Chem. 285, 1989–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Booy E. P., Henson E. S., Gibson S. B. (2011) Epidermal growth factor regulates Mcl-1 expression through the MAPK-Elk-1 signalling pathway contributing to cell survival in breast cancer. Oncogene 30, 2367–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kobayashi S., Werneburg N. W., Bronk S. F., Kaufmann S. H., Gores G. J. (2005) Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology 128, 2054–2065 [DOI] [PubMed] [Google Scholar]
- 38. Drake T. A., Morrissey J. H., Edgington T. S. (1989) Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am. J. Pathol. 134, 1087–1097 [PMC free article] [PubMed] [Google Scholar]
- 39. Camerer E., Huang W., Coughlin S. R. (2000) Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc. Natl. Acad. Sci. U.S.A. 97, 5255–5260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sorensen B. B., Rao L. V., Tornehave D., Gammeltoft S., Petersen L. C. (2003) Antiapoptotic effect of coagulation factor VIIa. Blood 102, 1708–1715 [DOI] [PubMed] [Google Scholar]
- 41. Versteeg H. H., Spek C. A., Richel D. J., Peppelenbosch M. P. (2004) Coagulation factors VIIa and Xa inhibit apoptosis and anoikis. Oncogene 23, 410–417 [DOI] [PubMed] [Google Scholar]
- 42. Slee E. A., Adrain C., Martin S. J. (2001) Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 276, 7320–7326 [DOI] [PubMed] [Google Scholar]
- 43. Sawamukai N., Yukawa S., Saito K., Nakayamada S., Kambayashi T., Tanaka Y. (2010) Mast cell-derived tryptase inhibits apoptosis of human rheumatoid synovial fibroblasts via ρ-mediated signaling. Arthritis Rheum. 62, 952–959 [DOI] [PubMed] [Google Scholar]
- 44. Buzza M. S., Netzel-Arnett S., Shea-Donohue T., Zhao A., Lin C. Y., List K., Szabo R., Fasano A., Bugge T. H., Antalis T. M. (2010) Membrane-anchored serine protease matriptase regulates epithelial barrier formation and permeability in the intestine. Proc. Natl. Acad. Sci. U.S.A. 107, 4200–4205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gratio V., Loriot C., Virca G. D., Oikonomopoulou K., Walker F., Diamandis E. P., Hollenberg M. D., Darmoul D. (2011) Kallikrein-related peptidase 14 acts on proteinase-activated receptor 2 to induce signaling pathway in colon cancer cells. Am. J. Pathol. 179, 2625–2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takeuchi T., Harris J. L., Huang W., Yan K. W., Coughlin S. R., Craik C. S. (2000) Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J. Biol. Chem. 275, 26333–26342 [DOI] [PubMed] [Google Scholar]
- 47. Darragh M. R., Schneider E. L., Lou J., Phojanakong P. J., Farady C. J., Marks J. D., Hann B. C., Craik C. S. (2010) Tumor detection by imaging proteolytic activity. Cancer Res. 70, 1505–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ma Y., Bao-Han W., Lv X., Su Y., Zhao X., Yin Y., Zhang X. (2013) MicroRNA-34a mediates the autocrine signaling of PAR2-activating proteinase and its role in colonic cancer cell proliferation. PLoS One 8, e72383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cottrell G. S., Amadesi S., Grady E. F., Bunnett N. W. (2004) Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J. Biol. Chem. 279, 13532–13539 [DOI] [PubMed] [Google Scholar]
- 50. Datta S. R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M. E. (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91, 231–241 [DOI] [PubMed] [Google Scholar]
- 51. Scheid M. P., Schubert K. M., Duronio V. (1999) Regulation of bad phosphorylation and association with Bcl-x(L) by the MAPK/Erk kinase. J. Biol. Chem. 274, 31108–31113 [DOI] [PubMed] [Google Scholar]
- 52. Tan Y., Demeter M. R., Ruan H., Comb M. J. (2000) BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J. Biol. Chem. 275, 25865–25869 [DOI] [PubMed] [Google Scholar]
- 53. Kim H., Rafiuddin-Shah M., Tu H. C., Jeffers J. R., Zambetti G. P., Hsieh J. J., Cheng E. H. (2006) Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol. 8, 1348–1358 [DOI] [PubMed] [Google Scholar]
- 54. Fang X., Yu S., Eder A., Mao M., Bast R. C., Jr., Boyd D., Mills G. B. (1999) Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene 18, 6635–6640 [DOI] [PubMed] [Google Scholar]
- 55. Zha J., Harada H., Yang E., Jockel J., Korsmeyer S. J. (1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X (L). Cell 87, 619–628 [DOI] [PubMed] [Google Scholar]
- 56. Wang J. M., Chao J. R., Chen W., Kuo M. L., Yen J. J., Yang-Yen H. F. (1999) The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol. Cell. Biol. 19, 6195–6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Townsend K. J., Zhou P., Qian L., Bieszczad C. K., Lowrey C. H., Yen A., Craig R. W. (1999) Regulation of MCL1 through a serum response factor/Elk-1-mediated mechanism links expression of a viability-promoting member of the BCL2 family to the induction of hematopoietic cell differentiation. J. Biol. Chem. 274, 1801–1813 [DOI] [PubMed] [Google Scholar]
- 58. Domina A. M., Vrana J. A., Gregory M. A., Hann S. R., Craig R. W. (2004) MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene 23, 5301–5315 [DOI] [PubMed] [Google Scholar]
- 59. Cardone M. H., Roy N., Stennicke H. R., Salvesen G. S., Franke T. F., Stanbridge E., Frisch S., Reed J. C. (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science 282, 1318–1321 [DOI] [PubMed] [Google Scholar]
- 60. Tsuruta F., Masuyama N., Gotoh Y. (2002) The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J. Biol. Chem. 277, 14040–14047 [DOI] [PubMed] [Google Scholar]
- 61. Gardai S. J., Hildeman D. A., Frankel S. K., Whitlock B. B., Frasch S. C., Borregaard N., Marrack P., Bratton D. L., Henson P. M. (2004) Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J. Biol. Chem. 279, 21085–21095 [DOI] [PubMed] [Google Scholar]
- 62. Ley R., Balmanno K., Hadfield K., Weston C., Cook S. J. (2003) Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J. Biol. Chem. 278, 18811–18816 [DOI] [PubMed] [Google Scholar]
- 63. Luciano F., Jacquel A., Colosetti P., Herrant M., Cagnol S., Pages G., Auberger P. (2003) Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene 22, 6785–6793 [DOI] [PubMed] [Google Scholar]