

Background: Selenoprotein H (SelH), a proposed redox-responsive DNA-binding protein, is little-studied.
Results: SelH shRNA cells display severe proliferation defects and accelerated senescence with abnormal responses to DNA damage and oxidative stress.
Conclusion: SelH suppresses senescence and may have important roles in gatekeeping genomic integrity.
Significance: Learning how SelH keeps senescence in check is crucial for advancing our understanding linking selenium and aging intervention.
Keywords: Ataxia Telangiectasia, p53, Reactive Oxygen Species (ROS), Selenium, Senescence
Abstract
Oxidative stress and persistent DNA damage response contribute to cellular senescence, a degeneration process critically involving ataxia telangiectasia-mutated (ATM) and p53. Selenoprotein H (SelH), a nuclear selenoprotein, is proposed to carry redox and transactivation domains. To determine the role of SelH in genome maintenance, shRNA knockdown was employed in human normal and immortalized cell lines. SelH shRNA MRC-5 diploid fibroblasts under ambient O2 displayed a distinct profile of senescence including β-galactosidase expression, autofluorescence, growth inhibition, and ATM pathway activation. Such senescence phenotypes were alleviated in the presence of ATM kinase inhibitors, by p53 shRNA knockdown, or by maintaining the cells under 3% O2. During the course of 5-day recovery, the induction of phospho-ATM on Ser-1981 and γH2AX by H2O2 treatment (20 μm) subsided in scrambled shRNA but exacerbated in SelH shRNA MRC-5 cells. Results from clonogenic assays demonstrated hypersensitivity of SelH shRNA HeLa cells to paraquat and H2O2, but not to hydroxyurea, neocarzinostatin, or camptothecin. While SelH mRNA expression was induced by H2O2 treatment, SelH-GFP did not mobilize to sites of oxidative DNA damage. The glutathione level was lower in SelH shRNA than scrambled shRNA HeLa cells, and the H2O2-induced cell death was rescued in the presence of N-acetylcysteine, a glutathione precursor. Altogether, SelH protects against cellular senescence to oxidative stress through a genome maintenance pathway involving ATM and p53.
Introduction
Cellular senescence restricts cell proliferation through permanent withdrawal from the cell cycle and plays dual physiological roles (1–4). While cellular senescence can curb tumorigenesis at the precancerous stage and control fibrosis during cutaneous healing early in life (5, 6), it contributes to cellular and tissue aging and age-related disorders later in life (7–10). Although replicative senescence is induced by gradual telomere dysfunction in proliferating cells, stress-induced senescence occurs in essentially any cell type. Reactive oxygen species (ROS)3 can induce the formation of oxidative and broken DNA, resulting in persistent activation of the DNA damage response and senescence if left unrepaired. Ataxia telangiectasia-mutated (ATM) is a key DNA damage response kinase coordinating checkpoint and senescence responses. ATM is activated by either DNA breaks or oxidative stress (11, 12), and plays an essential role in the senescence response by phosphorylating and stabilizing p53 (13–15).
In mammals, most selenoproteins carry antioxidative functions (16, 17). Selenoprotein expression requires the essential trace element, selenium. Selenoprotein H (SelH), glutathione peroxidase-1, selenoprotein W, and selenoprotein M are more sensitive than other selenoproteins to body selenium fluctuations (18–20). Tissue-specific knockout of selenocysteine tRNA for global suppression of selenoproteins in epidermal cells or osteo-chondroprogenitor cells renders the mice susceptible to age-related disorders including alopecia and bone abnormality (21, 22). These observations are consistent with an estimation linking eleven selenoproteins to aging or age-related disorders (23).
SelH is a thioredoxin-like nuclear protein exhibiting glutathione peroxidase activity (24). Furthermore, the homologue of human SelH in Drosophila is critical for embryogenesis through its antioxidative activity (25). Studies of human SelH in HT22 mouse neuronal cells have implicated this selenoprotein in the protection against UVB-induced apoptosis and as a transactivator for GSH biosynthesis (26–29). Nonetheless, a role of SelH in the senescence response to DNA damage and oxidative stress has not been explored. Because SelH expression is enriched in nucleoli, and this organelle has been proposed as a stress-sensing center in the nucleus (24, 30, 31), we hypothesized that SelH protects against oxidative stress through genome maintenance and the limitation of cellular senescence. Thus, we stably knocked down SelH expression in human normal diploid fibroblasts and cancerous cells to evaluate their cellular and biochemical responses to various DNA-damaging agents. Our results suggested a new role of SelH specifically in the cellular response to oxidative stress that suppresses senescence and gatekeeps genomic integrity in a manner depending on ATM and p53.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
The MRC-5 diploid lung fibroblasts (Coriell Institute, Camden, NJ), HeLa cervical cancer cells (ATCC, Manassas, VA), and HCT116 colorectal adenocarcinoma cells complemented with hMLH1 (HCT116+hMLH1) (32, 33) were cultured as described previously in 20% or 3% O2 incubators (34, 35). However, no additional selenium was supplemented in the current study. Because selenium inevitably appears in FBS, a typical cell culture medium containing 10–15% FBS can support selenoprotein expression at nutritional level. By analysis, the batch of FBS used in this study contains selenium at 355 nm. N-Acetylcysteine (NAC), a GSH precursor, was dissolved in water. KU 60019 and KU 55933 (Tocris, Ellisville, MO) were dissolved in DMSO. All chemicals were from Sigma-Aldrich unless otherwise indicated.
shRNA Knockdown
SelH and SelH2 shRNA sequences targeting the 3′ SelH mRNA at nucleotides 333–353 and 503–523 were designed based on Invitrogen Block-itTM RNAi designer, and the human non-target scrambled sequence (5′-CCTAAGGTTAAGTCGCCCTCGC-3′) was adapted from Addgene Organization. The lentiviral particles containing shRNA cassette were produced by BLOCK-iT™ Lentiviral RNAi Expression System (Life Technologies) and used to infect cells in culture. After viral infection (1 day), recovery (1 day), and blasticidin selection (14 days), 12 viable clones were picked from each viral infection and sub-cultured to confirm the knockdown efficiency by using quantitative RT-PCR. Human TaqMan probes (FAM-tagged) and primers were purchased from Applied Biosystems using inventoried TaqMan gene expression assays: SelH (Hs00415057_m1) and GAPDH (Hs99999905_m1). Cells containing SelH or SelH2 shRNA sequences expressed ∼80% less SelH mRNA as compared with scrambled shRNA cells. A viable colony is defined as one containing more than 50 cells. Passage 2 SelH shRNA MRC-5 cells were employed for experiments unless otherwise indicated. p53 shRNA knockdown was performed as described previously (36).
Detection of ROS, Immunofluorescence, and Immunoblotting
Levels of intracellular ROS were assessed by using 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA, Invitrogen). CM-H2DCFDA stock (1 m) was prepared in DMSO. MRC-5 cells on coverslips were washed twice with PBS, incubated in phenol red-free RPMI medium containing 5 μm CM-H2DCFDA for 15 min, washed twice with PBS, and then incubated in phenol red-free RPMI medium for an additional 15 min in an incubator. Cells were then imaged under a Zeiss AxioObserver 100 fluorescence microscope for image acquisition using the FITC 488-nm excitation spectra setting. Immunofluorescence and immunoblotting analyses were performed as described previously (34, 35). Focus positive cells were defined as those containing at least five foci within the nucleus (34, 35, 37). Briefly, permeabilized cells were incubated overnight at 4 °C with antibodies against phospho-H2AX on Ser-139 (γH2AX, 1:200; Abcam, Cambridge, MA), phospho-ATM on Ser-1981 (pATM Ser-1981, 1:500; Rockland, Gilbertsville, PA), H2AX (1:500; Abcam), and ATM (1:500; Epitomics, Burlingame, CA). γH2AX and pATM Ser-1981 are well-defined markers for DNA breaks and ATM pathway activation, respectively (38, 39). Six pictures were randomly taken from each slide. Nuclear fraction was prepared by using a Nuclear and Cytoplasmic Extraction Kit (G-Bioscience, MO), separated by SDS-PAGE, and transferred onto PVDF membranes. Blots were incubated with antibodies against phospho-Nrf2 on Ser-40 (pNrf2, 1:1000; Epitomics), Nrf2 (1:1000; Santa Cruz Biotechnology), and lamin B (1:1000; Santa Cruz Biotechnology), followed by incubation with HRP-conjugated secondary antibodies and chemiluminescent reagents (Super Signal, Pierce) for signal acquisition. All experiments were performed in duplicate and a minimum of three times.
Senescence and Autofluorescence Assays
The activity of senescence-associated β-galactosidase (SA-β-gal), a senescence marker undetectable in actively proliferating cells (10), was determined using a Senescence Detection Kit (BioVision, San Francisco, CA) according to manufacturer's instruction. MRC-5 cells were seeded onto 12-well plates (104 cells/well) in the presence or absence of H2O2 (20 μm) in a 3% or a 20% O2 incubator. Images were captured under a light microscope for quantification. Autofluorescence is another index of aging in cultured human diploid fibroblasts (40). Passage 4 SelH and scrambled shRNA MRC-5 cells were subjected to Zeiss AxioObserver 100 fluorescence microscope at 200× magnification. The fluorescence signal was acquired by using filter cube set 49 (excitation, 365 nm; filter, 395 nm; emission, 445 nm), and the intensity of autofluorescence was analyzed by Axiovision software.
Clonogenic Assay
After being seeded (750 cells/6-cm dish) for 24 h, SelH and scrambled shRNA HeLa and HCT116+hMLH1 cells were incubated with a gradient concentration of DNA-damaging agents, including hydroxyurea, neocarzinostatin, camptothecin, paraquat, and H2O2 (Fisher Scientific) for 24 h. Then, the drug-containing medium was replaced by a complete medium and cultured for additional 7 days. Cells were washed with 1× PBS, fixed in 90% methanol, and stained by 0.5% crystal violet (Alfa Aesar, MA) in 25% methanol. A viable colony is defined as one composing of more than 50 cells.
Laser Microirradiation and Live Cell Imaging
A mixture of oxidative and broken DNA damage was generated in live cell nuclei by laser-induced microirradiation using a pulsed nitrogen laser as previously described (41). SelH-GFP (24) and proliferating cell nuclear antigen (PCNA)-DsRed plasmids were transiently transfected into MRC-5 cells. The laser system was coupled to a Zeiss Axiovert microscope for live cell, time lapse image capture.
Detection of GSH and Apoptotic Cells
Total intracellular GSH was estimated using monochlorobimane (mBCL), which formed fluorescent adducts with GSH (42, 43). Adherent cells were collected and preloaded with mBCl (40 μm) in PBS for 10 min, followed by incubation with propyl iodide permeable only to dead cells. The stained cells were applied to FACSCanto II flow cytometer (BD Bioscience, CA) immediately to determine the content of GSH per 10,000 live cells. Apoptotic cells were determined by a Mitocapture kit (Biovision) on the basis of the disruption of mitochondrial membrane potential. The Mitocapture reagent accumulated in the mitochondria and showed red fluorescence only in live cells, but could not aggregate and showed green fluorescence in apoptotic cells. Cells on coverslips were incubated with 1:1000 Mitocapture reagent (diluted in pre-warmed incubation buffer) in an incubator for 15 min, and washed three times with incubation buffer. The stained cells were subjected to fluorescent microscopic analyses of GFP and DsRed signals. Eight pictures were randomly taken from each slide.
RESULTS
Essential Role of SelH in the Inhibition of Replicative Senescence and Oxidative Stress
Strikingly, SelH shRNA MRC-5 cells barely proliferated, accumulated non-dividing large and flat cells, and showed completely stalled growth by passage 4 or 36 days after clonal selection, whereas scrambled shRNA MRC-5 cells proliferated exponentially (Fig. 1, A and B). These results indicate that SelH deficiency restricts replicative lifespan in human diploid fibroblasts. Old or senescent cells in culture have previously been shown to exhibit elevated autofluorescence (40, 44). Consistent with these observations, microscopic analyses of passage 4, non-proliferating SelH shRNA MRC-5 cells showed 7-fold greater autofluorescence signal as compared with scrambled shRNA cells (Fig. 1B). Further microscopic analyses of CM-H2DCFDA fluorescence demonstrated that level of intracellular ROS was 2-fold greater in SelH than in scrambled shRNA MRC-5 cells at passage 4 when cultured under 20% O2 (Fig. 1C). Overall, these results implicate ROS in the slow proliferation and the formation of senescence-like phenotypes in SelH shRNA MRC-5 cells.
FIGURE 1.

Severe inhibition of proliferation, early onset of cellular senescence, and increased oxidative stress in SelH shRNA MRC-5 cells. SelH shRNA (two targeting sequences) and scrambled shRNA MRC-5 human diploid fibroblasts were seeded (104 cell/well) onto 12-well plates and cultured for 36 days in a 20% (A) or a 3% O2 incubator (E) (n = 3), followed by cell counting when approaching confluence. B, autofluorescence in the cells was assessed through the FITC channel of Zeiss AxioObserver 100 fluorescence microscope (n = 6). C, passage 2 cells were incubated with CM-H2DCFDA (5 μm) for 15 min at 37 °C, followed by FITC signal acquisition through a fluorescence microscope. The related ROS levels were presented as mean ± S.E. (n = 6) and representative pictures were shown. D, cells were cultured in a 20% or a 3% O2 incubator for 7 days, followed by identification of SA-β-gal-positive cells with their respective mean ± S.E. (n = 3) presented. *, p < 0.05, compared with scrambled shRNA cells; #, p < 0.05, compared with 3% O2.
Because fibroblasts cultured in a typical 20% O2 incubator is believed to be under chronic oxygen tension and can accelerate replicative senescence (45, 46), features of senescence in MRC-5 cells were also assessed under a 3% O2 (physiological level) culture condition. While SA-β-gal expression was significantly greater (p < 0.05) in SelH than in scrambled shRNA MRC-5 cells being cultured either in a 3% or a 20% O2 incubator for 7 days (Fig. 1D), the extent of which was significantly reduced when cultured under 3% O2 in both SelH and scrambled shRNA MRC-5 cells. Moreover, the complete growth inhibition of SelH shRNA MRC-5 cells maintained under 20% O2 for 5 weeks could be partially rescued when grown in a 3% O2 incubator, although SelH shRNA MRC-5 cells remained to proliferate poorly (∼300-fold slower) as compared with scrambled shRNA MRC-5 cells (Fig. 1, A and E). Similarly, scrambled shRNA MRC-5 cells proliferated 55-fold faster in a 3% than in a 20% O2 incubator. Altogether, these results suggest that SelH is required for cellular proliferation and the suppression of replicative senescence in a manner depending on ROS in human diploid fibroblasts.
SelH shRNA MRC-5 Cells Display Sustained DNA Damage Response and Exacerbated Senescence Induction after H2O2 Treatment
Senescence in human diploid fibroblasts is associated with persistent broken and oxidative DNA damage (47, 48), both of which can result in ATM pathway activation (11, 12, 49). Thus, SelH and scrambled shRNA MRC-5 cells were treated with H2O2 (20 μm) for 1 day, followed by 0–5 days recovery. The percent γH2AX and pATM Ser-1981 positive cells were greater (p < 0.05) in SelH than in scrambled shRNA cells before and 1 day after H2O2 treatment (Fig. 2, A and B). During the course of 5-day recovery, the abundance of γH2AX and pATM Ser-1981 positive cells subsided in the scrambled shRNA cells whereas these DNA damage markers accumulated further in SelH shRNA cells. Interestingly, the recovery of pATM Ser-1981 expression was complete and happened faster than that of γH2AX in scrambled shRNA MRC-5 cells, suggesting that 20 μm was a physiological dose of H2O2 in normal MRC-5 cells and that the ATM pathway activation preceded γH2AX formation in this scenario. The expression and distribution of H2AX and ATM did not differ before and after H2O2 treatment, as shown in the representative pictures (Fig. 2, A and B). Furthermore, the percent SA-β-gal-positive cells 5 days after recovery were 17 and 70% in scrambled shRNA and SelH shRNA MRC-5 cells, respectively, in a 20% O2 incubator (Fig. 2C). When maintained under 3% O2, they dropped (p < 0.05) to 2 and 34% in scrambled and SelH shRNA MRC-5 cells, respectively. Taken together, these results suggest that SelH plays an essential role in gatekeeping genomic integrity and suppressing senescence in the response of MRC-5 normal diploid fibroblasts to oxidative stress.
FIGURE 2.

Persistent DNA damage response and exacerbated cellular senescence in SelH shRNA MRC-5 cells after H2O2 treatment. Passage 2 SelH shRNA and scrambled shRNA MRC-5 cells seeded onto coverslips were treated with H2O2 (20 μm), followed by a course of 5-day recovery. The percent γH2AX (A) and pATM Ser-1981 (B)-positive cells were presented with mean ± S.E. (n = 6), and representative pictures taken at day 5 after recovery were shown. C, 5 days following the recovery, SA-β-gal-positive cells were determined as described in Fig. 1, and data are presented with mean ± S.E. (n = 6). *, p < 0.05, compared with scrambled shRNA cells; #, p < 0.05, compared with 3% O2.
SelH Deficiency Specifically Sensitizes Cells to DNA-damaging Agents That Directly Contribute to Oxidative Stress
Next, we asked whether SelH protected against genotoxic agents other than H2O2. Although clonogenic assay is considered a gold standard for assessing cell proliferation after DNA damage, not all cells, including MRC-5 cells, can effectively form colonies when seeded at very low density. To circumvent this limitation and to evaluate the protective role of SelH in other cell types, SelH shRNA and scrambled shRNA HeLa and HCT116 colorectal cancer cells were generated. Results from clonogenic assays showed that SelH shRNA HeLa cells displayed increased sensitivity to oxidative stress inducers paraquat and H2O2 (Fig. 3, A and B), but not to replication stress inducers hydroxyurea and camptothecin or a potent γ-irradiation mimicry, neocarzinostatin (Fig. 3, C–E). In addition, SelH shRNA HCT116+hMLH1 cells (35) displayed increased sensitivity to H2O2 exposure dose-dependently (data not shown). Interestingly, SelH mRNA levels were induced by H2O2 exposure in a dose-dependent manner (Fig. 3F). To further investigate the role of SelH in cellular response to DNA damage, live cell imaging was performed to track SelH translocation to an area containing a mixture of locally and freshly induced oxidative and broken DNA. Although DsRed-tagged PCNA, a sensitive marker of DNA synthesis and repair (50, 51), was rapidly recruited to and enriched at the site of DNA damage, SelH-GFP did not mobilize in the MRC-5 cells (Fig. 4). Therefore, SelH specifically protects against genotoxic agents that induce oxidative stress, but does not seem to be directly involved in the repair of or early response to the DNA damage.
FIGURE 3.

Increased sensitivity of SelH shRNA HeLa cells to H2O2 and paraquat, but not other DNA-damaging agents. Clonogenic assays were performed in SelH shRNA and scrambled shRNA HeLa cells following treatment with a gradient concentration of H2O2 (A), paraquat (B), hydroxyurea (C), camptothecin (D), and neocarzinostatin (E). The survival rates were presented with mean ± S.E. (n = 3). F, quantitative RT-PCR analyses of SelH mRNA expression in scrambled shRNA HeLa cells treated with a gradient concentration of H2O2 for 24 h. After normalization with 18 S rRNA, the expression level without H2O2 treatment was set as 1. *, p < 0.05, compared with no H2O2 treatment.
FIGURE 4.
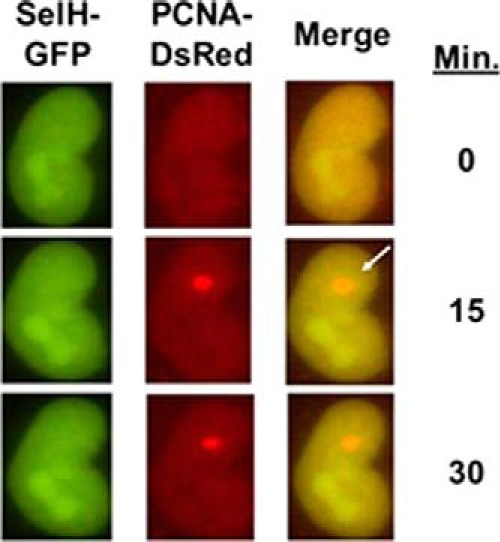
SelH does not mobilize to laser-induced oxidative and broken DNA in live cells. Localized DNA damage was generated and followed up to 30 min post-irradiation in live MRC-5 cells transiently expressing SelH-GFP and PCNA-DsRed. The localizations of SelH and PCNA were detected under a time-lapse fluorescence microscopy by employing GFP and DsRed channels, respectively. The open arrow indicates the site of localized laser damage.
The Slow Proliferation Phenotype in SelH shRNA MRC-5 Cells Depends on ATM Kinase Activity and p53 Expression
The ATM kinase and p53 protein are known to modulate the senescence response to oxidative stress in fibroblasts and endothelial cells (34, 52). Treatment with Ku 60019 (5 μm), a specific ATM kinase inhibitor (53), for 28 days alleviated the slow proliferation phenotype by 3-fold in SelH shRNA MRC-5 cells, but slightly suppressed the proliferation in scrambled shRNA MRC-5 cells (Fig. 5A). SelH shRNA MRC-5 cells proliferated 14-fold slower than scrambled shRNA cells, but such a difference was reduced to 3.6-fold in the presence of Ku 60019. Similar proliferation and senescence results were observed when the cells were treated with Ku 55933 (Fig. 5B), a popular but less specific ATM kinase inhibitor; however, the extent of slow proliferation alleviation in SelH shRNA MRC-5 cells was much greater compared with Ku 60019. Analyses of SA-β-gal staining confirmed that the ATM kinase is required to senesce SelH shRNA cells (40% versus 5%) after being cultured for 28 days (Fig. 5C). SelH shRNA MRC-5 cells significantly accumulated (p < 0.05) additional γH2AX and pATM Ser-1981 after 28 days in a 20% O2 incubator, but such induction was completely reversed or inhibited (p < 0.05) in the presence of Ku 60019 (Fig. 5, D and E). Next, SelH and p53 double shRNA MRC-5 cells were generated to test a role of p53 in the slow proliferation phenotype. After puromycin selection, the amount of colonies was 5-fold greater in SelH and p53 double shRNA than in SelH shRNA MRC-5 cells (Fig. 5F). In particular, the cell size was much smaller and the shape was rounded in the double shRNA cells, as opposed to those in SelH shRNA MRC-5 cells displaying enlarged and flattened senescence phenotypes, under a 40-fold light microscope. Altogether, the ATM kinase activity and p53 protein are necessary for the induction of replicative senescence in SelH shRNA MRC-5 diploid fibroblasts under chronic oxidative stress.
FIGURE 5.

The ATM kinase and p53 protein are involved in the protection of SelH against genome instability and replicative senescence in MRC-5 cells. SelH shRNA and scrambled shRNA MRC-5 cells in 12-well plates (2 × 104 cells/well) were cultured in the presence or absence of Ku 60019 (A) and Ku 55933 (B) at 5 μm for 28 days, followed by cell counting. Immunofluorescent analyses of γH2AX and pATM Ser-1981 expression and microscopic capturing of SA-β-gal appearance were performed in SelH shRNA MRC-5 cells (C–E, *, p < 0.05, compared with Day 0 or with Ku 60019 or Ku 55933 treatment). F, SelH and p53 double shRNA and the control MRC-5 cells were generated 14 days after puromycin selection, followed by counting viable colonies stained with crystal violet. A colony is defined as one containing at least 50 cells. Values are means ± S.E. (n = 3) (*, p < 0.05). Representative pictures taken under a 40-fold bright field were shown.
GSH Deficiency and Nuclear pNrf2 Accumulation in the Apoptotic Response of SelH shRNA HeLa Cells to H2O2 Treatment
The expression of glutamylcysteine synthetase, a key enzyme for de novo GSH biosynthesis, is increased in murine hippocampal HT22 cells overexpressing human SelH (27). Here we showed that the level of intracellular GSH was significantly lower (p < 0.05) in SelH shRNA than in scrambled shRNA HeLa cells before and after exposure with H2O2 for 24 h (Fig. 6A). H2O2 treatment linearly increased GSH level up to a concentration of 80 and 160 μm in the SelH and scrambled shRNA cells, respectively. While H2O2 treatment (160 μm) resulted in a time-dependent induction of mitochondrial membrane potential disruption, an indicator of apoptosis, in both SelH shRNA and scrambled shRNA cells, the extent of which was significantly greater (p < 0.05) in the former than the latter cells (Fig. 6B). Further statistical analyses indicated that the intracellular GSH level was inversely correlated (p < 0.05) with apoptotic death after H2O2 treatment. To further understand a role of GSH in the response of SelH shRNA cells to H2O2 treatment, clonogenic assays were performed in the presence or absence of NAC, a GSH precursor. Although SelH shRNA cells were more sensitive than scrambled shRNA cells to H2O2 treatment, the supplement of NAC (10 mm) rescued the retarded proliferation of SelH shRNA cells to a level similar to that of the scrambled shRNA cells (Fig. 6C). The Nrf2-Keap1 pathway also regulates GSH biosynthesis (54). Upon oxidative stress, Nrf2 is phosphorylated and disassociates from Keap1, followed by nuclear translocation and transactivation of genes assisting GSH synthesis. Results from Western and immunofluorescent analyses showed that levels of pNrf2 and Nrf2 in the nucleus were increased after treatment of the cells with a gradient concentration of H2O2 (0–80 μm, Fig. 7, A and B) and during a 5-day recovery (Fig. 7C), the extent of which was significantly greater (p < 0.05) in SelH shRNA than in scrambled shRNA cells. Nuclear lamin B level did not differ by H2O2 treatment or between cell types. Because nuclear Nrf2 level also increased as a result of the pNrf2 translocation (55), lamin B was used as a loading control in the nucleus. Altogether, SelH-deficient cells display declined intracellular GSH content and sustained nuclear pNrf2 accumulation in response to oxidative stress.
FIGURE 6.
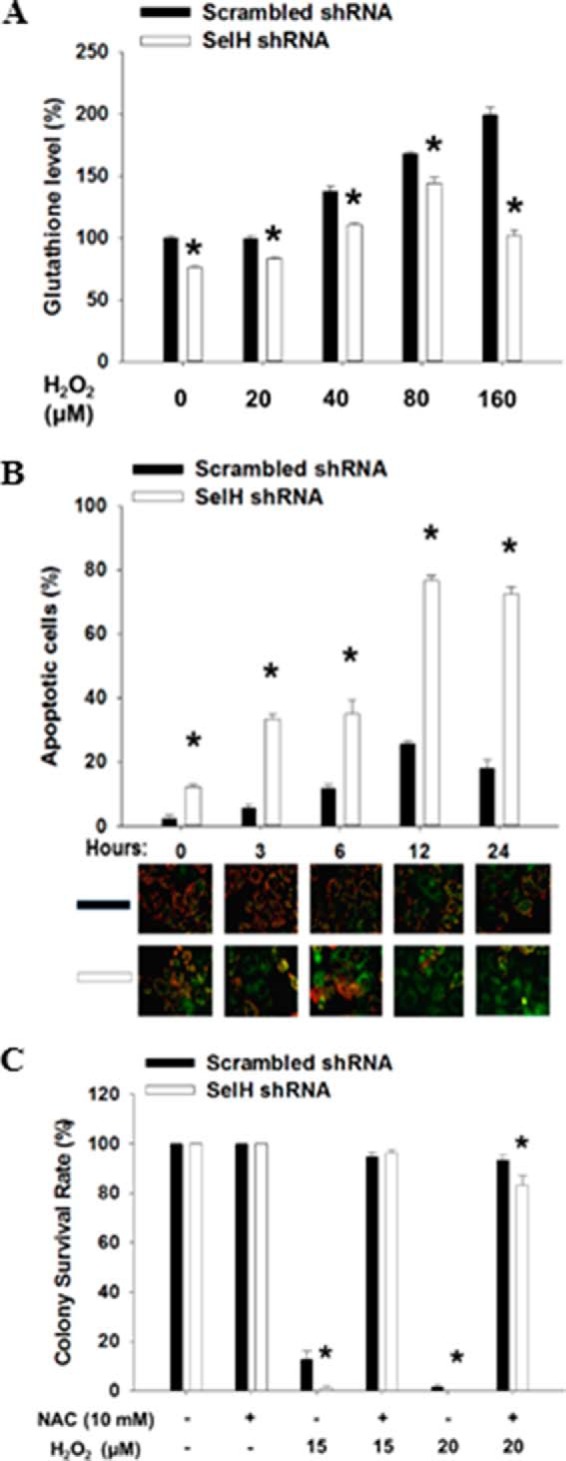
Roles of SelH in intracellular GSH content and apoptotic death in the response to H2O2 treatment in HeLa cells. A, early passage cells were treated with H2O2 (0–160 μm) for 24 h, followed by staining of trypsinized single cells with mBCL for flow cytometric analysis. B, cells were treated with H2O2 (160 μm) and harvested for Mitocapture staining to assess apoptosis by determining the disruption of mitochondrial membrane potential. Representative pictures were shown (green, apoptotic cells; red, live cells). The percent apoptotic cells were presented with mean ± S.E. (n = 8). C, clonogenic assay was performed after treatment with H2O2 alone or in combination with NAC. The number of colonies in SelH and scrambled shRNA cells without H2O2 or NAC treatment was set as 100%. Values are means ± S.E. (n = 3). *, p < 0.05, compared with scrambled shRNA cells.
FIGURE 7.
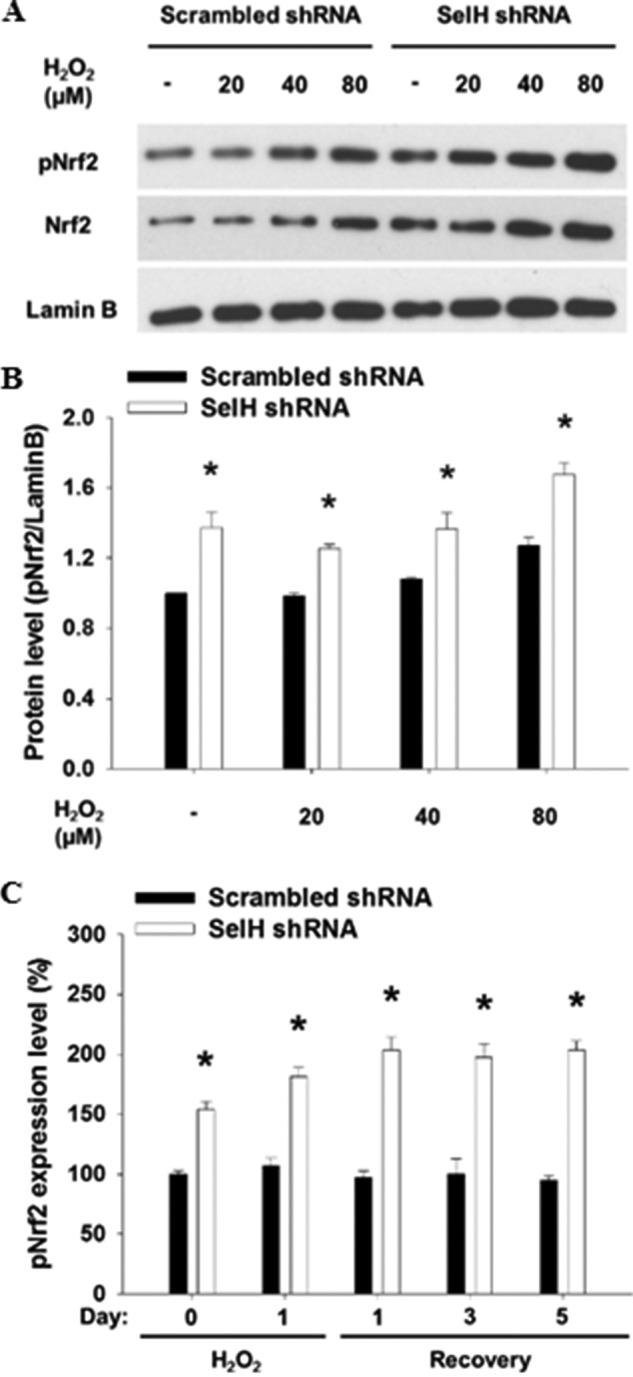
Roles of SelH in the nuclear accumulation of phosphorylated Nrf2 after H2O2 treatment. A, nuclear fractions from SelH shRNA and scrambled shRNA HeLa cells treated with H2O2 for 24 h were applied for Western analysis. B, immunoblotting intensity was quantified by ImageJ and nuclear pNrf2 expression was normalized by lamin B and presented with means ± S.E. (n = 3). C, SelH shRNA and scrambled shRNA MRC-5 cells were seeded onto coverslip, treated with H2O2 (20 μm), and recovered during the course of 5 days in fresh medium. Fluorescence intensity of nuclear pNrf2 was measured, set as 100% in scrambled shRNA cells without H2O2 treatment, and presented with means ± S.E. (n = 6). *, p < 0.05, compared with scrambled shRNA cells.
DISCUSSION
In this report, we provide cellular and biochemical evidence demonstrating that SelH plays an essential role in the protection against a senescence response to oxidative stress in a manner depending on ATM and p53. SelH-deficient cells display persistent DNA damage, increased oxidative stress and Nrf2 activation, and decreased GSH level. Previous reports suggest dual roles of human SelH as an oxidoreductase and a transactivator on genes involved in GSH synthesis and phase II detoxification (24, 27). Although these results are based on overexpressed SelH in immortalized mouse cells incapable of developing replicative senescence, they offer insightful clues to explain the new role of SelH demonstrated herein. As proposed in Fig. 8, the CXXU motif may confer the redox function of SelH (24) against genome instability and oxidative stress. Furthermore, the A-T hook motif enables SelH to bind DNA minor grooves and may up-regulate the expression of genes counteracting oxidative stress (27). Taken together, cellular senescence in SelH-deficient cells seems to be induced as a result of increased oxidative stress and the subsequent ATM-p53 pathway of DNA damage response, in parallel with deficiency in the expression of phase II antioxidant enzymes.
FIGURE 8.
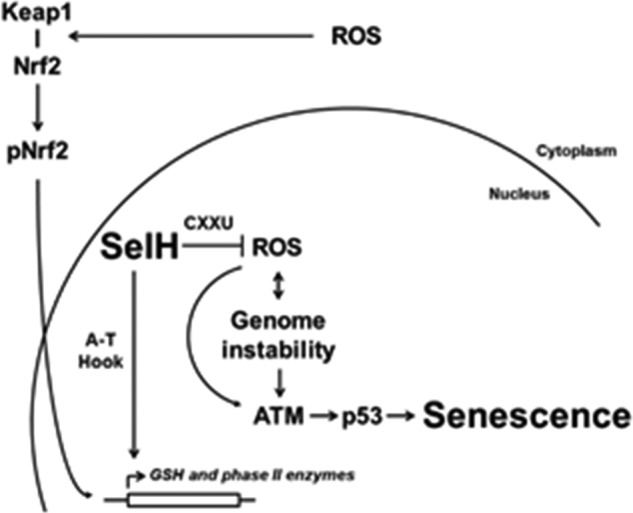
Schematic model of SelH-mediated suppression of the senescence response to oxidative stress. SelH is a dual function nuclear protein. First, SelH may serve as an antioxidant through the CXXU motif to directly clear ROS, especially H2O2 (24). Second, de novo synthesis of GSH and phase II detoxification enzymes may be transactivated by pNrf2 and SelH through the A-T hook motif (27), when Nrf2 is phosphorylated and dissociated from Keap1 and translocated to the nucleus under oxidative stress. Collectively, SelH protects the nucleus from an ATM- and p53-dependent senescent response to oxidative stress.
A key observation made in this study is the protective role of SelH against chronic oxidative stress and replicative senescence in diploid fibroblasts through genome maintenance. Culturing cells under ambient O2 level (20%) constitutively activates stress-induced senescence (56). SelH shRNA MRC-5 cells cultured in a 20% O2 incubator display ATM pathway activation and DNA breaks intrinsically, as well as persistent DNA damage after treatment with physiological concentration of H2O2. Nonetheless, other kinases such as DNA-PKcs and ATR may also participate in the senescence response, as treatment of SelH shRNA cells with the less-specific ATM kinase inhibitor Ku 55933 alleviates the growth retardation phenotype to a greater extent compared with Ku 60019 (Fig. 5, A and B). Because unrepaired DNA damage and persistent DNA damage response are hallmarks of cellular senescence (47, 57, 58), a fair question to ask is whether SelH plays a direct role in the repair of DNA damage. Results from analysis of live cell imaging of SelH mobilization to localized DNA damage do not support such a hypothesis. As the expression of recombinant wild-type selenoprotein (i.e. without substituting Sec with Cys) is becoming technically feasible (59), it is of future interest to purify SelH for enzymatic and kinetic analyses of oligonucleotide substrates representing various types of DNA damage.
SelH shRNA cells are hypersensitive to H2O2 and paraquat, but not hydroxyurea, camptothecin, and neocarzinostatin. As a pro-oxidant, paraquat initiates the generation of superoxide, which in turn is reduced to H2O2 in vivo. Interestingly, SelH siRNA cells display increased sensitivity to H2O2, but not other peroxides (24). Furthermore, although hydroxyurea is best known as an inhibitor of ribonucleotide reductase, it can also increase superoxide level and iron availability for the formation of hydroxyl radicals (60). Similarly, both camptothecin, an inhibitor of topoisomerase I (61), and neocarzinostatin, an ionizing radiation mimicry, have been reported to be capable of inducing ROS formation (62). Thus, it is surprising why SelH shRNA cells do not show increased sensitivity to these three DNA-damaging agents. They may indirectly induce the formation of ROS in forms not recognized by SelH, thus resulting in a “secondary burst” of DNA damage independent of SelH status. It is also possible that SelH does not play direct roles clearing H2O2 at later time points. Whatever the reason, data support a direct and upstream role of SelH in the dealing of H2O2 (24, 27), resulting in the protection against ROS-induced genome instability and senescence.
How does SelH protect against replicative senescence under chronic oxidative stress? Accumulation of ROS (63, 64) and reduction of antioxidant capacity, DNA repair efficacy and selenoprotein expression (65–72) have been linked to cellular and organismal aging. However, damage accumulation early in life can also contribute to or accelerate cellular senescence, as DNA breaks and ATM pathway activation are prominent and there are 3-fold more viable clones when SelH shRNA MRC-5 cells are maintained in 3% O2 than in 20% O2 incubators (data not shown) as early as two passages after clonal selection. Our data are in line with previous findings demonstrating that the ATM-p53 pathway contributes to persistent amplification of DNA damage signal and replicative senescence (15). Moreover, increased intracellular ROS may activate replicative senescence by guanine oxidation on telomeres, subsequently resulting in the disruption of telomeric T-loops and telomere dysfunction (73, 74). Although the expression of SelH, among other eight selenoproteins, has been shown to be decreased by senescence or by maintenance under a low Se medium in WI-38 cells (71), our data provide the first evidence of SelH in the direct protection against replicative senescence under chronic oxidative stress through the maintenance of genomic stability.
SelH may play a dual role in the response to oxidative stress. Burk et al. have demonstrated that the activation of Nrf2-ARE pathway in mouse liver is specifically induced by dietary deprivation of selenium, but not other dietary antioxidants such as vitamin E (75). SelH may confer the interaction between selenium and Nrf2 as this selenoprotein is among the few most sensitive ones to dietary selenium deficiency (19, 76). Consistent with this observation, SelH-deficient cells display increased nuclear pNrf2 expression endogenously or dose-dependently after H2O2 treatment. Furthermore, pNrf2 expression is known to be correlated with GSH content (77). We propose that, with increased concentrations of H2O2, GSH is dispensed faster than those synthesized de novo through Nrf2 translocation in SelH shRNA cells when ROS scavenge by SelH is hampered. Increased pNrf2 expression in SelH-deficient cells may further activate the compensatory expression of other antioxidant selenoproteins including thioredoxin reductase-1 and glutathione peroxidase-2 (78, 79). On the other hand, SelH may directly induce the de novo synthesis of GSH (27). Indeed, SelH may play an important role in sensing oxidative stress as the majority of SelH-GFP is localized in the nucleolus (Ref. 24 and Fig. 4), a nuclear region being proposed as a sensor of multiple forms of stress including H2O2 (30, 31).
How does SelH, assumed to be localized in the nucleolus (24), protect genomic integrity? As the nuclear domain accommodating the tandem repeat rDNA gene, nucleolus is a proposed sensor of nuclear oxidative stress (30, 31). First, JNK2 activation by oxidative stress promptly phosphorylates and inactivates a RNA polymerase I transcription factor TIF-IA in the nucleoli to down-regulate rRNA synthesis (31). Second, decreased rDNA copy numbers can hamper genomic integrity (81). Thus, part of the gatekeeper functions of SelH may be attributed to its proximity to rDNA and preventing TIF-IA phosphorylation by JNK2 such that rRNA down-regulation is limited. Through its antioxidative motif, SelH may also decompose local H2O2 and prevent rDNA oxidation in the nucleoli. Furthermore, there is no membrane boundary separating nucleolus from the surrounding nucleoplasm. Considering the topological and dynamic properties of a genome (82), long-range contacts of chromosomes in nucleoplasm with SelH in nucleolus may enable this selenoprotein to interact with and protect the genome distantly.
As depicted in Fig. 8, SelH is proposed to play dual roles in gatekeeping genomic integrity against a senescence response to oxidative stress. SelH may enzymatically decompose H2O2, as well as directly increase the expression of GSH as a transactivator and indirectly up-regulate other antioxidant enzymes through Nrf2. Considering the specialized nucleolar localization and function (24), SelH in principle can keep ROS levels in check, thus suppressing the accumulation of oxidative and incision damage on DNA. Such impact of SelH on the maintenance of genomic stability is consistent with the recent, refined damage theory of aging (80). In this regard, SelH could serve as a potential intervention target for the protection against genomic damage during the aging process.
Acknowledgments
We thank Dr. Vadim Gladyshev for SelH-GFP construct, Dr. Thomas Wang for sharing some of the reagents and equipment for performing quantitative RT-PCR, and Elliot Mattson and Erica Lee for helping with some cellular assays.
This is Manuscript No. J-12591 of the Mississippi Agricultural and Forestry Experimental Station, Mississippi State University.
- ROS
- reactive oxygen species
- PCNA
- proliferating cell nuclear antigen
- SelH
- selenoprotein H
- ATM
- ataxia telangiectasia-mutated
- NAC
- N-acetylcysteine
- CM-H2DCFDA
- 5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate
- SA-β-gal
- senescence-associated β-galactosidase
- mBCL
- monochlorobimane.
REFERENCES
- 1. Rodier F., Campisi J. (2011) Four faces of cellular senescence. J. Cell Biol. 192, 547–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fumagalli M., Rossiello F., Clerici M., Barozzi S., Cittaro D., Kaplunov J. M., Bucci G., Dobreva M., Matti V., Beausejour C. M., Herbig U., Longhese M. P., d'Adda di Fagagna F. (2012) Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mallette F. A., Ferbeyre G. (2007) The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle 6, 1831–1836 [DOI] [PubMed] [Google Scholar]
- 4. Takahashi A., Ohtani N., Yamakoshi K., Iida S., Tahara H., Nakayama K., Nakayama K. I., Ide T., Saya H., Hara E. (2006) Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 8, 1291–1297 [DOI] [PubMed] [Google Scholar]
- 5. Halazonetis T. D., Gorgoulis V. G., Bartek J. (2008) An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355 [DOI] [PubMed] [Google Scholar]
- 6. Jun J. I., Lau L. F. (2010) The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 12, 676–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ressler S., Bartkova J., Niederegger H., Bartek J., Scharffetter-Kochanek K., Jansen-Dürr P., Wlaschek M. (2006) p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 5, 379–389 [DOI] [PubMed] [Google Scholar]
- 8. Voghel G., Thorin-Trescases N., Farhat N., Nguyen A., Villeneuve L., Mamarbachi A. M., Fortier A., Perrault L. P., Carrier M., Thorin E. (2007) Cellular senescence in endothelial cells from atherosclerotic patients is accelerated by oxidative stress associated with cardiovascular risk factors. Mech Ageing Dev. 128, 662–671 [DOI] [PubMed] [Google Scholar]
- 9. Wang C., Jurk D., Maddick M., Nelson G., Martin-Ruiz C., von Zglinicki T. (2009) DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 8, 311–323 [DOI] [PubMed] [Google Scholar]
- 10. Dimri G. P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E. E., Linskens M., Rubelj I., Pereira-Smith O. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee J. H., Paull T. T. (2005) ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308, 551–554 [DOI] [PubMed] [Google Scholar]
- 12. Guo Z., Kozlov S., Lavin M. F., Person M. D., Paull T. T. (2010) ATM activation by oxidative stress. Science 330, 517–521 [DOI] [PubMed] [Google Scholar]
- 13. Canman C. E., Lim D. S., Cimprich K. A., Taya Y., Tamai K., Sakaguchi K., Appella E., Kastan M. B., Siliciano J. D. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677–1679 [DOI] [PubMed] [Google Scholar]
- 14. Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N., Vassiliou L. V., Kolettas E., Niforou K., Zoumpourlis V. C., Takaoka M., Nakagawa H., Tort F., Fugger K., Johansson F., Sehested M., Andersen C. L., Dyrskjot L., Ørntoft T., Lukas J., Kittas C., Helleday T., Halazonetis T. D., Bartek J., Gorgoulis V. G. (2006) Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637 [DOI] [PubMed] [Google Scholar]
- 15. Suzuki M., Suzuki K., Kodama S., Yamashita S., Watanabe M. (2012) Persistent amplification of DNA damage signal involved in replicative senescence of normal human diploid fibroblasts. Oxid. Med. Cell Longev. 2012:310534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kryukov G. V., Castellano S., Novoselov S. V., Lobanov A. V., Zehtab O., Guigó R., Gladyshev V. N. (2003) Characterization of mammalian selenoproteomes. Science 300, 1439–1443 [DOI] [PubMed] [Google Scholar]
- 17. Lu J., Holmgren A. (2009) Selenoproteins. J. Biol. Chem. 284, 723–727 [DOI] [PubMed] [Google Scholar]
- 18. Sunde R. A. (2010) Molecular biomarker panels for assessment of selenium status in rats. Exp. Biol. Med. 235, 1046–1052 [DOI] [PubMed] [Google Scholar]
- 19. Raines A. M., Sunde R. A. (2011) Selenium toxicity but not deficient or super-nutritional selenium status vastly alters the transcriptome in rodents. BMC Genomics 12, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kipp A. P., Banning A., van Schothorst E. M., Méplan C., Coort S. L., Evelo C. T., Keijer J., Hesketh J., Brigelius-Flohé R. (2012) Marginal selenium deficiency down-regulates inflammation-related genes in splenic leukocytes of the mouse. J. Nutr. Biochem. 23, 1170–1177 [DOI] [PubMed] [Google Scholar]
- 21. Sengupta A., Lichti U. F., Carlson B. A., Ryscavage A. O., Gladyshev V. N., Yuspa S. H., Hatfield D. L. (2010) Selenoproteins are essential for proper keratinocyte function and skin development. PLoS ONE 5, e12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Downey C. M., Horton C. R., Carlson B. A., Parsons T. E., Hatfield D. L., Hallgrímsson B., Jirik F. R. (2009) Osteo-chondroprogenitor-specific deletion of the selenocysteine tRNA gene, Trsp, leads to chondronecrosis and abnormal skeletal development: a putative model for Kashin-Beck disease. PLoS Genet 5, e1000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCann J. C., Ames B. N. (2011) Adaptive dysfunction of selenoproteins from the perspective of the triage theory: why modest selenium deficiency may increase risk of diseases of aging. FASEB J. 25, 1793–1814 [DOI] [PubMed] [Google Scholar]
- 24. Novoselov S. V., Kryukov G. V., Xu X. M., Carlson B. A., Hatfield D. L., Gladyshev V. N. (2007) Selenoprotein H is a nucleolar thioredoxin-like protein with a unique expression pattern. J. Biol. Chem. 282, 11960–11968 [DOI] [PubMed] [Google Scholar]
- 25. Morozova N., Forry E. P., Shahid E., Zavacki A. M., Harney J. W., Kraytsberg Y., Berry M. J. (2003) Antioxidant function of a novel selenoprotein in Drosophila melanogaster. Genes Cells 8, 963–971 [DOI] [PubMed] [Google Scholar]
- 26. Ben Jilani K. E., Panee J., He Q., Berry M. J., Li P. A. (2007) Overexpression of selenoprotein H reduces Ht22 neuronal cell death after UVB irradiation by preventing superoxide formation. Int. J. Biol. Sci. 3, 198–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Panee J., Stoytcheva Z. R., Liu W., Berry M. J. (2007) Selenoprotein H is a redox-sensing high mobility group family DNA-binding protein that up-regulates genes involved in glutathione synthesis and phase II detoxification. J. Biol. Chem. 282, 23759–23765 [DOI] [PubMed] [Google Scholar]
- 28. Mendelev N., Witherspoon S., Li P. A. (2009) Overexpression of human selenoprotein H in neuronal cells ameliorates ultraviolet irradiation-induced damage by modulating cell signaling pathways. Exp. Neurol. 220, 328–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mendelev N., Mehta S. L., Witherspoon S., He Q., Sexton J. Z., Li P. A. (2011) Upregulation of human selenoprotein H in murine hippocampal neuronal cells promotes mitochondrial biogenesis and functional performance. Mitochondrion 11, 76–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lewinska A., Wnuk M., Grzelak A., Bartosz G. (2010) Nucleolus as an oxidative stress sensor in the yeast Saccharomyces cerevisiae. Redox Rep. 15, 87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mayer C., Bierhoff H., Grummt I. (2005) The nucleolus as a stress sensor: JNK2 inactivates the transcription factor TIF-IA and down-regulates rRNA synthesis. Genes Dev. 19, 933–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koi M., Umar A., Chauhan D. P., Cherian S. P., Carethers J. M., Kunkel T. A., Boland C. R. (1994) Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 54, 4308–4312 [PubMed] [Google Scholar]
- 33. Yanamadala S., Ljungman M. (2003) Potential role of MLH1 in the induction of p53 and apoptosis by blocking transcription on damaged DNA templates. Mol. Cancer Res. 1, 747–754 [PubMed] [Google Scholar]
- 34. Wu M., Kang M. M., Schoene N. W., Cheng W. H. (2010) Selenium compounds activate early barriers of tumorigenesis. J. Biol. Chem. 285, 12055–12062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qi Y., Schoene N. W., Lartey F. M., Cheng W. H. (2010) Selenium compounds activate ATM-dependent DNA damage response via the mismatch repair protein hMLH1 in colorectal cancer cells. J. Biol. Chem. 285, 33010–33017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu M., Wu R. T., Wang T. T., Cheng W. H. (2011) Role for p53 in Selenium-Induced Senescence. J. Agric. Food Chem. 59, 11882–11887 [DOI] [PubMed] [Google Scholar]
- 37. Maude S. L., Enders G. H. (2005) Cdk inhibition in human cells compromises chk1 function and activates a DNA damage response. Cancer Res. 65, 780–786 [PubMed] [Google Scholar]
- 38. Löbrich M., Shibata A., Beucher A., Fisher A., Ensminger M., Goodarzi A. A., Barton O., Jeggo P. A. (2010) gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle 9, 662–669 [DOI] [PubMed] [Google Scholar]
- 39. Bakkenist C. J., Kastan M. B. (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 [DOI] [PubMed] [Google Scholar]
- 40. Wilhelm J., Vytásek R., Ostádalová I., Vajner L. (2009) Evaluation of different methods detecting intracellular generation of free radicals. Mol. Cell Biochem. 328, 167–176 [DOI] [PubMed] [Google Scholar]
- 41. Uematsu N., Weterings E., Yano K., Morotomi-Yano K., Jakob B., Taucher-Scholz G., Mari P. O., van Gent D. C., Chen B. P., Chen D. J. (2007) Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell Biol. 177, 219–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sebastià J., Cristòfol R., Martín M., Rodríguez-Farré E., Sanfeliu C. (2003) Evaluation of fluorescent dyes for measuring intracellular glutathione content in primary cultures of human neurons and neuroblastoma SH-SY5Y. Cytometry A 51, 16–25 [DOI] [PubMed] [Google Scholar]
- 43. Franco R., Cidlowski J. A. (2006) SLCO/OATP-like transport of glutathione in FasL-induced apoptosis: glutathione efflux is coupled to an organic anion exchange and is necessary for the progression of the execution phase of apoptosis. J. Biol. Chem. 281, 29542–29557 [DOI] [PubMed] [Google Scholar]
- 44. Kuilman T., Michaloglou C., Mooi W. J., Peeper D. S. (2010) The essence of senescence. Genes Dev. 24, 2463–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Parrinello S., Samper E., Krtolica A., Goldstein J., Melov S., Campisi J. (2003) Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 5, 741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Poulios E., Trougakos I. P., Chondrogianni N., Gonos E. S. (2007) Exposure of human diploid fibroblasts to hypoxia extends proliferative life span. Ann. N.Y. Acad. Sci. 1119, 9–19 [DOI] [PubMed] [Google Scholar]
- 47. Campisi J., d'Adda di Fagagna F. (2007) Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740 [DOI] [PubMed] [Google Scholar]
- 48. Chen Q., Fischer A., Reagan J. D., Yan L. J., Ames B. N. (1995) Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. U.S.A. 92, 4337–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tanaka T., Halicka H. D., Huang X., Traganos F., Darzynkiewicz Z. (2006) Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle 5, 1940–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Balajee A. S., Geard C. R. (2001) Chromatin-bound PCNA complex formation triggered by DNA damage occurs independent of the ATM gene product in human cells. Nucleic Acids Res. 29, 1341–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mortusewicz O., Fouquerel E., Amé J. C., Leonhardt H., Schreiber V. (2011) PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic Acids Res. 39, 5045–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhan H., Suzuki T., Aizawa K., Miyagawa K., Nagai R. (2010) Ataxia telangiectasia mutated (ATM)-mediated DNA damage response in oxidative stress-induced vascular endothelial cell senescence. J. Biol. Chem. 285, 29662–29670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Golding S. E., Rosenberg E., Valerie N., Hussaini I., Frigerio M., Cockcroft X. F., Chong W. Y., Hummersone M., Rigoreau L., Menear K. A., O'Connor M. J., Povirk L. F., van Meter T., Valerie K. (2009) Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 8, 2894–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moinova H. R., Mulcahy R. T. (1999) Up-regulation of the human gamma-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem. Biophys. Res. Commun. 261, 661–668 [DOI] [PubMed] [Google Scholar]
- 55. Huang H. C., Nguyen T., Pickett C. B. (2000) Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc. Natl. Acad. Sci. U.S.A. 97, 12475–12480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Moussavi-Harami F., Duwayri Y., Martin J. A., Buckwalter J. A. (2004) Oxygen effects on senescence in chondrocytes and mesenchymal stem cells: consequences for tissue engineering. Iowa Orthop J. 24, 15–20 [PMC free article] [PubMed] [Google Scholar]
- 57. Di Micco R., Cicalese A., Fumagalli M., Dobreva M., Verrecchia A., Pelicci P. G., di Fagagna F. (2008) DNA damage response activation in mouse embryonic fibroblasts undergoing replicative senescence and following spontaneous immortalization. Cell Cycle 7, 3601–3606 [DOI] [PubMed] [Google Scholar]
- 58. Rodier F., Coppé J. P., Patil C. K., Hoeijmakers W. A., Muñoz D. P., Raza S. R., Freund A., Campeau E., Davalos A. R., Campisi J. (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Han X., Fan Z., Yu Y., Liu S., Hao Y., Huo R., Wei J. (2013) Expression and characterization of recombinant human phospholipid hydroperoxide glutathione peroxidase. IUBMB Life 65, 951–956 [DOI] [PubMed] [Google Scholar]
- 60. Davies B. W., Kohanski M. A., Simmons L. A., Winkler J. A., Collins J. J., Walker G. C. (2009) Hydroxyurea induces hydroxyl radical-mediated cell death in Escherichia coli. Mol. Cell 36, 845–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wall M. E., Wani M. C., Cook C. E., Palmer K. H., McPhail A. I., Sim G. A. (1966) Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from camptotheca acuminate. J. Am. Chem. Soc. 88, 3888–3890 [Google Scholar]
- 62. Kang M. A., So E. Y., Simons A. L., Spitz D. R., Ouchi T. (2012) DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis 3, e249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Harman D. (1960) The free radical theory of aging: the effect of age on serum mercaptan levels. J. Gerontol 15, 38–40 [DOI] [PubMed] [Google Scholar]
- 64. Lawless C., Jurk D., Gillespie C. S., Shanley D., Saretzki G., von Zglinicki T., Passos J. F. (2012) A stochastic step model of replicative senescence explains ROS production rate in ageing cell populations. PLoS ONE 7, e32117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Williams J. B., Roberts S. P., Elekonich M. M. (2008) Age and natural metabolically-intensive behavior affect oxidative stress and antioxidant mechanisms. Exp Gerontol 43, 538–549 [DOI] [PubMed] [Google Scholar]
- 66. Sivonová M., Tatarková Z., Duracková Z., Dobrota D., Lehotský J., Matáková T., Kaplán P. (2007) Relationship between antioxidant potential and oxidative damage to lipids, proteins and DNA in aged rats. Physiol. Res. 56, 757–764 [DOI] [PubMed] [Google Scholar]
- 67. Kim J. W., No J. K., Ikeno Y., Yu B. P., Choi J. S., Yokozawa T., Chung H. Y. (2002) Age-related changes in redox status of rat serum. Arch Gerontol. Geriatr. 34, 9–17 [DOI] [PubMed] [Google Scholar]
- 68. Pieri C., Testa R., Marra M., Bonfigli A. R., Manfrini S., Testa I. (2001) Age-dependent changes of serum oxygen radical scavenger capacity and haemoglobin glycosylation in non-insulin-dependent diabetic patients. Gerontology 47, 88–92 [DOI] [PubMed] [Google Scholar]
- 69. Kimoto-Kinoshita S., Nishida S., Tomura T. T. (1999) Age-related change of antioxidant capacities in the cerebral cortex and hippocampus of stroke-prone spontaneously hypertensive rats. Neurosci. Lett. 273, 41–44 [DOI] [PubMed] [Google Scholar]
- 70. Pérez R., López M., Barja de Quiroga G. (1991) Aging and lung antioxidant enzymes, glutathione, and lipid peroxidation in the rat. Free Radic Biol. Med. 10, 35–39 [DOI] [PubMed] [Google Scholar]
- 71. Legrain Y., Touat-Hamici Z., Chavatte L. (2014) Interplay between selenium levels, selenoprotein expression, and replicative senescence in WI-38 human fibroblasts. J. Biol. Chem. 289, 6299–6310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Garm C., Moreno-Villanueva M., Bürkle A., Petersen I., Bohr V. A., Christensen K., Stevnsner T. (2013) Age and gender effects on DNA strand break repair in peripheral blood mononuclear cells. Aging Cell 12, 58–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Opresko P. L., Fan J., Danzy S., Wilson D. M., 3rd, Bohr V. A. (2005) Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 33, 1230–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Richter T., Proctor C. (2007) The role of intracellular peroxide levels on the development and maintenance of telomere-dependent senescence. Exp. Gerontol 42, 1043–1052 [DOI] [PubMed] [Google Scholar]
- 75. Burk R. F., Hill K. E., Nakayama A., Mostert V., Levander X. A., Motley A. K., Johnson D. A., Johnson J. A., Freeman M. L., Austin L. M. (2008) Selenium deficiency activates mouse liver Nrf2-ARE but vitamin E deficiency does not. Free Radic Biol. Med. 44, 1617–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kipp A., Banning A., van Schothorst E. M., Méplan C., Schomburg L., Evelo C., Coort S., Gaj S., Keijer J., Hesketh J., Brigelius-Flohé R. (2009) Four selenoproteins, protein biosynthesis, and Wnt signalling are particularly sensitive to limited selenium intake in mouse colon. Mol. Nutr. Food Res. 53, 1561–1572 [DOI] [PubMed] [Google Scholar]
- 77. Suh J. H., Shenvi S. V., Dixon B. M., Liu H., Jaiswal A. K., Liu R. M., Hagen T. M. (2004) Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. U.S.A. 101, 3381–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sakurai A., Nishimoto M., Himeno S., Imura N., Tsujimoto M., Kunimoto M., Hara S. (2005) Transcriptional regulation of thioredoxin reductase 1 expression by cadmium in vascular endothelial cells: role of NF-E2-related factor-2. J. Cell. Physiol. 203, 529–537 [DOI] [PubMed] [Google Scholar]
- 79. Singh A., Rangasamy T., Thimmulappa R. K., Lee H., Osburn W. O., Brigelius-Flohé R., Kensler T. W., Yamamoto M., Biswal S. (2006) Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am. J. Respir. Cell Mol. Biol. 35, 639–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gladyshev V. N. (2014) The free radical theory of aging is dead. Long live the damage theory! Antioxid. Redox Signal. 20, 727–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ide S., Miyazaki T., Maki H., Kobayashi T. (2010) Abundance of ribosomal RNA gene copies maintains genome integrity. Science 327, 693–696 [DOI] [PubMed] [Google Scholar]
- 82. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]