

Abstract
Polycystic liver disease (PLD) occurs in 75–90% of patients affected by autosomal dominant polycystic kidney disease (ADPKD), which affects 1∶400–1,000 adults and arises from inherited mutations in the PKD1 or PKD2 genes. PLD can lead to bile duct obstructions, infected or bleeding cysts, and hepatomegaly, which can diminish quality of life. At present, no effective, approved therapy exists for ADPKD or PLD. We recently showed that inhibition of the molecular chaperone heat shock protein 90 (HSP90) with a small molecule inhibitor, STA-2842, induced the degradation of multiple HSP90-dependent client proteins that contribute to ADPKD pathogenesis and slowed the progression of renal cystogenesis in mice with conditional deletion of Pkd1. Here, we analyzed the effects of STA-2842 on liver size and cystic burden in Pkd-/- mice with established PLD. Using magnetic resonance imaging over time, we demonstrate that ten weeks of STA-2842 treatment significantly reduced both liver mass and cystic index suggesting selective elimination of cystic tissue. Pre-treatment cystic epithelia contain abundant HSP90; the degree of reduction in cysts was accompanied by inhibition of proliferation-associated signaling proteins EGFR and others, and induced cleavage of caspase 8 and PARP1, and correlated with degree of HSP90 inhibition and with inactivation of ERK1/2. Our results suggest that HSP90 inhibition is worth further evaluation as a therapeutic approach for patients with PLD.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD), an inherited syndrome affecting 1∶400–1,000 individuals [1], [2], arises from mutations in the PKD1 or PKD2 genes, encoding the polycystins. ADPKD is invariably associated with the replacement of normal kidney parenchyma with fluid-filled cysts in middle-aged adults. For many individuals with APDKD, a secondary feature of the disease is the development of hepatic cysts [3], [4], [5], which can be symptomatic or asymptomatic. Polycystic liver disease (PLD) has been associated with mutations in both the PKD1 and PKD2 genes in patients, and is also observed in genetically engineered mice bearing these mutations [6], [7], [8]. Those who suffer from PLD and ADPKD typically develop renal failure and require dialysis and/or kidney transplantation, but rarely require hepatic transplantation. However, some individuals can experience PLD-associated complications including infected and bleeding cysts, bile duct obstruction and hepatomegaly that can require surgical intervention and diminish quality of life.
The polycystin proteins encoded by PKD1 and PKD2 regulate multiple signaling pathways that influence hepatic and renal growth and homeostasis. In ADPKD, renal cells have multiple anomalously activated signaling proteins relevant to these processes, including ribosomal protein S6 (S6), ribosomal S6 kinase (RSK/S6K), AKT, mammalian target of rapamycin (mTOR), SRC, ERK1/2, and RAF, among others [1], [2]. Therapeutics that have been evaluated for the treatment of ADPKD include targeted inhibitors of some of these proteins, such as SRC and mTOR [9], [10]. These have shown some potential for improvement of symptoms in preclinical models [11]. In clinical trials, mTOR inhibitors have demonstrated some effect in slowing kidney growth, although have had less pronounced effect on kidney function [12]. However, no highly effective therapy is currently available [13]. While many features of growth control in hepatic and renal cells are conserved and similarly affected by mutations associated with ADPKD, there is some evidence the biology of cyst formation differs in the two organs (reviewed in [14]). Somatostatin analogs have offered some benefit in reducing liver cystogenesis [15], as has inhibition of mTOR or VEGFR [7], [8].
In attempting to improve management of ADPKD, we considered that numerous studies of drug effectiveness in cancer have indicated that inhibiting a single signaling protein is generally insufficient for halting tumor growth because of functional redundancy in pathways [16], [17]. Many of the signaling proteins activated in ADPKD are also commonly activated in cancer [18], and notably, many of these proteins are dependent on the molecular chaperone heat shock protein 90 (HSP90) for stability and/or activity. HSP90 inhibition has recently demonstrated particular clinical efficacy in cancer, based on the simultaneous inhibition of multiple pro-proliferative proteins in the absence of this important chaperone [19]. In recent work, we found that inhibition of HSP90 significantly slowed renal cystogenesis and kidney growth in vivo in mice developing ADPKD because of a conditional knockout of the Pkd1 gene [20]. Because of this encouraging result, we hypothesized that HSP90 inhibition might also be beneficial for controlling the growth of hepatic cysts. In this study, we assessed the efficacy of the HSP90 inhibitor STA-2842 in limiting the development of PLD in conditional Pkd1-knockout mice. Our results, presented below, suggest that HSP90 inhibition may be both a useful and a well-tolerated therapeutic approach for patients with PLD.
Results and Discussion
To assess the value of HSP90-targeting inhibitors for PLD, we first analyzed HSP90 expression in cystic livers. Conditional Pkd1 knockout mice (Pkd1fl/fl;Cre/Esr1+, subsequently labeled Pkd1−/−) and control mice (Pkd1fl/fl;Cre/Esr1−, subsequently labeled wild type, wt) were injected with tamoxifen at post-natal days P37 and P38 of age to inactivate Pkd1, leading to significant hepatic cystogenesis [21], [22]. Immunohistochemical analysis of liver sections from 6.5-month-old mice with established PLD indicated consistent and intense expression of the inducible isoform HSP90α in the epithelial cells lining liver cysts in Pkd1−/− mice ( Fig. 1 ). No endothelial cells lining blood vessels (portal veins) in either Pkd1−/− or wt mice expressed HSP90α. Bile ducts observed in either genotype displayed heterogenous expression of HSP90α, with some having moderate to high staining, but others negative. In non-cystic tissue, low levels of HSP90α staining were observed in hepatocytes ( Fig. 1 ).
Figure 1. HSP90α is upregulated in epithelial cells lining liver cysts.
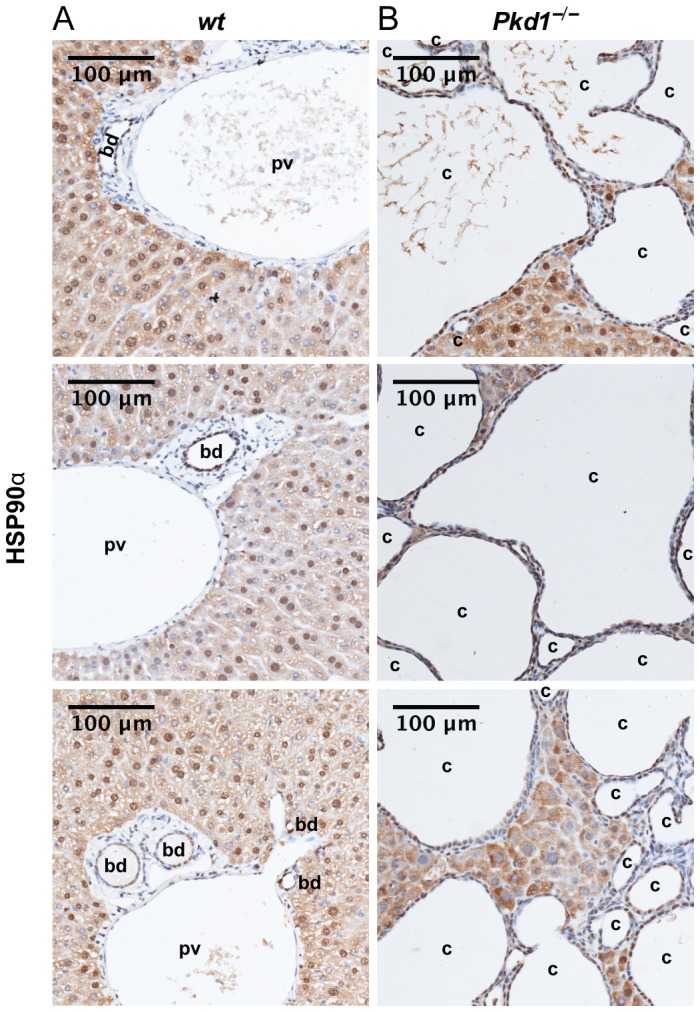
(A, B) Representative hematoxylin stained liver sections with immunohistochemical detection of HSP90α (brown) from three (A) wild type (wt) and (B) Pkd1 –/– mice. bd = bile duct, pv = portal vein, c = cyst. Scale bars = 100 µm; magnification, 20x.
We analyzed whether the HSP90 inhibitor STA-2842, a resorcinolic triazole molecule that selectively binds with the N-terminus on the ATP binding pocket of HSP90 [20], controlled the growth of liver cysts. After tamoxifen treatment at P37/P38, Pkd1−/− and wt mice were dosed weekly for 10 weeks with STA-2842 (50 mg/kg and 100 mg/kg) or vehicle (5% dextrose) starting at 4 months of age ( Fig.2A ). Magnetic resonance imaging (MRI) was performed at 0, 5, and 10 weeks after the commencement of treatment to longitudinally track the development of hepatic cysts ( Fig. 2B ). Hepatic cysts were already abundant in 4-month-old mice, and did not further increase in volume over the following 10 weeks in mice treated with vehicle ( Fig. 2C ). MRI analysis indicated no change in liver cyst volume in Pkd1−/− mice treated with 100 mg/kg STA-2842 for 5 weeks, however; a significant decrease in cyst volume was observed after 10 weeks of treatment (∼41%; p = 0.01). Low dose STA-2842 (50 mg/kg) had no statistically significant effect on liver cystogenesis at either time point ( Fig.2C ).
Figure 2. HSP90 inhibition reduces late stages of liver cyst formation in conditional Pkd1 –/– mice.

(A) Schematic outlining the timetable of the study. (B) Representative MRI images of mice, imaged at 0, 5, and 10 weeks of treatment with vehicle or drug (STA-2842) in wt (+) or Pkd1 –/– (–) mice. Scale bars = 0.5 cm. (C) Cyst volume estimated as a percentage of hepatic tissue at 0, 5, and 10 weeks of treatment of Pkd1 –/– or wt mice. n = 6–8 mice. * indicates comparisons to Pkd1 –/–, vehicle-treated mice: *, P≤0.05. All data were graphed as mean ± standard error of the mean (SEM). No cysts were detected in any wt mice.
After 10 weeks of treatment, mice were euthanized and livers analyzed ( Fig. 3A, B ). Liver weight of vehicle treated mice was significantly enlarged in vehicle treated Pkd1−/− mice in contrast to wt mice (179% increase; P = 0.002). However, the size of Pkd1−/− livers was very significantly decreased (36.5%; P = 0.0316) in mice that had been treated with 100 mg/kg of STA-2842, while no effect was seen with 50 mg/kg STA-2842 ( Fig. 3A, B ), while crude visual inspection indicated a qualitative difference in color and texture of the livers treated with 100 mg/kg STA-2842, versus the other two cohorts. Hematoxylin and eosin (H&E) staining of liver sections confirmed the reduced cyst burden predicted by MRI imaging ( Figs. 3C, D ). Quantification predicted a ∼39% decrease in the percentage of grid intersections with cystic tissue in H&E stained slides, as compared to vehicle treated Pkd1−/− mice, but no noticeable decrease with the 50 mg/kg dose.
Figure 3. Histopathological development of cysts.

(A) Representative livers collected from Pkd1 –/– or wt mice treated with vehicle or STA-2842 at the indicated doses, at 6.5 months of age. (B) Liver weights (lw) to body weight (bw) ratio of in wt (+) or Pkd1 –/– (–) mice after 10 weeks of treatment with STA-2842 (50, 100) or vehicle (–). n = 5–8 mice. * indicates comparisons to Pkd1 –/–, vehicle-treated mice; # indicates comparisons to wt vehicle-treated mice: *, P<0.05; ##, P<0.01; ###, P<0.001; ####, P<0.0001. (C) Representative H&E stained liver sections after 10 weeks of treatment with vehicle or drug in wt (+) or Pkd1 –/– (–) mice, indicating extent of cystogenesis. Scale bars = 100 µm; magnification = 10x. (D) Cystic indices quantified as a percentage of grid intersections that cross cysts on H&E slides. n = 6–8 mice. * indicates comparisons to Pkd1–/–, vehicle-treated mice: *, P<0.05. All data graphed as mean ± standard error of the mean (SEM). (E) Relative change in cyst burden by drug dose (6 month value divided by 4 month value), with simple linear fits of the relationship shown. None of the slopes were statistically significant (p>0.11 in all three cases). Since the range of values differed among the three doses, the X-axis is drawn on the log scale to better depict the relationships. F. Relative expression of cleaved PARP, normalized to total PARP, following treatment of Pkd1 –/– (–) mice with the indicated doses of STA-2842. **, P<0.01 to vehicle-treated mice; ##, P<0.01 relative to mice treated with 50 mg/kg STA-2842. G. Relative expression of cleaved caspase 8, normalized to GAPDH, following treatment of Pkd1 –/– (–) mice with the indicated doses of STA-2842. *, P<0.05, ***, P<0.001 in reference to vehicle-treated mice.
Activity of STA-2842 in reducing liver cysts could be independent of activity in the drug in controlling expansion of kidney cysts; alternatively, reduction of cysts in the two organs may be correlated. To evaluate these possibilities, we compared cyst burden in liver and kidney tissue from [20], across the three treatment groups ( Fig. 3E , Fig. S1). This analysis indicated that first, there is no correlation between the burden of cysts in the kidney and the liver cross-sectionally before dosing commences (Fig S1). Second, there is no correlation for drug response between 4 and 6 months at the 0 or 50 mg/kg doses. While at the 100 mg/kg dose a slight negative relationship is observed, it is not statistically significant (p = 0.12) ( Fig. 3E ). These results suggest that if there is a relationship of kidney cysts with liver cysts, the association is weak at best.
A reduction in cystic and liver volume likely reflects changes in proliferation or apoptosis induced by STA-2842. In the analysis of liver sections collected at the experiment endpoint and stained for the proliferation marker Ki-67 and the apoptotic marker cleaved caspase 3 (Fig. S2), less than 1% of nuclei were positive for either marker making it impossible to quantify an effect, and consistent with the fact that cystic diseases such as PLD are progressive and slow-acting. However, examination of tissue lysates by Western blot (Fig. S3) with antibodies to total and cleaved PARP ( Fig. 3F ) and cleaved caspase 8 ( Fig. 3G ) indicated STA-2842 increased basal levels of apoptosis, in a dose-dependent effect.
To gain insight into the role of STA-2842 in influencing proliferative signaling pathways in the context of cystogenesis, we analyzed liver lysates by Western analysis for the expression and/or activity of HSP90 and other relevant proteins (Fig S3). We first looked for evidence of HSP90 inhibition, which is typically reflected by compensatory upregulation of an alternative heat shock protein, HSP70 [23], and occasionally by elevated expression of total HSP90. We observed a trend toward upregulation of HSP90 at the 100 mg/kg dose level of STA-2842, and statistically significant, dose-dependent upregulation of HSP70 ( Fig. 4A ). We then compared expression of HSP70 ( Figs. 4B, C ) and HSP90 ( Figs. 4D, E ) with the cyst volume and liver weight to body weight ratio for all individual mice in the treatment groups. This indicated a striking negative correlation between the expression of either heat shock protein with both cystic volume and liver weight, indicating that degree of efficacy of the STA-2842 compound in individual mice predicted degree of therapeutic response, and compatible with the observation that response in kidney and liver cysts was correlated at the effective 100 mg/kg dose ( Fig. 3E ).
Figure 4. Changes in protein expression and activation of heat shock proteins associated with HSP90 inhibition.

(A) Quantification of HSP90α and HSP70 expression levels among the Pkd1 –/– groups. n = 6–8 mice. * indicates comparisons to Pkd1 –/–, vehicle-treated mice: **, P≤0.01; ***, P≤0.001. (B–E) Negative correlations between HSP70, HSP90α expression in the drug treatment groups and (B, D) cyst volume/region of interest (ROI) (P = 0.0046 for HSP70 and P = 0.0136 for HSP90) and (C, E) liver weight/body weight (lw/bw) ratios (P = 0.0141 for HSP70 P = 0.0138 for HSP90). Dots represent individual mice; lines represent linear regression functions. n = 6–8 mice.
We next analyzed by Western analysis the expression and activation of two proteins strongly linked to the increased proliferation of PLD cells, EGFR, ERK1/2, the ribosomal protein S6, and AKT ( Fig. 5 ; Figs. S3 and S4). As EGFR, Akt1, the ERK1/2 activator RAF, and the S6 activator mTOR are all direct clients of HSP90, STA-2842 would be expected to reduce expression and/or activity of these proteins [20]. STA-2842 reduced expression of total EGFR, and additionally reduced the ratio of phY1068/total and phY1173/total EGFR at the effective 100 mg/kg dose ( Fig. 5A ). Treatment with STA-2842 reduced the ratio of active (T202/Y204-phosphorylated) to total ERK1/2 at the 100 mg/kg dose level, although this was not statistically significant ( Fig. 5B ). However, in correlative analysis, mice with the lowest ration of phosphorylated to total ERK had a significantly reduced liver weight to body weight ratio ( Fig. 5C ), implying biological importance of the reduction. Subsequent analysis of phT202/Y204ERK1/2 in cystic epithelial linings versus total liver tissue indicated a significant reduction in the epithelial cells, although not in total liver tissue ( Fig 5D , Fig. S4). Based on all of these analyses, inhibition of the EGFR-ERK1/2 signaling axis at the 100 mg/kg dose corresponded well to biological inhibition of cyst growth. Finally, in further Western analysis, STA-2842 treatment significantly reduced activation (phS235/S236 phosphorylation) of the S6 protein, while not affecting total expression of S6, at both the 50 mg/kg and 100 mg/kg dose levels ( Fig. 5E ). Levels of active phS473 AKT and total AKT were somewhat elevated, as was the phS473/total AKT ratio, following STA-2842 treatment, but this response was also not dose-dependent and significant ( Fig. 5F ).
Figure 5. Changes in protein expression and activation of signaling proteins associated with HSP90 inhibition.

(A) Differences in expression of phosphorylated Y1068 and Y1173 EGFR and total EGFR normalized to actin, and Y1068 and Y1173 EGFR normalized to total EGFR, following treatment with the indicated doses of STA-2842. *, P<0.05; **, P<0.01. (B) Insignificant differences in expression of phosphorylated T202/Y204 ERK1/2 or total ERK1/2, normalized to tubulin, and phosphorylated T202/Y204 ERK1/2 normalized to total ERK1/2, following treatment with the indicated doses of STA-2842; differences are not significant (C) Positive correlations between the ratio of phosphorylated T202/Y204 ERK1/2 normalized to total ERK1/2 expression in the drug treatment groups and (left) cyst volume/region of interest (ROI) (P = 0.174) and (right) liver weight/body weight (lw/bw) ratio (P = 0.040). Dots represent individual mice; lines represent linear regression functions. n = 6–8 mice. All data graphed as mean ± standard error of the mean (SEM). (D) Expression of phosphorylated T202/Y204 ERK1/2 in cystic epithelia (left) or total liver (right) quantified from immunohistochemical staining of liver sections from Pkd1 –/– mice treated with vehicle or 100 mg/kg STA-2842 (see Fig S4). **, P<0.01. (E) Differences in expression of phosphorylated phS235/S236 S6 or total S6, normalized to tubulin, and phosphorylated phS235/S236 S6 normalized to total S6, following treatment with the indicated doses of STA-2842; *, P<0.05. P-values for comparisons between Pkd1 –/– vehicle- versus 100 mg/kg-treated groups bordering on significant (P = 0.060). (F) Differences in expression of phosphorylated phS473 AKT or total AKT, normalized to tubulin, and phosphorylated phS473 AKT normalized to total AKT, following treatment with the indicated doses of STA-2842; *, P<0.05.
In sum, these results indicate that HSP90 facilitates PLD pathogenesis and provide a proof of concept for the exploration of HSP90 inhibition to treat PLD. Importantly, this represents the first observation that treatment with an HSP90 inhibitor can reverse established disease. This was not observed in the response of renal cysts to STA-2842, where the main effect of the drug was to limit the rate at which new cysts were formed [20]. The likely reason for the difference in outcome is the greater regenerative capacity of hepatic versus renal tissue. Although the overall low levels of proliferation of hepatic cystic tissue make it difficult to quantify drug action, the significant inhibitory effect of HSP90 inhibition on ERK and EGFR activity at the effective dose, with ERK inhibition particularly concentrated in cystic epithelia, generally support this interpretation.
STA-2842 is a preclinical agent, which is not likely to move into the clinic in the near future. However, a related compound, ganetespib (formerly STA-9090), has demonstrated encouraging signs of clinical activity in breast cancer and ALK+ lung cancer [19], and is currently undergoing late stage clinical trials, with little evidence of toxic side effects. While particular care would be required to ensure HSP90 inhibition by ganetespib does not cause cytotoxicity specifically in the setting of PLD, where liver function is compromised, the data presented here suggest that it may be worth evaluating ganetespib in this disease. The range of therapeutic possibilities for HSP90 inhibitors extends beyond their sole use as a single inhibitor to remedy disease pathology. Some research suggests that HSP90 inhibition can help sensitize the body to chemotherapies, rendering it useful in treating cancers driven by multiple or indeterminate initiating factors [24], [25]. In a randomized study of docetaxel with or without ganetespib in advanced stage lung adenocarcinoma patients, the combination of ganetespib and docetaxel showed an acceptable safety profile and demonstrated substantial improvements in both progression free survival and overall survival [26]. Additionally, using HSP90 inhibitors in combination with other targeted protein inhibitors may be beneficial. In ADPKD and PLD pathogenesis, increased expression and activation of mTOR-related signaling proteins is important [7]. HSP90 inhibition has already been shown to combine productively with mTOR inhibitors in cancer settings [23]. This result suggests that pairing an HSP90 inhibitor with an mTOR inhibitor such as everolimus or temsirolimus may bring about an increased therapeutic effect, revivifying interest in an mTOR inhibitory strategy that was less promising than expected in initial clinical trials [27].
Materials and Methods
Ethics statement
All mouse work was approved by the Institutional Animal Care and Use Committee (IACUC) of Fox Chase Cancer Center (animal protocol #06-1). The Animal Welfare Assurance on file in the Office of Laboratory Animal Welfare is #A3285-01. All mice were handled by individuals with appropriate specialized training. Mice were anesthetized during MRI scanning and were euthanized by CO2 asphyxiation.
Mouse strains and drug treatment
Conditional Pkd1 knockout mice were created utilizing the Cre-flox regulatory system for specific inactivation of the Pkd1 gene in vivo and were kindly supplied by Gregory Germino (NIDDK, National Institutes of Health, Bethesda, MD) [21], [22]. To inactivate Pkd1, Pkd1fl/fl;Cre/Esr1+ (Pkd1−/−) mice and Pkd1fl/fl;Cre/Esr1− (wild type, wt) mice were intraperitoneally injected with a tamoxifen (250 mg/kg, resolved in corn oil) on postnatal days P37 and P38 to induce Pkd1 deletion in the non-control mice. Both male and female specimens were used in all mouse groups, and gender was determined not to affect the results of the experiments. Mice were dosed weekly for 10 weeks starting at 4 months of age with vehicle (5% dextrose), or 50 mg/kg or 100 mg/kg of STA-2842. Mice were euthanized at ∼6 months of age by CO2 asphyxiation. Livers were collected, weighed, and divided, with part placed in 10% phosphate-buffered formaldehyde, and part flash frozen.
MRI administration and image analysis
Mice were imaged at 0, 5, and 10 weeks of treatment in a vertical bore MR system, using methods described in detail previously [20]. The ImageJ (NIH, Bethesda, MD) program was used to quantify cyst volume measurements. A method of subtracting backgrounds then thresholding MRI images was used to approximate the area of the white (i.e. cystic) areas in the livers within an oval region of interest (ROI) of 0.178 cm2 on a single MRI. The ROI was selected so that cysts in hepatic tissue could be analyzed over 7 sequential images. To establish the % cystic tissue, the location of cysts between MRIs was assumed to be constant for the purposes of this analysis and the total cystic area was multiplied by the distance between the images (0.075 cm) and then divided by the total volume the cylindrical ROI (0.09345 cm3).
Protein expression analysis by fluorescent Western blotting
Flash frozen tissue from Pkd1-/- mice was lysed and protein concentrations of the resulting cystic liver lysates were measured using the Pierce BCA Protein Assay Kit (Thermo Scientific, Waltham, MA). Lysates were denatured at 98°C in sample buffer for 10 minutes, and 42 µg of protein was loaded in NuPAGE Novex 8% Bis-Tris 20 Well Midi Protein Gels (Life Technologies). Two SDS-PAGE gels containing all analyzed specimens were run in parallel in an XCell SureLock Mini-Cell Electrophoresis System (Life Technologies, Grand Island, NY), with 4 samples common to both gels as a reference group. The proteins were then transferred overnight at 20 V from to Immobilon-FL PVDF membranes (EMD Millipore, Billerica, MA). The membranes were incubated for 40 minutes at room temperature in a blocking solution of a 1∶1 mix of phosphate buffered saline (PBS) containing 0.1% Tween 20 (Sigma-Aldrich, St Louis, MO), to Odyssey blocking Buffer (LI-COR). Odyssey Blocking Buffer (LI-COR, Lincoln, NE). The membranes were then incubated for 1 hour at room temperature or overnight at 4°C in primary antibodies (1∶1000): anti-HSP70 (monoclonal, mouse, #ADI-SPA-820) or anti-HSP90α (polyclonal, rabbit, #ADI-SPS-771) (both from Enzo, Farmingdale, NY); or anti-pERK1/2Thr202/Tyr204 (polyclonal, rabbit, #9101), anti-ERK1/2 (monoclonal, mouse, #4696), anti-pS6Ser235/236 (monoclonal, rabbit, #4858), anti-S6 (monoclonal, mouse, #2317), anti-phospho AKTS473 (polyclonal, rabbit, #4060S) and anti-AKT (polyclonal, rabbit, #2920S) and anti-α/β-Tubulin (polyclonal, rabbit, #2148) (each from Cell Signaling, Danvers, MA). IRDye800CW Goat anti-Mouse and IRDye 680 RD Goat anti-Rabbit secondary antibodies (1∶10,000, LI-COR, city, state) were applied at room temperature for 1 hour. Membranes were imaged and quantified in separate channels (700 and 800 nm) in the Odyssey Infrared Imaging System using Odyssey V3.0 software (LI-COR).
To analyze the expression levels of phospho- and total EGFR, PARP and cleaved caspase 8, cells were lysed and resolved by SDS-Page. Western Blotting was performed using standard procedures, and blots developed by chemiluminescence using Luminata Western HRP substrates (Classico, Crescendo and Forte, EMD Millipore) and Immun-Star AP Substrate (Bio-Rad Laboratories). Primary antibodies included anti-phospho EGFRY1068 (polyclonal, rabbit, #3777S, Cell Signaling Technologies), anti-phospho EGFRY1173 (polyclonal, rabbit, #44794G, Invitrogen) and anti-EGFR (polyclonal, rabbit, #2646, Cell Signaling Technologies), anti-PARP (polyclonal, rabbit, #9542S, Cell Signaling Technologies), anti-cleaved caspase 8 (polyclonal, rabbit, #8592P, Cell Signaling Technologies), anti-GAPDH (mouse, monoclonal mAbcam 9484, #ab9482, Abcam) and anti-vinculin (mouse, monoclonal hVIN-1, #V9131, Sigma-Aldrich). Secondary anti-mouse and anti-rabbit HRP-conjugated antibodies (GE Healthcare) were used at a dilution of 1∶10,000 and secondary anti-mouse and anti-rabbit AP-conjugated antibodies (Jackson Immunoresearch Labs) were used at a dilution of 1∶5,000. Quantification of signals on Western blots was done using the NIH ImageJ Imaging and Processing Analysis Software with signaling intensity normalized to loading control.
Tissue preparation, histology, and immunohistochemistry
Sections of the mouse livers were suspended in formalin (10% phosphate-buffered formaldehyde) for 1–2 days and were subsequently dehydrated and embedded in paraffin blocks. Paraffin blocks were prepared into slides stained by hematoxylin and eosin (H&E), anti-Ki-67 (1∶100, monoclonal rat anti-mouse, #M7249, Dako, Carpinteria, CA), anti-HSP90α (1∶800, monoclonal rabbit, #NB120-2928, Novus Biologicals, Littleton, CO), anti-pERK1/2Thr202/Tyr204 (polyclonal, rabbit, #9101, Cell Signaling, Danvers, MA), or cleaved caspase-3 (1∶200, polyclonal, #9661 Cell Signaling, Danvers, MA) using standard procedures described previously [20]. All slides containing immunohistochemical staining were imaged using a ScanScope CS scanner (Aperio, Leica Biosystems, Buffalo Grove, IL). To quantify cystic indices, the ImageJ program was used to generate computerized grids with an area per point of 5000 µm2 and to superimpose them upon images of H&E-stained liver sections scanned at 10x magnification using an EVOS XL Core Cell Imaging System (Life Technologies, Grand Island, NY). Cystic indices were calculated as a percentage of grid intersections that crossed cysts, with ∼1450 grid intersections analyzed for per mouse. Clear areas with diameters >40 µm were defined as cysts; those with diameters >20 µm defined as dilated tubules.
Statistical analysis
We used t-tests, assuming unequal variances for hypothesis testing. Prism 6 (GraphPad Software, San Diego, CA) was used to identify statistical correlations between protein expression and physical results and then to plot regression lines. We used nonparametric Spearman correlations to examine relationships between variables of interest, such as relative change in kidney and liver cyst volumes between time points.
Supporting Information
Cyst burden in liver versus kidney tissue at the month 4 baseline (p = 0.57).
(TIF)
Absence of Ki-67 and cleaved caspase-3 among treated and non-treated Pkd1 –/–mice. Representative hematoxylin stained liver sections with immuno-histochemical detection of (A) Ki-67 (brown) and (B) cleaved caspase-3 (brown) from three independent Pkd1 –/–(–) mice treated with vehicle, 50 mg/kg STA-2842, or 100 mg/kg STA-2842. Magnification, 20x. Scale bars = 200 µm.
(TIF)
Western blotting for proteins indicated, from wt (+) or Pkd1 –/– (–) mice after 10 weeks of treatment with STA-2842 (50, 100 mg/kg) or vehicle (–).
(TIF)
Representative hematoxylin stained liver sections with immunohistochemical detection of phosphorylated T202/Y204 ERK1/2 (brown) from Pkd1 –/– mice treated with vehicle or 100 mg/kg STA-2842, as indicated. Scale bar – 50 µm.
(TIF)
Acknowledgments
We thank Dr. Gregory Germino for the gift of the Pkd1fl/fl;Cre/Esr1+/- mice. We are grateful to Simon Tarpinian and Justin Rambert of the Fox Chase Laboratory Animal Facility for assistance with mice. We thank Tim Beck for advice and assistance with experiments. We are very thankful to Catherine and Peter Getchell and the Bucks County Chapter of the Fox Chase Board of Associates for their support.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors are very thankful to Catherine and Peter Getchell and the Bucks County Chapter of the Fox Chase Board of Associates for their financial support. This work was further supported by funding from DOD PRMRP W81XWH-12-1-0437/PR110518 (to E. A. G.), from the German Research Foundation (DFG) SE2280/1, SE2280/2 and Köln Fortune 252/2013 (to T. S.-N.) and from the National Institutes of Health Core Grant CA06927 (to Fox Chase Cancer Center). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Torres VE, Harris PC (2006) Mechanisms of Disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol 2:40–55 quiz 55. [DOI] [PubMed] [Google Scholar]
- 2. Torres VE, Harris PC (2009) Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int 76:149–168. [DOI] [PubMed] [Google Scholar]
- 3. Milutinovic J, Fialkow PJ, Rudd TG, Agodoa LY, Phillips LA, et al. (1980) Liver cysts in patients with autosomal dominant polycystic kidney disease. The American journal of medicine 68:741–744. [DOI] [PubMed] [Google Scholar]
- 4. D'Agata ID, Jonas MM, Perez-Atayde AR, Guay-Woodford LM (1994) Combined cystic disease of the liver and kidney. Seminars in liver disease 14:215–228. [DOI] [PubMed] [Google Scholar]
- 5. Chauveau D, Fakhouri F, Grunfeld JP (2000) Liver involvement in autosomal-dominant polycystic kidney disease: therapeutic dilemma. Journal of the American Society of Nephrology: JASN 11:1767–1775. [DOI] [PubMed] [Google Scholar]
- 6. Kurbegovic A, Cote O, Couillard M, Ward CJ, Harris PC, et al. (2010) Pkd1 transgenic mice: adult model of polycystic kidney disease with extrarenal and renal phenotypes. Human molecular genetics 19:1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, et al. (2010) Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin-2-defective mice. Hepatology 51:1778–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, et al. (2010) ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin-2 defective mice. Gastroenterology 138:360–371 e367.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sweeney WE Jr, von Vigier RO, Frost P, Avner ED (2008) Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol 19:1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, et al. (2006) The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 103:5466–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shillingford JM, Leamon CP, Vlahov IR, Weimbs T (2012) Folate-conjugated rapamycin slows progression of polycystic kidney disease. Journal of the American Society of Nephrology: JASN 23:1674–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu YM, Shao YQ, He Q (2014) Sirolimus for treatment of autosomal-dominant polycystic kidney disease: a meta-analysis of randomized controlled trials. Transplantation proceedings 46:66–74. [DOI] [PubMed] [Google Scholar]
- 13. Wuthrich RP, Mei C (2014) Pharmacological management of polycystic kidney disease. Expert opinion on pharmacotherapy 15:1085–1095. [DOI] [PubMed] [Google Scholar]
- 14. Strazzabosco M, Somlo S (2011) Polycystic liver diseases: congenital disorders of cholangiocyte signaling. Gastroenterology 140:1855–1859 1859 e1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chrispijn M, Gevers TJ, Hol JC, Monshouwer R, Dekker HM, et al. (2013) Everolimus does not further reduce polycystic liver volume when added to long acting octreotide: results from a randomized controlled trial. Journal of hepatology 59:153–159. [DOI] [PubMed] [Google Scholar]
- 16. Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, et al. (2010) Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci Signal 3:ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chan DA, Giaccia AJ (2011) Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov 10:351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grantham JJ (1990) Polycystic kidney disease: neoplasia in disguise. American journal of kidney diseases: the official journal of the National Kidney Foundation 15:110–116. [DOI] [PubMed] [Google Scholar]
- 19. Proia DA, Bates RC (2014) Ganetespib and HSP90: Translating Preclinical Hypotheses into Clinical Promise. Cancer research 74:1294–1300. [DOI] [PubMed] [Google Scholar]
- 20. Seeger-Nukpezah T, Proia DA, Egleston BL, Nikonova AS, Kent T, et al. (2013) Inhibiting the HSP90 chaperone slows cyst growth in a mouse model of autosomal dominant polycystic kidney disease. Proceedings of the National Academy of Sciences of the United States of America 110:12786–12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG (2007) A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med 13:1490–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Piontek KB, Huso DL, Grinberg A, Liu L, Bedja D, et al. (2004) A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J Am Soc Nephrol 15:3035–3043. [DOI] [PubMed] [Google Scholar]
- 23.Acquaviva J, He S, Sang J, Smith DL, Sequeira M, et al. (2014) mTOR Inhibition Potentiates HSP90 Inhibitor Activity via Cessation of Heat Shock Protein Synthesis. Molecular cancer research: MCR. [DOI] [PubMed]
- 24.Liu H, Xiao F, Serebriiskii IG, O'Brien SW, Maglaty MA, et al. (2013) Network analysis identifies an HSP90-central hub susceptible in ovarian cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. [DOI] [PMC free article] [PubMed]
- 25. Proia DA, Zhang C, Sequeira M, Jimenez JP, He S, et al. (2014) Preclinical activity profile and therapeutic efficacy of the HSP90 inhibitor ganetespib in triple-negative breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research 20:413–424. [DOI] [PubMed] [Google Scholar]
- 26. Ramalingam S, Goss GD, Andric ZG, Bondarenko I, Zaric B, et al. (2013) A randomized study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel versus docetaxel alone for second-line therapy of lung adenocarcinoma (GALAXY-1). J Clin Oncol 33:18s (CRA8007).. [DOI] [PubMed] [Google Scholar]
- 27. Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, et al. (2010) Everolimus in patients with autosomal dominant polycystic kidney disease. The New England journal of medicine 363:830–840. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cyst burden in liver versus kidney tissue at the month 4 baseline (p = 0.57).
(TIF)
Absence of Ki-67 and cleaved caspase-3 among treated and non-treated Pkd1 –/–mice. Representative hematoxylin stained liver sections with immuno-histochemical detection of (A) Ki-67 (brown) and (B) cleaved caspase-3 (brown) from three independent Pkd1 –/–(–) mice treated with vehicle, 50 mg/kg STA-2842, or 100 mg/kg STA-2842. Magnification, 20x. Scale bars = 200 µm.
(TIF)
Western blotting for proteins indicated, from wt (+) or Pkd1 –/– (–) mice after 10 weeks of treatment with STA-2842 (50, 100 mg/kg) or vehicle (–).
(TIF)
Representative hematoxylin stained liver sections with immunohistochemical detection of phosphorylated T202/Y204 ERK1/2 (brown) from Pkd1 –/– mice treated with vehicle or 100 mg/kg STA-2842, as indicated. Scale bar – 50 µm.
(TIF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.