

Abstract
Different ethnic groups have distinct mutation spectrums associated with inheritable deafness. In order to identify the mutations responsible for congenital hearing loss in the Tibetan population, mutation screening for 98 deafness-related genes by microarray and massively parallel sequencing of captured target exons was conducted in one Tibetan family with familiar hearing loss. A homozygous mutation, TMPRSS3: c.535G>A, was identified in two affected brothers. Both parents are heterozygotes and an unaffected sister carries wild type alleles. The same mutation was not detected in 101 control Tibetan individuals. This missense mutation results in an amino acid change (p.Ala179Thr) at a highly conserved site in the scavenger receptor cysteine rich (SRCR) domain of the TMPRSS3 protein, which is essential for protein-protein interactions. Thus, this mutation likely affects the interactions of this transmembrane protein with extracellular molecules. According to our bioinformatic analyses, the TMPRSS3: c.535G>A mutation might damage protein function and lead to hearing loss. These data suggest that the homozygous mutation TMPRSS3: c.535G>A causes prelingual hearing loss in this Tibetan family. This is the first TMPRSS3 mutation found in the Chinese Tibetan population.
Introduction
Hearing impairment is a common birth defect. Epidemiologic studies have shown that 50% of childhood deafness cases are associated with genetic defects. Hearing loss caused by genetic defects is also characterized by high allelic and locus heterogeneity. Many deafness genes, including the well-known deafness genes GJB2 (MIM#121011), SLC26A4 (MIM#605646) and 12S rRNA (MIM#561000), have been identified [1]–[5]. In the Chinese deaf population, 21% have GJB2 mutations, 14.5% have SLC26A4 mutations, and 3.4% have the 12S rRNA:A1555G mutation [2]. High genetic heterogeneity suggests that hearing loss may be caused by variable mutations in hundreds of genes or that a deafness mutation may lead to different deafness phenotypes. Moreover, people with different ethnicities may have different mutation spectrums. Li et al. reported that different ethnic groups present different incidences of large vestibular aqueduct (LVA) disorders [6]. In China, Han Chinese has the highest incidence of LVA disorders, while minorities in southwest China have low incidences. In our previous study, two cases with heterozygous GJB2 mutations and two cases with homoplasmic 12S rRNA:A1555G mutations were identified from 92 Tibetan prelingual deafness students who underwent mutation screening for GJB2, SLC26A4, and mitochondria DNA 12S rRNA mutations [7]. These studies suggested that deafness mutation hotspots are variable in populations with different ethnicities. Consistent with those findings, Dai et al. showed that only the 12S rRNA:A1555G mutation was detected in four individuals from 114 Tibetan deafness patients [5]. They suggested that the different mutation spectrums in Han Chinese and Tibetan Chinese might have been related to distinct geographic features in their living areas. Tibetan Chinese live in high altitude regions, which may contribute to the increased incidence of deafness in Tibetan Chinese due to the low oxygen micro-environment in the inner ear.
The TMPRSS3 (Transmembrane protease, serine 3, MIM#605511) gene encodes an enzyme that is expressed in multiple tissues, including inner hair cells, spiral ganglion neurons, and stria vascularis of the cochlear duct. TMPRSS3 is involved in the development and maintenance of the inner ear perilymph and endolymph [8], [9]. Many mutant alleles of TMPRSS3 have been reported to be associated with nonsyndromic recessive deafness (DFNB8/B10) [8], [10], [11]. Although TMPRSS3 mutations seem to be less common than GJB2 mutations, TMPRSS3 mutations lead to inheritable deafness, especially when common GJB2, SLC26A4 and 12S rRNA mutations are excluded.
The TMPRSS3 protein contains several common functional domains, including a transmembrane domain, a low density lipoprotein receptor A domain (LDLRA), a scavenger receptor cysteine rich domain (SRCR), and a carboxyl terminal serine protease domain [9]. Many TMPRSS3 mutations have been reported as pathogenic mutations for inheritable deafness, including an 8-bp deletion, insertion of multiple beta-satellite repeat units, and a frameshift mutation [8].
In order to explore the distinct mutation spectrum associated with inheritable deafness in Tibetan Chinese, we performed mutation screening on 98 deafness genes using both microarray and massively parallel sequencing of captured exons in an affected Tibetan family. We defined the homozygous mutation of TMPRSS3: c.535G>A as the genetic etiology for two brothers. To our knowledge, this is the first report of a TMPRSS3 mutation causing deafness in Chinese.
Materials and Methods
Ethics Statement
The experimental protocol was established according to the ethical guidelines of the Helsinki Declaration and was approved by the Ethics Committee of the First Hospital of Jilin University in China. Written informed consents were obtained from all of the adult participants. If the participant was underage, the participants’ parents or guardians signed the consent.
Subjects
The proband, a 12-year old boy living at an altitude of 3560 meters, failed otoacoustic emission (OAE) tests bilaterally. His auditory brainstem response (ABR) test showed response threshold values more than 85 dBnHL bilaterally. These test results suggested profound sensorineural hearing loss. The proband’s 10-year old brother was also diagnosed with profound bilateral sensorineural hearing loss (BSNHL). General physical checkups and examinations for overall development were normal. In addition, eye and thyroid gland tests showed normal results, excluding the possibility of syndromic hearing loss. Deafness caused by otitis media, meningitis, viral encephalitis, parotitis, and general infection were also excluded for both siblings. The parents and sister have normal hearing. Parents claimed no familiar history of hearing loss or consanguinity.
DNA Sample Collection
Peripheral blood specimens (5 mL) were collected from each individual for DNA analysis. Genomic DNA was extracted using a TIANamp Blood DNA Kit (Tiangen Biotech, Beijing, China). Genomic DNA products (concentration = 100∼150 ng/µl; OD260/OD280 = 1.7∼1.9) were purified according to manufacturer’s instructions and stored in a −20°C freezer.
Mutation Detection by Microarray
A Deafness Gene Mutation Detection Array kit (CapitaBio, Beijing, China) was used to detect the following known hotspot mutations related to hereditary hearing loss: GJB2 (35 delG, 176 del16, 235 delC, and 299–300 delAT), GJB3 (538C>T) (MIM#603342), SLC26A4 (IVS7-2A>G and 2168A>G), and 12S rRNA (1494C>T and 1555A>G). The standard procedures recommended by the manufacturer were applied to detect nine mutations in the four genes. Target DNA fragments were amplified with nine pairs of primers in two multiplex PCR reactions with the following components: 150 ng of template genomic DNA, 1.5 µl of 10x Hotstart buffer, 2.5 mM dNTP, primer mix, 5 u/µl of HotStart HiFidelity DNA Polymerase and ddH2O in 15 µl of total volume. PCR reactions were run with the following conditions: initial incubation at 95°C for 15 min and at 96°C for 1 min, followed by 32 cycles of 94°C for 30 sec, 55°C for 30 sec, and 70°C for 45 sec. A final incubation step was performed at 60°C for 10 mins. Hybridization reactions were performed according to the manufacturer’s manual using a BioMixerTM II Microarray Hybridization Station (CapitaBio, Beijing, China). A LuxScanTM 10K Microarray Scanner (CapitaBio, Beijing, China) was used for microarray data capture.
Illumina Library Preparation
Sample DNA was extracted from patient blood using the DNA Extraction kit (DP319, Tiangen, Beijing, China). The DNA was first quantified with a Nanodrop 2000 Spectrophotometer (Thermal Fisher Scientific, DE). A minimum of 3 µg of DNA was used to create the indexed Illumina libraries according to the manufacturer’s protocol. The final library, which had a size ranging from 300 bp to 400 bp including adapter sequences, was finally selected.
Deaf Panel Gene Enrichment and Sequencing
All of the exons of the 98 deafness genes were enriched using the GenCap Human Deafness Genes enrichment kit (MyGenostics Inc., Beijing, China). The biotinylated 60-mer oligo baits were designed to tile all of the exons. The capture experiment was conducted according to the manufacturer’s protocol. In brief, 1 µg of DNA library was mixed with Buffer BL and biotinylated probes (MyGenostics Inc., Beijing, China) and then heated at 95°C for 7 min and 65°C for 2 min on a PCR machine. Next, 23 µl of the 65°C pre-warmed Buffer HY (MyGenostics Inc., Beijing, China) was added to the mixture, which was subsequently held at 65°C for 22 hours for hybridization. Next, 50 µl of MyOne beads (Life Technology, USA) were washed in 500 µL of 1X binding buffer three times and resuspended in 80 µl of 1X binding buffer. Then, 64 µl of 2X binding buffer was added to the hybrid mixture and transferred to the tube containing 80 µl of MyOne beads. The mixture was rotated for 1 hour on a rotator. The beads were then washed once with WB1 buffer at room temperature for 15 min and washed three times with WB3 buffer at 65°C for 15 min. The bound DNA was then eluted with Buffer Elute. The eluted DNA was finally amplified using the following conditions: 98°C for 30 sec (1 cycle); 98°C for 25 sec, 65°C for 30 sec, 72°C for 30 sec (15 cycles); and 72°C for 5 min (1 cycle). The PCR product was purified using SPRI beads (Beckman Coulter) according to the manufacturer’s protocol. The enrichment libraries were sequenced on an Illumina HiSeq 2000 sequencer for 100 bp paired reads.
Confirmation by Sanger Sequencing
The following primers were designed to amplify exon 6 of TMPRSS3: TMPRSS3-ex6-Forward, 5′-TCTCCCACCATCTTCCTA-3′; TMPRSS3-ex6-Reverse, 5′-ACTGATGCCAACACCAAC-3′. PCR amplification was performed with the following conditions: 95°C for 15 min; 96°C for 1 min; 32 cycles of 94°C for 30 sec, 55°C for 30 sec, and 72°C for 45 sec; 72°C for 5 min. Purified PCR products with a size of 420 bp were sent for Sanger sequencing (Sangon Biotech, Shanghai, China). Sequencing results were analyzed and aligned against the TMPRSS3 NM_032404 sequence shown in the NCBI database using the GeneTool 1.0 software (BioTools Inc.). Sequencing data from 101 normal Tibetan children were used as control.
Data Analysis
The SOAPaligner software was used for sequence alignment and data quality analysis (BGI, China). The SOAPsnp software was used for SNP genotyping (BGI, China). SNP data with poor quality (quality value<20) and low coverage (depth<10) were filtered. Annotations for SNP variants were completed using multiple databases, including Consensus CDS (CCDS) database, human genome builder NCBI 36.3, and dbSNP database V130. For all of the variants, the results were filtered using a quality value of single base sequencing ≧20. Online tools, SIFT, PolyPhen-2, and MutationTaster were used to predict the functional outcome of the detected SNP variants.
Results
Audiology Tests
Family pedigree and audiograms are shown in Figure 1. Pure tone audiology examinations showed that both brothers, III-2 and III-3 in the pedigree, had profound BSNHL. They both had a 70∼90 decibels hearing level (dB HL) at a frequency spectrum from 0.5 to 2.0 kHz. The hearing curve was flat and showed decreased hearing at all frequencies. The family members, II-1, II-2 and II-3, have normal hearing. III-2 and III-3 showed type A tympanograms (data not shown). Stapedial acoustic reflex and otoacoustic emission (OAE) were not detected at all frequencies (data not shown).
Figure 1. Family pedigree and relative audiograms.

Audiograms of family members were obtained using pure tone audiometery with air conduction at frequencies from 250 to 8000 Hz. The proband’s parents, II-2 at age 41 and II-3 at age 39, showed normal hearing audiograms, while the affected brothers, III-2 and III-3, showed severe to profound hearing loss.
Detection of Deafness Genes Mutations by Microarray
Mutations were not detected for the brothers with BSNHL by the microarray assay. These results ruled out the nine common deafness mutations of GJB2, GJB3, SLC26A4, and 12S rRNA in the two affected brothers.
Identification of Deafness Mutations by Target Capture and Massively Parallel Sequencing
Thereafter, deep sequencing data for exons of 98 deafness genes were analyzed. Overall, 98.0% of the targeted disease gene regions were sequenced and 94.1% of the targeted bases were sequenced with >10X depth, allowing us to accurately assess SNPs. In total, 188 variants were identified in the proband’s sample. Of the 188 variants, 96 were non-synonymous, missense, nonsense or splice variants. These were narrowed to three variants by excluding variants reported in the HapMap 28 and the SNP release of the 1000 Genome Project with a minor allele frequency >0.01. Further analyses showed that the proband III-2 had a homozygous mutation in exon 6 of TMPRSS3 (TMPRSS3: c.535G>A) (Table 1 and Figure 2). This proband also carried a heterozygous mutation in USH2A (USH2A:c.10246T>G) (MIM#608400).
Table 1. Candidate SNPs identified in the proband with BSNHL.
Figure 2. Deep sequencing data for the homozygous mutation in TMPRSS3 and the heterozygous mutation in USH2A.

Sequencing alignment data for the homozygous mutation TMPRSS3: c.535G>A is shown in the left panel. Sequencing alignment data for the heterozygous mutation USH2A: c.10246 T>G is shown in the right panel.
Mutation Confirmation by Sanger Sequencing
Sanger sequencing for the affected brothers and their direct relatives confirmed the mutations in the brothers. In addition, Sanger sequencing revealed that the parents, II-2 and II-3, are heterozygous carriers for the mutation TMPRSS3: c.535G>A. In addition, the normal sister, III-1, has two wild type alleles at this locus (Figure 3).
Figure 3. Data from Sanger sequencing for the two affected patients and the direct relatives in the family.

Homozygous mutation genotype A/A in the two affected brothers, III-2 and III-3. Wild type genotype G/G in the unaffected sister, III-1. Heterozygous genotype G/A in both parents, II-2 and II-3.
Functional Outcome Predicted by SIFT, Polyphen-2, and Mutationtaster
The TMPRSS3: c.535G>A mutation identified in this family is a missense mutation and leads to an amino acid change from Ala to Thr (p.Ala179Thr). Alanine (A) is a highly conserved amino acid in vertebrate species (Figure 4). This mutation was predicted to “affect protein function” by SIFT, “probably damaging” by PolyPhen-2, and “disease causing” by MutationTaster (Table 2).
Figure 4. Human TMPRSS3 protein domains and the analysis of evolutionary conservation for the sequence around the mutation site.
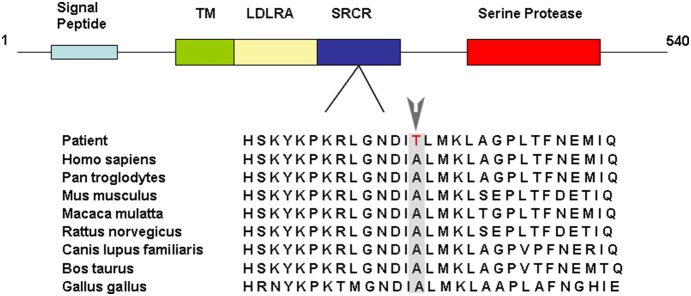
The TMPRSS3 protein consists of 540 amino acids and contains a signal peptide, a transmembrane (TM) domain, a low density lipoprotein receptor A (LDLRA) domain, a scavenger receptor cysteine rich (SRCR) domain, and a carboxyl terminal serine protease domain. The Alanine (179) in the SRCR domain, which is mutated to Threonine in the two patients, is highly conserved in vertebrates.
Table 2. Functional prediction for the TMPRSS3 missense mutation, c.535G>A.
| Algorithm | c.535G>A (p.A179T) |
| PolyPhen-2 | Probably damaging (Score: 0.99) |
| SIFT | Affect protein function (Score:0.00) |
| MutationTaster | Disease causing |
Discussion
In our study, new mutation detection techniques such as microarray and massive parallel sequencing of captured target exons were used to screen 98 known deafness genes in Tibetan patients with profound BSNHL. Our results demonstrated that a homozygous mutation of TMPRSS3 was associated with the congenital hearing loss in this Tibetan family. To our knowledge, this is the first report of a TMPRSS3 mutation causing hearing loss in a Chinese population. TMPRSS3: c.535G>A is a missense mutation in exon 6 of TMPRSS3 and leads to the substitution of Ala to Thr at amino acid 179 (p.Ala179Thr) (Figure 3). In addition, Sanger sequencing analyses of the direct relatives in this family revealed that only the two affected brothers have the homozygous mutation, while both parents are carriers of the heterozygous mutation and the unaffected sister does not carry the mutation. These results clearly showed the co-segregation of hearing loss with this mutation in this Tibetan family. Moreover, the TMPRSS3: c.535G>A mutation was not detected in 101 control Tibetans (data not shown), suggesting that TMPRSS3: c.535G>A is a disease causing mutation rather than a polymorphism. All three function prediction tools (SIFT, PolyPhen-2, and MutationTaster) suggest that this mutation affects protein function and is a disease causing mutation (Table 2). Overall, these data support that the hearing loss in the two affected brothers in this family is caused by the homozygous TMPRSS3: c.535G>A mutation.
After the first published report of a protease associated with hearing loss by Scott et al. [8], 23 TMPRSS3 mutations have been reported as pathogenic mutations for inheritable deafness (Table 3). Among these mutations, 15 mutations are located in the serine protease domain, three in the SRCR domain, and five in the LDLRA domain. The finding of the homozygous missense mutation (p.Ala179Thr) in our patients added one more case of BSNHL caused by a mutation in the SRCR domain of TMPRSS3.
Table 3. Pathogenic deafness mutations in TMPRSS3 identified in previous studies.
| Ethnicity | Nucleotide | AminoAcid Change | FunctionalDomain | Type ofVariant | Reference |
| British | c.413C>G | p.Ala138Glu | SRCR | mis | 13 |
| Dutch | c.595G>A | p.Val199Met | SRCR | mis | 13, 26 |
| Pakistani | c.207delC | p.Thr70fs | LDLRA | del | 17, 18 |
| Spanish | |||||
| Greek | |||||
| Newfoundlander | |||||
| German | c.916G>A | p.Ala306Thr | TM | mis | 12, 14,19 |
| Korean | |||||
| Dutch | c.1276G>A | p.Ala426Thr | TM | mis | 13 |
| Turkish | c.647G>T | p.Arg216Leu | TM | mis | 17 |
| Pakistani | c.325C>T | p.Arg109Trp | LDLRA | mis | 9, |
| Korean | |||||
| Pakistani | c.581G>T | p.Cys194Phe | SRCR | mis | 9 |
| Turkish | c.1192C>T | p.Gln398X | TM | mis | 20 |
| Tunisian | c.1211C>T | p.Pro404Leu | TM | mis | 20 |
| UK Caucasian | c.268G>A | p.Ala90Thr | LDLRA | mis | 21, 22 |
| Moroccan | |||||
| Greek | c.308A>G | p.Asp103Gly | LDLRA | mis | 18 |
| German | c.646C>T | p.Arg216Cys | TM | mis | 14 |
| Pakistani | c.1219T>C | p.Cys407Arg | TM | mis | 9, 16, 17 |
| Korean | c.743C>T | p.Thr248Met | TM | mis | 12 |
| Pakistani | c.767C>T | p.Arg256Val | TM | mis | 16 |
| Tunisian | c.753G>C | p.Trp251Cys | TM | mis | 10 |
| Pakistani | c.1273T>C | p.Cys425Arg | TM | mis | 16 |
| Pakistani | c.310G>A | p.Glu104Lys | LDLRA | mis | 16 |
| c.310G>T | p.Glu104Stop | non | |||
| Palestinian | c.1180_1187del8ins68 | _ | TM | _ | 8 |
| Italian | c.1019C>G | p.Thr340Arg | TM | mis | 23 |
| Italian | c.1291C>T | p.Pro431Ser | TM | mis | 23 |
| Newfoundlander | c.782+8insT | - | TM | - | 17 |
TM: transmembrane domain; LDLRA: low-density lipoprotein receptor A domain; SRCR: scavenger-receptor cysteine rich domain; Mis: missense; Del: delete; Non: nonsense.
Proteins containing SRCR domains have been proposed to function in the homeostasis of epithelia and the immune system and some have been reported to be associated with a number of diseases and pathogenic states such as atherosclerosis, Alzheimer’s disease, autoimmune diseases, and cancer. Thus, they exhibit promising potential as targets for diagnostic and/or therapeutic intervention [12], [13]. p.A138G, p.V199M and p.C194F are the mutations in the SRCR domain of TMPRRS3 that have been found to be associated with deafness [10], [14], [15]. The mutation found in our patients results in p.A179T in the SRCR domain of TMPRRS3. The SRCR domain includes amino acids from V110 to T205 and contains four cysteine rich motifs, which can bind negatively charged molecules such as lipoproteins and sulphate polysaccharides. Proteins that are predicted by the online software STRING 9.1 to interact with TMPRSS3 in humans include multiple known deafness proteins such as MYO7A (MIM#276903), GJB2, DFNB59 (MIM#610219), and SLC26A4 (Figure 5). Therefore, the amino acid change p.A179T in the SRCR domain in our patients likely affects the interaction of TMPRSS3 with other hearing-associated proteins, leading to hearing damage. Nevertheless, further studies with a large case volume are needed to establish the clear genotype and phenotype association for mutations in the SRCR domain.
Figure 5. The interactive protein network for human TMPRSS3 predicted by STRING 9.1.

Proteins predicted to interact with human TMPRSS3 include multiple known deafness proteins such as MYO7A, GJB2, DFNB59 and SLC26A4. The software tool is available in the website: http://string-db.org/.
Experiments using the Xenopus oocyte expression system conducted by Guipponi et al. have shown that TMPRSS3 cleaves epithelial sodium channel (ENaC) and promotes ENaC mediated currents [16]. This report also showed co-expression of ENaC and TMPRSS3 in Corti’s organ, stria vascularis and spiral ganglions, indicating that TMPRSS3 may play a role in signal transduction between spiral ganglions and hair cells. Molina et al. showed that TMPRSS3 in mice maintains normal expression of KCNMA1 potassium channels and normal outward K+ currents in inner hair cells (IHCs) [17]. The mutation identified in our study does not fall in the protease domain. However, it would be interesting to test whether this mutation indirectly interferes with protease activity and affects normal outward K+ currents in IHCs.
It has been shown by multiple studies that the detection rates of TMPRSS3 mutations are variable in different ethnic groups [10], [14], [18], [19], [20]. Thus far, TMPRSS3∶207delC is the most commonly reported deafness mutation for this gene. It has been detected in Spanish people, Grecians, Newfoundlanders, and Pakistan populations [19], [20]. TMPRSS3 mutations account for 5.9% of autosomal recessive non-syndromic hearing loss (arNSHL) cases and 8.3 of postlingual arNSHL cases in Korean patients [21]. The mutation TMPRSS3∶916G>A, which has been detected in German and Korean deaf patients, leads to an amino acid change A306T in the transmembrane domain [18], [21], [22]. This mutation results in decreased protease activity, which may explain its underlying molecular etiology for deafness. Moreover, the mutation pR109W in the LDLRA domain of TMPRSS3 has been detected in multiple Asian familiar deafness cases [10], [22]. Weegerink et al. reported that mutations in different domains of TMPRSS3 resulted in varied hearing loss phenotypes, likely due to the distinct influence of protease activity by various mutations [14]. Several other studies have also found a variety of TMPRSS3 mutations in patients with hearing loss (Table 3) [11], [23]–[27]. The genetic spectrum of arNSHL in Chinese patients mainly includes mutations in GJB2, SLC26A4, MYO7A, POU3F4, USH2A and TMC1. Mutations in GJB2, followed by mutations in SLC26A4, are the most commonly identified cause of sensorineural hearing loss in Chinese patients [2]. Dai et al. suggested that hotspots for hearing loss mutations are variable in different regions in China [5]. Their studies found distinct allele frequencies for GJB2∶235delC in different regions in China. It has also been reported that some common molecular etiologies, such as GJB2 and SLC26A4 mutations, in the general Chinese deaf population are rare in the Tibetan Chinese deaf population [28]. Consistent with these studies, our microarray screening of nine common deafness mutations in GJB2, GJB3, SLC26A4 and 12S rRNA in Tibetan Chinese patients with hearing loss resulted in a low positive detection rate.
In this study, we reported the first mutation in TMPRSS3 (TMPRSS3: c.535G>A in exon 6, resulting p.A179T in the SRCR domain) found in the Chinese Tibetan population. Our results provided a new example of prelingual deafness caused by a TMPRSS3 mutation.
Acknowledgments
We thank the members in the Chinese Tibetan family for their participation and cooperation of this study.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was supported by the National Science Foundation of China (Grant No. 8116360). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Eisen MD, Ryugo DK (2007) Hearing molecules: contributions from genetic deafness. Cell Mol Life Sci 64:566–580 doi: 10.1007/s00018-007-6417-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ouyang XM, Yan D, Yuan HJ, Pu D, Du LL, et al. (2009) The genetic bases for non-syndromic hearing loss among Chinese. J Hum Genet 54:131–140 doi: 10.1038/jhg.2009.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petersen MB, Willems PJ (2006) Non-syndromic, autosomal-recessive deafness. Clin Genet 69:371–392 doi: 10.1111/j.1399-0004.2006.00613.x. [DOI] [PubMed] [Google Scholar]
- 4. Kim H-J, Park C-H, Kim H-J, Lee K-O, Won H-H, et al. (2010) Sequence Variations and Haplotypes of the GJB2 Gene Revealed by Resequencing of 192 Chromosomes from the General Population in Korea. Clin Exp Otorhinolaryngol 3:65–69 doi: 10.3342/ceo.2010.3.2.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dai P, Stewart AK, Chebib F, Hsu A, Rozenfeld J, et al. (2009) Distinct and novel SLC26A4/Pendrin mutations in Chinese and U.S. patients with nonsyndromic hearing loss. Physiol Genomics 38:281–290 doi: 10.1152/physiolgenomics.00047.2009. [DOI] [PubMed] [Google Scholar]
- 6. Li Q, Dai P, Huang D, Yuan Y, Zhu Q, et al. (2007) Frequency of SLC26A4 IVS7-2A>G mutation in patients with severe to profound hearing loss from different area and ethnic group in China. Zhonghua er bi yan hou tou jing wai ke za zhi 42:893–897. [PubMed] [Google Scholar]
- 7. Fan D, Yu S, Jin P, Zhu W, De J, et al. (2013) The Deafness-Related Gene Mutation Survey of 92 Tibetan Students at Schools for the Deaf. Chinese Sci J Hear Speech Rehabil 11:271–274. [Google Scholar]
- 8. Scott HS, Kudoh J, Wattenhofer M, Shibuya K, Berry A, et al. (2001) Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet 27:59–63 doi: 10.1038/83768. [DOI] [PubMed] [Google Scholar]
- 9. Guipponi M, Toh M-Y, Tan J, Park D, Hanson K, et al. (2008) An integrated genetic and functional analysis of the role of type II transmembrane serine proteases (TMPRSSs) in hearing loss. Hum Mutat 29:130–141 doi: 10.1002/humu.20617. [DOI] [PubMed] [Google Scholar]
- 10. Ben-Yosef T (2001) Novel mutations of TMPRSS3 in four DFNB8/B10 families segregating congenital autosomal recessive deafness. J Med Genet 38:396–400 doi: 10.1136/jmg.38.6.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Masmoudi S, Antonarakis SE, Schwede T, Ghorbel AM, Gratri M, et al. (2001) Novel missense mutations of TMPRSS3 in two consanguineous Tunisian families with non-syndromic autosomal recessive deafness. Hum Mutat 18:101–108 doi: 10.1002/humu.1159. [DOI] [PubMed] [Google Scholar]
- 12. Martínez VG, Moestrup SK, Holmskov U, Mollenhauer J, Lozano F (2011) The conserved scavenger receptor cysteine-rich superfamily in therapy and diagnosis. Pharmacol Rev 63:967–1000 doi: 10.1124/pr.111.004523. [DOI] [PubMed] [Google Scholar]
- 13. Sarrias MR, Grønlund J, Padilla O, Madsen J, Holmskov U, et al. (2004) The Scavenger Receptor Cysteine-Rich (SRCR) domain: an ancient and highly conserved protein module of the innate immune system. Crit Rev Immunol 24:1–37. [DOI] [PubMed] [Google Scholar]
- 14. Weegerink NJD, Schraders M, Oostrik J, Huygen PLM, Strom TM, et al. (2011) Genotype-phenotype correlation in DFNB8/10 families with TMPRSS3 mutations. J Assoc Res Otolaryngol 12:753–766 doi: 10.1007/s10162-011-0282-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee YJ (2003) Pathogenic mutations but not polymorphisms in congenital and childhood onset autosomal recessive deafness disrupt the proteolytic activity of TMPRSS3. J Med Genet 40:629–631 doi: 10.1136/jmg.40.8.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guipponi M (2002) The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum Mol Genet 11:2829–2836 doi: 10.1093/hmg/11.23.2829. [DOI] [PubMed] [Google Scholar]
- 17. Molina L, Fasquelle L, Nouvian R, Salvetat N, Scott HS, et al. (2013) Tmprss3 loss of function impairs cochlear inner hair cell Kcnma1 channel membrane expression. Hum Mol Genet 22:1289–1299 doi: 10.1093/hmg/dds532. [DOI] [PubMed] [Google Scholar]
- 18. Elbracht M, Senderek J, Eggermann T, Thürmer C, Park J, et al. (2007) Autosomal recessive postlingual hearing loss (DFNB8): compound heterozygosity for two novel TMPRSS3 mutations in German siblings. J Med Genet 44:e81 doi: 10.1136/jmg.2007.049122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wattenhofer M, Di Iorio MV, Rabionet R, Dougherty L, Pampanos A, et al. (2002) Mutations in the TMPRSS3 gene are a rare cause of childhood nonsyndromic deafness in Caucasian patients. J Mol Med (Berl) 80:124–131 doi: 10.1007/s00109-001-0310-6. [DOI] [PubMed] [Google Scholar]
- 20. Ahmed ZM, Li XC, Powell SD, Riazuddin S, Young T-L, et al. (2004) Characterization of a new full length TMPRSS3 isoform and identification of mutant alleles responsible for nonsyndromic recessive deafness in Newfoundland and Pakistan. BMC Med Genet 5:24 doi: 10.1186/1471-2350-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee J, Baek J-I, Choi JY, Kim U-K, Lee S-H, et al. (2013) Genetic analysis of TMPRSS3 gene in the Korean population with autosomal recessive nonsyndromic hearing loss. Gene 532:276–280 doi: 10.1016/j.gene.2013.07.108. [DOI] [PubMed] [Google Scholar]
- 22. Chung J, Park SM, Chang SO, Chung T, Lee KY, et al. (2014) A novel mutation of TMPRSS3 related to milder auditory phenotype in Korean postlingual deafness: a possible future implication for a personalized auditory rehabilitation. J Mol Med (Berl). 10.1007/s00109-014-1128-3 [DOI] [PubMed] [Google Scholar]
- 23. Lee K, Khan S, Islam A, Ansar M, Andrade PB, et al. (2012) Novel TMPRSS3 variants in Pakistani families with autosomal recessive non-syndromic hearing impairment. Clin Genet 82:56–63 doi: 10.1111/j.1399-0004.2011.01695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wattenhofer M, Sahin-Calapoglu N, Andreasen D, Kalay E, Caylan R, et al. (2005) A novel TMPRSS3 missense mutation in a DFNB8/10 family prevents proteolytic activation of the protein. Hum Genet 117:528–535 doi: 10.1007/s00439-005-1332-x. [DOI] [PubMed] [Google Scholar]
- 25. Hutchin T, Coy NN, Conlon H, Telford E, Bromelow K, et al. (2005) Assessment of the genetic causes of recessive childhood non-syndromic deafness in the UK - implications for genetic testing. Clin Genet 68:506–512 doi: 10.1111/j.1399-0004.2005.00539.x. [DOI] [PubMed] [Google Scholar]
- 26. Charif M, Abidi O, Boulouiz R, Nahili H, Rouba H, et al. (2012) Molecular analysis of the TMPRSS3 gene in Moroccan families with non-syndromic hearing loss. Biochem Biophys Res Commun 419:643–647 doi: 10.1016/j.bbrc.2012.02.066. [DOI] [PubMed] [Google Scholar]
- 27. Vozzi D, Morgan A, Vuckovic D, D’Eustacchio A, Abdulhadi K, et al. (2014) Hereditary hearing loss: a 96 gene targeted sequencing protocol reveals novel alleles in a series of Italian and Qatari patients. Gene 542:209–216 doi: 10.1016/j.gene.2014.03.033. [DOI] [PubMed] [Google Scholar]
- 28. Yuan Y, Zhang X, Huang S, Zuo L, Zhang G, et al. (2012) Common molecular etiologies are rare in nonsyndromic Tibetan Chinese patients with hearing impairment. PLoS One 7:e30720 doi: 10.1371/journal.pone.0030720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.