

Abstract
Reversible protein thiol oxidation is an essential regulatory mechanism of photosynthesis, metabolism, and gene expression in photosynthetic organisms. Herein, we present proteome-wide quantitative and site-specific profiling of in vivo thiol oxidation modulated by light/dark in the cyanobacterium Synechocystis sp. PCC 6803, an oxygenic photosynthetic prokaryote, using a resin-assisted thiol enrichment approach. Our proteomic approach integrates resin-assisted enrichment with isobaric tandem mass tag labeling to enable site-specific and quantitative measurements of reversibly oxidized thiols. The redox dynamics of ∼2,100 Cys-sites from 1,060 proteins under light, dark, and 3-(3,4-dichlorophenyl)-1,1-dimethylurea (a photosystem II inhibitor) conditions were quantified. In addition to relative quantification, the stoichiometry or percentage of oxidation (reversibly oxidized/total thiols) for ∼1,350 Cys-sites was also quantified. The overall results revealed broad changes in thiol oxidation in many key biological processes, including photosynthetic electron transport, carbon fixation, and glycolysis. Moreover, the redox sensitivity along with the stoichiometric data enabled prediction of potential functional Cys-sites for proteins of interest. The functional significance of redox-sensitive Cys-sites in NADP-dependent glyceraldehyde-3-phosphate dehydrogenase, peroxiredoxin (AhpC/TSA family protein Sll1621), and glucose 6-phosphate dehydrogenase was further confirmed with site-specific mutagenesis and biochemical studies. Together, our findings provide significant insights into the broad redox regulation of photosynthetic organisms.
Reversible protein thiol oxidation has been recognized as a fundamental redox regulatory mechanism occurring throughout biology and plays essential roles in photosynthesis, cellular metabolism, gene expression, and other key biological processes (1, 2). Protein thiols serve as important redox switches in cells through reversible thiol oxidation by forming diverse post-translational modifications (PTMs),1 including disulfide formation (3), S-nitrosylation (4, 5), S-glutathionylation (6), and S-sulfenylation (7). In the case of photosynthesis, redox regulation is inextricably associated with the photosynthetic electron transport (PET) chain and regulation of gene expression. In light/dark cycles, thiol oxidation (e.g. disulfide formation) through the thioredoxin and glutaredoxin systems modulates the activation status of many enzymes linked to photosystem I and photosystem II (PSII), as well as many other redox-related processes in plants (1) and cyanobacteria (8). However, it is still largely unknown how broadly the redox process is involved beyond photosynthesis and, importantly, what specific cysteine (Cys) sites serve as redox switches in photosynthetic organisms.
Cyanobacteria are considered useful model organisms for photosynthesis research because of their evolutionary similarities to chloroplasts (9). These oxygenic photosynthetic prokaryotes are also increasingly recognized as microbial biofactories for the production of desired chemicals (10), and potentially for biofuels from solar energy and carbon dioxide (11, 12). Of particular interest are the thiol redox dynamics of proteins localized in the thylakoid lumen, as redox regulation directly affects the PET chain and photosystem stability, which in turn affects light energy utilization efficiency and biofuel production (13). In the cyanobacterium Synechocystis sp. PCC 6803 (hereinafter referred to as Synechocystis), modulation of the thiol redox states of the PET chain (14) and the redox environment (15) has been observed in response to a light/dark cycle. The oxidative environment of the dark phase, due to the decreased PSII activity and NADPH production (14, 16), alters the redox state of PET chain electron carriers by oxidizing the proteins, similar to changes exhibited in the presence of electron transport inhibitors (14, 16). This in turn suggests that the redox state of the PET chain is a key mechanism of light/dark gene transcript regulation, involving the stability and protein PTMs in response to environmental stresses (16, 17). In the current study, we applied a recently described quantitative redox proteomic approach (18) with the aim of obtaining a proteome-wide view of in vivo thiol-oxidation in Synechocystis as modulated by the light/dark cycle. Because of the complexity of the multiple forms of Cys oxidation (e.g. disulfide, S-nitrosylation, S-glutathionylation, S-sulfenylation), the current study focused on the quantification of total reversible thiol oxidation as a starting point for investigating the thiol redox proteome.
Measurements of in vivo thiol redox status at physiological conditions have traditionally been challenging because free thiols are unstable and prone to oxidation during sample preparation with labile reversible oxidized Cys-residues. Although many redox-sensitive proteins have been reported (19), in most cases it was unknown which specific Cys-residues were modified in vivo. To identify Cys modifications, most initial redox approaches employed differential chemical or isotopic labeling followed by biotin-based affinity enrichment. In these methods, free protein thiols were initially blocked by alkylation, and then specific oxidized Cys-residues were selectively reduced and labeled with biotin tags (20), fluorophores (21, 22), radioactive compounds (23), or isotope-coded affinity tags (24, 25) for Western blot, gel electrophoresis, or mass spectrometry (MS) analysis. Alternatively, thiol-reactive chemical probes coupled with click chemistry have recently been applied to identify redox-sensitive proteins or Cys-residues (26, 27) and in vivo thiol redox changes (28). However, most of these approaches do not provide broad site-specific identifications of Cys modifications with accurate quantification. Several groups have recently reported an alternative iodoTMT or cysTMT switch approach for multiplexed quantification of reversible thiol modifications (29–31). However, the enrichment specificity of the anti-TMT-based approach is still a potential factor of concern that might limit the achievable coverage of Cys-peptides; in one recent report only ∼21% of enriched peptides were observed to be TMT-labeled peptides using an optimized competitive elution buffer (30).
Recently, we and others have developed a resin-assisted enrichment to facilitate more sensitive proteomic identification and quantification of Cys-containing peptides (32) and Cys-based reversible modifications (18, 33). In this study, we extended the resin-assisted enrichment for proteome-wide quantification of in vivo reversible thiol oxidation on individual Cys sites under multiple conditions (light, dark, and in the presence of the PSII inhibitor 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU)) in Synechocystis. ∼2,100 Cys sites from 1,060 proteins were identified, with the vast majority of Cys sites displaying redox changes induced by the light/dark switch. Our results provide a broad quantitative picture of redox-mediated changes of the thiol proteome in cyanobacteria, allowing us to infer the extent of thiol-based redox regulation in photosynthetic organisms.
EXPERIMENTAL PROCEDURES
Materials
Synechocystis cells were obtained from Washington University (St. Louis, MO). Escherichia coli strains were purchased from New England Biolabs (Ipswich, MA). BCA protein assay reagents, a silver stain kit, spin columns, TMT reagents, and isopropyl β-d-1-thiogalactopyranoside were purchased from Thermo Fisher Scientific (Rockford, IL). Porcine trypsin was from Promega (Madison, WI). The SeeBlue Plus2 protein standard was from Invitrogen (Carlsbad, CA). Thiopropyl Sepharose 6B affinity resin was from GE Healthcare Bio-Sciences (Pittsburgh, PA). Tris/glycine/SDS buffer, Laemmli sample loading buffer, and Tris-HCl precast gel with a 4%–20% linear gradient were all from Bio-Rad Laboratories (Hercules, CA). Unless otherwise noted, all other chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Cyanobacterial Culture Conditions
Wild-type Synechocystis cultures were grown in 100 ml of liquid BG11 medium (34) in acid-washed 250-ml glass Erlenmeyer flasks topped with foam stoppers at 30°C. Cultures were maintained under constant shaking and illumination by cool-white fluorescent lights at low light (∼30 μmol photons m−2 s−1). Wild-type Synechocystis were grown to exponential phase from a master culture (OD 730 nm of ∼0.2). The cells were then exposed to light, dark (covered by foil), or 10 μm DCMU in light conditions for 2 h. To prevent exposure to light, samples from the dark conditions were wrapped in foil and harvested in a dark room. Cells were pelleted at 6,371g at 4°C for 10 min and flash frozen in liquid nitrogen for the following sample preparation.
Sample Preparation
Cell pellets were resuspended with 10% (w/v) trichloroacetic acid (TCA) and incubated on ice for 20 min, which led to partial lysis of the bacterial cells and precipitation of proteins and effectively stopped thiol-disulfide exchange reactions. After this incubation, proteins and cell debris were spun down at 4°C for 30 min at 13,000g, and then the pellet was carefully rinsed first with 500 μl of ice-cold 10% (w/v) TCA and then with 200 μl ice-cold 5% (w/v) TCA. Lysis buffer (pH 7.6) containing 100 mm N-ethylmaleimide (NEM), 200 mm Tris-HCI, 10 mm EDTA, 0.5% (w/v) SDS, and 8 m urea was added to dissolve the pellet, and brief intermittent sonication was applied until the pellet dissolved. The resultant mixture was incubated at 37°C for 2 h. Bead-beating was performed using 200 μl of 0.1-mm zirconia/silica beads to extract more proteins. Another 200 μl of lysis buffer was used to wash beads, and cell lysates were centrifuged at 10,000g for 10 min at 4°C to pellet cellular debris. Supernatant was transferred to a new 2-ml microcentrifuge tube, and excess NEM was removed by cold acetone precipitation. Proteins were re-dissolved in lysis buffer, and 10 mm DTT was added to the mixture to reduce the oxidized thiols. After reduction, excessive DTT reagents were removed via buffer exchange with 8 m urea and then Milli-Q water by means of centrifugation at 4,000g for 30 min at 4°C. Protein concentration was determined via BCA assay, and a total of 500 μg of proteins was used for the subsequent enrichment experiment. ∼100 μg of proteins from each sample was subjected to trypsin digestion without enrichment according to the previously described procedure (35) for global proteome profiling.
Selective Enrichment of Oxidized or Total Cys-peptides
The samples were transferred to Handee mini spin columns with 35 mg of prewetted and prewashed Thiopropyl Sepharose 6B resin and 50 mm HEPES buffer as described previously (18). To enrich the total Cys-peptides, NEM was not added during sample processing. Enrichment of free Cys-containing proteins was carried out by incubating the samples in a Thermomixer at room temperature with shaking at 850 rpm for 2 h. Nonspecifically bound proteins were removed by five washes with the following solutions: (i) 8 m urea, (ii) 2 m NaCl, (iii) 80% (v/v) acetonitrile with 0.1% (v/v) trifluoroacetic acid (TFA), and (iv) 25 mm HEPES (pH 7.7). To perform the on-resin digestion, 120 μl of solution containing 25 mm HEPES buffer (pH 7.7), 1 m urea, 5 mg trypsin, and 1 mm CaCl2 was added to the columns, and the samples were incubated at 37°C with shaking at 850 rpm for 3 h. Nonspecifically bound peptides were washed away using the same procedure as described above for removing nonspecifically bound proteins.
On-resin TMT Labeling
On-resin isobaric labeling using six-plex TMTs was performed to label the captured peptides in order to achieve relative quantification for oxidized Cys under light, dark, or DCMU. For TMT labeling, acetonitrile (40 μl) was added to the manufacturer-provided TMT reagents to dissolve/dilute them. Then, 80 μl of dissolution buffer and the TMT reagent solutions mentioned above were added to the spin columns. The labeling reaction was carried out at room temperature with shaking at 850 rpm on a Thermomixer for 1 h. The reaction was stopped by the addition of 8 μl of 5% (w/w) NH2OH in 200 mm triethylammonium bicarbonate buffer followed by incubation at room temperature with shaking at 850 rpm for 15 min. The excess TMT reagents were removed by five washes with the following solutions: (i) 80% (v/v) acetonitrile/0.1% (v/v) TFA and (ii) 25 mm ammonium bicarbonate (pH 7.8). Cys-peptides were eluted via incubation with 20 mm DTT in 100 μl of 25 mm ammonium bicarbonate for 30 min followed by rinsing with 100 μl of 80% acetonitrile and 0.1% TFA. Cys-peptide samples were then concentrated in a Thermo Scientific SpeedVac concentrator and adjusted to a final volume of 25 μl with water. The final Cys-peptide sample contained 20 mm DTT, which prevented free thiols from oxidation before LC-MS/MS analysis. Equal amounts of the samples from each labeling channel were then mixed to generate the final sample for LC-MS/MS analyses.
SDS-Polyacrylamide Gel Electrophoresis
SDS-PAGE was carried out with a 4%–20% (w/v) precast linear gradient Tris-HCl polyacrylamide gel (Bio-Rad) to assess enrichment efficiency and total oxidation levels. Equal volumes (5 μl) of the above Cys-peptide sample and Laemmli sample buffer (Bio-Rad) were mixed and then incubated at 95°C for 5 min. Meanwhile, 1 ml of See Blue Plus 2 protein standard was directly loaded onto the gel. Gel electrophoresis was run at 170 V for 30 min in Tris/glycine/SDS buffer (Bio-Rad). After electrophoresis, the gel image was developed with silver staining according to the manufacturer's standard protocol.
LC-MS/MS Analysis
All TMT-labeled samples were analyzed via LC-MS/MS with two technical replicates. All peptide samples were analyzed using a Waters nano-Aquity UPLC system (Waters Corporation, Milford, MA) with a homemade 75 μm inner diameter × 70 cm reversed-phase capillary column using 3-μm C18 particles (Phenomenex, Torrance, CA). The system was operated at a constant flow of 300 nl/min over 3 h with a gradient of 100% mobile phase A (0.1% (v/v) formic acid in water) to 60% (v/v) mobile phase B (0.1% (v/v) formic acid in acetonitrile). MS analysis was performed on a Thermo Scientific LTQ-Orbitrap Velos mass spectrometer coupled with an electrospray ionization interface using homemade 150 μm outer diameter × 20 μm inner diameter chemically etched electrospray emitters (36). The heated capillary temperature and spray voltage were 350°C and 2.2 kV, respectively. Full MS spectra were recorded at a resolution of 100 K (m/z 400) over the range of m/z 400–2,000 with an automated gain control value of 1 × 106. MS/MS was performed in the data-dependent mode with an automated gain control target value of 3 × 104. The most abundant 10 parent ions were selected for MS/MS using high-energy collision dissociation with a normalized collision energy setting of 45. Precursor ion activation was performed with an isolation width of 2 Da, a minimal intensity of 500 counts, and an activation time of 10 ms. A dynamic exclusion time of 45 s was used. Label-free analyses were performed with the same LC-MS platform from three biological replicates under data-dependent acquisition mode using collision-induced dissociation with a normalized collision energy setting of 35.
Data Analysis
LC-MS/MS raw data were converted into .dta files using Bioworks Cluster 3.2 (Thermo Fisher Scientific, Cambridge, MA), and an MSGF Plus algorithm (v.9979, released March 2014) (37) was used to search MS/MS spectra against the Synechocystis sp. PCC 6803 FASTA database (3,169 entries, Kazusa Genome Resources, September 9, 2011). The key search parameters used were 20-ppm tolerance for precursor ion masses, 0.5-Da tolerance for fragment ions, partial tryptic search with up to three miscleavages, dynamic oxidation of methionine (15.9949 Da), dynamic NEM modification of Cys (125.0477 Da), and static six-plex TMT modification of lysine and N termini of peptides (229.1629 Da). For the label-free global proteome analysis, dynamic oxidation of methionine, NEM modification of Cys, and carbamidomethylation of Cys-residues (57.0215 Da) were applied. Peptides were identified from database search results with the following criteria: MSGF E-value < 1E-8, Q-value < 0.01, and mass measurement error < 10 ppm. The decoy database searching methodology (38) was used to confirm the final false discovery rate at the unique peptide level as ∼0.3%. Because NEM blocked only the free Cys-residues, all originally oxidized Cys-residues were identified as unmodified Cys. The Cys-sites of oxidation were identified based on each peptide and its corresponding protein sequence using an in-house software tool, Protein Coverage Summarizer (available from the Pacific Northwest National Laboratory).
For TMT-based relative quantification, all MS/MS spectra were grouped based on individual Cys-sites, and their TMT reporter ion intensities were summed from all spectra corresponding to a given Cys-site. The relative oxidation levels for individual Cys-sites were calculated by dividing the summed intensities for each TMT channel in a given six-plex experiment by the average values across the three conditions. Following this conversion, the data from the two independent six-plex experiments were merged to provide data with four biological replicates for each condition (supplemental Table S1). We then compared the dynamic changes in relative oxidation for individual Cys-sites by calculating the ratio between dark and light and the ratio of DCMU and light. At least 50% change in the level of Cys oxidation was considered a confident redox change (p < 0.05, analysis of variance). To calculate the relative oxidation level of a given protein, we averaged the data from all significant Cys-sites from a specific protein as the relative oxidation level of the specific protein.
For TMT-based stoichiometric (percent of reversible oxidation) analysis, the ratios of reporter ion intensities between channels for oxidized thiols and channels for total thiols were calculated as a percentage. For the global proteome profiling, a label-free spectral counting strategy was applied. Peptides were chosen if they were observed in all three biological replicates, and spectral counts of each peptide were transformed to log2 values and normalized using the central tendency approach. The peptide level was considered significantly different when the p value was less than 0.05 (analysis of variance) with at least 50% change between conditions. Biological function categorization of the identified proteins was clustered based on Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway analyses using the DAVID functional annotation (39).
Strains and Culture Preparation for Recombinant Proteins
E. coli Top10 cells were used as a host for Gibson assembly (40). The E. coli strains BL2 (DE3) and Lemo21 (DE3) (New England Biolabs) were used for protein expression. All bacterial cultures were grown on Luria broth (LB) agar plates or in liquid culture media with 100 μg/ml ampicillin. For plasmid DNA purification, a single colony was grown in 10 ml of LB liquid culture medium in a 125-ml Pyrex glass Erlenmeyer flask overnight at 37°C at 250 rpm. Similarly, for E. coli protein expression, a single colony was inoculated in 10 ml of LB medium and grown overnight at 37°C at 250 rpm. An aliquot (2 ml) of overnight culture was transferred into 50 ml of LB medium with 100 μg/ml ampicillin to reach the initial optical density (OD 600) of 0.15. After the cultures were grown to mid-logarithmic phase with OD 600 of 0.5 to 0.6, isopropyl β-d-1-thiogalactopyranoside was added into the cultures to a final concentration of 0.5 mm. The cultures were further grown at 30°C and 250 rpm for 18 to 24 h. Bacterial cultures (10 ml) were transferred into 15-ml centrifuge tubes, and the rest into 50-ml centrifuge tubes. The bacteria were pelleted down via centrifugation at 4°C and 4,500g for 15 min. Cell pellets were stored at −80°C in a freezer until use.
Site-specific Mutagenesis and Protein Expression Vector Construction
The genomic DNA isolation of Synechocystis sp. PCC 6803 mainly followed the DNA extraction protocol from cyanobacteria described by Sinha et al. (83) at Protocol Online, with some modifications. Cyanobacterial cells (20 μl) were resuspended in 1 ml of 10 mm Tris buffer with 1 mm EDTA, pH 8.0, and pelleted by centrifugation for 2 min at 10,000g at room temperature. The pelleted cells were resuspended in 200 μl of 1% SDS and incubated at 70°C for 15 min. After heat treatment, 300 mg of glass beads (diameter of 0.4 to 0.6 mm) and 200 μl of water-saturated phenol (pH 8.0, Invitrogen) were added and vortexed for 1 min. The cell debris was pelleted by centrifugation at 15,000g for 5 min at room temperature. The supernatant was further treated with 1 mg/ml ribonuclease A for 30 min at 50°C, extracted twice with an equal volume of phenol-chloroform (1:1), and centrifuged at 15,000g for 5 min at room temperature. The genomic DNA in the supernatant was precipitated with 0.1 volume of 3 m sodium acetate (pH 5.2) and 2.5 volumes of ice-cold 95% ethanol at room temperature for 30 min and pelleted by centrifugation at 5,000g for 10 min at 4°C. Finally, the genomic DNA was resuspended in 100 μl of the 10 mm Tris buffer (pH 8.0). The site-specific mutagenesis was carried out for the replacement of selected cysteine with serine in cyanobacterial proteins of glucose-6-phosphate dehydrogenase (Zwf, C187S, C265S, and C445S), glyceraldehyde-3-phosphate dehydrogenase (Gap2, C154S, C154S/C158S, and C292S), and peroxiredoxin (Sll1621, C55S, C155S/C162S, and C162S). Cynaobacterial genomic DNA (50 ng) were used as DNA templates for polymerase chain reaction to generate a specific DNA fragment with one unit Phusion high-fidelity DNA polymerase (New England Biolabs), 50 μl of reaction mixture (Phusion HF buffer and dNTP), and a pair of corresponding oligonucleotides (listed in the supplemental material). DNA fragments were separated in 1% agarose gel and purified with a QIAquick gel extraction kit (Qiagen, Valencia, CA).
Protein expression vector construction was carried out with a Gibson assembly kit (New England Biolabs). Briefly, the plasmid DNA of protein expression vector pET6xHN-N (Clontech, Mountain View, CA) was linearized by digestion with restriction endonucleases StuI and XbaI. Proportional amounts of the linearized plasmid DNA of pET6xHN-N and corresponding DNA fragments were mixed and assembled at 50°C for 1 h. The assembled DNA mixtures were transferred into E. coli Top10 cells via chemical transformation for plasmid DNA replication. The plasmid DNA was purified from specific Top10 cells with a QIAprep Spin Miniprep Kit (Qiagen). DNA sequencing by Beckman Coulter Genomics (Danvers, MA) verified the site-specific mutation in the selected protein expression vector.
Expression and Purification of Recombinant G6PDH, Gap2, and Sll1621
The plasmid DNA of designated protein expression vectors was transformed into the E. coli BL2 ((DE3), G6PDH and Gap2) or Lemo21 ((DE3), Sll1621) according to the manufacturer's transformation protocol. Because all three proteins were in-frame assembled with six repeating His-Asn and enterokinase cleavage sites at their N termini, proteins were purified with His60 Ni superflow gravity columns (Clontech). Briefly, the E. coli cell pellets were fully resuspended in 1 ml of ice-cold Ni60 Ni xTractor buffer per 10 ml of the original cultures by being pipetted up and down gently. The resuspended cells were further homogenized with a Cole Parmer 4710 series homogenizer at the output of 3 and 70% cycle duty three times for 5 s each time on ice with a 1-min interval. The cell lysates were incubated on ice for 15 min and then centrifuged at 10,000g and 4°C for 20 min. Finally, the supernatants were passed through 1-ml His60 Ni gravity columns to purify the recombinant proteins according to the detailed instructions provided in the manufacturer's manual (Clontech). Purified proteins (30 μg) were separated on SDS-PAGE gels and visualized using the Gel DocTM EZ system (Bio-Rad).
Enzymatic Activity Measurement
G6PDH catalyzes the first step in the pentose phosphate pathway by oxidizing glucose-6-phosphate and reducing NADP+ to NADPH. The rate of NADPH formation is correlated to the G6PDH activity and can be measured based on the change in NADPH absorbance at 340 nm. G6PDH (35 μg) and its mutants were mixed with reaction buffer (50 mm phosphate buffer, 10 mm MgCl2, pH 7.5) and 0.67 mm NADP in a 1-ml final volume in a 1-ml plastic cuvette and kept at room temperature for 3 min. The reaction kinetics was initiated by the addition of 2 mm glucose-6-phosphate. G6PDH activity was calculated mainly by following the instructions for G6PDH activity calculation described by Sigma-Aldrich. The activities of Gap2 (GAPDH), peroxiredoxin (Sll1621, thioredoxin reductase), and their mutants were determined using a KDalertTM GAPDH assay kit (Invitrogen, Grand Island, NY) and a thioredoxin reductase assay kit (Sigma-Aldrich).
For DTT and H2O2 treatment, we treated wild-type Gap2 proteins with either 10 mm DTT or a specific amount of H2O2 by directly adding the reagent into the reaction mixture and incubating the mixture at room temperature for 2 min prior to the initiation of enzyme reactions. For the treatment of Sll1621, 10 mm DTT was mixed with 100 μl (250 μg) of enzyme and kept at room temperature for 15 min. Then, excess DTT was removed by Amicon Ultra-0.5 centrifugal filter devices (EMD Millipore, Billerica, MA) with 500 μl of 50 mm phosphate buffer, pH 7.5, in five passes. The same procedures for DTT treatment and washing were applied prior to an additional 15-min treatment with 1 mm H2O2. The activities were determined with a thioredoxin reductase assay kit.
RESULTS
Quantitative Site-specific Measurements of Reversible Thiol Oxidation
Resin-assisted thiol-affinity enrichment has recently been shown to be effective for the identification and quantification of S-nitrosylation (33, 41) and S-glutathionylation (42). Here we adapted this strategy for the quantitative profiling of reversible thiol oxidation in the Synechocystis proteome in response to light/dark modulation. Briefly, Synechocystis was grown in the presence of oxygen and continuous light to the mid-log phase, and then it was exposed to continuous light, darkness, or DCMU in continuous light (hereinafter referred to as DCMU) for 2 h before cell harvesting. All endogenous free thiols were initially trapped by acid trapping using TCA precipitation (23) followed by blocking with NEM (Fig. 1A). Following free thiol blocking, the oxidized Cys-residues (i.e. disulfide or other reversible oxidative modifications) were reduced back to free thiols by DTT. The newly formed free thiols from the originally oxidized proteins were then specifically captured by Thiopropyl Sepharose 6B resin through a disulfide exchange reaction (32). The enriched proteins were subjected to on-resin trypsin digestion. Resin-bound Cys-peptides were further labeled with amine-reactive isobaric TMT reagents (43) to facilitate quantification of the dynamics of thiol oxidation across multiple conditions. The six-plex TMT reagents allowed simultaneous quantification of six samples in a single experiment. This meant that two biological replicates of light, dark, and DCMU conditions could be quantified in a single experiment (Fig. 1B). The TMT-labeled Cys-peptides were subjected to LC-MS/MS analyses for site-specific identification and relative quantification of the levels of oxidation on individual Cys sites.
Fig. 1.

Overview of the enrichment and site-specific quantification strategy for thiol oxidation. A, enrichment and processing workflow by the resin-assisted approach. Note that S-X denotes oxidized thiols. Proteins were extracted from different conditions, and free thiols were blocked by NEM alkylation. Oxidized Cys was reduced by DTT and captured by Thiopropyl Sepharose resin. On-resin protein digestion and TMT isobaric labeling of enriched Cys-peptides were carried out and followed by DTT elution of the enriched peptides for LC-MS/MS analysis. B, six-plex quantitative strategy for profiling Cys redox dynamics. Synechocystis were cultured under continuous light, switched to dark for 2 h, or treated with 10 μm DCMU in light for 2 h. Proteins were extracted and processed to enrich the oxidized Cys-peptides accordingly, and enriched peptides were labeled with TMT reagents with different reporter tags (126–131). The six labeled samples were combined to facilitate MS/MS-based six-plex quantification. C, workflow for quantifying the percentage of reversible Cys oxidation. Equal amounts of protein samples were processed in parallel both with NEM blocking (oxidized thiols) and without NEM blocking (oxidized thiols). Total thiols (free plus reversibly oxidized thiols) were enriched in the sample reduced by DTT without NEM blocking. D, gel image of enriched total Cys-containing peptides and oxidized Cys-peptides for the samples processed with incubation with 100 mm NEM before bead-beating. Samples were from the light condition. Lane 1, standard protein ladder; Lane 2, total enriched Cys-peptides; Lane 3, enriched oxidized Cys-peptides. E, gel image of enriched total Cys-containing peptides and oxidized Cys-peptides for the samples processed with bead-beating prior to incubating with 100 mm NEM. Lane 1, standard protein ladder; Lane 2, total enriched Cys-peptides; Lane 3, enriched oxidized Cys-peptides.
In addition to relative quantification, our strategy can also be modified to quantify the percentage (or stoichiometry) of thiol oxidation (reversibly oxidized/total thiols) for individual Cys-residues (Fig. 1C). In this case, a given biological sample is divided into two identical aliquots, with one aliquot processed for oxidized thiols only and the other for total thiols without NEM blocking. With six-plex TMT labeling, the percentage of reversible oxidation of three replicates can be quantified in a single LC-MS/MS experiment (24). But several caveats associated with this method should be noted. First, the irreversible oxidation is excluded in the measurement of total thiols. Thus, overestimation of the percentage of reversible oxidation might occur if irreversible oxidation is present at high levels (44). Second, when a given peptide contains more than one Cys-residue, the percentage of oxidation cannot be accurately measured. We note that this is a common limitation shared by several previously reported methods (24, 45) due to the existence of multiple combinatory forms of oxidized Cys-sites for a given sequence.
In our approach, a key step for measuring oxidized thiols is the effective blocking of free thiols. To achieve this, we adapted the acid trapping method (23, 46) using 10% TCA followed by alkylation with a high concentration of NEM (100 mm) in lysis buffer to block free thiols and prevent artificial thiol oxidation during sample processing. To assess the efficiency of the free thiol blocking, we dissolved one set of sample pellets from the TCA precipitation in lysis buffer containing the alkylating agent, NEM, and another set was not exposed to NEM. The set of sample pellets in lysis buffer containing NEM was carefully dissolved with sonication so as to not introduce additional oxygen from air and incubated to block free thiols to prevent artificial oxidation. The final level of enriched oxidized Cys-peptides was observed to be very low (Fig. 1D) relative to the level of total Cys-peptides in the sample pellet not exposed to NEM. The results suggested that there was minimal artificial oxidation as a result of effective blocking with NEM, and evidence that the in vivo thiol redox state was being preserved. However, for comparison, cell pellet samples were alternatively subjected to bead-beating before NEM incubation. Simply altering the preparation by performing a bead-beating process prior to blocking introduced a sufficiently oxidative environment to artificially increase thiol oxidation. The level of oxidized Cys-peptides was nearly the same as the level of total Cys-peptides, abrogating the detection of any differential abundance (Fig. 1E).
Broad Redox Changes on Cys Sites Modulated by Light/Dark
To identify site-specific redox changes on protein thiols, we performed LC-MS/MS analyses for four biological replicates of light, dark, and DCMU conditions. ∼2,100 oxidized Cys sites from 1,060 proteins were identified and quantified based on TMT reporter ion intensities in MS/MS spectra (supplemental Table S1). Among all identified peptides, ∼98% were Cys-containing peptides, in line with the high efficiency of the resin-assisted enrichment (32, 41). The overall results (Fig. 2A and supplemental Fig. S1A) demonstrate that most protein thiols were less oxidized (or more reduced) under continuous light but became significantly more oxidized in the dark phase or in the presence of DCMU. DCMU is a specific inhibitor of PSII that blocks the plastoquinone binding site of PSII and stops the electron flow in the PET chain (47), simulating the dark condition in which no photosynthetic electron flow occurs. ∼80% of the identified Cys sites were observed with significant redox changes for light, dark, and DCMU conditions (p < 0.05, analysis of variance) (supplemental Table S1). After we applied cutoffs of >1.5 in abundance ratios (i.e. >50% change in oxidation levels) and p < 0.05, we observed a total of ∼1,100 Cys sites with substantial redox changes (Fig. 2B). This observation of increased oxidation under the dark condition is consistent with the understanding that cells under light, with active photosynthesis and electron flow through the PET chain, foster a reductive environment through the regulatory mechanisms of the reduced ferredoxin and thioredoxin systems, and hence are reflected in a more reduced form of thiols from thioredoxin target enzymes (1, 48). However, the dark conditions, or similarly DCMU inhibition of the PET chain, facilitates a shift to a more oxidative environment, leading to higher levels of oxidative modifications in target enzymes, which modulates enzyme activity across numerous cellular processes.
Fig. 2.

Broad Cys redox changes modulated by light/dark. A, the relative oxidation levels of ∼2,000 identified Cys sites under light, dark, or DCMU conditions. The relative oxidation levels were calculated by dividing the total reporter ion intensity from each TMT channel for a given Cys site by the average intensity across all conditions for the same Cys site. B, the redox sensitivity of individual Cys sites. The y-axis abundance ratios are expressed as the ratios of oxidation levels under dark or DCMU conditions divided by those under the light condition. Peptides were considered confidently redox-sensitive if all four data points were observed (p < 0.05 and fold change > 1.5-fold). C, distribution of Cys sites in terms of the percentage of Cys oxidation for individual Cys residues. Histograms show a shift to higher levels of oxidation for dark and DCMU relative to light.
Percentage of Reversible Oxidation of Individual Cys Sites
We sought to measure the percentage or stoichiometry of reversible oxidation of individual Cys sites using the strategy outlined in Fig. 1C. Initial gel-based results revealed a relatively high percentage of oxidation in the DCMU condition via comparison of the overall level of oxidized thiols to total thiols (supplemental Fig. S1B). LC-MS/MS analyses were performed to measure the percentages of oxidation of individual Cys sites in the DCMU condition only. The percentages of oxidation in light and dark conditions were calculated based on the data from the DCMU condition and the relative abundance changes shown in Fig. 2A. Using this strategy, we obtained stoichiometry data for 1,351 specific Cys sites (∼65% of all Cys sites from the dynamic measurements in Fig. 2A). Because of the limitation for stoichiometric measurements of peptides containing multiple Cys sites, these peptides were excluded from further analysis. Fig. 2C shows the distribution of Cys-site peptides in terms of the percentage of oxidation for individual Cys sites (supplemental Tables S1 and S2).
Again, one potential source of bias in calculating this percentage is the unknown level of irreversible oxidation. To address this issue, we compared the levels of total thiols (free and reversibly oxidized thiols) across light, dark, and DCMU conditions, and no obvious change in the levels of total thiols was observed in two independent experiments (left-hand lanes in supplemental Figs. S1C and S1D). The results suggested that no significant levels of irreversible oxidation occurred in either dark or DCMU conditions, as the levels of total thiols (excluding irreversibly oxidized thiols) should decrease if the levels of irreversible oxidation are high.
As shown in Fig. 2C, the majority of Cys residues under the light condition were mostly reduced, with only 5% to 20% in the oxidized form. However, the levels of oxidation were significantly increased in dark and DCMU conditions, with the majority of Cys sites at 20% to 40% oxidation. The overall levels of oxidation support the idea that protein thiols are mostly in a reduced state under physiological conditions because of the highly reducing intracellular environment (49); both darkness and DCMU induced a consistent pattern of increased thiol oxidation in the proteome relative to continuous light (Fig. 2C). The results suggest that a primary redox-mediated mechanism in a light/dark cycle can function by altering the thiol redox states. It is interesting to note that DCMU inhibition consistently resulted in a slightly higher level of thiol oxidation than the dark condition. We note that DCMU effectively inhibits electron flow under continuous light, leading to a state similar to the dark condition; however, the DCMU condition does not inhibit photo-oxidation of PSII and oxygen generation completely. The slightly higher level of thiol oxidation in the DCMU condition is presumably due to photo-oxidation, which is absent in the dark condition.
Total Protein Abundances
To verify that the observed redox changes were not due to the changes in total protein abundance, we performed global proteome profiling across these conditions. Aliquots of all protein samples before enrichment were digested and subjected to LC-MS/MS analyses. No significant abundance changes were observed among the vast majority of the 413 proteins with spectral count data (supplemental Table S3). 273 proteins overlapped with those detected with oxidized Cys residues (supplemental Table S1). Only seven proteins displayed a consistent increase in abundance (with p < 0.05) similar to the observed redox changes. The overall results suggest that the short 2-h dark incubation or DCMU inhibition was not long enough to induce significant protein abundance changes as compared with previous large-scale diurnal-cycle studies. Such global diurnal proteome effects in the cyanobacterium Cyanothece sp. ATCC 51142 (50) induced significant changes in the abundance of only ∼70 proteins among the ∼1,200 identified proteins in the first 1 to 3 h of the dark phase during the diurnal cycle. In contrast, genomic studies of the dynamic transcriptional profiling of the light-to-dark transition in Synechocystis has revealed that 387 genes of the full genome (3,169 genes) respond to light exposure within 2 h (51) and the expression of 64 genes significantly changes in response to DCMU (52). Interestingly, we noted a number of transcriptional regulators (supplemental Table S4) that were potentially redox-regulated, which might facilitate initial changes in gene expression followed by changes in protein expression at later time points.
Functional Implications of Redox-sensitive Proteins
To further narrow down the redox-sensitive Cys sites that displayed significant dynamic changes (>1.5-fold) between light and dark conditions, we applied >20% oxidation in dark or DCMU conditions as an additional criterion for selecting those Cys sites more likely to induce a physiological effect (24). This criterion was based on the general notion that the potential physiological effect is greater when the extent of the protein being modified is greater. This additional filtering decreased the original list of ∼1,100 sites with significant changes to ∼600 redox-sensitive sites (from 428 proteins) (supplemental Table S2). Among the 428 identified redox-sensitive proteins, only ∼100 proteins in Synechocystis were reported with evidence of redox relevance as interacting with thioredoxin (53, 54); however, direct evidence of oxidation and site information were not available for most of these proteins.
To gain an overall picture of the pathways and processes that are potentially redox- regulated, functional analysis based on Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway information was performed for these redox-sensitive proteins. These proteins were observed to be broadly involved in various biological processes or molecular functions, including amino acid biosynthesis, glycolysis, PET, carbon fixation, and many enzyme classes such as oxidoreductase, ligase, and hydrolase (supplemental Fig. S2). The identified redox-sensitive proteins within the context of several key biological processes including PET, carbon fixation, glycolysis, and the Krebs cycle are illustrated in Fig. 3. The observation of a number of enzymes as redox-sensitive in these processes is consistent with the existing knowledge on the functional significance of the disulfide proteome in these biological processes (55), and our quantitative site-specific data provide potential functional sites in the enzymes. Fig. 4 further illustrates the percentages of thiol oxidation of selected proteins in these biological processes, demonstrating the consistent increases in thiol oxidation in response to dark or DCMU and different degrees of relative changes for different proteins.
Fig. 3.

Redox-sensitive proteins involved in (A) PET, (B) carbon fixation, and (C) glycolysis and the Krebs cycle. The identified redox-regulated enzymes are labeled in red.
Fig. 4.
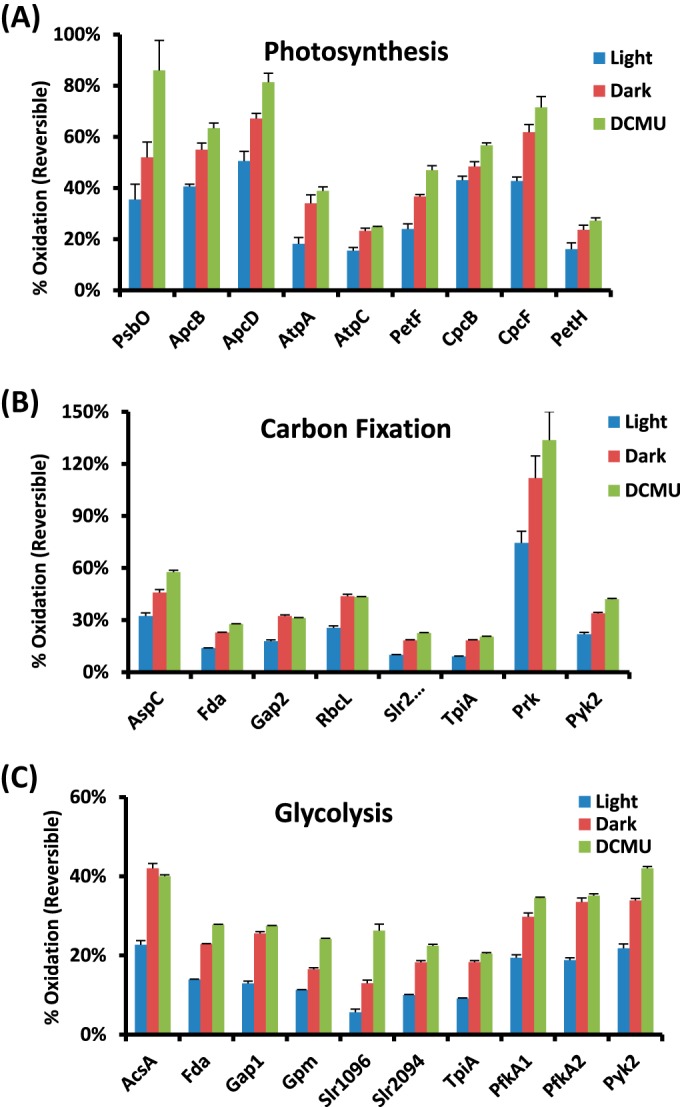
The levels of thiol oxidation for individual proteins in different biological processes. (A) PET, (B) carbon fixation, and (C) glycolysis. The percent of oxidation for each protein is represented by the Cys site with the highest stoichiometry of each protein. Error bars are the standard errors from four biological replicates.
Our results revealed that several proteins within the PET chain, including the phycobilisome complex and PSII, were redox-sensitive and were in a more reduced state under continuous light than under the other two conditions. This finding is consistent with the knowledge that protein thiols play a significant role in the redox regulation of the PET chain through the ferredoxin/thioredoxin system (1, 14). Moreover, the redox regulation of the PET chain is known to play a key role in modulating the redox homeostasis and gene expression of cyanobacteria (14, 56). Among the identified redox-sensitive proteins in photosynthesis (Figs. 3A and 4A), α-subunits of the ATP synthase complex were identified as thioredoxin targets in Synechocystis (8, 19). A number of the observed redox-sensitive proteins (e.g. PSII manganese-stabilizing polypeptide and F-type H+-transporting ATPase gamma subunit) were also reported as thioredoxin targets in Arabidopsis thaliana (19). The redox changes observed in several phycobiliproteins in this study suggest that the redox changes on Cys residues play a role in the assembly or stability of the phycobilisome complex.
A number of key enzymes involved in the Calvin cycle, glycolysis, and the Krebs cycle have been previously reported as potentially redox-regulated (55). Our study identified not only the putative redox-regulated enzymes in these processes, but also the potentially functional redox-sensitive Cys sites. As shown in Fig. 3B and Fig. 4B, most of the enzymes in the Calvin cycle were observed to be redox-sensitive, confirming previous reports that the entire Calvin cycle is under the control of thioredoxin in both chloroplast and cyanobacteria thylakoid (55). The thiols in most enzymes were more reduced in the light condition (Fig. 4B) than in the dark, supporting higher enzyme activities for carbon fixation in the light condition. Moreover, our results provide further insights into the redox regulation of these enzymes. For example, fructose-1,6-bisphosphatase in chloroplasts has been known to undergo light/dark redox transitions for many years (57). However, the redox regulation of its cyanobacterial counterpart, the bifunctional fructose 1,6-bisphosphatase/sedoheptulose-1,7-biphosphatase (Slr2094 in Synechocystis), has not been well established. For instance, one study reported that the FBPase activity was not regulated by light intensities (58). However, an earlier study reported that the fructose-1,6-bisphosphatase activity in Synechococcus sp. strain 6301 was under delicate control of the oxidizing and reducing conditions (59). Similarly, the enzyme phosphoribulokinase has been found to be regulated by light/dark and activated through the ferredoxin/thioredoxin system in the chloroplast of plants (60); however, it has not been confirmed to undergo light/dark modulation in cyanobacteria, although its activity in vitro was observed to be regulated by reversible thiol oxidation based on H2O2 and 5,5-dithiobis (2-nitrobenzoic acid) treatments (61). Our results clearly show light/dark modulation of the thiol oxidation on these enzymes, providing evidence of potential light/dark modulation of these enzyme activities. Additionally, a number of enzymes were observed as redox-sensitive in glycolysis and the Krebs cycle (Figs. 3C and 4C), supporting the role of redox-regulation in these metabolic processes. For example, aconitase and isocitrate dehydrogenase were found to be associated with thioredoxin in higher plants, indicating the potential redox regulation of their activities (62). Currently, it is still unclear whether redox-regulation of many potentially redox-sensitive enzymes exists in cyanobacteria under physiologically relevant conditions. Further studies need to be carried out in order to fully determine how redox regulation happens in the cells.
Site-specific Redox Sensitivity Predicts Functional Sites
A unique aspect of this study is the proteome-wide site-specific redox sensitivity information for Cys residues in response to physiological perturbations. One question is whether such site-specific data provide any predictive value for identifying potential functionally active Cys residues. To examine this predictive potential, we compiled the existing data from identified proteins with known functional Cys sites (either annotated active Cys sites or functional information based on site-directed mutagenesis) in Synechocystis as listed in supplemental Table S5. Out of 14 proteins, the functional sites of 12 proteins (8 selected proteins listed in Table I) were observed to be either the most sensitive or the second most sensitive Cys residues to light/dark modulation for that particular protein. This provides a level of confidence in linking site-specific redox sensitivity with functional Cys residues, implying the predictability of the site-specific redox sensitivity data for identifying potential functional sites. Such predictability is in good agreement with results from several recent reactivity profiling studies in other organisms (26, 27).
Table I. The levels of relative Cys oxidation on individual Cys sites for selected proteins with known active or functional Cys sites.
| Gene I.D. | Accession | Protein name | Cys sites | Ratios of oxidation levelsa |
||
|---|---|---|---|---|---|---|
| Percent oxidation (reversible) (%) | Dark/light | DCMU/light | ||||
| sll0080 | P54899 | N-acetyl-γ-glutamyl-phosphate reductase (argC) | 151 | 10.9 | 22.7 | |
| 151, 154b | 3.3 | 42.8 | ||||
| 154b | 3.9 | 5.0 | ||||
| 86 | 2.8 | 3.5 | ||||
| sll0427 | P10549 | Photosystem II manganese-stabilizing polypeptide (psbO) | 48c | 75 | 1.4 | 2.8 |
| 73 | 86 | 1.5 | 2.4 | |||
| sll1342 | P80505 | NAD(P)-dependent glyceraldehyde-3-phosphate dehydrogenase (gap2) | 154b | 3.4 | 4.1 | |
| 154,b 158 | 2.6 | 2.8 | ||||
| 158 | 2.6 | 2.7 | ||||
| 292 | 31 | 1.8 | 1.7 | |||
| slr0394 | P74421 | Phosphoglycerate kinase (pgk) | 216 | 11 | 2.2 | 2.3 |
| 314c | 19 | 3.0 | 4.5 | |||
| 94 | 1.8 | 1.8 | ||||
| 94, 97 | 1.3 | 1.3 | ||||
| 97 | 1.8 | 1.8 | ||||
| slr0506 | Q59987 | Light-dependent NADPH-protochlorophyllide oxidoreductase (por) | 37c | 22 | 2.3 | 2.7 |
| 133 | 21 | 1.3 | 1.5 | |||
| 226c | 21 | 3.7 | 7.5 | |||
| slr0527 | Q55468 | Transcription regulator ExsB homolog (queC) | 190c | 1.8 | 2.7 | |
| slr1463 | P28371 | Elongation factor EF-G (fusA) | 105c | 25 | 2.2 | 2.6 |
| 388 | 28 | 1.3 | 1.2 | |||
| 676 | 30 | 2.0 | 2.2 | |||
| 68 | 28 | 2.2 | 2.7 | |||
| slr1643 | Q55318 | Ferredoxin-NADP+ oxidoreductase (petH) | 213 | 21 | 1.8 | 2.4 |
| 231c | 3.4 | 4.6 | ||||
| 231,c 236 | 2.7 | 3.5 | ||||
| 236 | 3.2 | 3.1 | ||||
| 371 | 27 | 1.5 | 1.7 | |||
Several examples illustrating the correlation of redox sensitivity with functional sites are shown in Fig. 5. NADP+-dependent glyceraldehyde-3-phosphate dehydrogenase (Gap2 or GAPDH, GeneBank: X83564) is a classic redox-sensitive enzyme that is specific to the Calvin cycle and non-photosynthetic gluconeogenesis (63). The functional domain of GAPDH is highly conserved, and it is a well-known redox switch in cellular metabolism (64). Our site-specific redox sensitivity data imply that Cys-154 and Cys-158 were the most sensitive to light/dark modulation (Fig. 5A). The observed similar redox sensitivity for both the active site Cys-154 and the adjacent site Cys-158 is consistent with several previous reports on other organisms (26, 42). Gap2 in Synechococcus elongatus (PDB code: 2D2I) shares 80% homology with Synechocystis and is conserved at the region containing the active sites. As shown in Fig. 5B, the nicotinamide ring of cofactor NADP+ faces toward the catalytic Cys-154 in Gap2 (65). Previous mutational studies have shown that the catalytic Cys of Gap2 can be oxidized and inactivated by oxidative stress (26, 41). To confirm the functional Cys-residues in Synechocystis Gap2, recombinant Gap2 mutants C154S, C154S/C158S, and C292S were generated and tested for catalytic activity. Interestingly, all the mutations completely abolished Gap2 enzyme activity (Fig. 5C), demonstrating that all redox-active Cys sites are crucial for Gap2 enzymatic function. It is surprising to observe that C292S also abolished enzyme activity, as it has not been previously reported as a functional site. For this reason, we examined the protein expression levels for these mutants via SDS-PAGE and observed that mutant C292S was essentially not detected (Fig. 5C), suggesting that Cys-292 is structurally relevant for protein stability, with C292S possibly causing rapid degradation of the translated protein product. Purified recombinant Gap2 was also treated with various concentrations of H2O2 or DTT. The catalytic activity of Gap2 was significantly reduced after exposure to H2O2, whereas it was increased by DTT (Fig. 5D), which is in agreement with previous reports (26, 65).
Fig. 5.

Site-specific redox sensitivity and functional Cys sites. A, relative oxidation levels of individual Cys sites of Gap2 as modulated by light, dark, and DCMU. B, monomer crystal structure of Gap2 (PDB code: 2D2I) in Synechococcus elongatus PCC 7942. NADP is shown by a stick model; catalytic Cys154 is highlighted in a ball-and-stick model. C, site-specific mutants of Gap2 abolished its activity. Top: expressed levels of recombinant wild-type (WT), C154S, C154S/C158S, and C292S. Bottom: Gap2 activities of WT, C154S, C154S/C158S, and C292S. D, redox regulation of Gap2 activity as treated by 10 mm DTT or various concentrations of H2O2 for 2 min. The asterisks denote that the differences from the DTT-treated sample are statistically significant (p < 0.05). E, relative oxidation levels of individual Cys sites of PetH. F, monomer structure of ferredoxin NADP+ reductase (PetH, FNR) in Synechococcus sp. PCC 7002 (PDB code: 2B5O). FAD is shown in a stick model, and the FAD binding regions Cys231 and Cys236 are highlighted in a ball-and-stick model. Quantitative data are represented as the mean ± S.E.
Ferredoxin NADP+ reductase (PetH, FNR) is the last enzyme in the PET chain for transferring electrons from reduced ferredoxin to NADPH, which serves as the electron donor for the Calvin cycle. PetH is known to be bound to the phycobilisome complexes of Synechocystis (66). The activity of PetH is enhanced in light; however, the mechanism of the enzyme activation is not fully elucidated (67). Site-specific redox data (Fig. 5E) showed high redox sensitivity for Cys-231 and Cys-236, and both residues were found within the known FAD binding region. Mutation of Cys-231 and Cys-236 to serine in spinach PetH resulted in decreased enzyme activity due to the altered kinetic properties (68), supporting a prediction of redox alterations in Cys-231 and Cys-236 as functionally relevant mechanisms for regulating PetH activity in Synechocystis. Fig. 5F shows PetH in Synechococcus sp. PCC 7002 (PDB code: 2B5O), which shares 76% homology with Synechocystis and with a highly conserved FAD binding region. Cys-231 of PetH locates in the nucleotide binding region for FAD.
Novel Functional Cys Sites in Redox Regulatory Proteins
Based upon the predictive observations linking redox sensitivity and functional Cys residues (Table I), we then attempted to determine whether our data would provide valuable information for the identification of novel functional sites of redox regulatory proteins. One protein of interest is peroxiredoxin, encoded by sll1621 (GeneBank: Bak49948.1) in the AhpC/TSA family. Peroxiredoxins have been well recognized as major players in the antioxidant defense system and the redox regulatory network of plant and cyanobacterial cells (69), and they are conserved markers of circadian rhythms (70). It is known that reduced Sll1621 plays a critical role in protection against photo-oxidative stress, especially under high light conditions (71). A Synechocystis sll1621 deletion mutant showed a severely reduced growth rate relative to the wild type under light (71, 72). As Sll1621 is one of the most abundant and active Synechocystis peroxiredoxin isoforms (73), Sll1621 likely represents most of the peroxiredoxin activity in maintaining redox homeostasis for Synechocystis growth; however, its catalytic activity and functional sites are still unknown. We observed that Cys-155 and Cys-162 were the most redox-sensitive sites among the three oxidized Cys sites identified (Fig. 6A). Recombinant protein of Synechocystis Sll1621 and the mutants of C55S, C162S, and C155S/C162S were generated to determine the functions of selected Cys sites of Sll1621. The enzyme activity data (Fig. 6B) confirm that Cys-155 is a potentially novel functional site for peroxiredoxin activity, whereas both Cys-55 and Cys-162 had no effect on the enzymatic activity. The lack of activity reduction for mutant C162S implies that this residue is possibly exposed to capture redox sensitivity but is not directly related to (or required for) peroxiredoxin activity. Also, marked effects of DTT or H2O2 on its activity were also observed (Fig. 6C), confirming its roles in redox response. To further examine whether Cys-155 is a conserved residue across difference species, we blasted the sequence motif in the UniProt database and confirmed that the sequence motif “DNCP” containing Cys-155 in Sll1621 is indeed conserved among ∼50 cyanobacteria strains and up to 70 bacterial strains (supplemental Fig. S4), strongly supporting the functional importance of Cys-155.
Fig. 6.

Functional sites of peroxiredoxin (Sll1621) and G6PDH (Zwf). A, relative oxidation levels of individual Cys-sites of Sll1621 and Zwf in light, dark, and DCMU. B, enzymatic activity of purified peroxiredoxin (Sll1621) wild-type, C55S, C155S/C162S, and C162S mutants. Activity was indicated as mU/mg protein. C, enzymatic activity of purified peroxiredoxin (Sll1621) wild-type treated with water (control), 10 mm DTT for 15 min, and 1 mm H2O2 for 15 min after DTT treatment where DTT was removed via buffer exchange. H2O2 treatment was performed after the removal of DTT via buffer exchange with phosphate buffer. The same buffer exchange step was applied with DTT treatment prior to activity measurement. D, enzymatic activity of purified Zwf (G6PDH) wild-type, C187S, C265S, and C445S. All data are represented as the mean ± S.E. The asterisks denote that the differences from the parent type or untreated sample are statistically significant (p < 0.05).
Another protein of interest is G6PDH (Zwf) (GeneBank: NP 440771.1), which is the first enzyme in the dark-initiated pentose phosphate pathway. Zwf has been reported to be activated by Cys oxidation in dark conditions in plants (74); however, the specific Cys sites responsible for the activation of Zwf are not known in Synechocystis. Our data indicate that Cys-265 and Cys-445 are highly sensitive to oxidation and are potential functional sites of Zwf (Fig. 6A). Additionally, Cys-445 is a conserved Cys residue among multiple strains of cyanobacteria, such as Anabaena, Nostoc, and Synechococcus (75). We created Zwf mutants of C187S, C265S, and C445S for functional activity assays. Results showed that all three Cys sites played a role in maintaining Zwf activity, with Cys-445 playing the most significant role (Fig. 6D), which is consistent with the redox sensitivity data. Interestingly, further assays involving the addition of H2O2 or DTT did not affect the enzymatic activity of recombinant Zwf (data not shown), showing evidence that Zwf alone might not be sensitive to general redox status. It has been reported that Zwf is not sensitive to redox regulation without its allosteric activator (OpcA) present (76). This provides a potentially interesting scenario wherein Cys-445, -265, and/or -187 are likely functionally involved in Zwf/OpcA interactions facilitating redox regulation of the pentose phosphate pathway, and further studies will be necessary to confirm the role of these Cys residues in redox regulation.
DISCUSSION
Cyanobacteria are oxygenic photosynthetic prokaryotes that are believed to share a common ancestor with higher plant chloroplasts. As in all photosynthetic organisms, redox regulation via light/dark diurnal cycles is essential for photosynthesis, metabolism, and gene expression, from the PET chain through the ferredoxin/thioredoxin system to all downstream target proteins. Reversible oxidation of protein Cys residues, an important type of PTM, is a major mechanism of redox regulation in which thiols serve as redox switches (1, 55). Despite the significance, the overall knowledge of site-specific redox modifications on Cys, especially under physiological conditions, is still very limited because of the constraints of measurement technologies. A recent study on thiol-based redox modifications under physiological conditions of Caenorhabditis elegans only identified <200 Cys sites as oxidized using OxICAT (45), illustrating the challenge of identifying redox modification. This work provides a proteome-wide, quantitative, and site-specific analysis of light/dark-modulated changes of thiol oxidation in cyanobacteria covering ∼2,100 Cys residues, significantly expanding the current repertoire of thiol-based redox modifications under physiological conditions.
Our approach adapted an optimized resin-assisted enrichment approach for achieving site-specific quantification of Cys oxidation across multiple conditions. In addition to enabling the relative quantification of oxidation levels, our approach also was easily modified to quantify the stoichiometry or fractional occupancy of oxidation of specific Cys sites, which is particularly important for studying PTMs, as functional PTMs are likely to have higher fractional occupancies of modifications in certain biological conditions. We have shown the advantages of enrichment specificity, proteome coverage, and multiplexed quantification for measuring site-specific redox changes (Fig. 1B) relative to existing methods such as OxiICAT, other biotin-based approaches (24, 33), or anti-TMT antibody-based approaches (29–31). We, and others, have also shown the potential of applying this resin-assisted approach for studying other types of reversible Cys redox modifications (e.g. S-nitrosylation, S-glutathionylation) (18, 33, 42).
Significantly, the advantages of this approach enabled quantitative profiling of Cys oxidation in a physiological light-dark environment with in-depth proteome coverage of the Cys redox modifications in cyanobacteria. The observed landscape of dynamic redox changes of ∼2,100 Cys sites from 1,060 proteins (nearly one-third of the genome) is much broader than one would initially expect from light-dark modulation. However, given the fact that tens of thousands of modification sites have been identified in other PTMs such as phosphorylation (77), ubiquitylation (78), and acetylation (79), we assert that this study likely represents the beginning of the expansion on the potential sites of thiol-based redox modifications. Nevertheless, we recognize that the current approach still has several technical limitations. First, because this approach measures the level of total reversible thiol oxidation, the exact form of oxidation is not known. However, one can adapt our approach to measure exact forms of modification such as S-glutathionylation or S-nitrosylation by coupling selective reduction with resin-assisted enrichment, as recently described (18). Second, our approach does not measure the levels of irreversible oxidation such as S-sulfinic or S-sulfonic acids, which may exist in dark or DCMU conditions. The potential irreversible oxidation could complicate the data interpretation of reversible oxidation; however, the levels of irreversible oxidation under physiological conditions are most likely to be insignificant (based on the results shown in supplemental Figs. S1C and S1D).
It is important to note that the current findings demonstrate a redox rheostat in the cyanobacteria after light-to-dark transition on a global proteome scale—that is, oxidation and reduction of protein thiols do not function as all-or-nothing phenomena. Such dynamic redox changes are actually reflected more by partial activation or inactivation of specific proteins, given that the number of specific proteins reduced/oxidized differs between conditions. This is an interesting observation, as even in an environment so regimented as a light/dark cycle, we clearly still detect specific regulatory sites in both oxidized and reduced forms. The site-specific dynamic data provide important information on redox-sensitive sites for proteins involved in photosynthesis and metabolism (Figs. 3 and 4). Many specific enzymes in the Calvin and Krebs cycles, as well as other biosynthetic pathways, were previously identified as thioredoxin-regulated through disulfide formation (19). Most of these enzymes are mainly in the activated state with reduced thiols under light conditions and become inactivated by disulfide bond formation under dark conditions (1), consistent with our observations.
Similar to other PTM studies, significant challenges remain associated with deciphering which specific proteins and respective Cys sites are most likely to be directly (or indirectly) involved in protein function/regulation from such global proteomic data based upon the redox changes observed in such a relatively straightforward light/dark system. A broad coverage of thiol-based redox modifications is a prerequisite for identifying novel redox-regulated proteins or functional sites; however, the large number of available sites is also a challenge for determining functional redox sensitivity. One unique aspect of this study is that the redox sensitivity data for individual Cys residues provide important predictive information for identifying functional or key regulatory sites for given proteins. The examination of proteins with known functional sites showed good predictability (Fig. 5 and Table I). Moreover, the stoichiometry or fractional occupancy data on individual Cys sites provided additional evidence of functional relevance. Multiple site-mutagenesis and biochemical studies (Figs. 5 and 6) provided a range of redox regulatory scenarios showing proof-of-principle for both confirmation and identification of novel functional sites utilizing the redox data. Therefore, with the redox sensitivity and stoichiometry information on individual sites, this dataset should serve as a unique resource for identifying functional sites for redox-regulated proteins, as well as for novel redox proteins. Follow-up targeted site-specific mutagenesis and pertinent functional studies will help to elucidate the mechanisms of redox regulation and its impact on protein function and specific pathways in response to environmental stimuli.
Supplementary Material
Acknowledgments
Portions of experimental work were performed in the Environmental Molecular Science Laboratory, a DOE/BER national scientific user facility at PNNL in Richland, WA. PNNL is operated by Battelle for the DOE under Contract No. DE-AC05-76RLO-1830.
Footnotes
Author contributions: H.B.P. and W.Q. designed research; J.G., A.Y.N., Z.D., D.S., M.J.G., and R.J.M. performed research; R.D.S. and D.W.K. contributed new reagents or analytic tools; J.G., A.Y.N., J.M.J., M.E.M., and W.Q. analyzed data; J.G., A.Y.N., Z.D., H.B.P., and W.Q. wrote the paper.
* Portions of this work were supported by the DOE Early Career Research Award (to W.J.Q.), DOE Grant No. DE-FG02-99ER20350 (to H.B.P.), the Environmental Molecular Science Laboratory (EMSL) Research Campaign project, and the DOE Office of Biological and Environmental Research Genome Sciences Program under the Pan-omics project. A.N. has been supported by an NSF Graduate Research Fellowship.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PTM
- post-translational modification
- DCMU
- 3-(3,4-dichlorophenyl)-1,1-dimethylurea
- Cys
- cysteine
- PET
- photosynthetic electron transport
- PSII
- photosystem II
- NEM
- N-ethylmaleimide
- TCA
- trichloroacetic acid
- DTT
- dithiothreitol
- TMT
- tandem mass tag
- Gap2 or GAPDH
- NADP+-dependent glyceraldehyde-3-phosphate dehydrogenase
- PetH
- ferredoxin NADP+ reductase
- Zwf or G6PDH
- glucose-6-phosphate 1-dehydrogenase.
REFERENCES
- 1. Buchanan B. B., Balmer Y. (2005) Redox regulation: a broadening horizon. Annu. Rev. Plant Biol. 56, 187–220 [DOI] [PubMed] [Google Scholar]
- 2. Foyer C. H., Noctor G. (2009) Redox regulation in photosynthetic organisms: signaling, acclimation, and practical implications. Antiox. Redox Signal. 11, 861–905 [DOI] [PubMed] [Google Scholar]
- 3. Sato Y., Inaba K. (2012) Disulfide bond formation network in the three biological kingdoms, bacteria, fungi and mammals. FEBS J. 279, 2262–2271 [DOI] [PubMed] [Google Scholar]
- 4. Derakhshan B., Wille P. C., Gross S. S. (2007) Unbiased identification of cysteine S-nitrosylation sites on proteins. Nat. Protoc. 2, 1685–1691 [DOI] [PubMed] [Google Scholar]
- 5. Greco T. M., Hodara R., Parastatidis I., Heijnen H. F., Dennehy M. K., Liebler D. C., Ischiropoulos H. (2006) Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc. Natl. Acad. Sci. U.S.A. 103, 7420–7425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hess D. T., Matsumoto A., Kim S. O., Marshall H. E., Stamler J. S. (2005) Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166 [DOI] [PubMed] [Google Scholar]
- 7. Paulsen C. E., Truong T. H., Garcia F. J., Homann A., Gupta V., Leonard S. E., Carroll K. S. (2012) Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 8, 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lindahl M., Florencio F. J. (2003) Thioredoxin-linked processes in cyanobacteria are as numerous as in chloroplasts, but targets are different. Proc. Natl. Acad. Sci. U.S.A. 100, 16107–16112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Falcon L. I., Magallon S., Castillo A. (2010) Dating the cyanobacterial ancestor of the chloroplast. ISME J. 4, 777–783 [DOI] [PubMed] [Google Scholar]
- 10. Singh R. K., Tiwari S. P., Rai A. K., Mohapatra T. M. (2011) Cyanobacteria: an emerging source for drug discovery. J. Antibiotics 64, 401–412 [DOI] [PubMed] [Google Scholar]
- 11. Singh A. K., Elvitigala T., Cameron J. C., Ghosh B. K., Bhattacharyya-Pakrasi M., Pakrasi H. B. (2010) Integrative analysis of large scale expression profiles reveals core transcriptional response and coordination between multiple cellular processes in a cyanobacterium. BMC Syst. Biol. 4, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nogales J., Gudmundsson S., Knight E. M., Palsson B. O., Thiele I. (2012) Detailing the optimality of photosynthesis in cyanobacteria through systems biology analysis. Proc. Natl. Acad. Sci. U.S.A. 109, 2678–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rosgaard L., de Porcellinis A. J., Jacobsen J. H., Frigaard N. U., Sakuragi Y. (2012) Bioengineering of carbon fixation, biofuels, and biochemicals in cyanobacteria and plants. J. Biotechnol. 162, 134–147 [DOI] [PubMed] [Google Scholar]
- 14. Alfonso M., Perewoska I., Kirilovsky D. (2000) Redox control of psbA gene expression in the cyanobacterium Synechocystis PCC 6803. Involvement of the cytochrome b(6)/f complex. Plant Physiol. 122, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cameron J. C., Pakrasi H. B. (2010) Essential role of glutathione in acclimation to environmental and redox perturbations in the cyanobacterium Synechocystis sp. PCC 6803. Plant Physiol. 154, 1672–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pfannschmidt T., Brautigam K., Wagner R., Dietzel L., Schroter Y., Steiner S., Nykytenko A. (2009) Potential regulation of gene expression in photosynthetic cells by redox and energy state: approaches towards better understanding. Ann. Botany 103, 599–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lopez-Maury L., Sanchez-Riego A. M., Reyes J. C., Florencio F. J. (2009) The glutathione/glutaredoxin system is essential for arsenate reduction in Synechocystis sp. strain PCC 6803. J. Bacteriol. 191, 3534–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo J., Gaffrey M. J., Su D., Liu T., Camp D. G., 2nd, Smith R. D., Qian W. J. (2014) Resin-assisted enrichment of thiols as a general strategy for proteomic profiling of cysteine-based reversible modifications. Nat. Protoc. 9, 64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lindahl M., Kieselbach T. (2009) Disulphide proteomes and interactions with thioredoxin on the track towards understanding redox regulation in chloroplasts and cyanobacteria. J. Proteomics 72, 416–438 [DOI] [PubMed] [Google Scholar]
- 20. Jaffrey S. R., Erdjument-Bromage H., Ferris C. D., Tempst P., Snyder S. H. (2001) Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 3, 193–197 [DOI] [PubMed] [Google Scholar]
- 21. Izquierdo-Alvarez A., Martinez-Ruiz A. (2011) Thiol redox proteomics seen with fluorescent eyes: the detection of cysteine oxidative modifications by fluorescence derivatization and 2-DE. J. Proteomics 75, 329–338 [DOI] [PubMed] [Google Scholar]
- 22. Nelson K. J., Klomsiri C., Codreanu S. G., Soito L., Liebler D. C., Rogers L. C., Daniel L. W., Poole L. B. (2010) Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 473, 95–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leichert L. I., Jakob U. (2004) Protein thiol modifications visualized in vivo. PLoS Biol. 2, e333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leichert L. I., Gehrke F., Gudiseva H. V., Blackwell T., Ilbert M., Walker A. K., Strahler J. R., Andrews P. C., Jakob U. (2008) Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 8197–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sethuraman M., McComb M. E., Heibeck T., Costello C. E., Cohen R. A. (2004) Isotope-coded affinity tag approach to identify and quantify oxidant-sensitive protein thiols. Mol. Cell. Proteomics 3, 273–278 [DOI] [PubMed] [Google Scholar]
- 26. Deng X., Weerapana E., Ulanovskaya O., Sun F., Liang H., Ji Q., Ye Y., Fu Y., Zhou L., Li J., Zhang H., Wang C., Alvarez S., Hicks L. M., Lan L., Wu M., Cravatt B. F., He C. (2013) Proteome-wide quantification and characterization of oxidation-sensitive cysteines in pathogenic bacteria. Cell Host Microbe 13, 358–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weerapana E., Wang C., Simon G. M., Richter F., Khare S., Dillon M. B., Bachovchin D. A., Mowen K., Baker D., Cravatt B. F. (2010) Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sadler N. C., Melnicki M. R., Serres M. H., Merkley E. D., Chrisler W. B., Hill E. A., Romine M. F., Kim S., Zink E. M., Datta S., Smith R. D., Beliaev A. S., Konopka A., Wright A. T. (2013) Live cell chemical profiling of temporal redox dynamics in a photoautotrophic cyanobacterium. ACS Chem. Biol. 9, 291–300 [DOI] [PubMed] [Google Scholar]
- 29. Pan K. T., Chen Y. Y., Pu T. H., Chao Y. S., Yang C. Y., Bomgarden R. D., Rogers J. C., Meng T. C., Khoo K. H. (2014) Mass spectrometry-based quantitative proteomics for dissecting multiplexed redox cysteine modifications in nitric oxide-protected cardiomyocyte under hypoxia. Antiox. Redox. Signal. 20, 1365–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qu Z., Meng F., Bomgarden R. D., Viner R. I., Li J., Rogers J. C., Cheng J., Greenlief C. M., Cui J., Lubahn D. B., Sun G. Y., Gu Z. (2014) Proteomic quantification and site-mapping of S-nitrosylated proteins using isobaric iodoTMT reagents. J. Proteome Res. 13, 3200–3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murray C. I., Uhrigshardt H., O'Meally R. N., Cole R. N., Van Eyk J. E. (2012) Identification and quantification of S-nitrosylation by cysteine reactive tandem mass tag switch assay. Mol. Cell. Proteomics 11, M111.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu T., Qian W. J., Strittmatter E. F., Camp D. G., 2nd, Anderson G. A., Thrall B. D., Smith R. D. (2004) High-throughput comparative proteome analysis using a quantitative cysteinyl-peptide enrichment technology. Anal. Chem. 76, 5345–5353 [DOI] [PubMed] [Google Scholar]
- 33. Forrester M. T., Thompson J. W., Foster M. W., Nogueira L., Moseley M. A., Stamler J. S. (2009) Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 27, 557–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Allen M. M. (1968) Simple conditions for growth of unicellular blue-green algae on plates1, 2. J. Phycol. 4, 1–4 [DOI] [PubMed] [Google Scholar]
- 35. Qian W. J., Kaleta D. T., Petritis B. O., Jiang H., Liu T., Zhang X., Mottaz H. M., Varnum S. M., Camp D. G., 2nd, Huang L., Fang X., Zhang W. W., Smith R. D. (2008) Enhanced detection of low abundance human plasma proteins using a tandem IgY12-SuperMix immunoaffinity separation strategy. Mol. Cell. Proteomics 7, 1963–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kelly R. T., Page J. S., Luo Q., Moore R. J., Orton D. J., Tang K., Smith R. D. (2006) Chemically etched open tubular and monolithic emitters for nanoelectrospray ionization mass spectrometry. Anal. Chem. 78, 7796–7801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim S., Gupta N., Pevzner P. A. (2008) Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome Res. 7, 3354–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qian W. J., Liu T., Monroe M. E., Strittmatter E. F., Jacobs J. M., Kangas L. J., Petritis K., Camp D. G., 2nd, Smith R. D. (2005) Probability-based evaluation of peptide and protein identifications from tandem mass spectrometry and SEQUEST analysis: the human proteome. J. Proteome Res. 4, 53–62 [DOI] [PubMed] [Google Scholar]
- 39. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 40. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A., 3rd, Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 41. Su D., Shukla A. K., Chen B., Kim J. S., Nakayasu E., Qu Y., Aryal U., Weitz K., Clauss T. R., Monroe M. E., Camp D. G., 2nd, Bigelow D. J., Smith R. D., Kulkarni R. N., Qian W. J. (2013) Quantitative site-specific reactivity profiling of S-nitrosylation in mouse skeletal muscle using cysteinyl peptide enrichment coupled with mass spectrometry. Free Radic. Biol. Med. 57, 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Su D., Gaffrey M. J., Guo J., Hatchell K. E., Chu R. K., Clauss T. R., Aldrich J. T., Wu S., Purvine S., Camp D. G., Smith R. D., Thrall B. D., Qian W. J. (2014) Proteomic identification and quantification of S-glutathionylation in mouse macrophages using resin-assisted enrichment and isobaric labeling. Free Radic. Biol. Med. 67, 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thompson A., Schafer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Johnstone R., Mohammed A. K., Hamon C. (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 [DOI] [PubMed] [Google Scholar]
- 44. Held J. M., Danielson S. R., Behring J. B., Atsriku C., Britton D. J., Puckett R. L., Schilling B., Campisi J., Benz C. C., Gibson B. W. (2010) Targeted quantitation of site-specific cysteine oxidation in endogenous proteins using a differential alkylation and multiple reaction monitoring mass spectrometry approach. Mol. Cell. Proteomics 9, 1400–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Knoefler D., Thamsen M., Koniczek M., Niemuth N. J., Diederich A. K., Jakob U. (2012) Quantitative in vivo redox sensors uncover oxidative stress as an early event in life. Mol. Cell 47, 767–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Held J. M., Danielson S. R., Behring J. B., Atsriku C., Britton D. J., Puckett R. L., Schilling B., Campisi J., Benz C. C., Gibson B. W. (2010) Targeted quantitation of site-specific cysteine oxidation in endogenous proteins using a differential alkylation and multiple reaction monitoring mass spectrometry approach. Mol. Cell. Proteomics 9, 1400–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Trebst A. (2007) Inhibitors in the functional dissection of the photosynthetic electron transport system. Photosynth. Res. 92, 217–224 [DOI] [PubMed] [Google Scholar]
- 48. Dai S., Johansson K., Miginiac-Maslow M., Schurmann P., Eklund H. (2004) Structural basis of redox signaling in photosynthesis: structure and function of ferredoxin:thioredoxin reductase and target enzymes. Photosynth. Res. 79, 233–248 [DOI] [PubMed] [Google Scholar]
- 49. Bachi A., Dalle-Donne I., Scaloni A. (2013) Redox proteomics: chemical principles, methodological approaches and biological/biomedical promises. Chem. Rev. 113, 596–698 [DOI] [PubMed] [Google Scholar]
- 50. Stockel J., Jacobs J. M., Elvitigala T. R., Liberton M., Welsh E. A., Polpitiya A. D., Gritsenko M. A., Nicora C. D., Koppenaal D. W., Smith R. D., Pakrasi H. B. (2011) Diurnal rhythms result in significant changes in the cellular protein complement in the cyanobacterium Cyanothece 51142. PLoS One 6, e16680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gill R. T., Katsoulakis E., Schmitt W., Taroncher-Oldenburg G., Misra J., Stephanopoulos G. (2002) Genome-wide dynamic transcriptional profiling of the light-to-dark transition in Synechocystis sp. strain PCC 6803. J. Bacteriol. 184, 3671–3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hihara Y., Sonoike K., Kanehisa M., Ikeuchi M. (2003) DNA microarray analysis of redox-responsive genes in the genome of the cyanobacterium Synechocystis sp. strain PCC 6803. J. Bacteriol. 185, 1719–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mata-Cabana A., Florencio F. J., Lindahl M. (2007) Membrane proteins from the cyanobacterium Synechocystis sp. PCC 6803 interacting with thioredoxin. Proteomics 7, 3953–3963 [DOI] [PubMed] [Google Scholar]
- 54. Perez-Perez M. E., Florencio F. J., Lindahl M. (2006) Selecting thioredoxins for disulphide proteomics: target proteomes of three thioredoxins from the cyanobacterium Synechocystis sp. PCC 6803. Proteomics 6 Suppl 1, S186–S195 [DOI] [PubMed] [Google Scholar]
- 55. Lindahl M., Mata-Cabana A., Kieselbach T. (2011) The disulfide proteome and other reactive cysteine proteomes: analysis and functional significance. Antiox. Redox. Signal. 14, 2581–2642 [DOI] [PubMed] [Google Scholar]
- 56. Rochaix J. D. (2013) Redox regulation of thylakoid protein kinases and photosynthetic gene expression. Antiox. Redox. Signal. 18, 2184–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schurmann P. (2003) Redox signaling in the chloroplast: the ferredoxin/thioredoxin system. Antiox. Redox. Signal. 5, 69–78 [DOI] [PubMed] [Google Scholar]
- 58. Yan C., Xu X. (2008) Bifunctional enzyme FBPase/SBPase is essential for photoautotrophic growth in cyanobacterium Synechocystis sp. PCC 6803. Prog. Nat. Sci. 18, 149–153 [Google Scholar]
- 59. Udvardy J., Godeh M. M., Farkas G. L. (1982) Regulatory properties of a fructose 1,6-bisphosphatase from the cyanobacterium Anacystis nidulans. J. Bacteriol. 151, 203–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Buchanan B. B. (1991) Regulation of CO2 assimilation in oxygenic photosynthesis: the ferredoxin/thioredoxin system. Perspective on its discovery, present status, and future development. Arch. Biochem. Biophys. 288, 1–9 [DOI] [PubMed] [Google Scholar]
- 61. Kobayashi D., Tamoi M., Iwaki T., Shigeoka S., Wadano A. (2003) Molecular characterization and redox regulation of phosphoribulokinase from the cyanobacterium Synechococcus sp. PCC 7942. Plant Cell Physiol. 44, 269–276 [DOI] [PubMed] [Google Scholar]
- 62. Balmer Y., Vensel W. H., Tanaka C. K., Hurkman W. J., Gelhaye E., Rouhier N., Jacquot J. P., Manieri W., Schurmann P., Droux M., Buchanan B. B. (2004) Thioredoxin links redox to the regulation of fundamental processes of plant mitochondria. Proc. Natl. Acad. Sci. U.S.A. 101, 2642–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Koksharova O., Schubert M., Shestakov S., Cerff R. (1998) Genetic and biochemical evidence for distinct key functions of two highly divergent GAPDH genes in catabolic and anabolic carbon flow of the cyanobacterium Synechocystis sp. PCC 6803. Plant Mol. Biol. 36, 183–194 [DOI] [PubMed] [Google Scholar]
- 64. Brandes N., Schmitt S., Jakob U. (2009) Thiol-based redox switches in eukaryotic proteins. Antiox. Redox. Signal. 11, 997–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kitatani T., Nakamura Y., Wada K., Kinoshita T., Tamoi M., Shigeoka S., Tada T. (2006) Structure of NADP-dependent glyceraldehyde-3-phosphate dehydrogenase from Synechococcus PCC7942 complexed with NADP. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62, 315–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. van Thor J. J., Gruters O. W., Matthijs H. C., Hellingwerf K. J. (1999) Localization and function of ferredoxin:NADP(+) reductase bound to the phycobilisomes of Synechocystis. EMBO J. 18, 4128–4136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Talts E., Oja V., Ramma H., Rasulov B., Anijalg A., Laisk A. (2007) Dark inactivation of ferredoxin-NADP reductase and cyclic electron flow under far-red light in sunflower leaves. Photosynth. Res. 94, 109–120 [DOI] [PubMed] [Google Scholar]
- 68. Aliverti A., Piubelli L., Zanetti G., Lubberstedt T., Herrmann R. G., Curti B. (1993) The role of cysteine residues of spinach ferredoxin-NADP+ reductase As assessed by site-directed mutagenesis. Biochemistry 32, 6374–6380 [DOI] [PubMed] [Google Scholar]
- 69. Dietz K. J. (2011) Peroxiredoxins in plants and cyanobacteria. Antiox. Redox. Signal. 15, 1129–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Edgar R. S., Green E. W., Zhao Y., van Ooijen G., Olmedo M., Qin X., Xu Y., Pan M., Valekunja U. K., Feeney K. A., Maywood E. S., Hastings M. H., Baliga N. S., Merrow M., Millar A. J., Johnson C. H., Kyriacou C. P., O'Neill J. S., Reddy A. B. (2012) Peroxiredoxins are conserved markers of circadian rhythms. Nature 485, 459–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kobayashi M., Ishizuka T., Katayama M., Kanehisa M., Bhattacharyya-Pakrasi M., Pakrasi H. B., Ikeuchi M. (2004) Response to oxidative stress involves a novel peroxiredoxin gene in the unicellular cyanobacterium Synechocystis sp. PCC 6803. Plant Cell Physiol. 45, 290–299 [DOI] [PubMed] [Google Scholar]
- 72. Hosoya-Matsuda N., Motohashi K., Yoshimura H., Nozaki A., Inoue K., Ohmori M., Hisabori T. (2005) Anti-oxidative stress system in cyanobacteria. Significance of type II peroxiredoxin and the role of 1-Cys peroxiredoxin in Synechocystis sp. strain PCC 6803. J. Biol. Chem. 280, 840–846 [DOI] [PubMed] [Google Scholar]
- 73. Perez-Perez M. E., Mata-Cabana A., Sanchez-Riego A. M., Lindahl M., Florencio F. J. (2009) A comprehensive analysis of the peroxiredoxin reduction system in the Cyanobacterium Synechocystis sp. strain PCC 6803 reveals that all five peroxiredoxins are thioredoxin dependent. J. Bacteriol. 191, 7477–7489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Nee G., Zaffagnini M., Trost P., Issakidis-Bourguet E. (2009) Redox regulation of chloroplastic glucose-6-phosphate dehydrogenase: a new role for f-type thioredoxin. FEBS Lett. 583, 2827–2832 [DOI] [PubMed] [Google Scholar]
- 75. Newman J., Karakaya H., Scanlan D. J., Mann N. H. (1995) A comparison of gene organization in the zwf region of the genomes of the cyanobacteria Synechococcus sp. PCC 7942 and Anabaena sp. PCC 7120. FEMS Microbiol. Lett. 133, 187–193 [DOI] [PubMed] [Google Scholar]
- 76. Hagen K. D., Meeks J. C. (2001) The unique cyanobacterial protein OpcA is an allosteric effector of glucose-6-phosphate dehydrogenase in Nostoc punctiforme ATCC 29133. J. Biol. Chem. 276, 11477–11486 [DOI] [PubMed] [Google Scholar]
- 77. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 78. Mertins P., Qiao J. W., Patel J., Udeshi N. D., Clauser K. R., Mani D. R., Burgess M. W., Gillette M. A., Jaffe J. D., Carr S. A. (2013) Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 10, 634–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lundby A., Lage K., Weinert B. T., Bekker-Jensen D. B., Secher A., Skovgaard T., Kelstrup C. D., Dmytriyev A., Choudhary C., Lundby C., Olsen J. V. (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tsukamoto Y., Fukushima Y., Hara S., Hisabori T. (2013) Redox control of the activity of phosphoglycerate kinase in Synechocystis sp. PCC6803. Plant Cell Physiol. 54, 484–491 [DOI] [PubMed] [Google Scholar]
- 81. Townley H. E., Sessions R. B., Clarke A. R., Dafforn T. R., Griffiths W. T. (2001) Protochlorophyllide oxidoreductase: a homology model examined by site-directed mutagenesis. Proteins 44, 329–335 [DOI] [PubMed] [Google Scholar]
- 82. Menon B. R., Davison P. A., Hunter C. N., Scrutton N. S., Heyes D. J. (2010) Mutagenesis alters the catalytic mechanism of the light-driven enzyme protochlorophyllide oxidoreductase. J. Biol. Chem. 285, 2113–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sinha S., Dadheech P. K., KrishnaMo M. (2009) Rapid DNA extraction from cyanobacteria. Protocol Online, www.protocol-online.org
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.