

Abstract
Probably every cellular process is governed by protein-protein interaction (PPIs), which are often highly dynamic in nature being modulated by in- or external stimuli. Here we present KISS, for KInase Substrate Sensor, a mammalian two-hybrid approach designed to map intracellular PPIs and some of the dynamic features they exhibit. Benchmarking experiments indicate that in terms of sensitivity and specificity KISS is on par with other binary protein interaction technologies while being complementary with regard to the subset of PPIs it is able to detect. We used KISS to evaluate interactions between different types of proteins, including transmembrane proteins, expressed at their native subcellular location. In situ analysis of endoplasmic reticulum stress-induced clustering of the endoplasmic reticulum stress sensor ERN1 and ligand-dependent β-arrestin recruitment to GPCRs illustrated the method's potential to study functional PPI modulation in complex cellular processes. Exploring its use as a tool for in cell evaluation of pharmacological interference with PPIs, we showed that reported effects of known GPCR antagonists and PPI inhibitors are properly recapitulated. In a three-hybrid setup, KISS was able to map interactions between small molecules and proteins. Taken together, we established KISS as a sensitive approach for in situ analysis of protein interactions and their modulation in a changing cellular context or in response to pharmacological challenges.
A protein's function is largely mediated through its interactions with other proteins, hence the critical importance of protein-protein interaction (PPI)1 maps for understanding cellular mechanisms of action in health and disease. Whereas many proteins are organized in stable multi-protein complexes, the majority of cellular processes are governed by transient protein encounters, the dynamics of which are directed by a diversity of both intra- and extracellular signals. Our view of protein networks is still, however, mainly a static one (1). Current interactomes consist mainly of data generated by yeast 2-hybrid (Y2H) (2) and (tandem) affinity purification combined with mass spectrometry (3) and should be interpreted as scaffolds of potential PPIs that might occur at a certain time and place in the cell or as snapshots of PPIs taking place under a specific cellular condition. Although very robust and highly efficient, these approaches do not allow studying PPI modulation because they do not offer the proper context for mammalian PPI analysis, e.g. they operate in yeast cells (Y2H) or make use of cell lysates (affinity purification-based methods). Moreover, because these interactome mapping tools are biased against interactions that involve transmembrane proteins, the latter are underrepresented in current interactome network versions (4). Yet, membrane-associated proteins constitute around one third of the entire proteome and their significance is underscored by the fact that over half of currently marketed drugs target membrane proteins (5). These observations support the need for approaches that allow PPIs, including those involving transmembrane proteins, to be assayed in their native cellular environment.
Apart from the high-throughput methods mentioned above, a diverse arsenal of other PPI technologies has been developed, a number of which actually operate in mammalian cells. FRET and BRET, which rely on fluorescence or bioluminescence energy transfer between interacting fusion proteins, make assays with high spatiotemporal resolution (6, 7). A variety of PCAs have been reported, including split fluorescent protein or reporter enzyme technologies, that are able to capture aspects of PPI dynamics in a mammalian background (8, 9). A recent addition is an infrared fluorescent PCA that, unlike previous fluorescent PCAs, exhibits reversible complementation, thus enabling spatiotemporal analysis of dynamic PPIs (10). Another binary interaction assay, luminescence-based mammalian interactome mapping (LUMIER), has been applied to map TGFβ induced modulation of PPIs with components of the TGFβ signaling pathway (11). MaMTH, a mammalian version of the split ubiquitin approach, was designed particularly for the analysis of PPIs involving integral membrane proteins, also allowing the detection of functional PPI modulation (12). Efforts to apply purification-based methods for detecting context-dependent PPI modulation recently resulted in the development of AP-SRM (13) and AP-SWATH (14).
Our group previously conceived mammalian protein-protein interaction trap (MAPPIT) (supplemental Fig. S1A) (15, 16), a mammalian two-hybrid approach based on complementation of a cytokine receptor that was developed into a broad platform for PPI analysis (17, 18), screening for small molecule PPI disruptors (19, 20) and drug target profiling (21, 22). Although MAPPIT operates in intact human cells, thus providing the natural environment for human protein analysis, the interaction sensor is anchored to the plasma membrane, precluding the analysis of PPIs at their native subcellular localization. In addition, MAPPIT is incompatible with full size transmembrane proteins. Here we describe KInase Substrate Sensor (KISS), a novel binary PPI mapping approach that enables in situ analysis in living mammalian cells of protein interactions and their responses to physiological or pharmacological challenges.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
Preys were cloned in pMG1 and pMG2 vectors that have been described previously (23). The control prey plasmid expressing unfused gp130 and the MAPPIT pCLL-SKP1 bait vector were described elsewhere (23). KISS bait vectors were cloned by fusing the bait coding sequence of interest with a C-terminal fragment of human TYK2 (AA589–1187) and an HA-tag and inserting this into the pSVSport, pcDNA5, or pMet7 expression vector. Full size open reading frames were used for all bait and prey constructs except for p53 bait (MDM2-binding transactivation domain, AA1–71), BCL2 prey (cytoplasmic domain, AA1–213), HMGCR prey (statin-binding cytoplasmic domain, AA340–888), and ERN1cyt prey (cytoplasmic domain, AA571–977). All open reading frames were from human origin, except reverse transcriptase p66 and p51 (derived from HIV-1) and DHFR (derived from E. coli). ERN1 bait point mutants were generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The hsPRS-v1 and hsRRS-v1 reference sets (17) were transferred to a Gateway-compatible version of the KISS bait vector containing either a C- or an N-terminal TYK2 fusion using Gateway recombinational cloning (Invitrogen, Carlsberg, CA). The resulting C-terminal bait constructs encoded fusion of the following makeup and origin: ORF - PTFLYKVV (attB2 site) - GAA (cloning site) - GGS (hinge) - TYK2(AA589–1187) - HA-tag. For the N-terminal bait fusions, the resulting sequence was: HA-tag - TYK2(AA589–1187) - EL (cloning site) - TSLYKKVG (attB1 site) - ORF - PTFLYKVV (attB2 site). The GPCR bait constructs were also cloned using Gateway cloning and had the same makeup as the C-terminal bait fusions above. All other TYK2 bait fusions were cloned using classical restriction enzyme-based techniques. In the ERN1 bait plasmid, the TYK2 fragment was fused C-terminally, resulting in the following sequence: ERN1 - ASAAA (restriction enzyme site) - GGS (hinge) - TYK2(AA589–1187) - HA-tag. All other bait constructs contained an N-terminal fusion with the TYK2 fragment; the flanking sequence between the TYK2 fragment and the fused ORF encoded EFGSS (in the case of the HIV-1 RT bait plasmids), EF (for the p53 and BAD baits), or EL (for the DHFR bait). BiP was expressed as a genetic fusion with the Myc-tag from a pCMV vector (24) (Addgene plasmid 27164). The actin expression construct was generated in pMet7 by genetically fusing the full size open reading frame with a Flag-tag. The STAT3-dependent firefly luciferase reporter pXP2d2-rPAPI-luciferase has been described before (15).
MFC Synthesis
The chemical synthesis of the methotrexate-FK506 fusion compound has been described previously (21). The methotrexate-simvastatin compound was synthesized similarly, by coupling an acetylene-functionalized simvastatin to a methotrexate-polyethyleneglycol reagent equipped with an azide ligation handle via a copper mediated azide-alkyne cycloaddition reaction (see Supplementary Information).
MAPPIT and KISS Assays
HEK293-T cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, incubated at 37 °C, 8% CO2. In both MAPPIT and KISS assays, cells were transfected with bait, prey, and reporter plasmids applying a standard calcium phosphate transfection method, and luciferase activity was measured 48h after transfection using the Luciferase Assay System kit (Promega, Madison, WI) on a TopCount or Enspire luminometer (Perkin-Elmer, Waltham, MA). For MAPPIT, cells were stimulated with leptin (100 ng/ml) 24h after transfection. In KISS experiments that involved treatment with chemical compounds, agonists, or antagonists, these agents were added to the cells at 24h after transfection, and treatments lasted for 24h unless mentioned otherwise. Nutlin-3 was obtained from Cayman Chemical (Ann Arbor, MI), ABT-737 from Selleckchem (Houston, TX), losartan from Fluka (St. Louis, MO), somatostatin, angiotensin II, telmisartan, CYN154806, and tunicamycin from Sigma (St. Louis, MO). Luciferase data is represented as average relative light units (rlu) ± standard deviation of three technical transfection replicates. In Fig. 3B, 3F, 3G, and 3H, data is normalized by dividing the luciferase rlu obtained for a bait-prey combination by those obtained for the same bait combined with the control prey (unfused gp130). In Fig. 5C and 5D, luciferase activity measured in cells treated with methotrexate fusion compound is normalized against that obtained in untreated cells. Curves were fit using 4-parameter nonlinear regression in GraphPad Prism. In Fig. 2B and 2C, data for LUMIER, MAPPIT, Y2H, PCA, and wNAPPA were taken from Braun et al. (17). As in the case of the methods tested in Braun et al., also for KISS 2 assay configurations were tested (the TYK2 domain being fused either N- or C-terminally to the bait of interest, see Fig. 2A) and the data of both were combined. The ratios of the luciferase signal obtained for a bait-prey combination versus that obtained for the combination of the same bait with a negative control prey (unfused gp130) and versus that obtained for the combination of the same prey with a negative control bait (unfused TYK2 C-terminal fragment) were evaluated against a detection threshold. An interaction pair scored positive when both ratios exceeded that cut-off for either of the two configurations tested. A threshold was chosen that resulted in an optimal percentage of detected PRS versus RRS pairs (maximal detected PRS for minimal detected RRS). For configuration 1 (N-terminal TYK2 fusion) the threshold was 5, for configuration 2 (C-terminal TYK2 fusion) the cut-off was 7.
Fig. 3.

In situ monitoring of PPIs and their modulation in response to external stimuli or a changing physiological context. A, Bait and prey fusion proteins can be either soluble or transmembrane proteins. The combinations shown in the cartoon correspond to the cases illustrated in the text. B, Analysis of interactions between HIV-1 RT p51 and p66 subunits. Luciferase data is normalized for control prey (unfused gp130). For raw luciferase data and Western blot controls see supplemental Fig. S2A. C, D, Dose-dependent KISS induction in cells co-expressing somatostatin receptor 2 (SSTR2; C) or angiotensin receptor 1 (AGTR1; D) bait and beta arrestin 2 (ARRB2) prey and treated with increasing concentrations of somatostatin (SST-14) or angiotensin II (AngII), respectively. E, Subcellular localization of ERN1 bait and prey fusion proteins. The ER-resident protein calnexin is used as an ER marker. ERN1 bait and prey localize to the ER (upper panel), and cluster in that compartment upon tunicamycin treatment (1 μg/ml; lower panel). Scale bars indicate 20 μm. F, Dose-dependent detection of ERN1 oligomerization upon treatment with tunicamycin. G, Analysis of ERN1 mutants in the presence or absence of tunicamycin. Cells expressing ERN1 wild type or point mutant (K599 or D123) baits combined with ERN1 or empty control prey and the luciferase reporter plasmid were treated with tunicamycin (1 μg/ml) or vehicle (DMSO). H, Inhibition of tunicamycin-induced ERN1 clustering by overexpression of BiP. Cells co-expressing ERN1 bait and prey together with BiP or actin were treated with tunicamycin (1 μg/ml) or vehicle (DMSO). Expression controls for Fig. 3F, 3G, and 3H are shown in supplemental Fig. S4C, S4D, and S4E, respectively.
Fig. 5.

KISS three-hybrid analysis of interactions between small molecules and their protein targets. A, Schematic overview of the three-hybrid KISS setup. eDHFR (“D”) is tethered to a C-terminal kinase-domain-containing TYK2 fragment and co-expressed with a prey protein (“Y”) fused to a gp130 cytokine receptor fragment that contains STAT3 docking sites (black dots). When cells are treated with a fusion compound in which a small molecule of interest (asterisk) is linked to methotrexate (MTX) through a polyethyleneglycol linker, MTX binds to eDHFR with very high affinity, resulting in the small molecule being presented as bait. Interaction between the small molecule bait and the protein prey brings TYK2 in the proximity of the gp130 receptor tail, allowing the kinase to phosphorylate the STAT docking sites on the prey chimera (“P”). This leads to the recruitment of STAT3 molecules, which are in turn phosphorylated by the kinase, resulting in their activation. Activated STATs migrate to the nucleus where dimers of this transcription factor activate transcription of a reporter gene. B, KISS analysis of the interaction between the small molecules FK506 and simvastatin with their respective protein targets FKBP12 and HMGCR, respectively. A luciferase signal was generated specifically when eDHFR bait is co-expressed with FKBP12 or HMGCR prey fusions and treated with the appropriate MFC (MTX-FK506 and MTX-simvastatin, respectively; 5 μm). No signal was observed in unstimulated cells or in the presence of unfused gp130 as control prey. Control of bait and prey expression by Western blot is presented in Supplemental Fig. S5. C, D, Dose-dependent detection of the interaction between MTX-derivatized FK506 (C) and simvastatin (D) and their respective targets, FKBP12 and HMGCR. Structure of the MTX fusion compounds are shown below the corresponding curves.
Fig. 2.

Benchmarking KISS against other binary PPI methods. A, The PRS and RRS sets were tested in two complementary configurations with TYK2 fused either N- (configuration 1) or C-terminal (configuration 2) to the bait protein X; the prey fusions always contained the gp130 moiety fused N-terminally to the prey protein. B, KISS assay sensitivity (percentage of PRS positive; yellow bars) and assay specificity (percentage of RRS positive; blue bars) compared with that of orthogonal assays. C, Overview of the individual protein pairs of the PRS (top panel, yellow squares) and the RRS (bottom panel, blue squares) detected by KISS and the orthogonal assays. In B and C data for LUMIER, MAPPIT, Y2H, PCA, and wNAPPA have been extracted from Braun et al. (17). For details on thresholds used in the analysis of the KISS data, see Experimental Procedures.
Immunoprecipitation and Western blot Analysis
Transfected cells were lysed in modified RIPA buffer (200 mm NaCl, 50 mm Tris-HCl, pH 8.0, 0.05% SDS, 2 mm EDTA, 1% Nonidet P-40, 0.5% deoxycholic acid, and Complete protease inhibitor mixture (Roche, Basel, Switzerland)) and lysate supernatants were separated by SDS-PAGE. For immunoprecipitation, lysates were incubated with 1 μg/ml mouse anti-Flag M2 (Sigma) and Protein G Dynabeads (Invitrogen). Proteins were detected by immunoblotting using mouse anti-Flag M2 (Sigma), rabbit anti-Flag (Sigma), mouse anti-phosphotyrosine 4G10 Platinum (Millipore, Darmstadt, Germany), rabbit anti-β-actin (Sigma), rat anti-HA 3F10 (Roche), rabbit anti-gp130 (Santa Cruz, Santa Cruz, CA), rabbit anti-Myc (Upstate, Darmstadt, Germany), and rabbit anti-GAPDH (Abcam, Cambridge, UK) antibodies.
Confocal Microscopy
For confocal imaging, 105 HEK293-T cells/well (in 6-well plate) were seeded on glass coverslips (Zeiss, Oberkochen, Germany), coated with poly-l-lysine (Sigma). The next day, cells were transfected using a classic calcium phosphate protocol. After 48 h, cells were left untreated or treated for 3 h with vehicle (DMSO) or tunicamycin (1 μg/ml, Sigma). Next, cells were rinsed with 1× PBS and fixed for 15 min at room temperature in 4% paraformaldehyde. After three washes with 1× PBS, cells were permeabilized with 0.1% Triton X-100 in 1× PBS for 10 min and blocked in 1% BSA in 1× PBS for another 10 min at room temperature. Samples were then incubated for 1 h at 37 °C with primary antibody. After four washes in 1× PBS, cells were incubated for 1 h at room temperature with Alexa fluorochrome-conjugated secondary antibodies. The following antibodies were used: rat anti-HA, rabbit anti-gp130, goat anti-calnexin, anti-rat Alexa Fluor 488, anti-rabbit Alexa Fluor 568, and anti-goat Alexa Fluor 568. After secondary antibody incubation, cells were washed four times in 1x PBS and nuclei were stained with DAPI (2 μg/ml). After a final wash step in 1xPBS, coverslips were mounted using propyl gallate. Images were acquired using a 60 × 1.35 NA objective on an Olympus IX-81 laser scanning confocal microscope and analyzed using Fluoview 1000 software.
RESULTS
Origin of the KISS Approach
The origin of the KISS two-hybrid concept lays in the recurrent artifactual identification of TYK2-containing prey fusion proteins in MAPPIT cDNA library screens with unrelated bait proteins (supplemental Fig. S1B). In these preys, the gp130 cytokine receptor fragment that relays the MAPPIT signal was fused to a C-terminal portion of TYK2, invariably containing the intact kinase domain (supplemental Fig. S1C). Cells expressing such a prey exhibited a cytokine-independent MAPPIT reporter signal (Supplemental Fig. S1D), indicating that the TYK2 kinase domain in these fusion proteins is able to phosphorylate both the STAT3 docking sites of the gp130 chain and STAT3. We reasoned that if phosphorylation of these substrates would also occur in trans, this would yield a novel two-hybrid approach. Because it relies on complementation of a kinase and its substrate, the method was termed KISS, for KInase Substrate Sensor. In KISS, a fusion of the bait protein with the C-terminal (kinase domain containing) portion of TYK2 is co-expressed with a fusion between the prey protein and a gp130 cytokine receptor fragment containing TYK2 substrate motifs. Upon interaction between bait and prey, TYK2 comes into proximity of the gp130 receptor fragment thereby allowing tyrosine phosphorylation of the substrate motifs (supplemental Fig. S2B), enabling them to recruit STAT3 and to induce transactivation of a reporter gene (Fig. 1). Importantly, and in contrast to MAPPIT, both bait and prey can be either soluble or transmembrane proteins, enabling analysis of PPIs between or among both protein classes.
Fig. 1.
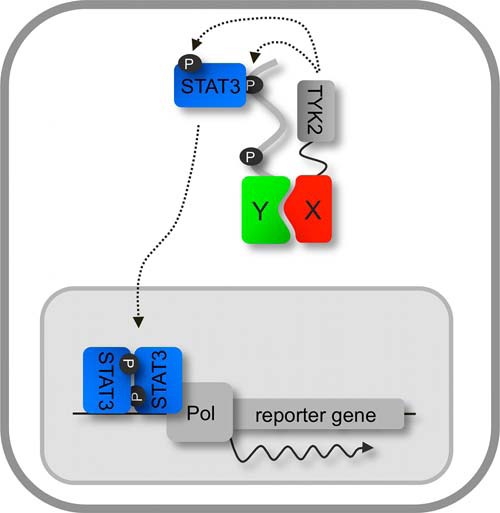
KISS concept. A bait protein (X) is fused to a kinase-containing portion of TYK2 and a prey (Y) is coupled to a gp130 cytokine receptor fragment. When bait and prey interact, TYK2 phosphorylates STAT3 docking sites on the prey chimera (P), which ultimately leads to activation of a reporter gene (see text for more details).
Benchmarking KISS Against Other Binary PPI Methods
We first set out for an unbiased assessment of assay sensitivity and specificity by benchmarking KISS against MAPPIT and other binary protein interaction assays through testing previously established human positive (PRS) and random (RRS) PPI reference sets. In an effort to establish a robust PPI toolkit, Braun et al. compiled a PRS and RRS set consisting of 94 well-documented pairs of interacting proteins and randomly chosen protein pairs, respectively (17). These reference sets were tested with five unrelated binary PPI mapping approaches, including MAPPIT, LUMIER, Y2H, a YFP-based PCA, and a variant of the nucleic acid programmable protein array (wNAPPA). Each of these methods was found to detect between 20 and 35% of the PRS, with 0 to 4% false positive interactions being detected in the RRS (17). To enable an accurate comparison with these data, which shows the combined result of testing two configurations of each of the assays, the reference sets were tested in two complementary configurations of KISS, the TYK2 moiety being fused N- or C-terminally to the bait (Fig. 2A). For each of the configurations, a threshold was set that maximized detection of PRS pairs while keeping the number of false positives from the RRS to a minimum. A protein pair was scored positive when it surpassed that threshold in either of the two configurations. The results shown in Fig. 2B indicate that the performance of KISS in terms of assay sensitivity (percentage of PRS positive) and assay specificity (percentage of RRS positive) is comparable with that of MAPPIT and the other assays documented in Braun et al., with KISS detecting 34% of the PRS and 1% of the RRS. When inspected at the level of the individual protein pairs, a partial overlap was observed with pairs detected by the other technologies (Fig. 2C).
In Situ Monitoring of PPIs and their Modulation in Response to External Stimuli or a Changing Physiological Context
Having validated overall assay performance, we next evaluated the use of KISS as an in situ sensor of physiological PPIs with proteins exhibiting different topological characteristics and occurring in different subcellular compartments (Fig. 3A). We first applied KISS for the detection of interactions between the cytoplasmic p51 and p66 subunits of the HIV-1 RT. Although the p51/p66 heterodimer corresponds to the biologically active configuration, homodimers can form as well. The binding affinity has been reported to vary significantly between dimers, ranging from submicromolar affinity for the heterodimer (Kd = 0.3 μm) to weak binding of the subunits in the p51 homodimer (Kd = 230 μm) (25). For each of the RT bait-prey combinations strong signals were obtained, whereas background signals were consistently low (supplemental Fig. S2A), yielding normalized induction windows of up to more than 300-fold (Fig. 3B). It is of note that although the reported Kd for the p66 homodimer (4.2 μm) is about a magnitude higher than that of the heterodimer, both PPIs exhibit a similar KISS signal. This observation suggests that saturation might be at play, e.g. at the level of endogenous STAT3 signaling or luciferase reporter enzyme buildup.
Next, we applied KISS to analyze PPIs involving GPCRs, using the interaction of somatostatin receptor 2 (SSTR2) or angiotensin receptor 1 (AGTR1) with beta arrestin 2 (ARRB2) as a model (26). A luciferase reporter signal was induced upon addition of the cognate ligand (supplemental Fig. S3), indicating that KISS can detect the ligand-dependent interaction between GPCRs and beta arrestin 2. The fact that this signal was induced using physiological GPCR agonists that do not penetrate the cell membrane suggests that the GPCR-TYK2 fusion protein is properly expressed at the plasma membrane. This also implies that the fusion does not significantly interfere with the receptor's ability to bind its ligand and to translate this binding event into intracellular signaling. Upon treatment with increasing concentrations of the ligand, a dose-dependent KISS signal was observed, which further supports the functional relevance of this experimental setup (Fig. 3C and 3D).
In a third case, we explored the utility of KISS to analyze binding among transmembrane proteins. Again, we took advantage of the fact that the method operates in intact cells, thus allowing the assessment of PPI modulation during complex physiological processes. We used KISS to evaluate ER stress-induced oligomerization of the transmembrane ER stress sensor ERN1 (alias IRE1α) (27). Accumulation of misfolded proteins in the ER results in stress induction, triggering an evolutionary conserved adaptive response named the unfolded protein respons (UPR) (28). According to the current model, human ERN1 is activated by oligomerization, which is prevented in unstressed cells by association of its luminal portion with the ER chaperone BiP (29). Misfolded proteins that accumulate in the ER titer BiP away from ERN1, thereby releasing the brake on ERN1 activation (30) (supplemental Fig. S4A). In KISS, full-length ERN1 bait or prey fusions localized to the ER (Fig. 3E, upper panel). In addition, treatment of the cells with tunicamycin, which induces ER stress by blocking N-linked glycosylation, promoted ERN1 bait-prey clustering in the ER (Fig. 3E, lower panel). As shown in Fig. 3F, KISS can be used to reveal dose-dependent ERN1 oligomerization in tunicamycin-treated cells. In accordance with the requirement of the ERN1 luminal domain to sense ER stress, no KISS signal was observed when the full length ERN1 bait was combined with a prey containing only the cytoplasmic portion of ERN1 (Fig. 3F), which loses its typical ER localization (supplemental Fig. S4B). As expected, ERN1 KISS baits carrying point mutations in either the luminal domain (D123P) or cytoplasmic ATP-binding pocket (K599A) that are expected to prevent ERN1 oligomerization and activation (30, 31) failed to generate a detectable KISS signal (Fig. 3G). Interestingly, KISS recapitulated BiP-mediated regulation of ERN1, as overexpression of BiP, but not of an irrelevant protein (actin), inhibited the KISS signal in a dose-dependent manner (Fig. 3H).
Analysis of Pharmacological Interference with PPIs
Because the above findings indicate that KISS assays can truthfully reproduce physiological behavior of PPIs, we sought to explore whether these assays can also be used to evaluate the effect of pharmacological interference with PPIs. We evaluated two prototypical examples of PPI disruptors, Nutlin-3 and ABT-737. By occupying the p53-binding pocket of MDM2, Nutlin-3 interferes with the interaction between p53 and MDM2, a well-validated oncology target (32). Dose-response analysis in a KISS assay consisting of p53 bait and MDM2 prey showed that this compound inhibited the signal with an IC50 (0.57 μm) close to that reported for in vitro binding experiments (0.1 μm (32); Fig. 4A). Consistent with the fact that Nutlin-3 specifically binds to MDM2 and not to MDM4, the compound inhibited the p53-MDM4 assay only at toxic doses. Similarly, no specific inhibition was observed of the background signal obtained for the p53 bait combined with a control prey.
Fig. 4.

Analysis of pharmacological interference with PPIs. A, B, Activity profile of the PPI inhibitors Nutlin-3 (A) and ABT-737 (B). Nutlin-3 and ABT-737 selectively disrupt the interaction between p53 and MDM2 (IC50 = 0.57 μm) or BAD and BCL2 (IC50 = 0.14 μm), respectively. MDM4 prey or unfused gp130 (ctrl) were used as control preys. C, D, Dose-dependent inhibition of the GPCR-beta arrestin 2 interaction by GPCR antagonists in cells expressing somatostatin receptor 1-beta arrestin 2 or angiotensin receptor 1-beta arrestin combinations as in Fig. 3C and 3D, and stimulated with the appropriate ligand (SST-14 or AngII; 10 μm).
The second compound, ABT-737, reportedly targets the interface between pro- and anti-apoptotic BCL2-family members, this way restoring the apoptotic capacity of cancer cells. In a KISS assay co-expressing BAD bait and BCL2 prey, ABT-737 exhibited a dose-dependent inhibition of the signal (Fig. 4B). The observed IC50 (0.14 μm) is in the range of what has previously been reported (33). The specificity of this inhibition was indicated by the absence of an effect on the signal for the BAD bait with a control prey.
In another example, KISS was used to evaluate indirect pharmacological effects on PPIs. Here, we applied the GPCR-beta arrestin 2 assays described above to analyze the effect of GPCR antagonists on GPCR activation as measured through their interaction with beta arrestin 2. Treatments with the ligand and antagonists revealed that the KISS luciferase signal was specifically inhibited by the proper antagonist(s), either the somatostatin receptor 2 peptide inhibitor CYN154806 (Fig. 4C) or the angiotensin receptor 1-selective small molecule inhibitors losartan and telmisartan (Fig. 4D). In the case of angiotensin receptor 1, the IC50 of telmisartan (0.6 nm) is markedly lower than that of losartan (15 nm), which is in line with previous observations (34).
KISS Three-hybrid Analysis of Interactions Between Small Molecules and their Protein Targets
In addition to the analysis of PPIs and their modulation by exogenous factors, KISS also enables the detection of physical interactions between small molecules and proteins. In the three-hybrid KISS setup, small molecules of interest are chemically tethered to MTX and recruited to an E. coli DHFR (eDHFR)-TYK2 fusion protein by virtue of the picomolar affinity between MTX and eDHFR (Fig. 5A). This way, a small molecule is displayed as bait inside the cell, and interactions with target protein prey fusions are detected through reporter induction as in the case of the KISS PPI assay. The concept was applied to the detection of interactions between the drugs FK506 and simvastatin and their respective protein targets. FK506 (or tacrolimus) is an immunosuppressant with clinical applications in organ transplantation. Through binding to its cellular target FKBP12, it inhibits the activity of the phosphatase calcineurin, which results in inhibition of T-cell activation and IL-2 production (35). Simvastatin is a member of the statin class of cholesterol-lowering drugs that act through binding to and inhibiting the activity of HMGCR, the rate-limiting enzyme in the cholesterol biosynthetic pathway (36). FK506 and simvastatin were coupled to MTX through a hexaethyleneglycol linker that served to reduce potential steric hindrance of binding to eDHFR and the drug target at either end of the chemical dimerizer (21) (Fig. 5C and 5D). MTX derivatives of FK506 and simvastatin induced a robust and fusion compound-specific KISS signal in cells co-expressing the eDHFR-TYK2 fusion protein together with the FKBP12 or HMGCR preys, respectively (Fig. 5B). A concentration gradient of the MTX fusion compounds induced KISS in a dose-dependent fashion, yielding a bell-shaped response curve in which the signal dropped at high concentrations because of saturation of the chemical dimerizer binding sites (the “hook effect”; Fig. 5C and 5D).
DISCUSSION
Driven by the critical importance of protein interaction information for the annotation of protein function, protein interactomics is a rapidly evolving field, both in terms of the amount of protein network data being published and with respect to the tools being developed to build these networks. A range of conceptually divergent approaches is currently available that differ in many aspects of the type of data they produce and the proteome subset they cover (8). In the quest for a more complete coverage of the interactome space, the value of such a diversified toolbox is underscored by the observation that different techniques detect different PPI subsets (17). Our data are in line with this finding, as KISS detected a subset of the tested PRS that overlaps only partially with that identified using any of the other methods compared by Braun et al. (17) (Fig. 2C). Even the overlap with the PPIs seen by the related MAPPIT approach is limited, which is reminiscent of reports on similar benchmarking studies for Y2H where different yeast strains or assay variants applied to the same reference sets yielded different results (17, 37). Remarkably, a significant portion of the PRS remains undetected by any of the methods. Possible underlying reasons have been previously investigated, identifying a combination of potential factors contributing to detection failure, including steric hindrance caused by the protein fusion, the use of other protein isoforms in the original reports describing the PPIs used in the PRS, and the fact that part of the PPIs involve extracellular proteins, which are not compatible with these assays (17).
Apart from the complementarity of KISS in terms of which PPIs it can detect, the benchmarking experiments also confirmed the overall sensitivity and specificity of the technique, which is similar to that of the best assays in the panel compared by Braun et al. (17) (Fig. 2B). For individual cases we have evidence that the technique exhibits exceptional sensitivity, for example, we obtained a robust KISS signal (64-fold induction, Fig. 3B) for the low affinity interaction between HIV-1 RT p51 subunits (Kd = 230 μm (25)), which cannot be detected in MAPPIT (38). We further validated another important feature of KISS, i.e. its compatibility with transmembrane proteins, a protein class difficult to address with many other PPI mapping approaches, including MAPPIT (4). The successful detection of interactions with GPCRs and ERN1, which take place at different submembranal compartments of the cell, illustrates the method's versatility toward protein topology and its ability for in situ detection of PPIs.
PPI networks are not static scaffolds but highly dynamic systems that embody a cell's potential to rapidly respond to environmental changes and challenges. Therefore, if one wants to understand the mechanisms underlying these cellular processes, it is crucial to gain insight into the dynamic aspects of the PPIs they involve. KISS operates in intact mammalian cells, which not only ensures proper folding and posttranslational modification of the tested proteins, but also enables monitoring the modulation of mammalian PPIs in their native environment. We show that the method can assess the effect on PPIs of external stimuli added to induce or mimic particular cellular processes. In particular, the example of the ER stress-induced ERN1 oligomerization nicely illustrates this potential. Even though, as any two-hybrid approach, KISS requires forced expression of fusion proteins, these proteins behave as their endogenous counterparts, both in terms of function (the ERN1 fusion proteins cluster upon induction of ER stress conditions, Fig. 3E and 3F) and localization (the fusions localize correctly to the ER, Fig. 3E). The BiP titration experiment (Fig. 3H) and the ERN1 mutant analysis (Fig. 3G) further illustrate the flexibility and utility of such live cell in situ sensor.
A limitation of the technology is the fact that the readout is indirect, precluding spatial or temporal analysis of PPIs, a feature that is provided by for example FRET and a number of the reported PCAs. The extent to which PPI modulation can be tracked using KISS is also limited by the use of luciferase as a reporter, which has a cellular half-life of around 3 h, precluding its use for kinetic measurements. Thus, the ability of the technology to analyze PPI dynamics is at the level of functional rather than temporal PPI modulation. The upside of the indirect readout, however, is that there is amplification at different levels of the signal generation cascade (substrate and STAT phosphorylation, reporter gene transcription, and luciferin substrate conversion), contributing to the method's sensitivity and robustness.
Because the KISS readout relies on endogenous STAT3, another limitation of the assay is that it is not suitable for studying interactions involving proteins or stimuli that modulate STAT3 signaling. For instance, several baits and preys from the PRS/RRS data set produced a background reporter signal in the absence of the interaction partner, potentially obscuring a PPI-dependent KISS signal. Also, we were unsuccessful in demonstrating ligand-induced association of the beta-2 adrenergic receptor with beta 2 arrestin because ligand triggering of this receptor activates STAT3 (data not shown). Nevertheless, including the proper controls allows for easy detection of false positives that occur independent of bait-prey interaction.
The importance of mapping PPIs and studying their functional modulation extends beyond fundamental biology. Therapeutic targeting of PPIs offers an attractive alternative to classical drug targets in cases where the target of interest does not exhibit any enzymatic activity or ligand binding site that can be easily targeted and because of the large number of potential PPI targets (39). Indeed PPIs represent an emerging class of drug targets and there is a growing interest in technologies that allow screening for small molecule disruptors of PPIs. Cellular assays such as KISS offer the advantage of supporting hit identification in the physiological context future drugs will operate in. Our data show that KISS can be used to evaluate both direct PPI inhibitors (e.g. Nutlin-3 and ABT737) and indirect pharmacological interference with PPIs (as in the case of the GPCR antagonists). Importantly, the IC50 values obtained for the different small molecules correspond well with prior reports, supporting the practical utility of the assay for example, with regard to in cell compound prioritization. Efforts are ongoing to turn the KISS assay into a high-throughput screening tool using previously established MAPPIT protocols (20).
Drug target profiling is a crucial element of any small molecule drug development program. Mapping the proteins a potential drug physically interacts with can shed light on that molecule's mechanism of action or its unwanted side effects. The former has become increasingly important in view of the recent tendency in the pharmaceutical industry to switch from target-based drug discovery to phenotypic screening where prior knowledge of the target is not a prerequisite. Although various approaches have been developed, target identification remains challenging (40, 41). In analogy to the three-hybrid assays that have been successfully applied to identify drug targets in yeast (42), we developed a three-hybrid variant of KISS that enables drug-target analysis in living mammalian cells. In this application again, the context of an intact mammalian (human) cell is extremely valuable, as it represents a physiologically relevant environment for the potential drug. We recently established an array-based screening platform for MASPIT (21), the three-hybrid MAPPIT setup, which we are currently adopting in KISS for high-throughput small molecule profiling. Because prey configuration in KISS is the same as in MAPPIT, the prey collections that have been compiled for MAPPIT and MASPIT screens (currently consisting of close to 15,000 full size ORFs) can be applied as such in KISS. Compared with MASPIT, the KISS three-hybrid approach will allow us to screen for integral membrane target proteins as well. Additionally, it will be interesting to expand the scope of the KISS three-hybrid assay to the detection of interactions with cellular metabolites including lipids or sugars.
In summary, we validated KISS as a sensitive and versatile sensor with a broad application range toward the detection of interactions among proteins and between small molecules and proteins and showed that the approach enables analyzing PPI modulation by small molecules and other exogenous stimuli in a physiological context. Taken together with the observed complementarity to existing technologies regarding the detected subset of PPIs, we consider KISS a valuable addition to the ever expanding PPI toolbox.
Supplementary Material
Acknowledgments
We thank Dr. R. Prywes for sharing the BiP expression plasmid.
Footnotes
Author contributions: S.L., S.G., I.L., D.J.D., M.D.R., S.V., and J.T. designed research; S.L., S.G., I.L., D.J.D., M.D.R., N.V., A.D., and E.R. performed research; S.L., D.J.D., M.D.R., and S.V. contributed new reagents or analytic tools; S.L., S.G., I.L., D.J.D., M.D.R., E.C., S.V., and J.T. analyzed data; S.L., S.G., I.L., D.J.D., M.D.R., E.C., S.V., and J.T. wrote the paper.
* This work was supported by grants from the Belgian government (Interuniversity Attraction Poles Project P6/36), the Fund for Scientific Research - Flanders (FWO-V Project G.0864.10), the GROUP-ID Multidisciplinary Research Partnership of Ghent University and the NIH (U01 Grant HG001715). S.G. and I.L. are postdoctoral fellows with the Fund for Scientific Research - Flanders (FWO-V). J.T. is the recipient of an ERC Advanced Grant (# 340941).
 This article contains Supplemental Figs. S1 to S5.
This article contains Supplemental Figs. S1 to S5.
1 The abbreviations used are:
- AGTR1
- angiotensin receptor 1
- AP-SRM
- affinity purification-selected reaction monitoring
- AP-SWATH
- affinity purification combined with sequential window acquisition of all theoretical spectra
- ARRB2
- beta arrestin 2
- ATP
- adenosine triphosphate
- BAD
- BCL2-associated agonist of cell death
- BCL2
- B-Cell CLL/Lymphoma 2
- BiP
- heat shock 70kDa protein 5
- BRET
- bioluminescence resonance energy transfer
- DAPI
- 4′,6-diamidino-2-phenylindole
- DHFR
- dihydrofolate reductase
- ER
- endoplasmic reticulum
- ERN1
- endoplasmic reticulum to nucleus signaling 1
- FKBP12
- FK506 binding protein
- FRET
- fluorescence resonance energy transfer
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- gp130
- membrane glycoprotein 130
- GPCR
- G-protein coupled receptor
- HIV-1
- human immunodeficiency virus 1
- HMGCR
- 3-hydroxy-3-methylglutaryl-CoA reductase
- IL-2
- interleukin 2
- IRE1α
- inositol-requiring enzyme 1α
- KISS
- kinase substrate sensor
- LEDGF
- lens epithelium-derived growth factor
- LUMIER
- luminescence-based mammalian interactome mapping
- MaMTH
- mammalian-membrane two-hybrid assay
- MAPPIT
- mammalian protein-protein interaction trap
- MDM2
- human homolog of mouse double minute 2
- MDM4
- human homolog of mouse double minute 4
- MTX
- methotrexate
- Myc
- avian myelocytomatosis viral oncogene homolog
- p53
- tumor protein p53
- PCA
- protein fragment complementation assay
- PPI
- protein protein interaction
- PRS
- positive reference set
- RIPA
- radioimmunoprecipitation assay
- rPAPI
- rat pancreatitis associated protein I
- RRS
- random reference set
- RT
- reverse transcriptase
- SKP1
- S-phase kinase-associated protein 1
- SSTR2
- somatostatin receptor 2
- STAT3
- signal transducer and activator of transcription 3
- TGFβ
- transforming growth factor
- TYK2
- tyrosine kinase 2
- UPR
- unfolded protein response
- wNAPPA
- nucleic acid programmable protein array
- Y2H
- yeast two-hybrid
- YFP
- yellow fluorescent protein.
REFERENCES
- 1. Ideker T., Krogan N. J. (2012) Differential network biology. Mol. Syst. Biol. 8, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hamdi A., Colas P. (2012) Yeast two-hybrid methods and their applications in drug discovery. Trends Pharmacol. Sci. 33, 109–118 [DOI] [PubMed] [Google Scholar]
- 3. Gavin A. C., Maeda K., Kuhner S. (2011) Recent advances in charting protein-protein interaction: mass spectrometry-based approaches. Curr. Opin. Biotechnol. 22, 42–49 [DOI] [PubMed] [Google Scholar]
- 4. Lam M. H., Stagljar I. (2012) Strategies for membrane interaction proteomics: no mass spectrometry required. Proteomics 12, 1519–1526 [DOI] [PubMed] [Google Scholar]
- 5. Overington J. P., Al-Lazikani B., Hopkins A. L. (2006) How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996 [DOI] [PubMed] [Google Scholar]
- 6. Piston D. W., Kremers G. J. (2007) Fluorescent protein FRET: the good, the bad, and the ugly. Trends Biochem. Sci. 32, 407–414 [DOI] [PubMed] [Google Scholar]
- 7. Pfleger K. D., Eidne K. A. (2006) Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 3, 165–174 [DOI] [PubMed] [Google Scholar]
- 8. Stynen B., Tournu H., Tavernier J., Van Dijck P. (2012) Diversity in genetic in vivo methods for protein-protein interaction studies: from the yeast two-hybrid system to the mammalian split-luciferase system. Microbiol. Mol. Biol. Rev. 76, 331–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Michnick S. W., Ear P. H., Manderson E. N., Remy I., Stefan E. (2007) Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat. Rev. Drug Discov. 6, 569–582 [DOI] [PubMed] [Google Scholar]
- 10. Tchekanda E., Sivanesan D., Michnick S. W. (2014) An infrared reporter to detect spatiotemporal dynamics of protein-protein interactions. Nat. Methods 11, 641–644 [DOI] [PubMed] [Google Scholar]
- 11. Barrios-Rodiles M., Brown K. R., Ozdamar B., Bose R., Liu Z., Donovan R. S., Shinjo F., Liu Y., Dembowy J., Taylor I. W., Luga V., Przulj N., Robinson M., Suzuki H., Hayashizaki Y., Jurisica I., Wrana J. L. (2005) High-throughput mapping of a dynamic signaling network in mammalian cells. Science 307, 1621–1625 [DOI] [PubMed] [Google Scholar]
- 12. Petschnigg J., Groisman B., Kotlyar M., Taipale M., Zheng Y., Kurat C. F., Sayad A., Sierra J. R., Mattiazzi Usaj M., Snider J., Nachman A., Krykbaeva I., Tsao M. S., Moffat J., Pawson T., Lindquist S., Jurisica I., Stagljar I. (2014) The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat. Methods 11, 585–592 [DOI] [PubMed] [Google Scholar]
- 13. Bisson N., James D. A., Ivosev G., Tate S. A., Bonner R., Taylor L., Pawson T. (2011) Selected reaction monitoring mass spectrometry reveals the dynamics of signaling through the GRB2 adaptor. Nat. Biotechnol. 29, 653–658 [DOI] [PubMed] [Google Scholar]
- 14. Collins B. C., Gillet L. C., Rosenberger G., Rost H. L., Vichalkovski A., Gstaiger M., Aebersold R. (2013) Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14–3-3 system. Nat. Methods 10, 1246–1253 [DOI] [PubMed] [Google Scholar]
- 15. Eyckerman S., Verhee A., Van der Heyden J. V., Lemmens I., Van Ostade X. V., Vandekerckhove J., Tavernier J. (2001) Design and application of a cytokine-receptor-based interaction trap. Nat. Cell Biol. 3, 1114–1119 [DOI] [PubMed] [Google Scholar]
- 16. Lievens S., Peelman F., De Bosscher K., Lemmens I., Tavernier J. (2011) MAPPIT: a protein interaction toolbox built on insights in cytokine receptor signaling. Cytokine Growth Factor Rev. 22, 321–329 [DOI] [PubMed] [Google Scholar]
- 17. Braun P., Tasan M., Dreze M., Barrios-Rodiles M., Lemmens I., Yu H., Sahalie J. M., Murray R. R., Roncari L., de Smet A. S., Venkatesan K., Rual J. F., Vandenhaute J., Cusick M. E., Pawson T., Hill D. E., Tavernier J., Wrana J. L., Roth F. P., Vidal M. (2009) An experimentally derived confidence score for binary protein-protein interactions. Nat. Methods 6, 91–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Simicek M., Lievens S., Laga M., Guzenko D., Aushev V. N., Kalev P., Baietti M. F., Strelkov S. V., Gevaert K., Tavernier J., Sablina A. A. (2013) The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response. Nat. Cell Biol. 15, 1220–1230 [DOI] [PubMed] [Google Scholar]
- 19. Eyckerman S., Lemmens I., Catteeuw D., Verhee A., Vandekerckhove J., Lievens S., Tavernier J. (2005) Reverse MAPPIT: screening for protein-protein interaction modifiers in mammalian cells. Nat. Methods 2, 427–433 [DOI] [PubMed] [Google Scholar]
- 20. Lievens S., Caligiuri M., Kley N., Tavernier J. (2012) The use of mammalian two-hybrid technologies for high-throughput drug screening. Methods 58, 335–342 [DOI] [PubMed] [Google Scholar]
- 21. Risseeuw M. D., De Clercq D. J., Lievens S., Hillaert U., Sinnaeve D., Van den Broeck F., Martins J. C., Tavernier J., Van Calenbergh S. (2013) A “clickable” MTX reagent as a practical tool for profiling small-molecule-intracellular target interactions via MASPIT. Chem. Med. Chem. 8, 521–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Caligiuri M., Molz L., Liu Q., Kaplan F., Xu J. P., Majeti J. Z., Ramos-Kelsey R., Murthi K., Lievens S., Tavernier J., Kley N. (2006) MASPIT: three-hybrid trap for quantitative proteome fingerprinting of small molecule-protein interactions in mammalian cells. Chem. Biol. 13, 711–722 [DOI] [PubMed] [Google Scholar]
- 23. Lievens S., Vanderroost N., Van der Heyden J., Gesellchen V., Vidal M., Tavernier J. (2009) Array MAPPIT: high-throughput interactome analysis in mammalian cells. J. Proteome Res. 8, 877–886 [DOI] [PubMed] [Google Scholar]
- 24. Shen J., Chen X., Hendershot L., Prywes R. (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111 [DOI] [PubMed] [Google Scholar]
- 25. Venezia C. F., Howard K. J., Ignatov M. E., Holladay L. A., Barkley M. D. (2006) Effects of efavirenz binding on the subunit equilibria of HIV-1 reverse transcriptase. Biochemistry 45, 2779–2789 [DOI] [PubMed] [Google Scholar]
- 26. Luttrell L. M., Gesty-Palmer D. (2010) Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol. Rev. 62, 305–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tirasophon W., Welihinda A. A., Kaufman R. J. (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jaeger P. A., Doherty C., Ideker T. (2012) Modeling transcriptome dynamics in a complex world. Cell 151, 1161–1162 [DOI] [PubMed] [Google Scholar]
- 29. Bertolotti A., Zhang Y., Hendershot L. M., Harding H. P., Ron D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332 [DOI] [PubMed] [Google Scholar]
- 30. Korennykh A., Walter P. (2012) Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 28, 251–277 [DOI] [PubMed] [Google Scholar]
- 31. Zhou J., Liu C. Y., Back S. H., Clark R. L., Peisach D., Xu Z., Kaufman R. J. (2006) The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. U. S. A. 103, 14343–14348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vassilev L. T., Vu B. T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., Liu E. A. (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 [DOI] [PubMed] [Google Scholar]
- 33. Oltersdorf T., Elmore S. W., Shoemaker A. R., Armstrong R. C., Augeri D. J., Belli B. A., Bruncko M., Deckwerth T. L., Dinges J., Hajduk P. J., Joseph M. K., Kitada S., Korsmeyer S. J., Kunzer A. R., Letai A., Li C., Mitten M. J., Nettesheim D. G., Ng S., Nimmer P. M., O'Connor J. M., Oleksijew A., Petros A. M., Reed J. C., Shen W., Tahir S. K., Thompson C. B., Tomaselli K. J., Wang B., Wendt M. D., Zhang H., Fesik S. W., Rosenberg S. H. (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 [DOI] [PubMed] [Google Scholar]
- 34. Kakuta H., Sudoh K., Sasamata M., Yamagishi S. (2005) Telmisartan has the strongest binding affinity to angiotensin II type 1 receptor: comparison with other angiotensin II type 1 receptor blockers. Int. J. Clin. Pharmacol. Res. 25, 41–46 [PubMed] [Google Scholar]
- 35. Tobert J. A. (2003) Lovastatin and beyond: The history of the HMG-CoA reductase inhibitors. Nat. Rev. Drug Discov. 2, 517–526 [DOI] [PubMed] [Google Scholar]
- 36. Guenole A., Srivas R., Vreeken K., Wang Z. Z., Wang S., Krogan N. J., Ideker T., van Attikum H. (2013) Dissection of DNA damage responses using multiconditional genetic interaction maps. Mol. Cell 49, 346–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen Y. C., Rajagopala S. V., Stellberger T., Uetz P. (2010) Exhaustive benchmarking of the yeast two-hybrid system. Nat. Methods 7, 667–668, author reply, 668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pattyn E., Lavens D., Van der Heyden J., Verhee A., Lievens S., Lemmens I., Hallenberger S., Jochmans D., Tavernier J. (2008) MAPPIT (MAmmalian Protein-Protein Interaction Trap) as a tool to study HIV reverse transcriptase dimerization in intact human cells. J. Virol. Methods 153, 7–15 [DOI] [PubMed] [Google Scholar]
- 39. Jin L., Wang W., Fang G. (2014) Targeting protein-protein interaction by small molecules. Annu. Rev. Pharmacol. Toxicol. 54, 435–456 [DOI] [PubMed] [Google Scholar]
- 40. Terstappen G. C., Schlupen C., Raggiaschi R., Gaviraghi G. (2007) Target deconvolution strategies in drug discovery. Nat. Rev. Drug Discov. 6, 891–903 [DOI] [PubMed] [Google Scholar]
- 41. Chan J. N., Nislow C., Emili A. (2010) Recent advances and method development for drug target identification. Trends Pharmacol. Sci. 31, 82–88 [DOI] [PubMed] [Google Scholar]
- 42. Chidley C., Haruki H., Pedersen M. G., Muller E., Johnsson K. (2011) A yeast-based screen reveals that sulfasalazine inhibits tetrahydrobiopterin biosynthesis. Nat. Chem. Biol. 7, 375–383 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.