

Abstract
An extensive mass spectrometry analysis of the human milk peptidome has revealed almost 700 endogenous peptides from 30 different proteins. Two in-house computational tools were created and used to visualize and interpret the data through both alignment of the peptide quasi-molecular ion intensities and estimation of the differential enzyme participation. These results reveal that the endogenous proteolytic activity in the mammary gland is remarkably specific and well conserved. Certain proteins—not necessarily the most abundant ones—are digested by the proteases present in milk, yielding endogenous peptides from selected regions. Our results strongly suggest that factors such as the presence of specific proteases, the position and concentration of cleavage sites, and, more important, the intrinsic disorder of segments of the protein drive this proteolytic specificity in the mammary gland. As a consequence of this selective hydrolysis, proteins that typically need to be cleaved at specific positions in order to exert their activity are properly digested, and bioactive peptides encoded in certain protein sequences are released. Proteins that must remain intact in order to maintain their activity in the mammary gland or in the neonatal gastrointestinal tract are unaffected by the hydrolytic environment present in milk. These results provide insight into the intrinsic structural mechanisms that facilitate the selectivity of the endogenous milk protease activity and might be useful to those studying the peptidomes of other biofluids.
Peptidomics is defined as the systematic, comprehensive, and quantitative analysis of the low-molecular-weight fraction of proteins present in a biological sample at a defined time point (1). This protein fraction includes biologically active peptide sequences, protein degradation products, and small proteins such as cytokines and signaling peptides (2). Endogenous peptides are produced from their corresponding proteins through the action of proteases naturally present in the same biological system. Consequently, the peptidome and proteome are intrinsically linked, and their balance is controlled by the presence of proteases and modulated by the levels of protease activators and inhibitors. This relationship between proteins and their hydrolytic products has fueled the emergence of peptidomics as a subdiscipline of proteomics. Human biofluids such as blood (3), cerebrospinal fluid (4), saliva (5, 6), tears (7), and urine (8) have been analyzed for endogenous peptides. As naturally occurring peptides reflect both the protein content of a tissue and a specific configuration of the proteolytic machinery, they represent a promising target for biomarker discovery (9–13). From a functional perspective, a number of peptidomic studies have revealed different bioactivities in endogenous sequences (14–17).
Peptidomic research has revealed that the endogenous low-molecular-weight protein fraction is generally composed of overlapping ladder peptide products originating from a few regions of specific proteins. This proteolytic pattern is explained as a result of the action of endopeptidases cleaving in specific protein regions and the subsequent partial degradation of these initial fragments by exopeptidases (18). The presence and abundance of the resulting endogenous peptides has been correlated with the amounts of both substrate proteins and proteolytic components (9, 19, 20); however, the determinants of the peptidase selectivity are still a matter of scientific debate. It is accepted that four factors determine the specificity of the proteolysis: (i) the coexistence of protease and substrate protein in the same space and time; (ii) the presence of exosites that, although not involved in the proteolysis itself, increase the affinity of the protease for specific substrates; (iii) the presence of the correct amino acid motif; and (iv) the structural context of the excisable bond (21). The last factor is related to the accessibility of the enzyme to the cleaving site, and it is commonly accepted that proteolysis happens in solvent-exposed, flexible substrate regions (22, 23). However, recent investigations have demonstrated that limited proteolysis frequently happens also in helix and b-sheet secondary structures (21, 24).
Milk is a unique fluid for peptidomics. The proteins in milk are well characterized, as are many of the proteases that are present. However, milk has been little studied from a peptidomic viewpoint. The vast majority of studies have focused on the discovery of bioactive milk peptides released from isolated milk proteins via in vitro digestion processes. In these studies, milk proteins were degraded by bacteria cultures (25–27) or commercial proteases (28) in environments that might or might not mimic biological conditions (e.g. stomach conditions (29, 30)), and the resulting released peptides were analyzed for function. Through this approach, dozens of protein fragments, mostly from bovine milk, but also from human milk, have been shown to have different functions (31), including antimicrobial (32, 33), antihypertensive (34, 35), immunomodulatory (36, 37), and opioid-like (38) actions. However, such in vitro digestion approaches fail to reveal the peptide content present endogenously in milk. Only a few attempts have been reported to characterize the naturally occurring peptide content in human milk, but those have focused on the description of peptides produced from a small number of milk proteins (20, 39). More recently, our group developed an analytical procedure to purify and analyze the endogenous peptide content in human milk (40).
In this study, we analyzed the milk peptidome as a whole and confirmed that the endogenous sequences produced in milk derive from specific regions of selected proteins. By combining degradation maps, we revealed that the proteolysis exhibited in the mammary gland is governed by the protease specificities and by the degree of disorder of the digested proteins. This study is the most extensive to date on the underlying mechanisms of proteolytic control for in vivo degradation of human milk. The conclusions regarding milk peptides are generalizable and provide insights about the formation of endogenous peptides in other biofluids.
EXPERIMENTAL PROCEDURES
Chemicals and Sample Set
Acetonitrile, formic acid, and trifluoroacetic acid were obtained from Thermo Fisher Scientific (Waltham, MA), and trichloroacetic acid was obtained from EMD Millipore (Darmstadt, Germany). Bovine serum albumin and bicinchoninic acid were obtained from Sigma-Aldrich.
Mature milk samples from 15 mothers were collected at day 90 of lactation. Milk was collected as part of a University of California, Davis Institutional Review Board–approved observational study. Samples were taken from milk expressed with breast milk pumps, transferred into sterile plastic containers, and immediately stored in home freezers. Milk samples were transported on dry ice to the laboratory and stored at −80 °C until the moment of sample preparation. Keeping the samples frozen was previously shown to be as effective as boiling in preventing further protein hydrolysis (40). The peptide purification procedure has been previously described (40).
Mass Spectrometry Analysis
Samples were analyzed in positive mode on an Agilent (Santa Clara, CA) nano-LC-chip-Q-TOF-MS/MS instrument (Chip-Q-TOF) with a chip C18 column at a flow rate of 0.3 μl/min. The gradient elution solvents were (A) 3% acetonitrile/0.1% formic acid and (B) 90% acetonitrile/0.1% formic acid. The gradient employed was ramped from 1% to 8% B from 0–5 min, 8% to 26.5% B from 5–24 min, and 26.5% to 99% B from 24–48 min, and then it was held at 99% B for 10 min and 99% A for 10 min to re-equilibrate the column. The drying gas was 325 °C, and the flow rate was 5 l/min. The required chip voltage for consistent spray varied from 1700 to 1850 V. Automated precursor selection based on abundance was employed to select peaks for tandem fragmentation. The collision energy was set using the formula (Slope) × (m/z)/100 + Offset, with slope = 3.6 and offset = −4.8. Mass calibration was performed during data acquisition based on an infused calibrant ion.
All ion molecules fragmented in an MS/MS experiment were incorporated into an exclusion list for the subsequent round of mass spectrometry. The exclusion list was composed of mass-to-charge signals, charge states, and retention times. Ions on the exclusion list were thus ignored by the instrument and were not fragmented again. Each sample was analyzed in the MS/MS mode using the iterative exclusion lists method four times. This methodology helps the instrument to fragment peaks of lesser abundance, which in turn allows deeper exploration of the samples. Finally, each sample was analyzed once in the MS mode using the same parameters to obtain ion counting information.
Protein Quantification
An aliquot of 5 μl from each human milk sample was used to determine the protein content using the BCA method (41). 10 μl of ethanol were added to each sample to precipitate out the proteins. The liquid phase was carefully removed and discarded. The remaining protein pellet was then sonicated in 5% SDS for 30 min and diluted 20-fold for BCA analysis. Bovine serum albumin was used as the standard and serially diluted to build a standard curve. The samples were incubated with the BCA reagent at 37 °C for 30 min, and absorbances were measured using a Genesys 20 spectrophotometer (Thermo Scientific). The protein content was finally determined by means of interpolation of the absorbance values with the bovine serum albumin standard curve.
Data Analysis
Database Search
Data files were exported as MGF files using MassHunter Work Station B.05.00 (Agilent). Peptide identification was accomplished using the database searcher X! Tandem included on GMP Manager 2.2.1 (42) against a human milk protein library composed of 975 entries constructed on the basis of previous proteomic studies (43–45) and compiled from UniProt. Masses were allowed 60 ppm error. No complete peptide modifications were allowed. Potential modifications allowed included serine, threonine, and tyrosine phosphorylation; methionine and tryptophan oxidation; asparagine and glutamine deamidation; and glutamine dehydration. A nonspecific cleavage ([X] [X]) (where X is any amino acid) was used to search against the protein sequences. No model refinement was employed. Peptide matches were accepted if e-values were less than or equal to 0.01, corresponding to a 99% confidence level.
Library Construction and Application
The results from X! Tandem were processed computationally in a library that included retention times, peptide sequence, neutral mass, protein of origin, and the number and nature of modifications that the peptide contained. Duplicate peptide entries were removed, and their corresponding retention times were averaged. The library was used to identify and quantitate, via ion counting, the peptides present in each sample. MS experiments were used for this purpose. Quasimolecular ion signals corresponding to different charge states of the same compound were grouped and searched against the library using both retention time and mass. The intensity of each signal matching an entry from the library was calculated as the area under the curve of its elution time.
Grouping and Visualizing Peptide Signals
Peptide signal intensities were normalized to the total protein content of each sample. Peptide sequences and their normalized intensities were aligned over their corresponding proteins of origin using an in-house script written in Python (PepEx). Protein sequences were analyzed with the Disprot VL3E neural network (46) to identify intrinsically disordered regions. The predicted degree of disorder was plotted with the proteolytic maps obtained with PepEx.
Determining Differential Enzymatic Participation
A custom script written in Python was used to estimate the activity of selected enzymatic systems. The program locates the position of each peptide on its corresponding protein. The termini of each peptide were compared with a selected set of proteolytic enzyme rules. As a measure of simplification, rules were assumed to only act on P1 and P1′. P1 is the amino acid directly before the cleavage site on the N-terminal side, and P1′ is the amino acid directly after the cleavage site on the C-terminal side. Enzymatic cleavage rules were derived from a list published on ExPASy (47). Peptides having termini that pass a comparison to an enzymatic rule have their mass spectral intensity (peak volume) added to the sum of the respective enzyme. If a peptide matches multiple enzyme rules simultaneously, the full intensity of that peptide is added to each enzymatic sum; thus the intensity value output represents potential activity rather than uniquely specific activity. Peptides failing all enzymatic comparisons have their intensity added to a list of remainders whose purpose is to assist in the identification of new enzymatic systems.
RESULTS
Nano-LC Mass Spectrometry Analysis of Endogenous Peptides in Milk
Peptides were identified using MS/MS. However, the duty cycle associated with MS/MS means that the number of peptides identified in a single run will be limited. To obtain a greater number of peptide identifications, we performed multiple analyses using an iterative-exclusion list for MS/MS. The samples were analyzed four times, increasing the number of peptide identifications 4-fold relative to a single MS/MS analysis (supplemental Fig. S1). After the results were compiled, a library composed of nearly 700 peptide sequences from 30 different proteins was obtained (supplemental Table S1).1 Peptides' accurate masses and retention times in the library were matched with the results of a final MS analysis performed on each sample. Ion abundances were used for label-free quantitation, variations in ionization efficiency and suppression effects notwithstanding. Finally, abundances were normalized to the total protein content of each sample measured via the BCA method (supplemental Table S2).
The identified peptides originated from 30 human milk proteins; however, 95% of the peptides were proteolytic products from only four proteins, namely, β-casein, αs1-casein, osteopontin, and polymeric immunoglobulin receptor (pIgR)2 (Fig. 1). Although ion intensities are not strictly quantitative, the results obtained were essentially similar for all milk samples. β-casein and αs1-casein are the most abundant milk proteins and were expected to contribute significantly to the peptide products. Osteopontin and pIgR, in contrast, are typically found in significantly lower abundances in human milk. Additionally, other abundant proteins in milk such as lactoferrin and α-lactalbumin were not represented in the peptide fragments in any appreciable amounts. These results strongly suggest a large degree of selectivity during the in vivo proteolysis leading to the production of the endogenous milk peptides from specific proteins.
Fig. 1.

Average (15 samples) normalized ion intensities of the total endogenous peptides by protein of origin. Error bars show the standard deviation from the average.
Proteolytic Mapping with PepEx
To determine the site specificity of the proteolysis, we developed a computational program called Peptide Extractor (PepEx) in our laboratory. PepEx uses a list of peptide entries and their corresponding abundances as input. The program localizes the position of each peptide in the respective proteins and plots the abundance over the sequence. The output of the software or proteolytic map is illustrated for β-casein in Fig. 2. In the horizontal axis, the sequence of the protein is represented from the N terminus to the C terminus (left to right). In the vertical axis, ion intensities (as a number of counts) are plotted. Each color area represents a peptide with its associated intensity aligned over the protein sequence. For example, the β-casein peptide RETIESLSSSEESITEYKQKVEK (Fig. 2, blue band) was the most abundant peptide from the N terminus of this protein. Shorter sequences such as RETIESLSSSEESITEYK or ETIESLSSSEESITEYK (red and green bands, respectively) were also observed, but in slightly lower abundances. Mapping the peptide intensities in this way allows visualization of the overall proteolytic activity toward the protein. The results shown in Fig. 2 indicate that β-casein was preferentially digested at the N and the C termini. The middle protein region also generated endogenous peptides, but in significantly lower abundances.
Fig. 2.

Endogenous proteolytic map of β-casein in milk determined by PepEx. The height of each area represents the ion intensity of the corresponding peptides.
The proteolytic characteristics of the same protein can also be compared over several samples using PepEx.3 PepEx compiles the total abundances associated with each amino acid of the protein sequence by summing the endogenous peptides that contain them. In this way, one can readily compare proteolytic maps of different samples. The proteolytic maps of β-casein for the 15 milk samples are shown in Fig. 3. Each colored line represents the map of a different sample. A strict quantitative comparison between milk samples, which is out of the scope of this study, would require steps like the use of internal standards. Nonetheless, the mere collation of the ion intensities revealed a remarkably constant proteolytic pattern for β-casein. In all the samples, the protein termini seemed to generate the majority of the endogenous fragments, and low-intensity peptides were found from internal regions of β-casein. Furthermore, the shape of the proteolytic map is highly conserved between samples so that the changes in the ion intensities along the protein sequence overlap. The arrows in Fig. 3 indicate specific proteolytic cleavages that can be assigned to the activity of distinct enzymes.
Fig. 3.
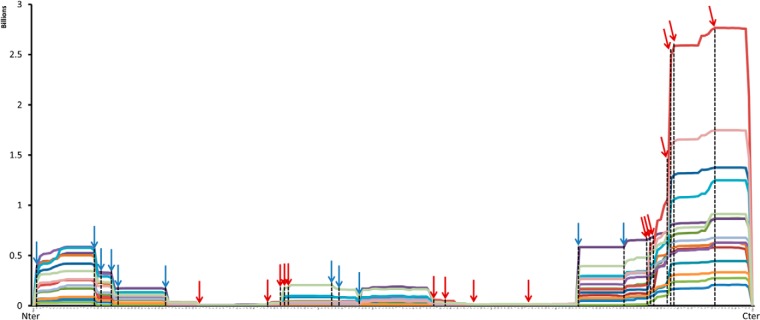
Endogenous proteolytic map of β-casein (15 samples) determined with PepEx. Arrows indicate the positions of cleavage sites for plasmin/trypsin (blue) and cathepsin-D (red).
The selectivity of the in vivo proteolysis was not limited to β-casein. Similar results were obtained for the other three proteins, namely, osteopontin, αs1-casein, and pIgR (Fig. 4). Three different regions of osteopontin generated endogenous milk peptides. Both the N- and the C-terminal regions were digested, but the majority of the peptides derived from an internal region of the protein, mostly between residues S169 and K203 (Fig. 4A). Although pIgR contains 746 amino acid residues, all the endogenous peptides found in human milk from this protein arose from a single region in all the samples: between residues A598 and V650 (Fig. 4C). Endogenous peptides from exactly the same region of pIgR have been previously found in tears (7), suggesting that the specificity of the proteolysis of this protein is not exclusive to breast milk. Similarly, only one region of αS1-casein generated endogenous peptides, namely, the N-terminal region of the protein (Fig. 4B). Analyses of the peptides of other proteins (supplemental Table S1) were also performed. Without exception, peptides were localized on specific regions of their respective proteins.
Fig. 4.

Proteolytic maps of (A) osteopontin, (B) αs1-casein, and (C) pIgR (15 samples) determined with PepEx. Arrows indicate the positions of cleavage sites for plasmin/trypsin (blue) and cathepsin-D (red).
Determination of Enzyme Participation
To determine what proteases acted on what proteins, we developed a computer program called Peptidomics Enzyme Tabulator (PEnTab) in our laboratory (this program is available online).3 PEnTab uses a list of peptide entries and their corresponding abundances as input. The program localizes the position of each peptide in the respective proteins and determines the amino acid residues that flank it. The termini of each peptide were compared with a selected set of proteolytic enzyme rules. Peptides having termini that pass a comparison to an enzymatic specificity have their abundances added to the sum of the respective enzyme. Proteases known to be present in milk include plasmin (48), trypsin (49, 50), elastase (50), cathepsin-D (51), and thrombin (39). In addition, undefined carboxy/amino peptidases have been proposed to act in milk (20). Based on the protease specificity (cleavage rules), PEnTab groups these enzymes in the systems plasmin/trypsin and cathepsin-D/elastase. Peptides failing all enzymatic comparisons are added and listed as “others.” Thrombin, which is known to specifically cleave osteopontin (39), was included as a separate heading in the analysis of this protein. Fig. 5 shows the results of the PEnTab analysis for pIgR, β-casein, αs1-casein, and osteopontin.
Fig. 5.

Relative participation of the proteases in the formation of endogenous peptides by protein (15 samples) estimated with EnTab. Error bars indicate standard deviation from the mean.
A strict peptide-based study of enzyme participation cannot be achieved without quantification of the peptides. Ion intensities were used for this purpose but might overestimate the enzymatic participation of some enzymes such as the system trypsin/plasmin. These proteases generate peptides with basic amino acids at the C terminus (R and K residues) that might show a higher ionization response on the electrospray. Nevertheless, the results in Fig. 5 show a rather low standard deviation between samples and may be discussed from a qualitative point of view. Both trypsin/plasmin and elastase/cathepsin-D acted on pIgR and β-casein. For the milk protein osteopontin, thrombin and plasmin/trypsin were the main enzymes involved; the low participation of elastase/cathepsin-D may be explained by the low number of potential cleavage sites for these enzymes in osteopontin. Enzyme miscleavages and in-source fragmentation can partially explain the high proportion of unknown cleavages, but the participation of other enzymes might be involved too. αS1-casein is an interesting case based on this analysis. Most of the endogenous fragments identified for αS1-casein are not associated with the enzymes included in this analysis. These results clearly indicate the activity of one or more additional enzymes that selectively generate the endogenous peptides from this protein.
DISCUSSION
Intrinsic Disorder and Proteolysis
Both β-casein and osteopontin are “naturally unfolded” proteins (52), as they lack a tight and stable tertiary structure. Because of their loose structure, more cleavage sites are exposed to proteolytic enzymes. The same reasoning can be applied for pIgR and αS1-casein. To understand the correlation between local disorder and the formation of peptide products, we calculated the local disorder of the pIgR and αS1-casein sequences. The local degree of disorder of these two proteins was calculated with a computational program, the VLE3 predictor (46). The VLE3 predictor scores the likelihood of local structural disorder based on the amino acid sequence. Regions with a score higher than 0.5 (on a 0–1 scale) are more likely to be unfolded. The result of the VLE3 predictor calculation for pIgR and αS1-casein superimposed with their proteolytic map is shown in Fig. 6.
Fig. 6.

Proteolytic map of (A) pIgR and (B) αs1-casein overlaid with the predicted degree of disorder for each part of the sequence. Values of disorder higher than 0.5 represent naturally unfolded regions.
Two major regions of disorder were present in pIgR, both at the C-terminus of the protein sequence, separated by a few amino acid residues (Fig. 6A). pIgR is involved in the transportation of polymeric immunoglobulins IgA and IgM across epithelial cells. At the end of the transcytosis process, pIgR is proteolytically cleaved from its transmembrane anchor (Fig. 6A), releasing the secretory component into milk (53). The domain where the proteolytic cleavage takes place corresponds to an unstructured sequence of the protein called the linker motif (53). The peptides from pIgR are formed from this linker motif region. Consequently, they are the result of the proteolytic release of the secretory component into the milk and represent side-products of the transcytosis of pIGR. The second disordered region, at or near the C-terminal region, does not produce detectable peptides. pIgR is a transmembrane protein with the C-terminal region residing in the cytosolic domain (53). Fragments formed from this region of the protein will therefore likely remain inside the cell, rather than be secreted in the milk.
A similar comparison between the proteolytic map and the degree of disorder was performed for αs1-casein (Fig. 6B). This protein contrasts with pIgR in that a large fraction of the protein is determined to be disordered (around 70%). Again, the peptide products were localized and focused near the N terminus. No peptides were found from the ordered region.
All abundant peptides identified (>99% of identified signal intensity) in this study derived only from regions of predicted disorder. These results strongly indicate that intrinsic protein disorder is a crucial requisite for in vivo proteolysis of milk proteins within the mammary gland. A similar relationship between protein disorder and proteolysis has been observed for the ubiquitin-independent degradation of proteins mediated by proteasomes (54). Our results extend this structure-based mechanism of proteolytic control to an extracellular biofluid.
Intramolecular Specificity of the Proteolysis
Although intrinsic disorder explains why some proteins are selectively digested in the mammary gland, our results show that not all unstructured regions inside the protein are equally digested. For example, the entire β-casein sequence is disordered, but regions close to the termini generated more peptides than internal regions (Fig. 3). Similarly, αs1-casein (Fig. 6B) was preferentially digested at the N terminal, even though 70% of the protein is intrinsically unfolded and hence accessible to the proteases. The higher abundance of sequences near the termini may be explained by the fact that only one cleavage is necessary to generate these species, whereas releasing an internal fragment requires two cleavages. Interestingly, the terminal regions of the casein proteins are known to exert different bioactivities: the C-terminal domain of β-casein is known to generate peptides with antibacterial properties (55), and peptides from the N-terminal regions of both β-casein and αs1-casein, abundantly phosphorylated (supplemental Table S1), are known to have a role as calcium carriers because of their abundant phosphorylation (56).
Osteopontin is also an intrinsically disordered protein and, similarly to β-casein, yields peptides from the terminal regions. The predominant digested region, however, is the internal region. The action of thrombin, which has high proteolytic specificity, explains the particular endogenous proteolytic pattern of osteopontin in human milk. Thrombin is known to specifically cleave between osteopontin residues R168 and S169, exposing the integrin binding domain SVVYGLR (57) and thereby regulating the ability of osteopontin to interact with different targets. Thrombin is known to be present in human milk (39). We may conclude that the exposure of cleavage sites in disordered regions, the requirement of a single cleavage for the generation of peptides from the termini, and the participation of specific enzymes such as thrombin explain the specificity of the endogenous proteolytic activity in the mammary gland.
Limited Proteolysis in Human Milk
The similarity between human milk and other biofluid peptidomes is remarkable, especially when compared with serum. Peptidomic studies of serum have shown that the most abundant peptides from this biofluid, similar to those from human milk, collapsed in a small group of clusters derived from a few proteins, and not necessarily the more abundant ones (18). The serum peptidome is mostly the consequence of a couple of well-described proteolytic cascades: coagulation and complement activation. However, coagulation does not occur in human milk, and although in this study we found peptides from complement C4-A (Fig. 1), their abundance was negligible. It has also been shown in analyses of serum peptides not proceeding from coagulation or complement activation that proteolysis takes place in exposed regions with special incidence at the N and C termini (58). Similar results have been obtained in analyses of cell line peptides not proceeding from the caspase cascade (59). The human milk peptidome seems more in line with these proteolytic processes in serum masked by the predominant proteolytic cascades. Unstructured proteins are abundant in milk, and in the absence of a known tissue-specific proteolytic cascade, proteolysis happens at the most accessible sites. Nevertheless, the products of this process are, at least partially, known bioactive products (e.g. β-casein phosphopeptides) or side-products of activation processes (e.g. pIgR-derived peptides). The barrier between what seems to be accidental good substrates for the peptidases present in milk and a limited proteolysis targeted to the production of specific products becomes narrow. Not all the peptides found in this study can be tracked to bioactive compounds, and further investigation of these peptides for functional properties is warranted.
Supplementary Material
Footnotes
Author contributions: A.G., D.C.D., J.B.G., and C.B.L. designed research; A.G., S. Contreras, S. Chee, and L.D. performed research; A.G., E.A.P., X.S., D.B., and C.B.L. contributed new reagents or analytic tools; A.G. and S. Contreras analyzed data; A.G., D.C.D., and C.B.L. wrote the paper.
* Funding provided by the National Institutes of Health (GM049077) is gratefully acknowledged.
 This article contains supplemental material.
This article contains supplemental material.
1 Data uploaded at the Peptide Atlas repository, www.peptideatlas.org/PASS/PASS00495.
3 E. A. Parker (2014), Peptide Extractor, Github Repository, https://github.com/eparker05/Peptidomics-enzyme-estimator.
2 The abbreviation used is:
- pIgR
- polymeric immunoglobulin receptor.
REFERENCES
- 1. Schulz-Knappe P., Schrader M., Zucht H.-D. (2005) The peptidomics concept. Combinat. Chem. High Throughput Screening 8, 697–704 [DOI] [PubMed] [Google Scholar]
- 2. Cunningham R., Ma D., Li L. (2012) Mass spectrometry-based proteomics and peptidomics for systems biology and biomarker discovery. Front. Biol. 7, 313–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shen Y., Liu T., Tolicì N., Petritis B. O., Zhao R., Moore R. J., Purvine S. O., Camp D. G., Smith R. D. (2010) Strategy for degradomic-peptidomic analysis of human blood plasma. J. Proteome Res. 9, 2339–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wiltfang J., Esselmann H., Smirnov A., Bibl M., Cepek L., Steinacker P., Mollenhauer B., Buerger K., Hampel H., Paul S., Neumann M., Maler M., Zerr I., Kornhuber J., Kretzschmar H. A., Poser S., Otto M. (2003) β-amyloid peptides in cerebrospinal fluid of patients with Creutzfeldt–Jakob disease. Ann. Neurol. 54, 263–267 [DOI] [PubMed] [Google Scholar]
- 5. Huang C.-M., Zhu W. (2009) Profiling human saliva endogenous peptidome via a high throughput MALDI-TOF-TOF mass spectrometry. Combinat. Chem. High Throughput Screening 12, 521–531 [DOI] [PubMed] [Google Scholar]
- 6. Messana I., Cabras T., Pisano E., Sanna M. T., Olianas A., Manconi B., Pellegrini M., Paludetti G., Scarano E., Fiorita A., Agostino S., Contucci A. M., Calò L., Picciotti P. M., Manni A., Bennick A., Vitali A., Fanali C., Inzitari R., Castagnola M. (2008) Trafficking and postsecretory events responsible for the formation of secreted human salivary peptides: a proteomics approach. Mol. Cell. Proteomics 7, 911–926 [DOI] [PubMed] [Google Scholar]
- 7. Hayakawa E., Landuyt B., Baggerman G., Cuyvers R., Lavigne R., Luyten W., Schoofs L. (2013) Peptidomic analysis of human reflex tear fluid. Peptides 42, 63–69 [DOI] [PubMed] [Google Scholar]
- 8. Jurgens M., Appel A., Heine G., Neitz S., Menzel C., Tammen H., Zucht H.-D. (2005) Towards characterization of the human urinary peptidome. Combinat. Chem. High Throughput Screening 8, 757–765 [DOI] [PubMed] [Google Scholar]
- 9. Shen Y., Tolić N., Liu T., Zhao R., Petritis B. O., Gritsenko M. A., Camp D. G., Moore R. J., Purvine S. O., Esteva F. J., Smith R. D. (2010) Blood peptidome-degradome profile of breast cancer. PLoS One 5, e13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ling X. B., Mellins E. D., Sylvester K. G., Cohen H. J. (2010) Urine peptidomics for clinical biomarker discovery. Advances in Clinical Chemistry (Gregory S. M., Ed), Vol. 51, pp. 181–213, Elsevier, London, UK: [DOI] [PubMed] [Google Scholar]
- 11. Wen Q., Liu L. Y., Yang T., Alev C., Wu S., Stevenson D. K., Sheng G., Butte A. J., Ling X. B. (2013) Peptidomic identification of serum peptides diagnosing preeclampsia. PLoS One 8, e65571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quintana L. F., Campistol J. M., Alcolea M. P., Bañon-Maneus E., Sol-González A., Cutillas P. R. (2009) Application of label-free quantitative peptidomics for the identification of urinary biomarkers of kidney chronic allograft dysfunction. Mol. Cell. Proteomics 8, 1658–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Villanueva J., Martorella A. J., Lawlor K., Philip J., Fleisher M., Robbins R. J., Tempst P. (2006) Serum peptidome patterns that distinguish metastatic thyroid carcinoma from cancer-free controls are unbiased by gender and age. Mol. Cell. Proteomics 5, 1840–1852 [DOI] [PubMed] [Google Scholar]
- 14. Sasaki K., Takahashi N., Satoh M., Yamasaki M., Minamino N. (2010) A peptidomics strategy for discovering endogenous bioactive peptides. J. Proteome Res. 9, 5047–5052 [DOI] [PubMed] [Google Scholar]
- 15. Svensson M., Sköld K., Svenningsson P., Andren P. E. (2003) Peptidomics-based discovery of novel neuropeptides. J. Proteome Res. 2, 213–219 [DOI] [PubMed] [Google Scholar]
- 16. Tamvakopoulos C. (2007) Mass spectrometry for the quantification of bioactive peptides in biological fluids. Mass Spectrom. Rev. 26, 389–402 [DOI] [PubMed] [Google Scholar]
- 17. Bernay B., Gaillard M.-C., Guryča V., Emadali A., Kuhn L., Bertrand A., Detraz I., Carcenac C., Savasta M., Brouillet E., Garin J., Elalouf J.-M. (2009) Discovering new bioactive neuropeptides in the striatum secretome using in vivo microdialysis and versatile proteomics. Mol. Cell. Proteomics 8, 946–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Villanueva J., Shaffer D. R., Philip J., Chaparro C. A., Erdjument-Bromage H., Olshen A. B., Fleisher M., Lilja H., Brogi E., Boyd J., Sanchez-Carbayo M., Holland E. C., Cordon-Cardo C., Scher H. I., Tempst P. (2006) Differential exoprotease activities confer tumor-specific serum peptidome patterns. J. Clin. Invest. 116, 271–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Diamandis E. P. (2006) Peptidomics for cancer diagnosis: present and future. J. Proteome Res. 5, 2079–2082 [DOI] [PubMed] [Google Scholar]
- 20. Ferranti P., Traisci M. V., Picariello G., Nasi A., Boschi V., Siervo M., Falconi C., Chianese L., Addeo F. (2004) Casein proteolysis in human milk: tracing the pattern of casein breakdown and the formation of potential bioactive peptides. J. Dairy Res. 71, 74–87 [DOI] [PubMed] [Google Scholar]
- 21. Timmer J. C., Zhu W., Pop C., Regan T., Snipas S. J., Eroshkin A. M., Riedl S. J., Salvesen G. S. (2009) Structural and kinetic determinants of protease substrates. Nat. Struct. Mol. Biol. 16, 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hubbard S. J. (1998) The structural aspects of limited proteolysis of native proteins. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1382, 191–206 [DOI] [PubMed] [Google Scholar]
- 23. Novotný J., Bruccoleri R. E. (1987) Correlation among sites of limited proteolysis, enzyme accessibility and segmental mobility. FEBS Lett. 211, 185–189 [DOI] [PubMed] [Google Scholar]
- 24. Kazanov M. D., Igarashi Y., Eroshkin A. M., Cieplak P., Ratnikov B., Zhang Y., Li Z., Godzik A., Osterman A. L., Smith J. W. (2011) Structural determinants of limited proteolysis. J. Proteome Res. 10, 3642–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seppo L., Jauhiainen T., Poussa T., Korpela R. (2003) A fermented milk high in bioactive peptides has a blood pressure–lowering effect in hypertensive subjects. Am. J. Clin. Nutrition 77, 326–330 [DOI] [PubMed] [Google Scholar]
- 26. Matar C., Amiot J., Savoie L., Goulet J. (1996) The effect of milk fermentation by Lactobacillus helveticus on the release of peptides during in vitro digestion. J. Dairy Sci. 79, 971–979 [DOI] [PubMed] [Google Scholar]
- 27. Smacchi E., Gobbetti M. (2000) Bioactive peptides in dairy products: synthesis and interaction with proteolytic enzymes. Food Microbiol. 17, 129–141 [Google Scholar]
- 28. Argyri K., Miller D. D., Glahn R. P., Zhu L., Kapsokefalou M. (2007) Peptides isolated from in vitro digests of milk enhance iron uptake by caco-2 cells. J. Agric. Food Chem. 55, 10221–10225 [DOI] [PubMed] [Google Scholar]
- 29. Ye A., Cui J., Singh H. (2011) Proteolysis of milk fat globule membrane proteins during in vitro gastric digestion of milk. J. Dairy Sci. 94, 2762–2770 [DOI] [PubMed] [Google Scholar]
- 30. Picariello G., Ferranti P., Fierro O., Mamone G., Caira S., Di Luccia A., Monica S., Addeo F. (2010) Peptides surviving the simulated gastrointestinal digestion of milk proteins: biological and toxicological implications. J. Chromatogr. B 878, 295–308 [DOI] [PubMed] [Google Scholar]
- 31. Clare D. A., Swaisgood H. E. (2000) Bioactive milk peptides: a prospectus. J. Dairy Sci. 83, 1187–1195 [DOI] [PubMed] [Google Scholar]
- 32. Bellamy W., Takase M., Wakabayashi H., Kawase K., Tomita M. (1992) Antibacterial spectrum of lactoferricin B, a potent bactericidal peptide derived from the N-terminal region of bovine lactoferrin. J. Appl. Bacteriol. 73, 472–479 [DOI] [PubMed] [Google Scholar]
- 33. Benkerroum N. (2010) Antimicrobial peptides generated from milk proteins: a survey and prospects for application in the food industry. A review. Int. J. Dairy Technol. 63, 320–338 [Google Scholar]
- 34. FitzGerald R. J., Murray B. A., Walsh D. J. (2004) Hypotensive peptides from milk proteins. J. Nutrition 134, 980S–988S [DOI] [PubMed] [Google Scholar]
- 35. Jäkälä P., Vapaatalo H. (2010) Antihypertensive peptides from milk proteins. Pharmaceuticals 3, 251–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Politis I., Chronopoulou R. (2008) Milk peptides and immune response in the neonate. Bioactive Components of Milk (Bösze Z., Ed), Vol 606, pp. 253–269, Springer, New York: [DOI] [PubMed] [Google Scholar]
- 37. Huang S.-M., Chen K.-N., Chen Y.-P., Hong W.-S., Chen M.-J. (2010) Immunomodulatory properties of the milk whey products obtained by enzymatic and microbial hydrolysis. Int. J. Food Sci. Technol. 45, 1061–1067 [Google Scholar]
- 38. Meisel H., FitzGerald R. J. (2000) Opioid peptides encrypted in intact milk protein sequences. Br. J. Nutrition 84 (Supplement S1), 27–31 [DOI] [PubMed] [Google Scholar]
- 39. Christensen B., Schack L., Kläning E., Sørensen E. S. (2010) Osteopontin is cleaved at multiple sites close to its integrin-binding motifs in milk and is a novel substrate for plasmin and cathepsin D. J. Biol. Chem. 285, 7929–7937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dallas D. C., Guerrero A., Khaldi N., Castillo P. A., Martin W. F., Smilowitz J. T., Bevins C. L., Barile D., German J. B., Lebrilla C. B. (2013) Extensive in vivo human milk peptidomics reveals specific proteolysis yielding protective antimicrobial peptides. J. Proteome Res. 12, 2295–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., Klenk D. C. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85 [DOI] [PubMed] [Google Scholar]
- 42. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 43. Molinari C. E., Casadio Y. S., Hartmann B. T., Livk A., Bringans S., Arthur P. G., Hartmann P. E. (2012) Proteome mapping of human skim milk proteins in term and preterm milk. J. Proteome Res. 11, 1696–1714 [DOI] [PubMed] [Google Scholar]
- 44. Mange A., Bellet V., Tuaillon E., Van de Perre P., Solassol J. (2008) Comprehensive proteomic analysis of the human milk proteome: contribution of protein fractionation. J. Chromatogr. B 876, 252–256 [DOI] [PubMed] [Google Scholar]
- 45. Liao Y., Alvarado R., Phinney B., Lonnerdal B. (2011) Proteomic characterization of human milk fat globule membrane proteins during a 12 month lactation period. J. Proteome Res. 10, 3530–3541 [DOI] [PubMed] [Google Scholar]
- 46. Peng K., Vucetic S., Radivojac P., Brown C. J., Dunker A. K., Obradovic Z. (2005) Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinformat. Comput. Biol. 03, 35–60 [DOI] [PubMed] [Google Scholar]
- 47. Gasteiger E., Hoogland C., Gattiker A., Wilkins M. R., Appel R. D., Bairoch A. (2005) Protein identification and analysis tools on the ExPASy server. The Proteomics Protocols Handbook, Humana Press, 571–607 [Google Scholar]
- 48. Korycha-Dahl M., Dumas B. R., Chene N., Martal J. (1983) Plasmin activity in milk. J. Dairy Sci. 66, 704–711 [Google Scholar]
- 49. Monti J. C., Mermoud A. F., Jollès P. (1986) Trypsin in human milk. Experientia 42, 39–41 [DOI] [PubMed] [Google Scholar]
- 50. Borulf S., Lindberg T., Månsson M. (1987) Immunoreactive anionic trypsin and anionic elastase in human milk. Acta Pædiatr. 76, 11–15 [DOI] [PubMed] [Google Scholar]
- 51. Vetvicka V., Vagner J., Baudys M., Tang J., Foundling S. I., Fusek M. (1993) Human breast milk contains procathepsin D—detection by specific antibodies. Biochem. Mol. Biol. Int. 30, 921–928 [PubMed] [Google Scholar]
- 52. Tompa P. (2002) Intrinsically unstructured proteins. Trends Biochem. Sci. 27, 527–533 [DOI] [PubMed] [Google Scholar]
- 53. Kaetzel C. S. (2005) The polymeric immunoglobulin receptor: bridging innate and adaptive immune responses at mucosal surfaces. Immunol. Rev. 206, 83–99 [DOI] [PubMed] [Google Scholar]
- 54. Asher G., Reuven N., Shaul Y. (2006) 20S proteasomes and protein degradation “by default.” Bioessays 28, 844–849 [DOI] [PubMed] [Google Scholar]
- 55. Minervini F., Algaron F., Rizzello C. G., Fox P. F., Monnet V., Gobbetti M. (2003) Angiotensin I-converting-enzyme-inhibitory and antibacterial peptides from Lactobacillus helveticus PR4 proteinase-hydrolyzed caseins of milk from six species. Appl. Environ. Microbiol. 69, 5297–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lönnerdal B. (2013) Bioactive proteins in breast milk. J. Paediatr. Child Health 49, 1–7 [DOI] [PubMed] [Google Scholar]
- 57. Lund S., Giachelli C., Scatena M. (2009) The role of osteopontin in inflammatory processes. J. Cell Commun. Signal. 3, 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Koomen J. M., Li D., Xiao L.-c., Liu T. C., Coombes K. R., Abbruzzese J., Kobayashi R. (2005) Direct tandem mass spectrometry reveals limitations in protein profiling experiments for plasma biomarker discovery. J. Proteome Res. 4, 972–981 [DOI] [PubMed] [Google Scholar]
- 59. Gelman J. S., Sironi J., Castro L. M., Ferro E. S., Fricker L. D. (2011) Peptidomic analysis of human cell lines. J. Proteome Res. 10, 1583–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.