

Abstract
Activin receptor type II (ACVR2) is a member of the transforming growth factor type II receptor family and controls cell growth and differentiation, thereby acting as a tumor suppressor. ACVR2 inactivation is known to drive colorectal tumorigenesis. We used an ACVR2-deficient microsatellite unstable colon cancer cell line (HCT116) to set up a novel experimental design for comprehensive analysis of proteomic changes associated with such functional loss of a tumor suppressor. To this end we combined two existing technologies. First, the ACVR2 gene was reconstituted in an ACVR2-deficient colorectal cancer (CRC) cell line by means of recombinase-mediated cassette exchange, resulting in the generation of an inducible expression system that allowed the regulation of ACVR2 gene expression in a doxycycline-dependent manner. Functional expression in the induced cells was explicitly proven. Second, we used the methionine analog azidohomoalanine for metabolic labeling of newly synthesized proteins in our cell line model. Labeled proteins were tagged with biotin via a Click-iT chemistry approach enabling specific extraction of labeled proteins by streptavidin-coated beads. Tryptic on-bead digestion of captured proteins and subsequent ultra-high-performance LC coupled to LTQ Orbitrap XL mass spectrometry identified 513 proteins, with 25 of them differentially expressed between ACVR2-deficient and -proficient cells. Among these, several candidates that had already been linked to colorectal cancer or were known to play a key role in cell growth or apoptosis control were identified, proving the utility of the presented experimental approach. In principle, this strategy can be adapted to analyze any gene of interest and its effect on the cellular de novo proteome.
Human tumors acquire a large number of genetic and epigenetic alterations that arise during progression from preneoplastic lesions to metastatic disease. However, the diversity of these alterations reflects the intratumoral heterogeneity and represents the genomic landscape of tumors. Among a high background number of irrelevant passenger alterations, only a limited number of genetic alterations are considered to be driving events that confer a selective advantage to tumor cells. Major signaling pathways affected by such driver mutations include the TGFβ, BMP, Activin, Wnt, and Notch pathways, abrogating normal regulation of key cellular processes such as cell fate, cell survival, and genome maintenance.
Both tumor-relevant driver mutations in a major signaling receptor and tumor-irrelevant passenger mutations can cause changes at the proteomic level. Passenger-mutation-associated proteomic patterns are propagated randomly and do not represent generic tumor-associated changes (1). Therefore, a focus on proteome alterations associated with single driver mutations is necessary in order for specific changes that underlie tumor development to be identified. However, such analyses encounter two major limitations at different levels.
At the molecular level, the genetic heterogeneity of tumors—especially those of the microsatellite unstable and mutator phenotype—poses a significant problem in determining mutation-specific effects. Two principal strategies for detecting cellular consequences of a single mutation have been applied. First, targeted gene knock-out in target-gene-proficient cell lines by means of homologous recombination, adeno-associated viral delivery, or zinc finger nucleases has been used successfully (2–4). However, these approaches are often limited by their low efficiency, are laborious and time-consuming, and bear the potential for confounding off-target effects. Second, transfer of the target gene into deficient cell lines via gene insertion or gene targeting methods has been extensively applied. Unfortunately, insertion methods are often affected by random insertion, a variable number of integrated gene copies per cell, and inconsistent integration sites, eventually resulting in unpredictable expression patterns (5). However, many non-integrating vectors, such as adenoviral DNA, are not often replicated during cell division, which limits their use in basic research.
At the protein level, sample complexity is a major limiting factor. In addition to prefractionation methods, metabolic labeling is a versatile tool in work focusing on proteomic changes induced by gene activation. Because the activation of tumor suppressor pathways directly regulates target gene expression, analysis of tumor-suppressor-dependent alterations of newly synthesized proteins via metabolic labeling is a reasonable approach for restricting proteomic complexity. Conventional methods for metabolic labeling usually rely on amino acids containing either radioactive or stable isotopes. Although radioactive labeling enables extremely sensitive detection methods, its use for proteomic analysis is limited because of the need for special handling and precautions against contamination of the analytical instrumentation. Stable isotopic labeling, in particular the SILAC methodology, is currently the preferred method for most metabolic labeling approaches in proteomic analyses, and especially for cell lines (6). However, when applying the SILAC technology, mass spectrometric detection of labeled peptides has to be conducted in the presence of numerous irrelevant, unlabeled peptides, which hampers the detection of labeled low-abundance peptides. A relatively new method, termed Click-iT labeling, that enables labeling of nascent proteins comparable to that by a radioactive compound can overcome this problem, because upon incorporation of the labeled compound a handle for specific extraction of the labeled protein is worked in. The click reaction makes use of a methionine derivative that is functionalized with an azide (l-azidohomoalanine (AHA)).1 During protein synthesis AHA is incorporated into proteins, which can then be tagged with a biotin alkyne (PEG4 carboxamide-propargyl biotin), resulting in the specific biotinylation of the metabolically labeled proteins (7). The final step is extraction of the labeled proteins by streptavidin beads.
In the present study we pursued a completely new strategy to determine proteomic changes caused by a tumor-specific mutation in a major signaling pathway exemplified by the activin receptor type II (ACVR2) tumor suppressor. Disruption of activin signaling is a frequent event during colorectal tumorigenesis and can be caused by different molecular mechanisms. In colon tumors with chromosomal instability but microsatellite stability, ACVR2 inactivation mainly occurs via epigenetic mechanisms, whereas in colon cancers with a high level of microsatellite instability (MSI) loss of ACVR2 emerges from frameshift mutation (8). MSI has been defined as a change in length (insertions/deletions) of small repetitive DNA sequences (microsatellites) that arises specifically in tumor cells with impaired DNA mismatch repair functions (mutator phenotype) but not in corresponding normal tissue (9, 10). The A8 coding microsatellite in exon 10 of the ACVR2 gene has been identified as one of the most frequent mutation targets in high-MSI colorectal tumors that abrogates normal ACVR2 protein expression (11–13). As a consequence, phosphorylation of downstream signaling mediators such as Smad2/3, as well as regulation of several target genes such as SERPINE, SMAD7, and MYC, is significantly impaired, and this contributes to MSI colorectal tumorigenesis.
Our strategy relied on a combination of two established technologies. First, we employed a recombination-mediated genomic targeting approach (14–16) to generate a genetically modified MSI colorectal tumor cell line that enabled doxycycline-inducible and reversible expression of an ACVR2 transgene and activation of ligand-dependent signaling in an isogenic background. Second, we performed metabolic labeling via a click-chemistry approach (7) in this MSI cell line to uncover alterations in the pattern of newly synthesized proteins (de novo proteome). In particular, we determined the constituents of the total cellular de novo proteome and subsets derived thereof that were regulated in an ACVR2-dependent manner. Our combinatorial strategy represents a versatile approach allowing one to overcome the limitations inherent in the genetic heterogeneity and proteomic complexity of MSI tumor cells. Moreover, this model system allows one to address any tumor target gene and thereby possesses broad applicability for studies of cancer proteome complexity, which is the logical starting point for identifying diagnostic biomarkers and therapeutic targets for cancer.
MATERIALS AND METHODS
Plasmids
S2F-cLM2CG-FRT3 (14) contains a tet-controlled bidirectional transcription unit for concurrent regulation of the two reporter genes firefly luciferase and red fluorescent protein mCherry. This expression cassette is flanked by two hetero-specific FLP-recognition sites, a mutated F3 and a wild-type F site (17). Recombination was mediated by the enzyme Flpo-recombinase, encoded by the plasmid pCAGGS-Flpo-IRES-Puro obtained from Michael Hahn (DKFZ, Heidelberg, Germany). The plasmid pE11.F3.HygTK.F (14), encoding a hygromycin B phosphotransferase-thymidine kinase (HygTK) translational fusion protein, was used for antibiotic selection and generation of the HCT116-HygTK master cell line (15). For generation of the retroviral vector S2FcLM2CG-FRT3-ACVR2, the wild-type ACVR2 cDNA was amplified via PCR from the expression plasmid pcDNA3.1/His-ACVR2 (18), and a FLAG-tag was added at the C terminus directly before the stop-codon of ACVR2 via PCR. After restriction digestion, the resulting EcoRI/NotI fragment was cloned into S2F-cLM2CG-FRT3, replacing the EcoRI/NotI mCherry fragment. Correct genomic targeting and insertion of the wild-type ACVR2 gene was confirmed by DNA sequence analysis (supplemental Table S1).
Cell Lines
All cell lines were cultured in DMEM (PAA, Cölbe, Germany) supplemented with 10% heat-inactivated FBS Gold (PAA), 100 U/ml penicillin, and 100 mg/ml streptomycin (PAA) and grown under standard conditions. The human hepatocellular liver carcinoma cell line HepG2 was purchased from ECACC (Salisbury, SP4 0JG, United Kingdom). The generation of the HCT116-HygTK master cell line has been described recently (15). These cells (i) are hygromycin resistant and ganciclovir sensitive, (ii) constitutively express the reverse transcriptional transactivator gene and the EGFP protein under the control of the chicken β-actin promoter (18), (iii) are mismatch-repair-deficient because of a homozygous nonsense mutation in the MLH1 DNA mismatch gene, and (iv) carry biallelic coding repeat frameshift mutations in the ACVR2 gene. Transfection experiments were carried out using Fugene HD Transfection Reagent according to the manufacturer's instructions (Roche Diagnostics, Mannheim, Germany). Hygromycin B (100 μg/ml; PAA), puromycin (1.5 μg/ml; Sigma, Taufkirchen, Germany), and ganciclovir (40 μm; Roche Diagnostics) were used for antibiotic selection. To examine signaling effects, we starved cells for 18 h in the presence and absence of 0.5 to 1 μg/ml doxycycline (Sigma) and subsequently incubated them with 10 ng/ml recombinant Activin A (Sigma) for 2 h. For metabolic labeling experiments using AHA (Invitrogen, Karlsruhe, Germany), cells were cultured in methionine-free RPMI medium (Sigma) supplemented with 10% heat-inactivated FBS Gold, 100 U/ml penicillin, and 100 mg/ml streptomycin (PAA) using standard conditions.
Generation of HCT116-ACVR2 Cells
We used a genomic targeting approach described previously (14, 15). 5 × 106 HCT116-HygTK cells were cotransfected with 2 μg of pCAGGS-Flpo-IRES-Puro and 2 μg of S2FcLM2CG-FRT3-ACVR2, and recombinant clones were selected for ganciclovir resistance (Fig. 1). A single cell clone of the resulting HCT116-ACVR2 cells was isolated. Correct genomic targeting of the ACVR2-luciferase cassette was confirmed by primers located in the ACVR2 and flanking genomic DNA sequence (supplemental Table S1). Doxycycline inducibility of this expression cassette was quantitatively determined via luciferase assay.
Fig. 1.
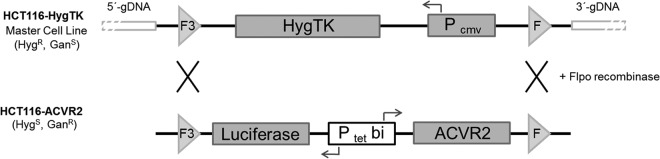
Recombination-mediated cassette exchange strategy. The previously described HCT116-HygTK master cell line (HygR, GanS) carries a HygTK expression cassette that is regulated by a constitutive promoter (PCMV) and is flanked by mutated (F3) and wild-type (F) sites. Co-transfection of these cells with the targeting vector S2F-cLM2CG-FRT3-luc-ACVR2 and an Flpo recombinase encoding plasmid leads to recombination-mediated replacement of the HygTK expression cassette by the dox-regulated bidirectional luciferase-ACVR2 expression cassette. The resulting HCT116-ACVR2 cells (GanR) were chosen for subsequent analyses.
Luciferase Assay
Luciferase activity was measured in duplicate according to the manufacturer's instructions (Luciferase Assay System, Promega, Mannheim, Germany). Luciferase activity was indicated as relative light units normalized to protein concentration as measured via Bradford assay (Bio-Rad, Munich, Germany).
Isolation of Genomic DNA, PCR, and Sequencing
Genomic DNA was isolated from cancer cell lines using the DNeasy Tissue kit (Qiagen, Hilden, Germany). For a standard PCR approach, TaqDNA polymerase and standard buffers were used (Invitrogen, Karlsruhe, Germany). PCR was performed in RoboCycler Gradient 96 (Invitrogen) using the following conditions: initial denaturation, 15 min at 95 °C; 35 30-s cycles of denaturation at 95 °C; annealing, 30 s at 60 °C; elongation, 30 s to 1 min at 72 °C; and final extension, 10 min at 72 °C. PCR products were purified using the High Pure PCR Product Purification Kit according to the manufacturer's protocol (Roche Diagnostics). The BigDye Terminator v1.1 sequencing kit (Invitrogen) was used for DNA sequence analysis. For data analysis we used Sequencing Analysis software (Version 3.7, Applied Biosystems, Darmstadt, Germany).
Real-time PCR Analysis
For cDNA synthesis, 1 μg of total RNA was isolated with the RNeasy Kit (Qiagen) and reverse transcribed using oligo-dT primers and SuperScript II reverse transcriptase according to the manufacturer's instructions (Invitrogen). For real-time PCR experiments, specific primers (supplemental Table S1) and PowerSYBR Green Master Mix (Applied Biosystems) were used. Real-time PCR analysis was performed in triplicate on different cDNA samples (-dox versus +dox) using an ABI StepOnePlus Cycler (Applied Biosystems) with the following conditions: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 60 s. Quantitative analysis of gene expression was performed relative to the expression of GAPDH in corresponding samples using standard amplification curves for each transcript.
Immunoprecipitation and Western Blotting
Cells were harvested with PBS, and cell pellets were lysed in RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton-X-100, 1% sodium deoxycholate, 0.1 mm CaCl2, 0.1% SDS, 0.01 mm MgCl2) supplemented with protease inhibitor mixture (Complete EDTA-free, Roche Diagnostics) and phosphatase inhibitor mixture (Roche Diagnostics). After sonication and incubation for 1 h at 4 °C on a rotator, cell lysates were centrifuged at 12,000 × g for 20 min at 4 °C. For immunoprecipitation of FLAG-labeled ACVR2, anti-FLAG M2 Affinity Gel (Sigma) was applied according to the manufacturer's instructions. A total volume of 600 μl containing 5 mg of protein was mixed with 130 μl of affinity gel slurry and incubated overnight on a rotor mixer. After three washes with 1 ml of TBS, the immunoprecipitated ACVR2-FLAG protein was eluted with 70 μl of 2× SDS-PAGE sample buffer at 99 °C for 5 min. After centrifugation at 1000 × g, 20 μl of the supernatant was subjected to Western blotting. For Western blot analysis, 50 μg of cell extracts or the immunoprecipitate was separated on NuPAGE Bis-Tris gradient mini gels (4–12%, Invitrogen) and blotted onto nitrocellulose via semi-dry electroblotting. Subsequently, blots were blocked for 30 min in blocking buffer (20 mm Tris-HCl, pH 7.5, 0.5 m NaCl, 0.1% Tween-20, and 5% non-fat dry milk) prior to the addition of primary antibodies in blocking buffer. Rabbit anti-ACVR2A antibody (kindly provided by B. Jung, Northwestern University, Chicago, IL; 1:500 at 4 °C overnight), anti-phosho-Smad2 (1:1000 at 4 °C overnight; Ser465/467, Cell Signaling, Danvers, MA), anti-Smad2 (1:1000 at 4 °C overnight; 86F7, Cell Signaling), and anti-β-Actin (1:30,000 at room temperature for 1 h; MP Biomedicals, Solon, Ohio) were used as primary antibodies. After five washes in 0.1% Tween-20, horseradish-peroxidase-coupled secondary rabbit (1:2500, Promega, Madison, WI) or mouse (1:5000, GE Healthcare, Munich, Germany) antibodies were added in blocking buffer for 1 h at room temperature. After three washing steps in 0.1% Tween-20, signals were detected using Western Lightning Plus ECL (PerkinElmer). As a transfer and loading control, the blotting membranes were stained with 0.1% Ponceau S (w/v in 5% acetic acid).
Labeling and Enrichment of Newly Synthesized Proteins
2 × 106 cells per dish were plated in triplicate onto 100-mm dishes suspended in 10 ml of RPMI medium containing ±0.5 μg/ml doxycycline. When cells reached ∼80% confluency (usually after 24 h), they were metabolically labeled with 40 μm Click-iT AHA (Invitrogen) in methionine-free RPMI medium (Sigma) supplemented with 10 ng/ml Activin A in the absence or presence of 0.5 μg/ml doxycycline for 4 h. As a control for the detection of newly labeled proteins, cells were also grown under the same culture conditions but in the absence of the labeling reagent AHA. Then cells were lysed with 1% SDS in 50 mm Tris-HCl, pH 8, supplemented with protease inhibitor mixture and phosphatase inhibitor mixture. After sonication and incubation for 30 min at 4 °C on a rotator, cell lysates were centrifuged at 12,000 × g for 20 min at 4 °C. Aliquots from the supernatants corresponding to 200 μg of protein were reacted with biotin alkyne (PEG4 carboxamide-propargyl biotin, Invitrogen) according to the manufacturer's instructions. After methanol/chloroform precipitation (500 μl of methanol, 150 μl of chloroform, and 400 μl of H20 per 200-μl sample), the precipitates were redissolved in 200 μl of RIPA buffer. Finally, labeled proteins were extracted with 40 μl of streptavidin-coated magnetic beads (Dynabeads MyOne Streptavidin T1, Invitrogen) for 2 h at 4 °C on a rotator. Then the beads were washed three times with 1 ml of Phosphate-Buffered Saline, Tween-20 containing 2% SDS and finally with PBS and 40 mm ammonium bicarbonate.
Sample Preparation for Mass Spectrometry
Protein-loaded beads were suspended in 100 μl of 10 mm DTT in 40 mm ammonium bicarbonate and reduced at 45 °C for 1 h in a thermomixer (Eppendorf, Hamburg, Germany) at 800 rpm. Free cysteine residues were alkylated with 100 μl of iodoacetamide (55 mm in 40 mm ammonium bicarbonate) for 30 min at room temperature in the dark. Between each step, beads were washed with 100 μl of 40 mm ammonium bicarbonate. Then samples were digested at 37 °C overnight using 50 μl of trypsin solution (5 μl of 0.5 μg/μl trypsin (Promega) in 1 mm HCl diluted in 495 μl of 40 mm ammonium bicarbonate). The resulting peptides were transferred to PCR tubes, and after the addition of 5 μl of 1% trifluoroacetic acid (Thermo Scientific, Karlsruhe, Germany), samples were sonicated for 5 min (Transsonic 310, Elma, Singen, Germany). After centrifugation at 12,000 × g, 5 μl of each sample were analyzed via nano-LC electrospray ionization MS/MS.
Electrospray Ionization MS/MS Analysis and Database Search
Tryptic peptide mixtures were separated using a nanoAcquity ultra-high-performance LC system (Waters GmbH, Eschborn, Germany). Peptides were trapped on a nanoAcquity C18 column (180 μm × 20 mm, 5-μm particle size; Waters GmbH). The liquid chromatography separation was performed on a C18 column (BEH 130 C18, 100 μm × 100 mm, 1.7-μm particle size; Waters GmbH) with a flow rate of 350 nl/min. For all samples, the chromatography was carried out using a 3-h gradient of solvent A (98.9% water, 1% acetonitrile, 0.1% formic acid) and solvent B (99.9% acetonitrile and 0.1% formic acid) in the following sequence: from 0% to 4% B in 1 min, from 4% to 30% B in 140 min, from 30% to 45% B in 15 min, from 45% to 90% B in 5 min, 10 min at 90% B, from 90% to 0% B in 0.1 min, and 9.9 min at 0% B. The ultra-high-performance LC system was coupled online to an LTQ Orbitrap XL mass spectrometer (Thermo Scientific). The mass spectrometer was operated in the sensitive mode with the following parameters: capillary voltage, 2400 V; capillary temperature, 200 °C; normalized collision energy, 35 V; activation time, 30,000 ms. Data were acquired in scan cycles of one Fourier transform MS scan with a resolution of 60,000 at m/z 400 and a range from 370 to 2000 m/z in parallel with six MS/MS scans in the ion trap of the most abundant precursor ions. The MGF files generated by Xcalibur software (Thermo Scientific, Waltham, MA) were used for database searches with the MASCOT search engine (version 2.4, Matrix Science, London, W1U 7GB, United Kingdom) against the Swiss-Prot database (version 2013 02, 539,165 sequences, 191,456,931 residues). The taxonomy was set to “human,” except for the bead proteome, which was searched with the taxonomy “all” to identify non-human proteins used for blocking. The peptide mass tolerance for database searches was set to 5 ppm, and the fragment mass tolerance was set at 0.4 Da. Carbamidomethylation of C was set as a fixed modification. Variable modifications included oxidation of M and deamidation of N and Q. One missed cleavage site in the case of incomplete trypsin hydrolysis was allowed. Furthermore, proteins were considered as identified if more than one unique peptide had an individual ion score exceeding the MASCOT identity threshold (ion score cutoff of 30). Identification under the applied search parameters refers to a false discovery rate between 2% and 4% and a match probability of p < 0.01, where p is the probability that the observed match is a random event. Candidate proteins were classified as ACVR2-deficient or -proficient if detected in at least two out of three biological replicates. In order to control for the unspecific binding and detection of proteins, a sample containing unlabeled proteins (in the absence of AHA) was also subjected to MS analysis and selected according to the same criteria as mentioned above.
RESULTS
HCT116-ACVR2 Cells as a Model System for Inducible ACVR2 Gene Expression
High-MSI CRC cell lines carrying an ACVR2 frameshift mutation represent an excellent model system for investigating the effects of perturbations of ACVR2 expression and/or signaling on protein expression. As recipient cells we used the recently described colorectal cancer master cell line HCT116-HygTK (15), which exhibits several important features. First, it is MMR-deficient and high-MSI, and thus reflects the molecular phenotype of the corresponding tumors. Second, these cells lack normal ACVR2 function as a result of homozygous frameshift mutations in the A8 coding microsatellite of the endogenous ACVR2 gene. Third, this cell line constitutively expresses a reverse transcriptional transactivator that enables dox-regulated expression of target genes driven by a tet-responsive promoter (14). Finally, at a specific genomic site these cells carry a HygTK marker gene flanked by two recombination sites enabling integration of any gene of interest at this particular site. Applying a genomic targeting approach (Fig. 1), we inserted a dox-regulated ACVR2-luciferase expression cassette into this specific genomic site of HCT116-HygTK cells via a single recombination step, replacing the HygTK expression cassette and conferring ganciclovir resistance to the recombinant clones. The resulting HCT116-ACVR2 cells showed bidirectional expression of the ACVR2 gene and luciferase reporter gene in a dox-dependent manner. Because this reversible expression occurred in an otherwise isogenic background, ACVR2-dependent effects on the cellular proteome could be specifically detected. These genetically modified HCT116-ACVR2 cells were further characterized and used for subsequent analyses.
Genetic and Functional Characterization of HCT116-ACVR2 Cells
We next examined the genetic integrity and functional reconstitution of the ACVR2 gene in these HCT116-ACVR2 cells. Because the cells lack normal mismatch repair function, the A8 coding microsatellite of the reconstituted ACVR2 gene might be affected by frameshift mutations during the selection and growth of HCT116-ACVR2 cells. In order to prevent such a setback, we performed DNA fragment length analysis on genomic DNA from HCT116-HygTK and HCT116-ACVR2 cells, which allowed amplicon length resolution at the single-nucleotide level. Capillary electrophoresis revealed two peaks in HCT116-ACVR2 cells that indicated amplicons of different lengths (Fig. 2A). The first peak corresponded to an amplicon harboring the ACVR2 mutant (A7) coding microsatellite, whereas the second peak originated from an amplicon comprising the ACVR2 wild-type (A8) repeat. In contrast, HCT116-HygTK cells showed only a single peak due to A7 coding repeat frameshift mutations in both alleles of the endogenous ACVR2 gene. The existence of the two alleles was also confirmed by DNA sequence analysis (data not shown). These results demonstrate that HCT116-ACVR2 cells have successfully integrated a wild-type copy of the ACVR2 transgene into their genome.
Fig. 2.

Genetic and functional reconstitution of ACVR2. A, PCR-based fragment analysis of the ACVR2 A7/A8 coding repeat alleles in parental HCT116-HygTK cells and ACVR2-reconstituted HCT116-ACVR2 cells. Electropherograms show a single peak in HCT116-HygTK cells (homozygous mutant A7 alleles of the endogenous ACVR2 gene), in contrast to HCT116-ACVR2 cells that carry a wild-type A8 allele (marked by asterisk) in addition to the endogenous mutant allele. DNA fragment size is given by nucleotide numbers. B, inducible ACVR2 expression using immunoprecipitation (anti-Flag) followed by Western blot analysis (anti-ACVR2A) (upper section) and Ponceau S staining as a loading control (lower section). In the presence of doxycycline for 24 h, the expression of the receptor protein was detected after immunoprecipitation in the established HCT116-ACVR2 cell line, whereas in the absence of dox no protein was visible, just as in the parental HCT116 cell line. 293T cells were transiently transfected using the pcDNA3.1/His-ACVR2 construct and used as a positive control (50 μg of total lysate). C, detection of Smad2 protein phosphorylation (pSmad2) via Western blot analysis. Treatment with dox (1 μg/ml) and ActA (10 ng/ml) led to higher levels of pSmad2 than in cells grown in the absence of dox. TGF-β1-responsive HepG2 cells served as a positive control. Total Smad2 and Actin were used as a loading control. D, ACVR2-dependent regulation of target gene transcription. Real-time PCR experiments demonstrated dox-dependent up-regulation (Smad 7, Serpine) or down-regulation (cMyc) of ACVR2 target genes. Bars represent the mean and S.D. of three independent experiments.
In order to quantify the range of dox-inducible ACVR2 transgene expression, we determined induction levels of luciferase activity as a surrogate marker. In the presence of dox, HCT116-ACVR2 cells showed a ∼250-fold increase of luciferase activity relative to the same cells grown in the absence of dox. The ability to induce the ACVR2/luciferase expression cassette was maintained in cultured cells for at least one year. This demonstrates the robustness and genetic stability of this heterologous expression system in a background of mismatch repair deficiency.
In order to verify the successful induction of ACVR2 protein expression, we immunoprecipitated the induced ACVR2 protein by targeting its FLAG-tag and performed subsequent immunoblotting. The ACVR2 protein was detected as a pattern of bands with an apparent molecular weight in the range of 62 KDa in immunoprecipitates from induced (+dox) HCT116-ACVR2 cells, but not from uninduced (−dox) HCT116-ACVR2 or parental HCT116 cells (Fig. 2B). HEK293 cells transiently transfected with an ACVR2 expression plasmid showed the same band pattern and served as a positive control. In addition to proving the induction of ACVR2 protein expression, we also examined the functional reconstitution of the ACVR2 signaling pathway in HCT116-ACVR2 cells. Canonical ACVR2-mediated signaling is initiated by ligand-induced (activin A (ActA)) ACVR1–ACVR2 receptor heterodimerization that leads to the recruitment and phosphorylation of receptor-regulated Smad proteins (Smad2, Smad3). These phospho-proteins (pSmad2, pSmad3) associate with the co-mediator Smad4 and, upon translocation to the nucleus and in cooperation with nuclear cofactors, regulate the transcription of target genes such as MYCC, SMAD7, and SERPINE (19). Thus, we analyzed pSmad2 protein by immunoblotting lysates of HCT116-ACVR2 cells that had been grown in the presence or absence of doxycycline and/or ligand (Fig. 2C). In the absence of ActA and independent of ACVR2 reconstitution, HCT116-ACVR2 cells expressed a very low basal amount of pSmad2. This basal pSmad2 level was increased upon ActA exposure even in the absence of functional ACVR2 protein (+ActA, −dox), a well-known phenomenon that has been reported previously (20). However, a significant additional increase in pSmad2 was observed upon dox-induced reconstitution of ACVR2 expression in the presence of ActA (+ActA, +dox). This ACVR2-dependent signaling activity, as well as proteomic changes derived therefrom, can thus be interrogated through comparative analysis of HCT116-ACVR2 cells grown in the presence of ligand and in the presence or absence of dox. The observed alterations are pSmad2 specific because overall Smad2 protein levels remained unaffected. As final proof of reconstituted ACVR2 signaling, we examined the regulation of three known target genes of this pathway. Dox and ligand-induced ACVR2 signaling caused an up-regulation of Smad7 and Serpine transcripts and modest down-regulation of the MycC transcript as assessed via real-time PCR analysis (Fig. 2D). These results confirm the genetic and functional reconstitution of ACVR2-dependent signaling in HCT116-ACVR2 cells.
ACVR2-dependent Alterations of the de Novo Proteome
In recent work with ACVR2 transiently transfected cells, we obtained preliminary evidence that ACVR2 might affect the pattern of newly (de novo) synthesized cellular glycoproteins (18). Using a novel combinatorial approach, we have now used our genetically modified cell line model system together with a click-chemistry approach to determine ACVR2-dependent alterations of the de novo cellular proteome that might contribute to MSI colorectal tumorigenesis. As a basic principle, the click-chemistry approach relies on metabolic labeling with azidohomoalanine, an azide-bearing analog of methionine that, upon incorporation into the nascent polypeptide chain, can form a stable triazole conjugate with alkyne-activated biotin, thereby enabling selective capture and covalent binding of de novo synthesized proteins to streptavidin-conjugated magnetic beads and subsequent identification of tryptic peptides via mass spectrometry (Fig. 3). By applying this approach to ActA-stimulated HCT116-ACVR2 cells grown in the presence (+ACVR2) or absence (−ACVR2) of doxycycline and using several filter and selection criteria, we identified a set of 513 proteins referred to herein as the “de novo proteome” (supplemental Table S2). Human keratins detected within this set are well-known contaminants resulting from sample processing. By analyzing the magnetic beads used to capture the modified proteins, we identified five proteins resulting from preblocking of the beads by the manufacturer (supplemental Table S3). Because of the quite harsh washing conditions, nonspecific binding as tested in a control without azidohomoalanine was almost negligible. Apart from keratin contaminations, only seven proteins of the de novo proteome were found in this control (supplemental Table S4). Only proteins recognized by at least two unique peptides and with an ion score of at least 30 were considered valid candidates. The majority of these de novo synthesized proteins, including the non-specifically bound proteins (94%, 480/513), were detected in both dox-treated and -untreated cells and thus remained unaffected by the ACVR2 expression status (Fig. 4). However, a subset of proteins (5%, 25/513) showed a clear ACVR2 dependence and thus defined the ACVR2-dependent de novo proteome. Most of these proteins (20/25, 80%) were detected exclusively in dox-treated ACVR2-proficient HCT116-ACVR2 cells (Fig. 4, Table I). For some of these proteins, such as CALU (21), DEK (22), LETM1 (23), IBP2 (24), IF4B (25), KI67 (26), MCM2 (27), NP1L1 (28), RBM8A (29), and S10AB (30), a link to tumorigenesis has been reported. In contrast, a much smaller number of de novo synthesized proteins (5/25, 20%) was associated with a lack of ACVR2 expression in HCT116-ACVR2 cells (Fig. 4, Table I). De novo synthesized proteins under these conditions might be of particular clinical relevance, because a loss of normal ACVR2 signaling is one of the most frequent events that occur during the development of MSI colorectal tumors. Candidate proteins of this subset include Flap endonuclease 1 (31–33), a histone H2A variant (H2A1C) (34), a Rho-related GTP-binding protein (RhoC) (35, 36), a 60S ribosomal protein (RL10a) (37), and a subunit of the facilitator of chromatin transcription complex (SPT16H) (38–40). Based on current knowledge, several of the de novo protein candidates identified in ACVR2-deficient as well as in ACVR2-proficient HCT116 cells (30–40) can be functionally assigned to hallmark processes of cancer development (supplemental Fig. S1) (41) Overall, these results illustrate the potential of our approach to correlate a tumor-relevant driver mutation (ACVR2) with specific changes in the tumor cell proteome. Moreover, the combination of our cellular model system with specific proteomic analysis represents a powerful novel approach for identifying gene-specific cellular proteomic states, and it also can easily be adapted for other genes or modulators of cellular growth in a time- and dose-dependent manner.
Fig. 3.
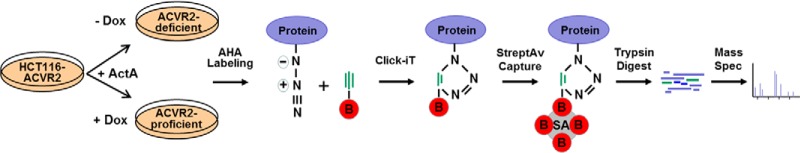
Differential analysis of ACVR2-dependent alterations affecting de novo synthesized proteins. Upon exposure to the ligand activin A (ActA), HCT116-ACVR2 cells were grown in the presence (+) or absence (−) of doxycycline (Dox). Cells were labeled with l-azidohomoalanine (AHA) in methionine-depleted (−Meth) medium for 4 h. AHA-labeled proteins were detected with biotin-alkyne (B). Subsequent to the click reaction, biotinylated proteins were captured with streptavidin (SA)-coated magnetic beads, subjected to tryptic digestion, and analyzed via mass spectrometry.
Fig. 4.
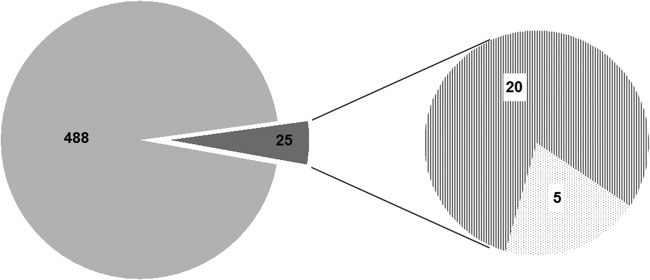
De novo proteome of HCT116-ACVR2 cells. Individual metabolically labeled protein species detected via mass spectrometry in both ACVR2-proficient and ACVR2-deficient cells (light gray area). The subset of differentially expressed proteins is indicated (dark gray) and splits up into protein species expressed exclusively in ACVR2-proficient (dashed area) or ACVR2-deficient cells (dotted area).
Table I. De novo proteome of ACVR2-proficient and ACVR2-deficient cells.
| Accession number | Protein description | Mass (Da) | Score | Peptides | Coverage (%) |
|---|---|---|---|---|---|
| ACVR2 proficient | |||||
| CALU_HUMAN | Calumenin | 37,198 | 188/308 | 2/7 | 26.3/49.8 |
| CC124_HUMAN | Coiled-coil domain-containing protein 124 | 25,820 | 122/121 | 2/2 | 26/16.1 |
| DEK_HUMAN | Protein DEK | 42,933 | 79/81 | 2/2 | 5.1/5.9 |
| DX39B_HUMAN | Spliceosome RNA helicase DDX39B | 49,416 | 270/301 | 4/4 | 16.6/16.6 |
| IBP2_HUMAN | Insulin-like growth factor-binding protein 2 | 35,875 | 105/177 | 2/2 | 7.7/7.7 |
| IF4B_HUMAN | Eukaryotic translation initiation factor 4B | 69,167 | 92/81/107 | 2/2/3 | 6.5/6.5/7.9 |
| KI67_HUMAN | Antigen KI-67 | 360,698 | 91/112 | 2/2 | 0.9/1 |
| LAP2B_HUMAN | Lamina-associated polypeptide 2, isoforms β/γ | 50,696 | 306/303 | 5/6 | 24/21.1 |
| LETM1_HUMAN | LETM1 and EF-hand domain-containing protein 1, mitochondrial | 83,986 | 80/122 | 2/3 | 3.2/6.1 |
| MCM2_HUMAN | DNA replication licensing factor MCM2 | 102,516 | 66/70 | 2/2 | 2.8/3.9 |
| NP1L1_HUMAN | Nucleosome assembly protein 1-like 1 | 45,631 | 155/127 | 2/2 | 11.8/9.7 |
| PDIA6_HUMAN | Protein disulfide-isomerase A6 | 48,490 | 163/143 | 2/3 | 12. Dez |
| PRS8_HUMAN | 26S protease regulatory subunit 8 | 45,768 | 168/100 | 3/2 | 12.0/12.0 |
| RBM8A_HUMAN | RNA-binding protein 8A | 19,934 | 71/78/92 | 2/2/2 | 10.9/10.9/10.9 |
| RL17_HUMAN | 60S ribosomal protein L17 | 21,611 | 126/145 | 3/3 | 21.7/21.7 |
| S10AB_HUMAN | Protein S100-A11 | 11,847 | 116/103 | 2/2 | 30.5/32.4 |
| SF3B3_HUMAN | Splicing factor 3B subunit 3 | 136,575 | 114/89 | 2/2 | 2.1/2.1 |
| SMD2_HUMAN | Small nuclear ribonucleoprotein Sm D2 | 13,632 | 109/82 | 2/2 | 32.2/16.1 |
| TBB4A_HUMAN | Tubulin β-4A chain | 50,010 | 675/794 | 11/13 | 33.3/44.4 |
| TNPO1_HUMAN | Transportin-1 O | 103,771 | 62/128 | 2/2 | 2.3/3.6 |
| ACVR2 deficient | |||||
| FEN1_HUMAN | Flap endonuclease 1 | 42,908 | 92/79 | 2/2 | 9.7/8.2 |
| H2A1C_HUMAN | Histone H2A type 1-C | 14,097 | 159/189 | 3/3 | 32.3/32.3 |
| RHOC_HUMAN | Rho-related GTP-binding protein RhoC | 22,334 | 196/229 | 4/2 | 46.6/46.6 |
| RL10A_HUMAN | 60S ribosomal protein L10a | 24,987 | 73/80 | 2/2 | 11.1/11.1 |
| SP16H_HUMAN | FACT complex subunit SPT16 | 120,409 | 97 | 2/2 | 4.5/5.2 |
Occurrence under these conditions is mutually exclusive. Only protein hits with at least two unique peptides and with a minimal ion score of 30 were considered as identified. Experiments were performed in biological triplicate (experiments 1–3). Protein molecular weights (“Mass”), Mascot scores (“Score”) of at least two samples, the number of unique peptides (“Peptides”), and protein sequence coverage (“Coverage”) are displayed for each protein annotated in UniProt.
DISCUSSION
The development of colorectal cancer is a multistep process usually occurring in a timeframe of many years. During that time, healthy colon tissue homeostasis is disrupted predominantly by a shift in the balance among several key signal transduction pathways, including TGFβ, BMP, ACVR2, Wnt, and Notch pathways (42, 43). Intensive molecular characterization of CRC tumors at the DNA and RNA level has shaped our current knowledge about the molecular mechanisms that drive the formation of CRC. The next step is to assign molecular, cellular, and biochemical functions to these predicted gene products and to explain how these products regulate complex physiological processes in cancer. Screening the proteome of tumors is a complex issue because the proteome is dynamic and constantly changing as a result of a combination of several factors (44). These include differential splicing of the respective mRNAs, post-translational modifications, and temporal and functional regulation of gene expression (45). Proteome analysis of CRC tumors can be performed using cultured cell lines or clinical material, whereas for basic functional analyses cell lines are more frequently used. This is mainly because of (i) the well-defined sample material; (ii) the wide range of practicability in analysis of the effects of individual genes; (iii) the effects of different drugs; (iv) subcellular cell compartments such as the nuclei, exosome, cell surface, and secretome; (v) the comparison of different methodologies; and (vi) the option to perform metabolic labeling (46–54). Many studies have identified colon-cancer-associated proteins; however, only a few studies have focused on the functional protein interaction networks derived from the proteomics data. The long-term goal is to understand the cellular protein networks at a level that will permit a totally new paradigm of diagnosis and allow therapy tailored to individual patients and situations. Therefore, the strength of our approach lies in studying the consequences of a major signaling pathway on the proteomic assembly with emphasis on the de novo proteome. By combining two approaches—recombination-mediated cassette exchange and Click-iT chemistry—we established a CRC cell line that provides a regulated target gene expression system and enables analysis of gene-specific effects on newly synthesized protein patterns. To the best of our knowledge, there are no studies published in this field of de novo proteomics in CRC.
In order to analyze ACVR2-mediated downstream signaling effects, we used the HCT116-ACVR2 cell line model system, which is a versatile tool for studying the functional consequences and biological importance of a given MSI tumor-associated mutation. Because the inducible expression of the ACVR2 transgene can be regulated by dox and the ACVR2 transgene has been integrated as a single copy at a defined site in the HCT116 genome, physiological levels of the wild-type ACVR2 protein can be achieved. It is important to note that results obtained from this ACVR2-reconstituted model system should reflect the inverse situation of the ACVR2-deficient status in MSI primary colorectal tumors, as induction with dox activates the wild-type ACVR2 signaling pathway. Our model system allows the ACVR2-dependent incorporation of the azide-containing amino acid and thereby the simple and efficient isolation of newly synthesized, labeled proteins from a complex proteomic background via click chemistry. The incorporation of azidohomoalanine can be compared with metabolic labeling using [35S]-methionine, but it avoids the use of radioactive compounds and enables the specific isolation of the labeled proteins. On-bead tryptic digestion of the streptavidin-bound proteins was chosen for the preparation of peptides for mass spectrometric analysis because initial experiments showed that even very harsh conditions did not allow quantitative elution of proteins from the beads (data not shown). An additional advantage is the possibility of applying very stringent washing conditions to reduce nonspecific binding to the beads. This was demonstrated with the control sample prepared without label.
Our model system not only possesses broad and easy applicability, but given that we were able to identify more than 500 metabolically labeled proteins, it also enables highly sensitive analyses. However, we also obtained evidence that we might have missed extremely low-abundant proteins. For example, we were not able to identify the ACVR2 protein in the dox-induced cells with this approach. Although our strategy aims to detect physiological expression levels, this detection limit is apparently not reached for low-abundant proteins such as the ACVR2 receptor, which is present in fewer than 500 copies per cell (55). However, we were able to verify ACVR2 induction by combining immunoprecipitation with Western blotting. Additionally, our proof of reconstituted ACVR2 signaling in the dox-induced cells demonstrates that functional amounts of the receptor are expressed.
We succeeded in detecting a subset of 25 proteins that showed clear ACVR2 dependence. Five of these proteins were specifically identified in ACVR2-deficient cells and are involved in key cellular processes such as DNA repair and chromatin dynamics (FEN1, HIST1H2AC, SPTH16), protein translation (RPL10A), and signal transduction (RHOC). Some of these candidates have already been linked to colorectal cancer or play a key role in cell growth or apoptosis control. For example, RhoC, a member of the Ras homologous subfamily of low-molecular-weight GTP-binding proteins, is known to be overexpressed in colon cancer and various other malignancies. It can promote post-epithelial-to-mesenchymal-transition cell migration and, because of its correlation with tumor progression, has been proposed as a prognostic marker for colon carcinoma (35, 36). Also, the DNA repair protein FEN1 has been reported to show high expression levels in proliferative tissues, and its expression in tumor tissues has been correlated with elevated tumor grade and increased invasiveness (31, 32). Moreover, several studies suggest that expression of the histone chaperone SUPT16H, as part of the heterodimeric FACT complex, can act in a pro-tumorigenic manner because it modulates nucleosome assembly and chromatin architecture, including the exchange of histone variants. Given that the SUPT16H subunit of FACT preferentially binds to the core histone complex H2A-H2B (56), there might be a causal link to the core histone H2A family member H2A1C that we also found to be expressed in ACVR2-deficient cells. Other studies indicate that SUPT16H/FACT expression is restricted to cells at the crypt bottom in normal colon epithelium, and it appears to be associated with a stem/progenitor cell expression signature, suggesting its involvement in maintaining an undifferentiated status (38). This is well in line with the known “stem-like” phenotype of the MSI colorectal cancer cell line HCT116 (34) upon which our model system is based. Given these correlations, it is tempting to speculate that the de novo synthesized candidate proteins identified in ACVR2-deficent HCT116 cells might help to establish their tumorigenic phenotype, a hypothesis that requires further investigation.
In contrast to what we observed in ACVR2-deficient HCT116 cells, a much larger number of de novo synthesized candidate proteins were observed upon reconstitution of ACVR2 signaling. The resulting proteomic changes are expected to reflect the growth-suppressing and apoptosis-promoting effects of this signaling pathway. In fact, several of the candidate proteins, such as CALU, IBP2, LETM1, PRS8, SF3B3, and TNPO1, have been reported to confer such growth-inhibitory effects. For example, the calcium-binding protein CALU can interfere with cytokinesis when added exogenously and also has been found to be associated with mitotic arrest and late apoptosis upon translocation from the membrane to the cytoplasm (57, 58). Similarly, the delicate balance among insulin-like growth factor receptors (IGF-IR, IGF-IIR), ligands (IGF1/2), and binding proteins (IGFBP1–6) usually ensures proper survival factor signaling in epithelial cells (59), but it would be severely disturbed by, for instance, high levels of IBP2 and the resulting inadequate levels of IGF1/2, thereby restricting tumor cell growth and proliferation (60). LETM1, another ACVR2-dependent candidate protein, is known to exhibit a very complex pathophysiology because of its involvement in Wolf–Hirschhorn Syndrome, ion homeostasis, and mitochondrial biogenesis (61), but it also can induce programmed tumor cell death upon overexpression (23, 62). Likewise, the 26S proteasomal component PRS8, also known as PSMC5, has been demonstrated to suppress colon cancer stem cell growth in mouse xenografts, and its yeast ortholog is able to initiate caspase-8-dependent cell death (63, 64). Moreover, evidence suggests that the spliceosomal SF3B3 protein is able to extend the half-life of the cell cycle inhibitor p27 (65). Finally, it has been demonstrated that the TNPO1 protein can mediate nuclear import of selected proteins and, in an oxidative environment with high levels of reactive oxygen species, translocates the transcriptional regulator FOXO4, thereby causing cell cycle arrest (66).
Overall, these examples illustrate the potential of our approach to detect tumor-relevant proteomic changes. Nevertheless, it is also important to emphasize that candidate proteins not yet linked to tumor-relevant functions should not be completely ignored, as some yet unknown mechanism or interaction with other proteins might be effective.
Although we were able to demonstrate the principal utility of our approach to detect tumor-relevant proteomic changes, there is also potential for further development and refinement of the methodology. In the present study we focused on soluble proteins. Because the extraction of the labeled proteins with high concentrations of detergents or chaotropic substances would not interfere with subsequent binding of biotin-labeled proteins to streptavidin beads, it would be possible to apply it to membrane-bound or other difficult-to-solubilize proteins. An attractive refinement would be the combination of our strategy with quantitative proteomic analytical methods. Thus our approach could be combined with SILAC or ITRAQ quantification. Also, a focus could be laid on tumor-gene-related changes in glycoprotein expression through the application of azido- or alkyne-derivatized building blocks of protein-bound oligosaccharide chains instead of azidohomoalanine (67). Finally, combination with various prefractionation methods, including electrophoretic and chromatographic methods, is feasible.
As a whole, this study, based on a combination of two technologies, provides a versatile platform for analysis of the proteomic de novo assembly of any MSI-relevant target gene of interest. This isogenic MSI tumor cell line model system allowed us to analyze the ACVR2 signaling mechanisms that contribute to the de novo protein synthesis in MSI tumor cells and to identify the proteomic assembly. Thus, we suggest that this approach might facilitate the identification of tumor markers that could be used for diagnostic and therapeutic applications.
Supplementary Material
Acknowledgments
We gratefully acknowledge S. Himmelsbach and M. Karl for excellent technical assistance. The anti-ACVR2 polyclonal serum was kindly provided by J. Bauer and B. Jung, Northwestern University, Chicago, IL.
Footnotes
Author contributions: J.G. and J.K. designed research; S.B. and J.L. performed research; M.S. contributed new reagents or analytic tools; S.B., J.L., U.W., and M.S. analyzed data; S.B., J.G., and J.K. wrote the paper.
* Financial support was provided by DFG (GE592/6–2 to J.K. and J.G.).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- CRC
- colorectal cancer
- AHA
- l-azidohomoalanine
- ACVR2
- activin receptor type II
- MSI
- microsatellite instability
- HygTK
- hygromycin B phosphotransferase-thymidine kinase
- ActA
- activin A.
REFERENCES
- 1. Vogelstein B., Papadopoulos N., Velculescu V. E., Zhou S., Diaz L. A., Jr., Kinzler K. W. (2013) Cancer genome landscapes. Science 339, 1546–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rahn J. J., Adair G. M., Nairn R. S. (2012) Use of gene targeting to study recombination in mammalian cell DNA repair mutants. Methods Mol. Biol. 920, 445–470 [DOI] [PubMed] [Google Scholar]
- 3. Kohli M., Rago C., Lengauer C., Kinzler K. W., Vogelstein B. (2004) Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 32, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Santiago Y., Chan E., Liu P. Q., Orlando S., Zhang L., Urnov F. D., Holmes M. C., Guschin D., Waite A., Miller J. C., Rebar E. J., Gregory P. D., Klug A., Collingwood T. N. (2008) Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc. Natl. Acad. Sci. U.S.A. 105, 5809–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baum C., Kustikova O., Modlich U., Li Z., Fehse B. (2006) Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum. Gene Ther. 17, 253–263 [DOI] [PubMed] [Google Scholar]
- 6. Ong S. E. (2012) The expanding field of SILAC. Anal. Bioanal. Chem. 404, 967–976 [DOI] [PubMed] [Google Scholar]
- 7. Dieterich D. C., Lee J. J., Link A. J., Graumann J., Tirrell D. A., Schuman E. M. (2007) Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat. Protoc. 2, 532–540 [DOI] [PubMed] [Google Scholar]
- 8. Jung B., Gomez J., Chau E., Cabral J., Lee J. K., Anselm A., Slowik P., Ream-Robinson D., Messer K., Sporn J., Shin S. K., Boland C. R., Goel A., Carethers J. M. (2009) Activin signaling in microsatellite stable colon cancers is disrupted by a combination of genetic and epigenetic mechanisms. PLoS One 4, e8308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boland C. R., Goel A. (2010) Microsatellite instability in colorectal cancer. Gastroenterology 138, 2073–2087, e2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lawes D. A., SenGupta S., Boulos P. B. (2003) The clinical importance and prognostic implications of microsatellite instability in sporadic cancer. Eur. J. Surg. Oncol. 29, 201–212 [DOI] [PubMed] [Google Scholar]
- 11. Duval A., Hamelin R. (2002) Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res. 62, 2447–2454 [PubMed] [Google Scholar]
- 12. Hempen P. M., Zhang L., Bansal R. K., Iacobuzio-Donahue C. A., Murphy K. M., Maitra A., Vogelstein B., Whitehead R. H., Markowitz S. D., Willson J. K., Yeo C. J., Hruban R. H., Kern S. E. (2003) Evidence of selection for clones having genetic inactivation of the activin A type II receptor (ACVR2) gene in gastrointestinal cancers. Cancer Res. 63, 994–999 [PubMed] [Google Scholar]
- 13. Jung B., Doctolero R. T., Tajima A., Nguyen A. K., Keku T., Sandler R. S., Carethers J. M. (2004) Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology 126, 654–659 [DOI] [PubMed] [Google Scholar]
- 14. Weidenfeld I., Gossen M., Low R., Kentner D., Berger S., Gorlich D., Bartsch D., Bujard H., Schonig K. (2009) Inducible expression of coding and inhibitory RNAs from retargetable genomic loci. Nucleic Acids Res. 37, e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee J., Ballikaya S., Schonig K., Ball C. R., Glimm H., Kopitz J., Gebert J. (2013) Transforming growth factor beta receptor 2 (TGFBR2) changes sialylation in the microsatellite unstable (MSI) colorectal cancer cell line HCT116. PLoS One 8, e57074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Welman A., Barraclough J., Dive C. (2006) Generation of cells expressing improved doxycycline-regulated reverse transcriptional transactivator rtTA2S-M2. Nat. Protoc. 1, 803–811 [DOI] [PubMed] [Google Scholar]
- 17. Schlake T., Bode J. (1994) Use of mutated FLP recognition target (FRT) sites for the exchange of expression cassettes at defined chromosomal loci. Biochemistry 33, 12746–12751 [DOI] [PubMed] [Google Scholar]
- 18. Patsos G., Andre S., Roeckel N., Gromes R., Gebert J., Kopitz J., Gabius H. J. (2009) Compensation of loss of protein function in microsatellite-unstable colon cancer cells (HCT116): a gene-dependent effect on the cell surface glycan profile. Glycobiology 19, 726–734 [DOI] [PubMed] [Google Scholar]
- 19. Deacu E., Mori Y., Sato F., Yin J., Olaru A., Sterian A., Xu Y., Wang S., Schulmann K., Berki A., Kan T., Abraham J. M., Meltzer S. J. (2004) Activin type II receptor restoration in ACVR2-deficient colon cancer cells induces transforming growth factor-beta response pathway genes. Cancer Res. 64, 7690–7696 [DOI] [PubMed] [Google Scholar]
- 20. Jung B. H., Beck S. E., Cabral J., Chau E., Cabrera B. L., Fiorino A., Smith E. J., Bocanegra M., Carethers J. M. (2007) Activin type 2 receptor restoration in MSI-H colon cancer suppresses growth and enhances migration with activin. Gastroenterology 132, 633–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Galamb O., Gyorffy B., Sipos F., Spisak S., Nemeth A. M., Miheller P., Dinya E., Molnar B., Tulassay Z. (2007) [Identification of colorectal cancer, adenoma, and inflammatory bowel disease specific gene expression patterns using whole genomic oligonucleotide microarray system]. Orvosi. Hetilap. 148, 2067–2079 [DOI] [PubMed] [Google Scholar]
- 22. Lin L., Piao J., Gao W., Piao Y., Jin G., Ma Y., Li J., Lin Z. (2013) DEK over expression as an independent biomarker for poor prognosis in colorectal cancer. BMC Cancer 13, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Piao L., Li Y., Kim S. J., Byun H. S., Huang S. M., Hwang S. K., Yang K. J., Park K. A., Won M., Hong J., Hur G. M., Seok J. H., Shong M., Cho M. H., Brazil D. P., Hemmings B. A., Park J. (2009) Association of LETM1 and MRPL36 contributes to the regulation of mitochondrial ATP production and necrotic cell death. Cancer Res. 69, 3397–3404 [DOI] [PubMed] [Google Scholar]
- 24. Diehl D., Hessel E., Oesterle D., Renner-Muller I., Elmlinger M., Langhammer M., Gottlicher M., Wolf E., Lahm H., Hoeflich A. (2009) IGFBP-2 overexpression reduces the appearance of dysplastic aberrant crypt foci and inhibits growth of adenomas in chemically induced colorectal carcinogenesis. Int. J. Cancer 124, 2220–2225 [DOI] [PubMed] [Google Scholar]
- 25. Chen H. J., Jiang Y. L., Lin C. M., Tsai S. C., Peng S. F., Fushiya S., Hour M. J., Yang J. S. (2013) Dual inhibition of EGFR and c-Met kinase activation by MJ-56 reduces metastasis of HT29 human colorectal cancer cells. Int. J. Oncol. 43, 141–150 [DOI] [PubMed] [Google Scholar]
- 26. Oshima C. T., Iriya K., Forones N. M. (2005) Ki-67 as a prognostic marker in colorectal cancer but not in gastric cancer. Neoplasma 52, 420–424 [PubMed] [Google Scholar]
- 27. Giaginis C., Georgiadou M., Dimakopoulou K., Tsourouflis G., Gatzidou E., Kouraklis G., Theocharis S. (2009) Clinical significance of MCM-2 and MCM-5 expression in colon cancer: association with clinicopathological parameters and tumor proliferative capacity. Dig. Dis. Sci. 54, 282–291 [DOI] [PubMed] [Google Scholar]
- 28. Line A., Slucka Z., Stengrevics A., Silina K., Li G., Rees R. C. (2002) Characterisation of tumour-associated antigens in colon cancer. Cancer Immunol. Immunother. 51, 574–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Solier S., Barb J., Zeeberg B. R., Varma S., Ryan M. C., Kohn K. W., Weinstein J. N., Munson P. J., Pommier Y. (2010) Genome-wide analysis of novel splice variants induced by topoisomerase I poisoning shows preferential occurrence in genes encoding splicing factors. Cancer Res. 70, 8055–8065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang G., Wang X., Wang S., Song H., Sun H., Yuan W., Cao B., Bai J., Fu S. (2008) Colorectal cancer progression correlates with upregulation of S100A11 expression in tumor tissues. Int. J. Colorectal Dis. 23, 675–682 [DOI] [PubMed] [Google Scholar]
- 31. Kim I. S., Lee M. Y., Lee I. H., Shin S. L., Lee S. Y. (2000) Gene expression of flap endonuclease-1 during cell proliferation and differentiation. Biochim. Biophys. Acta 1496, 333–340 [DOI] [PubMed] [Google Scholar]
- 32. Lam J. S., Seligson D. B., Yu H., Li A., Eeva M., Pantuck A. J., Zeng G., Horvath S., Belldegrun A. S. (2006) Flap endonuclease 1 is overexpressed in prostate cancer and is associated with a high Gleason score. BJU Int. 98, 445–451 [DOI] [PubMed] [Google Scholar]
- 33. Liu L., Zhou C., Zhou L., Peng L., Li D., Zhang X., Zhou M., Kuang P., Yuan Q., Song X., Yang M. (2012) Functional FEN1 genetic variants contribute to risk of hepatocellular carcinoma, esophageal cancer, gastric cancer and colorectal cancer. Carcinogenesis 33, 119–123 [DOI] [PubMed] [Google Scholar]
- 34. Yeung T. M., Gandhi S. C., Wilding J. L., Muschel R., Bodmer W. F. (2010) Cancer stem cells from colorectal cancer-derived cell lines. Proc. Natl. Acad. Sci. U.S.A. 107, 3722–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bellovin D. I., Simpson K. J., Danilov T., Maynard E., Rimm D. L., Oettgen P., Mercurio A. M. (2006) Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene 25, 6959–6967 [DOI] [PubMed] [Google Scholar]
- 36. Wang H. B., Liu X. P., Liang J., Yang K., Sui A. H., Liu Y. J. (2009) Expression of RhoA and RhoC in colorectal carcinoma and its relations with clinicopathological parameters. Clin. Chem. Lab. Med. 47, 811–817 [DOI] [PubMed] [Google Scholar]
- 37. Lu B., Xu J., Zhu Y., Zhang H., Lai M. (2007) Systemic analysis of the differential gene expression profile in a colonic adenoma-normal SSH library. Clin. Chim. Acta 378, 42–47 [DOI] [PubMed] [Google Scholar]
- 38. Garcia H., Fleyshman D., Kolesnikova K., Safina A., Commane M., Paszkiewicz G., Omelian A., Morrison C., Gurova K. (2011) Expression of FACT in mammalian tissues suggests its role in maintaining of undifferentiated state of cells. Oncotarget 2, 783–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Winkler D. D., Luger K. (2011) The histone chaperone FACT: structural insights and mechanisms for nucleosome reorganization. J. Biol. Chem. 286, 18369–18374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heo K., Kim H., Choi S. H., Choi J., Kim K., Gu J., Lieber M. R., Yang A. S., An W. (2008) FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol. Cell 30, 86–97 [DOI] [PubMed] [Google Scholar]
- 41. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 42. Crosnier C., Stamataki D., Lewis J. (2006) Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat. Rev. Genet. 7, 349–359 [DOI] [PubMed] [Google Scholar]
- 43. Levy L., Hill C. S. (2006) Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 17, 41–58 [DOI] [PubMed] [Google Scholar]
- 44. Tyers M., Mann M. (2003) From genomics to proteomics. Nature 422, 193–197 [DOI] [PubMed] [Google Scholar]
- 45. Van Eyk J. E. (2001) Proteomics: unraveling the complexity of heart disease and striving to change cardiology. Curr. Opin. Mol. Ther. 3, 546–553 [PubMed] [Google Scholar]
- 46. Gu S., Liu Z., Pan S., Jiang Z., Lu H., Amit O., Bradbury E. M., Hu C. A., Chen X. (2004) Global investigation of p53-induced apoptosis through quantitative proteomic profiling using comparative amino acid-coded tagging. Mol. Cell. Proteomics 3, 998–1008 [DOI] [PubMed] [Google Scholar]
- 47. Volmer M. W., Stuhler K., Zapatka M., Schoneck A., Klein-Scory S., Schmiegel W., Meyer H. E., Schwarte-Waldhoff I. (2005) Differential proteome analysis of conditioned media to detect Smad4 regulated secreted biomarkers in colon cancer. Proteomics 5, 2587–2601 [DOI] [PubMed] [Google Scholar]
- 48. Tan H. T., Tan S., Lin Q., Lim T. K., Hew C. L., Chung M. C. (2008) Quantitative and temporal proteome analysis of butyrate-treated colorectal cancer cells. Mol. Cell. Proteomics 7, 1174–1185 [DOI] [PubMed] [Google Scholar]
- 49. Fung K. Y., Lewanowitsch T., Henderson S. T., Priebe I., Hoffmann P., McColl S. R., Lockett T., Head R., Cosgrove L. J. (2009) Proteomic analysis of butyrate effects and loss of butyrate sensitivity in HT29 colorectal cancer cells. J. Proteome Res. 8, 1220–1227 [DOI] [PubMed] [Google Scholar]
- 50. Turck N., Richert S., Gendry P., Stutzmann J., Kedinger M., Leize E., Simon-Assmann P., Van Dorsselaer A., Launay J. F. (2004) Proteomic analysis of nuclear proteins from proliferative and differentiated human colonic intestinal epithelial cells. Proteomics 4, 93–105 [DOI] [PubMed] [Google Scholar]
- 51. Mathivanan S., Lim J. W., Tauro B. J., Ji H., Moritz R. L., Simpson R. J. (2010) Proteomics analysis of A33 immunoaffinity-purified exosomes released from the human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Mol. Cell. Proteomics 9, 197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xue H., Lu B., Zhang J., Wu M., Huang Q., Wu Q., Sheng H., Wu D., Hu J., Lai M. (2010) Identification of serum biomarkers for colorectal cancer metastasis using a differential secretome approach. J. Proteome Res. 9, 545–555 [DOI] [PubMed] [Google Scholar]
- 53. Slebos R. J., Brock J. W., Winters N. F., Stuart S. R., Martinez M. A., Li M., Chambers M. C., Zimmerman L. J., Ham A. J., Tabb D. L., Liebler D. C. (2008) Evaluation of strong cation exchange versus isoelectric focusing of peptides for multidimensional liquid chromatography-tandem mass spectrometry. J. Proteome Res. 7, 5286–5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boyce M., Bertozzi C. R. (2011) Bringing chemistry to life. Nat. Methods 8, 638–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Beck M., Schmidt A., Malmstroem J., Claassen M., Ori A., Szymborska A., Herzog F., Rinner O., Ellenberg J., Aebersold R. (2011) The quantitative proteome of a human cell line. Mol. Syst. Biol. 7, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Winkler D. D., Muthurajan U. M., Hieb A. R., Luger K. (2011) Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. J. Biol. Chem. 286, 41883–41892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ostergaard M., Hansen G. A., Vorum H., Honore B. (2006) Proteomic profiling of fibroblasts reveals a modulating effect of extracellular calumenin on the organization of the actin cytoskeleton. Proteomics 6, 3509–3519 [DOI] [PubMed] [Google Scholar]
- 58. Bull V. H., Fargestad E. M., Strozynski M., Thiede B. (2010) Temporal proteome profiling of taxol-induced mitotic arrest and apoptosis. Electrophoresis 31, 1873–1885 [DOI] [PubMed] [Google Scholar]
- 59. Samani A. A., Yakar S., LeRoith D., Brodt P. (2007) The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocrine Rev. 28, 20–47 [DOI] [PubMed] [Google Scholar]
- 60. Michell N. P., Langman M. J., Eggo M. C. (1997) Insulin-like growth factors and their binding proteins in human colonocytes: preferential degradation of insulin-like growth factor binding protein 2 in colonic cancers. Br. J. Cancer 76, 60–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nowikovsky K., Pozzan T., Rizzuto R., Scorrano L., Bernardi P. (2012) Perspectives on: SGP symposium on mitochondrial physiology and medicine: the pathophysiology of LETM1. J. Gen. Physiol. 139, 445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hwang S. K., Piao L., Lim H. T., Minai-Tehrani A., Yu K. N., Ha Y. C., Chae C. H., Lee K. H., Beck G. R., Park J., Cho M. H. (2010) Suppression of lung tumorigenesis by leucine zipper/EF hand-containing transmembrane-1. PLoS One 5, e12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhao Y., Zhang W., Guo Z., Ma F., Wu Y., Bai Y., Gong W., Chen Y., Cheng T., Zhi F., Zhang Y., Wang J., Jiang B. (2013) Inhibition of the transcription factor Sp1 suppresses colon cancer stem cell growth and induces apoptosis in vitro and in nude mouse xenografts. Oncol. Rep. 30, 1782–1792 [DOI] [PubMed] [Google Scholar]
- 64. Kumar Y., Radha V., Swarup G. (2010) Interaction with Sug1 enables Ipaf ubiquitination leading to caspase 8 activation and cell death. Biochem. J. 427, 91–104 [DOI] [PubMed] [Google Scholar]
- 65. Cordero-Espinoza L., Hagen T. (2013) Regulation of Cullin-RING ubiquitin ligase 1 by Spliceosome-associated protein 130 (SAP130). Biol. Open 2, 838–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Putker M., Madl T., Vos H. R., de Ruiter H., Visscher M., van den Berg M. C., Kaplan M., Korswagen H. C., Boelens R., Vermeulen M., Burgering B. M., Dansen T. B. (2013) Redox-dependent control of FOXO/DAF-16 by transportin-1. Mol. Cell 49, 730–742 [DOI] [PubMed] [Google Scholar]
- 67. Laughlin S. T., Bertozzi C. R. (2009) Imaging the glycome. Proc. Natl. Acad. Sci. U.S.A. 106, 12–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.