

Abstract
Overwintering plants are capable of exhibiting high levels of cold tolerance, which is acquired through the process of cold acclimation (CA). In contrast to CA, the acquired freezing tolerance is rapidly reduced during cold de-acclimation (DA) and plants resume growth after sensing warm temperatures. In order to better understand plant growth and development, and to aid in the breeding of cold-tolerant plants, it is important to decipher the functional mechanisms of the DA process. In this study, we performed comparative transcriptomic and proteomic analyses during CA and DA. As revealed by shotgun proteomics, we identified 3987 peptides originating from 1569 unique proteins and the corresponding mRNAs were analyzed. Among the 1569 genes, 658 genes were specifically induced at the transcriptional level during the process of cold acclimation. In order to investigate the relationship between mRNA and the corresponding protein expression pattern, a Pearson correlation was analyzed. Interestingly, 199 genes showed a positive correlation of mRNA and protein expression pattern, indicating that both their transcription and translation occurred during CA. However, 226 genes showed a negative correlation of mRNA and protein expression pattern, indicating that their mRNAs were transcribed during CA and were stored for the subsequent DA step. Under this scenario, those proteins were specifically increased during DA without additional transcription of mRNA. In order to confirm the negative correlation of mRNA and protein expression patterns, qRT-PCR and western blot analyses were performed. Mitochondrial malate dehydrogenase 1 (mMDH1) exhibited a negative correlation of mRNA and protein levels, which was characterized by CA-specific mRNA induction and protein accumulation specifically during DA. These data indicate that the expression of specific mRNAs and subsequent accumulation of corresponding proteins are not always in accordance under low temperature stress conditions in plants.
Low temperature stress is one of the major environmental stress factors that affect plant distribution, growth, and productivity. It is well-known that overwintering plants are capable of adapting to cold temperatures via an orchestrated expression of several sets of genes (1–4). This process is termed as cold acclimation (CA)1 and through this mechanism, plants are capable of acquiring freezing tolerance (5, 6). Especially, overwintering plants acquire freezing tolerance and are capable of surviving under persistent freezing conditions. Previous studies have demonstrated that CA is a complex process that is comprised of multiple steps such as the alteration of membrane fluidity and lipid composition, accumulation of compatible solutes and the regulation of gene expression via the DREB/CBF regulon (1–3, 7–9). Plants are also known to suspend growth under stress conditions as an adaptive measure to avoid damage from freezing temperature. In relation to the observed effect of growth suspension under cold conditions, overexpression of DREB/CBF, which is an important transcription factor for freezing tolerance, exhibits growth retardation under normal conditions (10, 11).
In contrast to CA, cold de-acclimation (DA) is an important regulatory mechanism as a result of the perception of warm temperatures that enables overwintering plants to resume growth in Spring. In comparison to CA, the DA response is more rapid under both field and experimental conditions. In the case of Solanum species, 15 days are required in order to acquire maximum cold tolerance during CA, however, this acquired cold tolerance is lost within only 1 day during DA (12). Similarly, cabbage seedlings have also been reported to lose freezing tolerance and to decrease sugar contents within 24 h of DA (13). In order to better understand the interrelated processes of freezing tolerance and plant growth and development, it is important to decipher the functional mechanisms of regulated gene and protein expression during CA and DA.
RNA regulation is a highly coordinated mechanism that functions to directly affect and regulate the cellular levels of mRNAs and proteins. Previous studies have investigated the global changes of stabilization and/or degradation of mRNA in plants (14, 15). These RNA regulatory mechanisms were shown to be controlled by RNA particles such as stress granules (SGs) and processing bodies (PBs) that are involved in mRNA stabilization and degradation, respectively (16, 17). Functional analyses with defective mutants for various components of these particles revealed sensitive phenotypes under abiotic stress conditions and confirmed the association of these particles to abiotic stress tolerance (18, 19). These data indicate that the regulatory processes of RNA, such as mRNA stabilization or degradation, are important functional components for plant stress responses.
In the case of CA in plants, many cold-inducible mRNAs have been identified by transcriptome analyses with microarray (2, 4). In addition, cold-inducible proteins have also been identified with proteome analyses using 2D electrophoresis or high throughput shotgun proteomics (20–22). However, the resultant data from these two approaches are not completely concordant with one another. It is also known that gene and protein expression patterns might not be identical because of post-transcriptional or translational regulation (23, 24). As a result, we hypothesized that a specific type of RNA regulation, such as post-transcriptional or translational regulation, occurs in plants in response to stress. During CA, many cold-inducible mRNAs are transcribed, however, not all of these transcripts are translated. Some mRNAs are degraded and others are stored in an untranslated state for anticipatory preparation to subsequent environmental alterations. These stored mRNAs are expressed during CA but their corresponding proteins are not translated during CA and are saved for subsequent translation at a later point in time (23, 24).
In order to identify the specific regulatory targets during the processes of CA and DA, we performed comparative transcriptomic and proteomic analyses during CA and DA. The present study revealed several putative targets of translational regulation under low temperature stress in plants. The identification of these targets provides a foundation of evidence to enable us to more completely understand the mechanism of RNA regulation in CA and DA in plants.
EXPERIMENTAL PROCEDURES
Plant Materials, Growth Conditions, and Cold Treatment
Arabidopsis thaliana ecotype Columbia-0 was used as the plant source material for the majority of the experiments. Plants were grown on Murashige and Skoog medium plates containing a final concentration of 1% sucrose at a normal growth temperature of (22 °C) under long day (16h/8h light/dark) conditions at 50–75 μmolm−2s−1. Two week-old Arabidopsis samples (NA, nonacclimated) were treated with cold (2 °C) for 7 days (CA7d) under 12 h/12 h light/dark conditions at 75–100 μmolm−2s−1. Subsequent to cold treatment, DA-treated samples (DA6h, DA12h, and DA24h) were grown at 22 °C for 24 h under long day conditions and samples were harvested at five time points; NA, CA7d, DA6h, DA12h, and DA24h for further experiments.
Plate-based Freezing Tolerance Analysis
A temperature controlled assessment of freezing tolerance was performed essentially as previously described (9, 25) under dark conditions. Plates containing NA, CA, or DA-treated Arabidopsis plants were pretreated at −2 °C for 2 h. Plates were nucleated for freezing by dipping the edge of plates into liquid nitrogen. Nucleated plates were subsequently exposed to a controlled freezing program at a cooling rate of (−1 °C/2 h). After reaching desired set-points of sub-zero temperatures, plates were then transferred to 4 °C and thawed overnight in dark conditions. The plates were then returned to normal growth temperature for a period of 2 weeks and the number of surviving plants were counted. A total of three biological replicates were performed.
Shotgun Proteomics
Arabidopsis plants were grown on MS plates and treated for CA and DA as described above with three biological replicates. 15N isotope-labeled standard proteins were prepared in order to normalize each sample. Standard plants were grown in 15N substituted MS medium using a stable isotope of 15N of KNO3 and NH4NO3 (SI science, Saitama, Japan) and treated for CA and DA. Total proteins were extracted from CA and DA-treated samples (three biological replicates) using extraction buffer (10 mm Tris-HCl pH 9.0 containing 8 m of urea). Details of sample preparation and nano-LC-MS/MS analysis database searching were previously described (26). Protein samples were digested by trypsin after reduction with dithiothreitol and alkylation with iodoacetamide. Desalted peptide samples were analyzed with a LTQ-Orbitrap XL (Thermo Fisher Scientific, Waltham, MA) that was coupled with a Dionex Ultimate3000 pump and an HTC-PAL autosampler (CTC analytics, Zwingen, Switzerland). A self-pulled needle (150 mm length × 100 μm i.d., 6-μm opening) packed with ReproSil C18 materials (3 μm; Dr. Maisch GmbH) was used as an analytical column with a “stone-arch” frit. A spray voltage of 2400 V was applied and the injection volume was 1 μg/5 μl with a flow rate of 500 nL min−1. The mobile phases consisted of 0.5% acetic acid (A) and 0.5% acetic acid and 80% acetonitrile (B). A three-step linear gradient of 5% to 10% B in 5 min, 10% to 40% B in 60 min, 40% to 100% B in 5 min, and 100% B for 10 min was employed. The MS scan range was m/z 300 to 1500. The top-10 precursor ions were selected in the MS scan by Orbitrap with resolution = 60,000. Subsequent MS/MS scans were performed with an ion trap in the automated gain control mode, where the automated gain control values of 5.00e + 05 and 1.00e + 04 were set for full MS and MS/MS, respectively. The normalized collision-induced dissociation was set to 35.0. A lock mass function was used for the LTQ-Orbitrap XL in order to obtain constant mass accuracy during the gradient analysis.
Database Searching and Quantitative Analysis
The Mass Navigator version 1.2 (Mitsui Knowledge Industry, Tokyo, Japan) with the default parameters for LTQ-Orbitrap XL was used to create peak lists on the basis of the recorded fragmentation spectra. The m/z values of the isotope peaks were converted to the corresponding monoisotopic peaks when the isotope peaks were selected as the precursor ions. In order to improve the quality of MS/MS spectra, the Mass Navigator software discarded all peaks that were less than 10 absolute intensity and with less than 0.1% of the most intense peaks in the MS/MS spectra. Peptides and proteins were identified by means of automated database searching using Mascot version 2.2 (Matrix Science, London, UK) in The Arabidopsis Information Resource database (TAIR8_pep_20080412, ftp://ftp.arabidopsis.org/home/tair/Sequences/blast_datasets/TAIR8_blastsets/, the number of sequences is 32825) with a precursor mass tolerance of 3 ppm and a fragment ion mass tolerance of 0.8 Da. Furthermore, strict trypsin specificity was employed, which allowed for up to a total of two missed cleavages. Carbamidomethylation of Cys was set as a fixed modification and oxidation of Met was allowed as a variable modification. Peptides were considered identified if the Mascot score was over the 95% confidence limit based on the “identity” score of each peptide.
For peptide quantification, we used a 15N isotope labeling method as shown in supplemental Fig. S1. 15N isotope-labeled samples were prepared for all five points of the time course samples: NA, CA7d, DA6h, DA12h, and DA24h. A standard sample (which is referred to as “Standard”) was prepared by mixing an equal amount of the labeled samples. All nonlabeled samples (which are referred to as “Target”) were spiked with an equal amount of the standard sample and analyzed by LC-MS/MS. The peak heights of the identified peptides (Target) and the corresponding 15N isotope-labeled peptide (Standard) pairs in all samples were determined using Mass Navigator v1.2. Relative peptide abundances were calculated by using the peak heights of 15N isotope-labeled peptides as standards. In order to quantify protein expression levels, unique peptides were selected under the conditions where the corresponding peaks were detected in more than 36/45 experiments (three biological and three technical replicates with five time points of samples) after the “Quantitation score” (Mass Navigator) was cut off at 0.7. In order to remove any error from peaks, we assessed the value of “Peak intensity at MSMS triggered retention time”, which represents the confidence of peptide peaks. When the value of “Peak intensity (Standard)” was “-1”, the “Peak top height (Target)” was calculated as a “nondetected” peak. When the value of the Peak intensity (Target) was “-1”, the, Peak top height (Target) was calculated as “0”. The average values of peptide “Peak top heights” were calculated in each experiment using the peptide values of “Peak top height (Target)” divided by “Peak top height (Standard)”. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (27) via the PRIDE partner repository with the dataset identifier PXD001216.
Microarray data were utilized for comparative analyses of expression levels of each peptide to respective mRNA's corresponding to those proteins. In order to detect a relationship between mRNA and peptide levels, Pearson product-moment correlation coefficients for each gene were calculated from Z-scores of mRNAs and peptides of all five data points. These Z-scores were separately obtained from mRNA and peptide levels. In the event that multiple peptides were detected from a single protein, peptides with the highest Pearson correlation values were used as relative expression of its protein and for further analysis.
Microarray Analysis
Total RNA was extracted from CA and DA-treated samples (three biological replicates) using the Plant RNA reagents (Invitrogen, Carlsbad, CA). After isolation, RNAs were synthesized into cDNAs using 500 ng of total RNA. cRNA was labeled with a single color (Cy3) using Quick Amp labeling kit (Agilent Technologies, Palo Alto, CA) and hybridized to an Arabidopsis oligo DNA microarray Ver. 3.0 (Agilent Technologies). The microarray data are available on the GEO website (GEO ID: GSE49796). Arrays were scanned with a microarray scanner (G2505B, Agilent Technologies) and analyzed using GeneSpring Ver.7 (Agilent Technologies). Raw signals less than 0.01 were adjusted to 0.01 and a 75 percentile normalization was performed for each chip. These data were used for reference data of mRNA expression for corresponding proteins, which was analyzed by shotgun proteomics.
Protein Identification of Spots from 2D-PAGE Gel
CA7d or DA6h protein samples were separated on 2D-PAGE and visualized using a SilverSNAP for Mass Spectrometry kit (Thermo Fisher Scientific). Protein spots were cut out from gels and destained using the same SilverSNAP kit. Details pertaining to the sample preparation were previously described (28) and the analysis and identification of peptides by the LTQ-Orbitrap XL system (Thermo Fisher Scientific) was described above.
qRT-PCR and Western Blot Analysis
Total RNAs were prepared from CA and DA-treated samples (three biological replicates) using the Plant RNA Reagent (Invitrogen) and cDNAs were synthesized using Quantitech cDNA synthesis kit (Qiagen, Venlo, Netherlands). The generated cDNAs were subsequently used as templates for qRT-PCR analyses that were performed with StepOne Plus (Applied Biosystems, Foster City, CA) using a Fast SYBR Green MasterMix (Applied Biosystems). The ACT2 gene was used as a reference for CA and DA-treated sample analyses because ACT2 expression was not significantly altered within our microarray analyses under cold and de-acclimated conditions. The following primer sets were used for amplification of mMDH1: 5′-TCAACGGTGTTCCAGATGTC-3′ and 5′-CACCTTCGAGGCAAAGAAAG-3′ and ACT2: 5′-TGCCAATCTACGAGGGTTTCT-3′ and 5′-CTTACAATTTCCCGCTCTGC-3′. Data were subsequently analyzed by StepOne Plus software (Applied Biosystems).
Total proteins were extracted from CA and DA-treated samples and 10 μg of total proteins were separated with SDS-PAGE (29). After transferring to nitrocellulose membrane, Hybond-C (GE Healthcare, Little Chalfont, England), anti-MDH2 antibody (Abcam, Cambridge, MA) and anti-rabbit IgG, which was conjugated with HRP (Santa Cruz Biotechnology, Santa Cruz, CA), were used for the detection of mMDH1. Protein signals were detected using SuperSignal West Pico (Thermo Fisher Scientific) and LAS-4000 (Fuji film, Tokyo, Japan) and image analyses were performed with ImageJ software. Data were analyzed from three biological replicates.
Polysomal Profiling Analysis
A polysomal fraction was prepared using a sucrose density gradient ultracentrifugation method (23, 30). CA and DA-treated seedling samples were ground to a fine powder and 300 μg samples were hydrated in 1 ml of extraction buffer [200 mm Tris pH 9.0 containing 200 mm KCl, 36 mm MgCl2, 25 mm ethylene glycol-bis (β-aminoethylether)-N,N,N',N'-tetraacetic acid (EGTA), 100 mm 2-mercaptoethanol, 50 mg/ml cycloheximide, 50 mg/ml chloramphenicol, 1% (v/v) Triton X-100, 1% (v/v) Brij-35, 1% (v/v) Tween-40, and 1% (v/v) Nonidet P-40]. After removal of cell debris by centrifugation, 1 ml of the supernatant was loaded onto 9 ml of sucrose gradient. Linear 10–50% (w/v) sucrose gradients were prepared according to the methods described by Abe and Davies (31). After centrifugation for 90 min at 4 °C at 40,000 rpm (275,000 × g) using a SW41Ti rotor (Beckman Coulter, Brea, CA), the gradient was loaded into Monitor UV-900 (GE Healthcare) for scanning at 254 nm using a perista pump (ATTO, Tokyo, Japan).
RESULTS
Freezing Tolerance of Arabidopsis
Similar to plants such as winter wheat, rye, barley, and spinach; Arabidopsis thaliana is also capable of cold acclimation and acquiring significant levels of freezing tolerance. Under our experimental conditions, cold acclimated Arabidopsis plants were confirmed to survive at temperatures that were below −12 °C (Fig. 1A). At least 3 days of exposure to low temperature conditions were required for Arabidopsis to acquire a high level of freezing tolerance. It is known that Arabidopsis requires a 1 week period of cold acclimation in order to stimulate a maximum level of freezing tolerance (32). Conversely, when plants were transferred to normal growth temperature conditions after exposure to cold treatments, plants began to rapidly de-acclimate within 24 h under our experimental conditions and freezing tolerance quickly disappeared (Fig. 1B, 1C). In good accordance to previous studies, these data suggest that Arabidopsis is capable of rapidly perceiving warm temperature and quickly transitions away from a CA state (12, 13, 33). In order to better understand the CA and DA mechanism and rapid transitions, we designed a comparative study that incorporated the usage of both CA and DA-treated samples for protein and mRNA analyses.
Fig. 1.

Freezing tolerance of Arabidopsis plants during nonacclimated, cold-acclimated, and de-acclimated conditions. A, Two week-old nonacclimated (NA) and 7 days cold-acclimated (CA) plants were frozen at −8, −10, and −12 °C or B, CA7d. De-acclimated plants (DA6h, DA12h, DA18h, and DA24h) were frozen at −16 °C and then grown at normal temperature for an additional 2 weeks. C, Survival rate was measured using a controlled freezing test at −16 °C with three biological replicates.
Protein Expression and Corresponding mRNA Expression Analysis
To understand the gene and protein regulatory mechanism during CA and DA, protein expression analyses were performed with shotgun proteomics. Corresponding mRNAs were analyzed with microarray analyses using protein and RNA that were extracted from CA and DA-treated plant materials. Two week-old seedlings were used as a control (NA) and were then subsequently treated at 2 °C for 7 days (CA7d). For the DA treatment, plants were harvested at 6, 12, and 24 h after exposure to 22 °C (DA6h, DA12h, and DA24h). For transcriptome analyses, DA4h and DA10h samples were collected and mRNAs were prepared with consideration for the time lag of translation.
According to the shotgun proteomics, 6945 peptides were detected within all of the NA, CA and DA-treated samples and 6307 unique peptides were used for further analysis (supplemental Table S1, annotated MS/MS spectra of the identified peptides were summarized in supplemental Fig. S2). To obtain the expression pattern of mRNA corresponding to the proteins that were identified from the shotgun proteomics approach, we performed microarray analyses with the same CA and DA-treated samples (supplemental Table S2). In order to verify that the experimental conditions were properly implemented for the study, we analyzed the expression patterns for well-known cold-inducible genes (RD29A, COR15a, and KIN2) at both the mRNA and protein level. Results from the analysis of these control genes revealed clear up-regulation in response to low temperature (supplemental Fig. S3) and therefore confirmed that our cold treatment and subsequent transcriptome and proteome analyses were reliable under our experimental conditions. Finally, expression levels of 3987 peptides corresponding 1569 proteins and mRNAs corresponding those protein were used for comparative quantitative analyses during CA and DA (supplemental Table S2).
Identification of Putative Targets of Translational Regulation
To clarify how mRNA expression and the accumulation of protein changes during CA and DA, the ratio of CA/NA or DA/CA of mRNA and proteins were calculated and plotted (Fig. 2). Plotting of the CA/NA data revealed alterations of mRNA and protein during CA (Fig. 2A) and DA/CA showed these changes early within the DA step (Fig. 2B). It was apparent that the ratio values of mRNA and protein expression were different and the mRNA ratio was higher than that of proteins (Fig. 2). This is most likely because of the fact that the induction rate of mRNA and translation speed could not be correlated with each other in all of the cases (34).
Fig. 2.
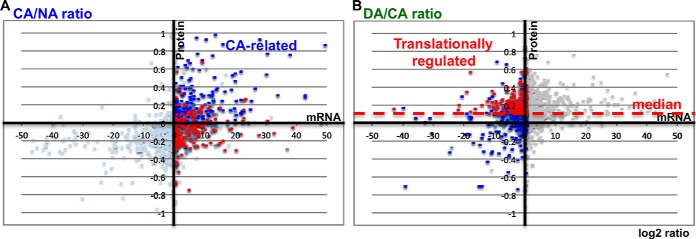
Comparative analysis of mRNA expression and protein expression during CA and DA. mRNA and protein expression patterns were assessed with microarrays and shotgun proteomics, respectively. The log2 ratio of mRNAs and proteins from CA/NA, A, and DA/CA, B, were plotted. In the case of mRNA and protein plots, DA4h/CA7d and DA6h/CA7d were calculated, respectively. Horizontal and vertical plots showed alteration of expression patterns for mRNAs and proteins, respectively. Blue spots indicate the genes that showed a positive correlation of mRNAs and proteins on CA/NA and expression patterns of mRNA and protein showed positive correlation during all points of samples. Red spots indicate the genes that showed values greater than the median (red dotted line) of protein but 0 or negative values of mRNA on DA/CA and expression patterns of mRNA and protein showed negative values for all points of samples.
In order to identify genes that were specifically induced by low temperature, we focused our analysis on the plots where mRNA levels were up-regulated under CA conditions and down-regulated during DA. Among 1569 genes, 658 genes showed positive values of mRNA in CA/NA (Fig. 2A, right half) and negative values of mRNA in DA/CA (Fig. 2B, left half); indicating that these mRNAs were specifically expressed during CA. To distinguish between CA-related genes and putative targets of translational regulation, we performed a Pearson correlation analysis of mRNA and protein expression patterns using all points of samples including NA, CA, and DA. These data were summarized and presented in the supplemental Table S2. When both translational data and correlation data were analyzed among 658 cold-inducible genes, only 199 genes were translated in CA and these mRNA and protein expression patterns were positively correlated (Fig. 2, blue spots). Both mRNA and protein expression patterns of blue spots were up-regulated during the NA to CA step (Fig. 2A) and down regulated during the CA to DA step (Fig. 2B), respectively. In fact, the expression of these 199 genes was specific to CA and their respective proteins also increased during CA. Therefore, it is reasonable to consider that these are CA-related genes/proteins and that they might possess putative functions for freezing tolerance. In contrast, 226 genes were translated specifically in DA (red spots, Fig. 2A, and above median of Fig. 2B) and showed a negative correlation between mRNA and protein in CA and DA-treated samples. This result indicated that the mRNA and protein expression patterns of red spots differed between CA and DA samples. To analyze protein levels in DA/CA, the median of all protein expression data was calculated and used instead of the zero line because the translational speed is increased in DA. When the plots of total mRNAs between CA/NA and DA/CA were compared, the medians of the horizontal direction (mRNAs) were similar. However, when we compared the median of the vertical direction (proteins), the DA/CA total plots shifted upward in comparison to CA/NA in our experimental conditions. It is plausible that these data resulted from a downshift of translational speed under cold temperature conditions. Therefore, it might be deduced that total protein synthesis was activated during the CA to DA step but that the synthesis of mRNA was not significantly altered during the same transition period. In order to address the general shift of translation during CA to DA transition, we used the median of ratio (DA/CA) of total detectable protein instead of a zero line to analyze the DA-specific translational targets in this study. These data indicated that these 226 mRNAs were expressed during CA (Fig. 2A, red spots) but that their corresponding proteins were expressed during DA (Fig. 2B, red spots). According to this comparative analysis, approximately half of the cold-inducible mRNAs were transcribed, translated and functional in CA. However, the other half were transcribed during CA and might be stored for a subsequent step where they gained functionality during DA.
We performed a gene ontology analysis of CA-related and translationally regulated targets as a means to identify the function of these targets of translational regulation. An enrichment test showed that the genes within the functional category of protein metabolism were significantly enriched in translationally regulated targets (Table I). This category contained ribosomal proteins (46 genes out of 226 genes) and translational initiation factors, such as translation initiation factor 3 subunit H1 (TIF3H1), eukaryotic translation elongation factor 1B (EEF1B), eukaryotic translation initiation factor 4A-2 and 3C (EIF4A-2 and EIF3C) (supplemental Table S3). When the detailed expression patterns of these 46 ribosomal genes were analyzed, mRNAs were induced in CA and decreased in DA. However, their corresponding proteins were not CA-inducible and increased during DA (Fig. 3A). In contrast to translationally regulated targets, the expression pattern of CA-related genes such as RD29A, COR15a, and KIN2 showed that both mRNAs and proteins were CA-inducible (Fig. 3B, supplemental Fig. S3, and supplemental Table S4). These data suggested that translational machinery was translated during an early step of DA and that those mRNA were already prepared in a previous step.
Table I. Gene ontology analysis of CA-related and translationally regulated target genes.
| GO Biological process, functional categorya | All genes |
CA-related genes |
Translationally regulated targets |
||
|---|---|---|---|---|---|
| Gene count | Gene count | Enrichmentb | Gene count | Enrichmentb | |
| Protein metabolism | 506 | 75 | 1.86E-01 | 99 | 1.10E-04 |
| DNA or RNA metabolism | 59 | 6 | 8.23E-01 | 14 | 2.71E-02 |
| Other cellular processes | 1176 | 162 | 3.65E-01 | 178 | 7.81E-02 |
| Other metabolic processes | 1147 | 147 | 7.65E-01 | 170 | 1.45E-01 |
| Response to stress | 527 | 78 | 1.84E-01 | 73 | 5.08E-01 |
| Unknown biological processes | 210 | 15 | 9.99E-01 | 29 | 5.31E-01 |
| Response to abiotic or biotic stimulus | 509 | 77 | 1.35E-01 | 68 | 6.41E-01 |
| Cell organization and biogenesis | 426 | 44 | 9.81E-01 | 55 | 7.32E-01 |
| Other biological processes | 448 | 69 | 1.16E-01 | 57 | 7.75E-01 |
| Signal transduction | 123 | 12 | 9.15E-01 | 13 | 8.85E-01 |
| Transport | 323 | 45 | 4.19E-01 | 32 | 9.88E-01 |
| Transcription, DNA-dependent | 109 | 13 | 7.21E-01 | 8 | 9.88E-01 |
| Developmental processes | 318 | 47 | 2.57E-01 | 30 | 9.94E-01 |
| Electron transport or energy pathways | 247 | 33 | 5.43E-01 | 20 | 9.98E-01 |
a Genes were functionally categorized using GO slim in TAIR.
b The p value for overlap was calculated using the hypergeometric distribution.
Fig. 3.

Expression patterns of mRNAs and proteins of regulated targets and cold inducible genes during CA and DA. A, Relative expression patterns of ribosomal protein genes and B, well-known CA-related genes. Left panel shows mRNA expression patterns and right panel contains protein expression patterns. mRNAs were analyzed using microarray data and proteins were evaluated using shotgun proteomics data.
mMDH1 is a Putative Target of Translational Regulation
In addition to the aforementioned experimental analyses, we also compared protein expression patterns using the same time course samples during CA and DA using 2D-DIGE (two dimensional fluorescence difference gel electrophoresis analysis) (supplemental Fig. S4 and supplemental text). Protein spots were quantified by measuring the intensity of fluorescence and 115 protein expression patterns during NA, CA7d, DA6h, DA12h, and DA24h were analyzed. 2D-DIGE analyses revealed that 51 proteins specifically increased during DA and 46 selected protein spots were identified by LC-MS/MS analysis (supplemental Table S5). Corresponding mRNA expression patterns of these identified proteins during CA and DA were also obtained and analyzed from microarray data. Among the proteins that were identified, mitochondrial malate dehydrogenase1 (mMDH1), aspartate aminotransferase 1 (ASP1), and nucleosome assembly protein 1;2 (NAP1;2) represented interesting target candidates of translational regulation. These candidates were contained in the overall list of targets as described above (supplemental Table S3). Because these genes are related to plant development, it is possible that they might function to rapidly resume growth during DA step. In order to confirm that mMDH1 is indeed a target of translational regulation, we analyzed mMDH1 mRNA and protein expression with qRT-PCR and western blot analyses, respectively. qRT-PCR data revealed that mMDH1 mRNA expression increased during CA and these data were in good accordance to those obtained by microarray analysis (Fig. 4). For protein expression analysis, a single band of ∼35.8 kDa was detected, which was consistent with the calculated molecular weight of mMDH1. Similar to the data obtained from the shotgun proteomics, it was confirmed that the mMDH1 protein accumulated during DA (Fig. 4). Taken together, these data demonstrated that mMDH1 mRNA was transcribed during CA but its corresponding protein was not concomitantly accumulated; and was subsequently translated during the de-acclimation period. Therefore, we hypothesize that mMDH1 is one of the translationally regulated targets within Arabidopsis that may function for rapidly resuming growth in de-acclimated plants.
Fig. 4.

mRNA and protein expression patterns of mMDH1 during CA and DA. Total RNA was extracted and converted into cDNA to serve as a template for qRT-PCR analyses. mRNA expression was analyzed using qRT-PCR and protein expression was analyzed by western blot analysis. After normalization to ACT2 mRNA, the relative expression pattern of mMDH1 was presented (left bottom). Data are shown as the means of three biological replicates. Total protein extracts were separated by SDS-PAGE and signal was detected using a MDH antibody (MDH2, Abcam). Relative expression was analyzed using ImageJ software. Data are shown as the means of three biological replicates. Microarray data (left) and shotgun data (right) are presented in the top panel.
Polysomal Profiling Analysis in CA and DA
We performed polysomal profiling with sucrose gradient analysis to examine how the translation state was altered during CA and DA. Messenger ribonucleoprotein (mRNP) complexes were separated into subpolysomes, which contain free RNA and 40S, 60S, and 80S complexes, and polysomes or other large complex fractions (supplemental Fig. S5). A subpolysome fraction containing 40S, 60S, and 80S complexes indicates that this fraction contains the translation initiation step of mRNPs, whose mRNAs were scanned by small subunits of ribosomes and followed by joining the large subunits of ribosomal proteins (Fig. 5). When this subpolysomal fraction was compared in CA and DA, the subpolysome peak decreased in CA5d and CA7d samples and increased in DA6h and DA12h (Fig. 5). These data indicate that translational initiation was decreasing in CA but increasing again during the early step of DA. On the other hand, the polysomal fraction was accumulated in CA compared with NA or DA6h; indicating that CA-related translation was promoted in CA (Fig. 5). Although these data are consistent with previous reports (35, 36), our proteomics data detected approximately the same number of CA-related and translationally regulated targets; indicating that many cold-inducible mRNAs were not translated and stored during CA. Thus, it is possible that this accumulated polysomal peak contains other large complexes such as inactive mRNPs.
Fig. 5.
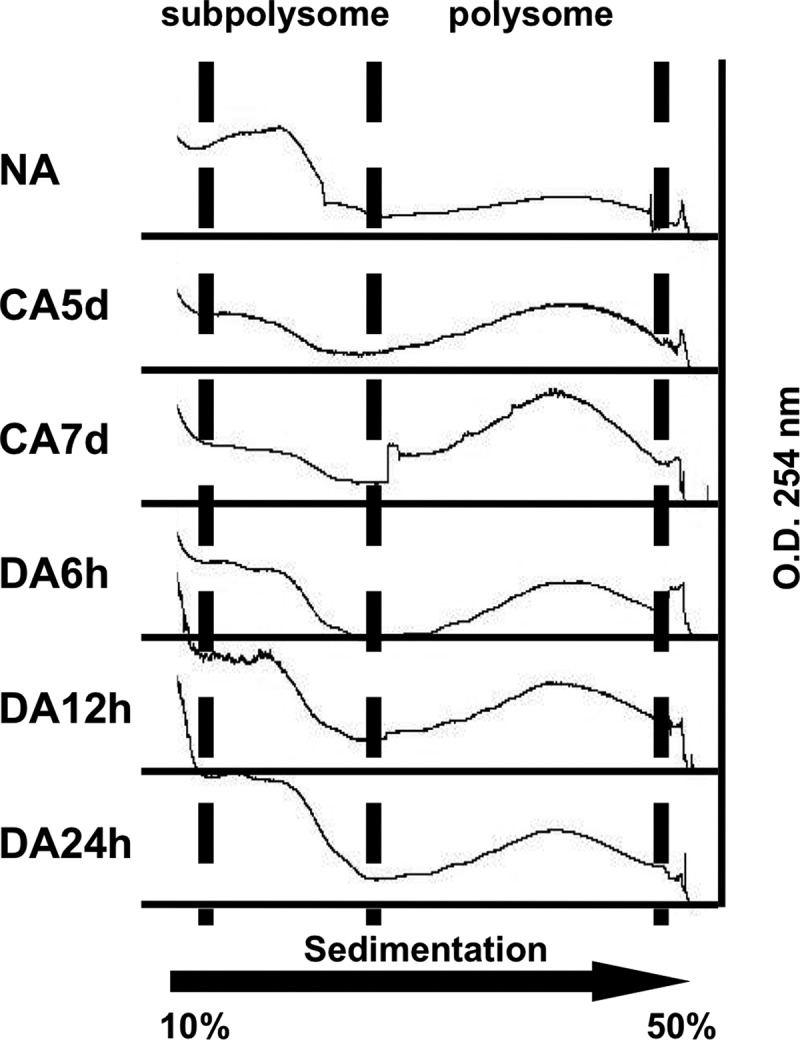
Polysomal profiling during CA and DA. Polysomal fractionation was prepared from NA, CA5d, CA7d, DA6h, DA12h, and DA24h samples and separated through 10–50% sucrose density gradients by ultracentrifugation. Separated complexes were scanned at 254 nm and recorded. The absorbance peaks represent subpolysome containing single ribosome subunits (40S and 60S) or monosomes (80S) and polysome complexes.
DISCUSSION
The process and mechanisms of cold acclimation (CA) have been very well studied at both the physiological and molecular levels; however, the process of de-acclimation (DA) is not as well understood. In order increase our understanding of DA on a molecular level, we designed comparative transcriptome and proteome analyses to characterize the expression profiles of genes and proteins during CA and DA. In this study, we demonstrated that many cold-inducible genes were transcribed and translated, however a similar number of corresponding cold-inducible genes were not translated during the CA stage and were translated at a later stage such as de-acclimation. According to a correlation analysis of mRNA and protein expression patterns, over 200 genes showed negative correlation when their mRNA and protein expression patterns were compared during CA and DA (Fig. 2 and supplemental Table S3). In addition to these gene expression patterns, we identified 286 DA-specific genes, which were translated during DA with de novo synthesis of mRNAs (supplemental Table S6). These data imply that these are likely regulated by translational regulation, which alters protein expression patterns in response to environmental alterations or developmental changes. In animals, Y-box binding protein (YB-1) or frog Y-box protein 2 (FRGY2) repress mRNA translation and enable the storage of mRNAs for subsequent use during embryogenesis or to resume translation by growth stimulation (37, 38). Thus, we hypothesize that these translationally regulated mRNAs were expressed and then stored during CA until the early steps of de-acclimation where they could be rapidly translated for rapid production of corresponding proteins with specific functions during the early stages of the DA process. Because of the disjunction between the up-regulation of specific genes and the lack of corresponding protein accumulation during CA, is likely that the translation of proteins during DA was primed with stored mRNA and occurred without requiring de novo mRNA synthesis. Similar regulation was observed in the case of seed germination; stored mRNA were used during germination without requiring de novo transcription (39).
Interestingly, approximately half of the cold-inducible genes were not translated during the same period and were stored for the subsequent step. Although these are still candidates of translationally regulated targets, many genes might function for the DA process but were not related to freezing tolerance. It was intriguing that the expression of so many proteins were regulated during the cold response of plants, although we knew from experience that the expression of specific mRNAs and subsequent accumulation of corresponding proteins are not always in accordance. These data suggested that transcriptome analysis only informs the expression level of mRNA and should not be used to predict and inform corresponding levels of proteins under low temperature stress conditions in plants.
We performed an analysis of gene ontology in order to elucidate the types of genes that were stored and function in DA. Because ribosomal proteins and translational initiation factors represented the major targets of translational regulation, it is plausible that they possess important functional roles for the DA process. It is apparent that plants possess mechanisms to enable them to rapidly change from a cold acclimated to a de-acclimated state; a process that is initiated by producing translational machinery during the early steps of DA. In contrast to the de-acclimated state, the mRNA that is required for encoding translational machinery is already produced during cold acclimation (Fig. 6). We hypothesize that this mechanism facilitates the rapid resumption of normal cellular functions and metabolism during DA. Polysomal profiling of CA and DA samples revealed that the subpolysome fraction increased during early points of the DA process. These data indicate that translational initiation was increased in DA and this may also be the reason why translational machinery was increased at an early point in the DA process. This is supported by the data pertaining to target candidates that encoded genes related to ribosomal proteins and translational initiation factors. mRNAs encoding ribosomal proteins were stored during CA and translated in DA, but additional ribosomal proteins were identified as CA-related genes in our list of candidates (Functional category: protein metabolism) (Table I and supplemental Table S4). Although an enrichment test did not show significant values compared with translationally regulated targets of protein metabolism (Table I), these data support the hypothesis that plants use different sets of translational machinery for various environmental conditions or stress responses.
Fig. 6.

Possible model of translational regulation during CA and DA in Arabidopsis. CA genes are translated to CA proteins and have potential functional roles in freezing tolerance. Specific sets of CA genes are masked during CA and translated in the early stage of DA. These specific subsets of genes may have specialized roles for resuming plant growth.
Comparative analyses of 2D-DIGE and microarrays revealed additional regulated target candidates encoding primary metabolism related enzymes and plant development related proteins such as mMDH1 and NAP1 (Fig. 4 and supplemental Table S3). Previous studies have documented the functional importance of mMDH1 and confirmed that it is essential for plant growth and development. Mutants of mmdh1 and mmdh2 showed slow growth and altered photorespiration (40). NAP1 is functionally important for plant growth because of its relation to cell cycles via the regulation of transcription or DNA repair (41, 42). Under stress conditions, plants are known to suspend growing as an adaptive mechanism to avoid damage and to promote the resumption of growth after nonstress conditions are sensed (43, 44). We hypothesize that these regulated targets function to rapidly resume plant growth during DA. As a result, plants appear to possess a mechanism to resume plant growth during DA by providing energy or promoting cell cycles during the early step of the DA process (Fig. 6).
As a result of our comparative analysis, we identified a large number of candidates that are targets of translational regulation. It is likely that the mRNAs were stored within inactive translational complexes. RNA granules, such as stress granules, or RNA masking complexes have been studied for their relationship to RNA regulation with a special emphasis on their roles in mRNA stabilization or storage (45, 46). These stress granules or RNA masking complexes consist of untranslated mRNAs and many RNA-binding proteins. It is thought that the size of the complexes might increase under stress conditions (47, 48). Protein synthesis is also thought to be increasing during CA because of the increased polysomal fraction (36). Although our data also showed that the polysomal fraction increased during CA, it is possible that these large complexes contain large inactive complexes such as a RNA masking system or stress granules; in addition to translationally active polysomes.
In this study, we demonstrated that translational regulation is one of the major steps to determine protein levels under stress conditions in plants and that comparative analysis is a powerful technique to identify the RNA regulation mechanism. Although putative targets of translation regulation were identified in our study, the precise mechanism of regulation remains unclear. Further analyses are required to identify and characterize complexes of stress granules or RNA masking components to better understand their functional roles during the CA and DA processes.
Database Accession
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (27) with the dataset identifier PXD001216.
The microarray data are available on the GEO website (GEO ID: GSE49796).
Supplementary Material
Acknowledgments
We thank Dr. Ko Kato (NAIST) and Dr. Hideyuki Matsuura (Osaka University) for experimental advice pertaining to the execution of the polysomal analysis. We would also like to thank Dr. Shinjiro Yamaguchi (Tohoku University) for technical advice and for supporting the 2D-DIGE system. Lastly, we would like to acknowledge Dr. Khurram Bashir (RIKEN CSRS) for critical reading of this manuscript, members of the laboratory, Dr. Jong-Myong Kim and Dr. Taiko Kim To for helpful discussions.
Footnotes
Author contributions: K.N., M.U., K.S., and M.S. designed research; K.N., H.N., A. Minami, Y.N., M.T., and J.I. performed research; K.N., A. Matsui, H.N., Y.N., T.M., and S.T. analyzed data; K.N., A. Matsui, H.N., A. Minami, and M.S. wrote the paper.
* This project was supported by the Special Postdoctoral Researcher's Program from RIKEN and Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 25850247 to K.N., by grants from RIKEN and Japan Science and Technology Agency (JST), Core Research for Evolutionary Science and Technology (CREST) to M.S., and lastly by JSPS KAKENHI Grant Numbers 24228008 to K.S., 24688007 and 26650106 to H.N.
 This article contains supplemental Tables S1 to S6, Figs S1 to S5, and Text.
This article contains supplemental Tables S1 to S6, Figs S1 to S5, and Text.
1 The abbreviations used are:
- CA
- cold acclimation
- DA
- cold de-acclimation
- NA
- nonacclimation
- DREB/CBF
- dehydration-responsive element binding protein/C-repeat-binding factor
- SGs
- stress granules
- PBs
- processing bodies
- 2D-DIGE
- 2 dimensional fluorescence difference gel electrophoresis.
REFERENCES
- 1. Fowler S., Thomashow M. F. (2002) Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell 14, 1675–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maruyama K., Sakuma Y., Kasuga M., Ito Y., Seki M., Goda H., Shimada Y., Yoshida S., Shinozaki K., Yamaguchi-Shinozaki K. (2004) Identification of cold-inducible downstream genes of the Arabidopsis DREB1A/CBF3 transcriptional factor using two microarray systems. Plant J. 38, 982–993 [DOI] [PubMed] [Google Scholar]
- 3. Zhang J. Z., Creelman R. A., Zhu J. K. (2004) From laboratory to field. Using information from Arabidopsis to engineer salt, cold, and drought tolerance in crops. Plant Physiol. 135, 615–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oono Y., Seki M., Satou M., Iida K., Akiyama K., Sakurai T., Fujita M., Yamaguchi-Shinozaki K., Shinozaki K. (2006) Monitoring expression profiles of Arabidopsis genes during cold acclimation and deacclimation using DNA microarrays. Funct. Integr. Genomics 6, 212–234 [DOI] [PubMed] [Google Scholar]
- 5. Guy C. L., Niemi K. J., Brambl R. (1985) Altered gene expression during cold acclimation of spinach. Proc. Natl. Acad. Sci. U.S.A. 82, 3673–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gilmour S. J., Hajela R. K., Thomashow M. F. (1988) Cold acclimation in Arabidopsis thaliana. Plant Physiol. 87, 745–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Palta J. P., Whitaker B. D., Weiss L. S. (1993) Plasma membrane lipids associated with genetic variability in freezing tolerance and cold acclimation of solanum species. Plant Physiol. 103, 793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Uemura M., Steponkus P. L. (1994) A Contrast of the plasma membrane lipid composition of oat and rye leaves in relation to freezing tolerance. Plant Physiol. 104, 479–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xin Z., Browse J. (1998) Eskimo1 mutants of Arabidopsis are constitutively freezing-tolerant. Proc. Natl. Acad. Sci. U.S.A. 95, 7799–7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kasuga M., Liu Q., Miura S., Yamaguchi-Shinozaki K., Shinozaki K. (1999) Improving plant drought, salt, and freezing tolerance by gene transfer of a single stress-inducible transcription factor. Nat. Biotechnol. 17, 287–291 [DOI] [PubMed] [Google Scholar]
- 11. Gilmour S. J., Sebolt A. M., Salazar M. P., Everard J. D., Thomashow M. F. (2000) Overexpression of the Arabidopsis CBF3 transcriptional activator mimics multiple biochemical changes associated with cold acclimation. Plant Physiol. 124, 1854–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen H. H., Li P. H. (1980) Characteristics of cold acclimation and deacclimation in tuber-bearing solanum species. Plant Physiol. 65, 1146–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sasaki H., Ichimura K., Oda M. (1996) Changes in sugar content during cold acclimation and deacclimation of cabbage seedlings. Ann. Bot. 78, 365–369 [Google Scholar]
- 14. Gutierrez R. A., Ewing R. M., Cherry J. M., Green P. J. (2002) Identification of unstable transcripts in Arabidopsis by cDNA microarray analysis: rapid decay is associated with a group of touch- and specific clock-controlled genes. Proc. Natl. Acad. Sci. U.S.A. 99, 11513–11518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chiba Y., Mineta K., Hirai M. Y., Suzuki Y., Kanaya S., Takahashi H., Onouchi H., Yamaguchi J., Naito S. (2013) Changes in mRNA stability associated with cold stress in Arabidopsis cells. Plant Cell Physiol. 54, 180–194 [DOI] [PubMed] [Google Scholar]
- 16. Bailey-Serres J., Sorenson R., Juntawong P. (2009) Getting the message across: cytoplasmic ribonucleoprotein complexes. Trends Plant Sci. 14, 443–453 [DOI] [PubMed] [Google Scholar]
- 17. Xu J., Chua N. H. (2011) Processing bodies and plant development. Curr. Opin. Plant Biol. 14, 88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCue A. D., Nuthikattu S., Reeder S. H., Slotkin R. K. (2012) Gene expression and stress response mediated by the epigenetic regulation of a transposable element small RNA. PLoS Genet. 8, e1002474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu J., Chua N. H. (2012) Dehydration stress activates Arabidopsis MPK6 to signal DCP1 phosphorylation. EMBO J. 31, 1975–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bae M. S., Cho E. J., Choi E. Y., Park O. K. (2003) Analysis of the Arabidopsis nuclear proteome and its response to cold stress. Plant J. 36, 652–663 [DOI] [PubMed] [Google Scholar]
- 21. Amme S., Matros A., Schlesier B., Mock H. P. (2006) Proteome analysis of cold stress response in Arabidopsis thaliana using DIGE-technology. J. Exp. Bot. 57, 1537–1546 [DOI] [PubMed] [Google Scholar]
- 22. Lee D. G., Ahsan N., Lee S. H., Kang K. Y., Lee J. J., Lee B. H. (2007) An approach to identify cold-induced low-abundant proteins in rice leaf. C. R. Biol. 330, 215–225 [DOI] [PubMed] [Google Scholar]
- 23. Kawaguchi R., Williams A. J., Bray E. A., Bailey-Serres J. (2003) Water-deficit-induced translational control in Nicotiana tabacum. Plant Cell Environ. 26, 221–229 [Google Scholar]
- 24. Kawaguchi R., Girke T., Bray E. A., Bailey-Serres J. (2004) Differential mRNA translation contributes to gene regulation under nonstress and dehydration stress conditions in Arabidopsis thaliana. Plant J. 38, 823–839 [DOI] [PubMed] [Google Scholar]
- 25. To T. K., Nakaminami K., Kim J. M., Morosawa T., Ishida J., Tanaka M., Yokoyama S., Shinozaki K., Seki M. (2011) Arabidopsis HDA6 is required for freezing tolerance. Biochem. Biophys. Res. Commun. 406, 414–419 [DOI] [PubMed] [Google Scholar]
- 26. Nakagami H., Sugiyama N., Mochida K., Daudi A., Yoshida Y., Toyoda T., Tomita M., Ishihama Y., Shirasu K. (2010) Large-scale comparative phosphoproteomics identifies conserved phosphorylation sites in plants. Plant Physiol. 153, 1161–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vizcaino J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Rios D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolome S., Apweiler R., Omenn G. S., Martens L., Jones A. R., Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 29. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 30. Mustroph A., Juntawong P., Bailey-Serres J. (2009) Isolation of plant polysomal mRNA by differential centrifugation and ribosome immunopurification methods. Methods Mol. Biol. 553, 109–126 [DOI] [PubMed] [Google Scholar]
- 31. Abe S., Davies E. (1986) Quantitative analysis of polysomes using a baseline from uncentrifuged blank gradients. Memoir Col. Agr. 31, 187–199 [Google Scholar]
- 32. Uemura M., Joseph R., Steponkus P. (1995) Cold Acclimation of Arabidopsis thaliana (Effect on plasma membrane lipid composition and freeze-induced lesions). Plant Physiol. 109, 15–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wanner L. A., Junttila O. (1999) Cold-induced freezing tolerance in Arabidopsis. Plant Physiol. 120, 391–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chino A., Makanae K., Moriya H. (2013) Relationships between cell cycle regulator gene copy numbers and protein expression levels in Schizosaccharomyces pombe. PloS One 8, e73319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Laroche A., Hopkins W. G. (1987) Polysomes from winter rye seedlings grown at low temperature: I. Size class distribution, composition, and stability. Plant Physiol. 85, 648–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Antikainen M., Pihakaski S. (1993) Cold-induced changes in the polysome pattern and protein-synthesis in winter rye (Secale-Cereale) Leaves. Physiol. Plant. 89, 111–116 [Google Scholar]
- 37. Ranjan M., Tafuri S. R., Wolffe A. P. (1993) Masking mRNA from translation in somatic cells. Genes Dev. 7, 1725–1736 [DOI] [PubMed] [Google Scholar]
- 38. Evdokimova V., Ruzanov P., Anglesio M. S., Sorokin A. V., Ovchinnikov L. P., Buckley J., Triche T. J., Sonenberg N., Sorensen P. H. (2006) Akt-mediated YB-1 phosphorylation activates translation of silent mRNA species. Mol. Cell. Biol. 26, 277–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sano N., Permana H., Kumada R., Shinozaki Y., Tanabata T., Yamada T., Hirasawa T., Kanekatsu M. (2012) Proteomic analysis of embryonic proteins synthesized from long-lived mRNAs during germination of rice seeds. Plant Cell Physiol. 53, 687–698 [DOI] [PubMed] [Google Scholar]
- 40. Tomaz T., Bagard M., Pracharoenwattana I., Linden P., Lee C. P., Carroll A. J., Stroher E., Smith S. M., Gardestrom P., Millar A. H. (2010) Mitochondrial malate dehydrogenase lowers leaf respiration and alters photorespiration and plant growth in Arabidopsis. Plant Physiol. 154, 1143–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhu Y., Dong A., Meyer D., Pichon O., Renou J. P., Cao K., Shen W. H. (2006) Arabidopsis NRP1 and NRP2 encode histone chaperones and are required for maintaining postembryonic root growth. Plant Cell 18, 2879–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu Z., Zhu Y., Gao J., Yu F., Dong A., Shen W. H. (2009) Molecular and reverse genetic characterization of NUCLEOSOME ASSEMBLY PROTEIN1 (NAP1) genes unravels their function in transcription and nucleotide excision repair in Arabidopsis thaliana. Plant J. 59, 27–38 [DOI] [PubMed] [Google Scholar]
- 43. Munns R. (2002) Comparative physiology of salt and water stress. Plant Cell Environ. 25, 239–250 [DOI] [PubMed] [Google Scholar]
- 44. Rymen B., Fiorani F., Kartal F., Vandepoele K., Inze D., Beemster G. T. (2007) Cold nights impair leaf growth and cell cycle progression in maize through transcriptional changes of cell cycle genes. Plant Physiol. 143, 1429–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mussgnug J. H., Wobbe L., Elles I., Claus C., Hamilton M., Fink A., Kahmann U., Kapazoglou A., Mullineaux C. W., Hippler M., Nickelsen J., Nixon P. J., Kruse O. (2005) NAB1 is an RNA binding protein involved in the light-regulated differential expression of the light-harvesting antenna of Chlamydomonas reinhardtii. Plant Cell 17, 3409–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weber C., Nover L., Fauth M. (2008) Plant stress granules and mRNA processing bodies are distinct from heat stress granules. Plant J. 56, 517–530 [DOI] [PubMed] [Google Scholar]
- 47. Kedersha N., Stoecklin G., Ayodele M., Yacono P., Lykke-Andersen J., Fritzler M. J., Scheuner D., Kaufman R. J., Golan D. E., Anderson P. (2005) Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169, 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kedersha N., Anderson P. (2002) Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 30, 963–969 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.