

Abstract
Database search programs are essential tools for identifying peptides via mass spectrometry (MS) in shotgun proteomics. Simultaneously achieving high sensitivity and high specificity during a database search is crucial for improving proteome coverage. Here we present JUMP, a new hybrid database search program that generates amino acid tags and ranks peptide spectrum matches (PSMs) by an integrated score from the tags and pattern matching. In a typical run of liquid chromatography coupled with high-resolution tandem MS, more than 95% of MS/MS spectra can generate at least one tag, whereas the remaining spectra are usually too poor to derive genuine PSMs. To enhance search sensitivity, the JUMP program enables the use of tags as short as one amino acid. Using a target-decoy strategy, we compared JUMP with other programs (e.g. SEQUEST, Mascot, PEAKS DB, and InsPecT) in the analysis of multiple datasets and found that JUMP outperformed these preexisting programs. JUMP also permitted the analysis of multiple co-fragmented peptides from “mixture spectra” to further increase PSMs. In addition, JUMP-derived tags allowed partial de novo sequencing and facilitated the unambiguous assignment of modified residues. In summary, JUMP is an effective database search algorithm complementary to current search programs.
Peptide identification by tandem mass spectra is a critical step in mass spectrometry (MS)-based1 proteomics (1). Numerous computational algorithms and software tools have been developed for this purpose (2–6). These algorithms can be classified into three categories: (i) pattern-based database search, (ii) de novo sequencing, and (iii) hybrid search that combines database search and de novo sequencing. With the continuous development of high-performance liquid chromatography and high-resolution mass spectrometers, it is now possible to analyze almost all protein components in mammalian cells (7). In contrast to rapid data collection, it remains a challenge to extract accurate information from the raw data to identify peptides with low false positive rates (specificity) and minimal false negatives (sensitivity) (8).
Database search methods usually assign peptide sequences by comparing MS/MS spectra to theoretical peptide spectra predicted from a protein database, as exemplified in SEQUEST (9), Mascot (10), OMSSA (11), X!Tandem (12), Spectrum Mill (13), ProteinProspector (14), MyriMatch (15), Crux (16), MS-GFDB (17), Andromeda (18), BaMS2 (19), and Morpheus (20). Some other programs, such as SpectraST (21) and Pepitome (22), utilize a spectral library composed of experimentally identified and validated MS/MS spectra. These methods use a variety of scoring algorithms to rank potential peptide spectrum matches (PSMs) and select the top hit as a putative PSM. However, not all PSMs are correctly assigned. For example, false peptides may be assigned to MS/MS spectra with numerous noisy peaks and poor fragmentation patterns. If the samples contain unknown protein modifications, mutations, and contaminants, the related MS/MS spectra also result in false positives, as their corresponding peptides are not in the database. Other false positives may be generated simply by random matches. Therefore, it is of importance to remove these false PSMs to improve dataset quality. One common approach is to filter putative PSMs to achieve a final list with a predefined false discovery rate (FDR) via a target-decoy strategy, in which decoy proteins are merged with target proteins in the same database for estimating false PSMs (23–26). However, the true and false PSMs are not always distinguishable based on matching scores. It is a problem to set up an appropriate score threshold to achieve maximal sensitivity and high specificity (13, 27, 28).
De novo methods, including Lutefisk (29), PEAKS (30), NovoHMM (31), PepNovo (32), pNovo (33), Vonovo (34), and UniNovo (35), identify peptide sequences directly from MS/MS spectra. These methods can be used to derive novel peptides and post-translational modifications without a database, which is useful, especially when the related genome is not sequenced. High-resolution MS/MS spectra greatly facilitate the generation of peptide sequences in these de novo methods. However, because MS/MS fragmentation cannot always produce all predicted product ions, only a portion of collected MS/MS spectra have sufficient quality to extract partial or full peptide sequences, leading to lower sensitivity than achieved with the database search methods.
To improve the sensitivity of the de novo methods, a hybrid approach has been proposed to integrate peptide sequence tags into PSM scoring during database searches (36). Numerous software packages have been developed, such as GutenTag (37), InsPecT (38), Byonic (39), DirecTag (40), and PEAKS DB (41). These methods use peptide tag sequences to filter a protein database, followed by error-tolerant database searching. One restriction in most of these algorithms is the requirement of a minimum tag length of three amino acids for matching protein sequences in the database. This restriction reduces the sensitivity of the database search, because it filters out some high-quality spectra in which consecutive tags cannot be generated.
In this paper, we describe JUMP, a novel tag-based hybrid algorithm for peptide identification. The program is optimized to balance sensitivity and specificity during tag derivation and MS/MS pattern matching. JUMP can use all potential sequence tags, including tags consisting of only one amino acid. When we compared its performance to that of two widely used search algorithms, SEQUEST and Mascot, JUMP identified ∼30% more PSMs at the same FDR threshold. In addition, the program provides two additional features: (i) using tag sequences to improve modification site assignment, and (ii) analyzing co-fragmented peptides from mixture MS/MS spectra.
EXPERIMENTAL PROCEDURES
JUMP is a database search algorithm (version 1.0.55) used to convert tandem MS raw files to a list of peptides and proteins. The software architecture is illustrated in Fig. 1, and a detailed scheme is shown in supplemental Fig. S1. JUMP was written in Perl via a modular approach that can be readily edited for further improvement. The program was also designed for high-performance parallel computing systems. JUMP source codes and detailed documents are freely available at the Peng Lab website.
Fig. 1.

The JUMP algorithm. A, overview of the JUMP algorithm. JUMP starts with a raw data file or an mzXML file and then performs MS1 and MS2 preprocessing for each spectrum. Both MS1 precursor ion mass and sequence tags derived from an MS2 spectrum are used to filter peptide sequences in the database. After pattern matching, JUMP generates a final score, the J score, to rank all identified peptides. B, an example to illustrate two-step filtering of theoretical peptides in the JUMP algorithm: (i) filtered by a precursor mass of 1402.651 Da within a mass tolerance range (e.g. 10 ppm); (ii) filtered by a sequence tag. C, an example of JUMP output. The J score is generated by integrating the tag E score and peptide E score.
Preprocessing of Precursor Ions
For high-resolution MS data, the precursor ion in MS scans is analyzed to determine its charge state and monoisotopic mass. Three steps are performed: (i) JUMP defines the precursor ion in the MS scan preceding each MS/MS spectrum when the data are collected via automated data-dependent acquisition. (ii) The charge state is computed by comparing the precursor ion peak to all other peaks within one mass unit window. If there is no isotopic peak that can be used to interpret the charge state, the program assigns two most common charge states (+2 and +3) to the precursor ion, which allows the algorithm to determine the appropriate charge state by means of PSM scoring. (iii) To identify the monoisotopic mass, JUMP first constructs a theoretical isotopic pattern based on the precursor ion mass and then uses the theoretical pattern to designate the monoisotopic peak in the MS scan.
Preprocessing of MS/MS Spectra
JUMP deconvolutes multiple charged peaks and isotope clusters, removes background noise, and normalizes the intensity of all peaks in a tandem MS spectrum. (i) The intact precursor ion and its related neutral loss peaks (e.g. H2O or NH3) are removed. (ii) The charge state and monoisotopic mass of fragment ions are identified as described above. In high-resolution MS/MS scans (e.g. Orbitrap), the isotopic peaks and differently charged peaks of the same product ion are merged with the monoisotopic peak of a singly charged peak, and their intensities are summed, assuming that ion intensity is proportional to charge state. (iii) The program filters weak peaks to reduce noise. In each window of 100 m/z (Fig. 2), the top peaks (e.g. n = 6) are retained, and the number of peaks can be edited by users. (iv) As different product ions are not transferred and detected by MS at the same efficiency over the full m/z range, the program normalizes peak intensity in each 100 m/z window using the equation
where M′i is the normalized intensity of peak i, Mi is its original intensity, SP is the strongest peak intensity in the entire spectrum, and SPi is the strongest peak intensity in the 100 m/z window containing peak i. To avoid equal intensity of peaks, all windows are ranked according to their strongest peak intensities, and the rank for each window (Rspi) is implemented in the normalization.
Fig. 2.

Tag inferred from MS/MS spectra of high quality (A) and low quality (B). The top panel shows original MS/MS spectra, the middle panel shows preprocessed spectra after peak filtering and intensity normalization, and the bottom panel shows the top six tags. The JUMP program assembles the tag in two reading orders, corresponding to possible b or y ion series.
Tag Generation and Scoring
To extract all potential tag sequences from an MS/MS spectrum, JUMP first labels pairs of peaks that have mass difference of one specific residue. These peak pairs are linked together to construct long tag sequences with flanking masses. The tags may be generated from either N-terminal fragments (e.g. b ions) or C-terminal fragments (e.g. y ions). The program recursively enumerates all possible tags from one spectrum and then sorts these tags according to any of the three ranking methods: tag intensity-based Wilcoxon rank sum test, tag position-based hypergeometric test, or a combined method of the two algorithms.
The Wilcoxon rank sum test generates a p value for each tag based on whether there is significant difference in intensities between the tag-corresponding peaks and the remaining peaks in an MS/MS spectrum. The Wilcoxon rank sum test was previously used in the DirecTag program (40). For example, if a tag is derived from the 1st, 4th, 5th, 11th, and 18th peaks in a spectrum consisting of 50 peaks, the rank sum of the tag is 39, corresponding to a p value of 0.001. The p value is converted to a tag E (expectation) score (Etr) that is equal to −log10 of the p value. Tags generated from strong peaks are more likely to be authentic and are thus scored better than those from weak peaks. In contrast to the Wilcoxon rank sum test, which covers the intensities of all ions in a full spectrum, the hypergeometric test produces a p value based on the occurrence probability of tag-corresponding peaks within a local range.
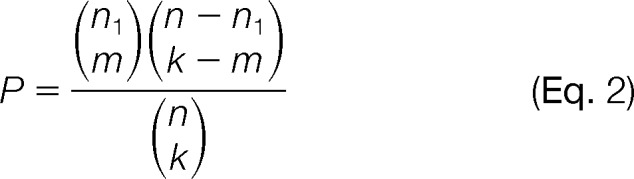 |
where n is all possible ion locations, calculated as the mass range of the tag divided by the mass tolerance; k is the total number of theoretical product ions of this tag; n1 is the number of detected product ions within the local mass range; and m is the number of matched product ions for generating the tag. In general, long tags are associated with low p values. Similarly, the p values are converted to an E score (Eth).
We propose a combining score (Etc) based on the intensity (“rank”, Etr) and the position of tags (“hyper”, Eth). To compare and combine these two scores in the same scale, JUMP standardizes the scores to a 0–1 range as follows:
 |
where Ei′ is a standardized E score of the ith tag from one MS/MS spectrum, Ei is either the rank or the hyper E score before standardization, and Emin and Emax are the minimal and maximal E scores of all tags generated from the same spectrum, respectively. For each tag, JUMP generates a standardized rank score (Etr′) and a standardized “hyper” score (Eth′) to calculate its combined score (Etc′).
The combined, standardized E score (Etc′) in the 0–1 range is converted back to an E score (Etc) in the hyper score (Eth) scale, as the tag E score is also merged with hypergeometric test scores during peptide matching (see below).
Finally, the tags from the same spectrum are ranked by the combined scores (Etc). The users can define how many top-ranked tags are selected for a database search.
Database Indexing
To rapidly retrieve candidate peptides from a protein database, JUMP generates three indexed files: protein index, peptide index, and mass index (supplemental Fig. S2). Decoy proteins are generated by reversing target protein sequences and switching Arg and Lys residues with the preceding amino acid (42). Theoretical peptides are derived from protein sequences according to the specified mass range, enzymatic digestion, miscleavage sites, and static modifications and are then sorted by mass for storage. The search for dynamic modifications is programmed separately, as the peptide number exponentially increases with multiple dynamic modifications. For instance, in a phosphoproteome study of triple SILAC labeling, one may simultaneously search for eight dynamic modifications: Met oxidation; Ser, Thr, and Tyr phosphorylation; two Lys modifications; and two Arg modifications. A number of strategies are employed to speed up the search. (i) All peptide masses are scaled by a factor of 1000 to remove non-integer mass, and the factor is adjustable, depending on mass tolerance. (ii) The program uses indexed files. (iii) The binary search algorithm is used to retrieve mass, peptide, and protein in the index files. (iv) The maximal modification number in one peptide is six in the current version.
Database Searching and Peptide Scoring
JUMP initially retrieves candidate peptides based on precursor ion mass and then filters the peptides by top-ranked tag sequences and flanking masses. While the isobaric residues Leu and Ile are exchangeable in the tags, the summed mass of two adjacent residues may be isobaric to that of a single residue within mass tolerance. The program takes into account all possible five isobaric cases: Gly-Gly and Asn, Gly-Ala and Gln, Gly-Val and Arg, Gly-Glu and Trp, and Ala-Asp and Trp. Peptide candidates that pass the tag-filtering step are subjected to peptide scoring.
Peptide scoring frequently relies on probability models (10), such as the Poisson distribution (11), the binomial distribution (18), and the hypergeometric test (15, 43). We used the hypergeometric test and the Wilcoxon rank sum test to compare theoretical product ions (e.g. b and y ions) to the detected MS/MS ions, similar to the computation of the hypergeometric test tag hyper score (Eth) and tag rank score (Etr). These two tests generate the peptide hyper E score (Eph) and rank E score (Epr). The peptide E score (Ep) is then calculated by combining these two scores (Eph + Epr). Then JUMP generates a J score to rank the identified peptides, which integrates the tag E score (Etc) and the peptide E score (Ep).
Like the SEQUEST program, JUMP generates a δ correlation score (dJn) to measure the distance between the best J score (Jscore1) and the second J score (Jscore2) from the same MS/MS spectrum.
The target-decoy strategy is commonly used to estimate global FDRs in protein identification (23–26) and to derive individual q values associated with PSMs (44). The FDR is calculated as the ratio between the numbers of decoy and target matches (45, 46). The JUMP program also provides the q value for each PSM based on this strategy.
Software Input and Output
JUMP processes MS data in either raw or mzXML format. The raw files are converted to mzXML files by free software such as ReAdW or msConvert. JUMP generates pepXML files for post-search filtering or integration with other software. In addition, JUMP produces a tab-delimited Excel file displaying basic search parameters, MS/MS data summary, and a PSM summary, as well as multiple charts for data visualization.
Sample Datasets
Four datasets were used to evaluate the JUMP algorithm. The first dataset was collected from a typical LC-MS/MS run of a digested protein sample of a human myeloma cell line (ANBL6, provided by Dr. Kenneth C. Anderson at Harvard Medical School) using our previously optimized protocol (47). Trypsinized cell lysate (∼1 μg) was loaded on a reverse phase column (75 mm × 10 cm, 2.7-μm HALO C18 resin) and eluted during a 60-min gradient of 10% to 40% solvent B (solvent A, 0.1% formic acid; solvent B, 0.1% formic acid, 70% acetonitrile; flow rate of 300 nl/min). The second dataset was obtained from a phosphopeptide-enriched sample of the same starting cell lysate. Phosphopeptides were enriched from the desalted total peptides (∼100 μg) by TiO2 (48) and analyzed in a 30-min LC-MS/MS run. The third dataset was from a long-gradient LC/LC-MS/MS analysis of a human sample. The tissue lysates were digested, and the resulting peptides were subjected to basic pH reverse phase chromatography to generate 10 fractions. Each fraction was then analyzed via long-gradient acidic-pH LC-MS/MS. The forth dataset was from mouse samples labeled with tandem mass tag reagents (Thermo Fisher Scientific). Phosphopeptides were isolated from the pooled sample by TiO2 (48) and analyzed in a 6-h LC-MS/MS run. The eluted peptides in the four experiments were analyzed on a Q Exactive mass spectrometer (Thermo Fisher Scientific) with one MS scan (35,000 resolution and 1 × 106 automatic gain control (AGC) target) and up to 20 data-dependent MS/MS scans. Instrument methods with parameters are available in raw files that can be downloaded from our lab's website.
Data Analysis
The four datasets were used for tuning JUMP parameters and comparing the performance of JUMP (version 1.0.55), SEQUEST (Proteome Discoverer 1.3), Mascot (version 2.3.0), PEAKS DB (PEAKS 7.0, build 20140321), and InsPecT (version 20120109). MS raw files were converted into mzXML format as input for all programs. The first three datasets were searched against the UniProt human database with 71,809 protein entries, and the forth dataset used the UniProt mouse database with 55,744 entries. Common parameters included a precursor ion mass tolerance of 15 ppm, a product ion mass tolerance of 0.02 Da, fully tryptic restriction, up to two miscleavages, and dynamic modifications of oxidized Met (+15.99492 Da). For the two phosphorylation datasets, additional dynamic modifications on serine, threonine, and tyrosine (+79.96633 Da) were used. For the tandem-mass-tag-labeled dataset, fixed modifications on the N terminus and lysine (+229.162932 Da) were also used. During MS/MS preprocessing, the top 10 peaks in each window of 100 m/z were selected for SEQUEST, Mascot, and JUMP. The function of mass correction was selected for PEAKS DB. For other parameters, the default settings were used. After searching, the PSMs were processed by filtering procedures provided by individual programs (e.g. Peptide Validator for SEQUEST, Percolator for Mascot, PEAKS studio for PEAKS DB, and ComputeFDR for InsPecT).
RESULTS AND DISCUSSION
Evaluation of False Discovery Rate in JUMP via the Target-Decoy Strategy
JUMP is designed as a hybrid algorithm that performs de novo tag sequencing and matches tandem MS spectra to a theoretical database (Fig. 1). To assess false PSMs, we adapted the commonly used target-decoy strategy (23–26) in which random PSMs are assumed to have equal probabilities of being assigned to target and decoy databases. We tested whether this strategy is suitable for JUMP by searching a null dataset. The null dataset was falsified by increasing the precursor ion mass of each MS/MS spectrum by 100 Da. As expected, JUMP generated almost equal targets (n = 6588) and decoys (n = 6599) (supplemental Fig. S3A) and similar distributions of PSM scores (i.e. J scores) in these targets and decoys (supplemental Fig. S3B). These results demonstrate that the target-decoy strategy is applicable for evaluating the FDR of JUMP-derived PSMs.
De Novo Tag Generation from MS/MS Spectra
The JUMP algorithm generates de novo amino acid tags from high-resolution MS/MS spectra (Fig. 2). The tag number derived from one spectrum varies depending on the spectral quality. In our first dataset, a standard 1-h LC-MS/MS run of total cell lysate, the tag number was in the range from 0 to hundreds, with a median of 53 (Fig. 3A). It should be noted that overlapping amino acid sequences were counted as different tags (Fig. 2A). The vast majority of spectra (98.8%) were able to generate at least one sequence tag.
Fig. 3.

Tag derived by JUMP. A, distribution of the tag number for individual MS/MS spectra in the first dataset. B, pseudo–receiver operating characteristic curves for the comparison of three tag scoring methods: the Wilcoxon rank sum test (“rank”), the hypergeometric test (“hyper”), and the combined method (“comb”). C, the number of expected true PSMs influenced by the number of top tags selected for the database search.
JUMP ranks these tags in the same MS/MS scan for subsequent selection during the database search. An ideal tag scoring method should sort the most promising tags to the top of the list, which reduces database search time and improves peptide scoring, because the tag scores are also considered during peptide scoring. Two previously reported methods were examined: the Wilcoxon rank sum test (40), which scores a tag based on the signal intensity rank of its related ions, and the hypergeometric test (15, 43), based on the mass of the ions. As the two parameters (i.e. ion intensity and mass) are orthogonal to each other, we proposed a combined score to utilize both parameters and compared its performance with that of the two reported methods. Although the hypergeometric test showed a better result than the Wilcoxon rank sum test, our combined method yielded the best outcome, identifying 1.8% and 6.1% more PSMs than the other two methods, respectively, at a 1% FDR (Fig. 3B).
We then determined how many top tags are needed to achieve maximal sensitivity in the peptide identification (Fig. 3C). Interestingly, the use of the top five tags was sufficient to reach the plateau, suggesting that our tag scoring function works efficiently. Relative to the non-tag search (e.g. tag number set to 0), the inclusion of tags dramatically increased the anticipated number of true PSMs (from 4800 to 6000), suggesting that the inclusion of tags improves the performance of the database search.
To further dissect the role of tags in database searches, we analyzed the relationship between the lengths of amino acid tags and the target-decoy PSMs (Fig. 4A) or J score distribution (Fig. 4B). When no tag was identified, the related spectra were of low quality, resulting in nearly equal numbers of target and decoy PSMs and indistinguishable distribution of target and decoy J scores, suggesting that JUMP can judge MS/MS spectral quality by tags, and the non-tag spectra may be discarded without a loss of sensitivity. As expected, the PSMs supported by long tags were likely to be correct and have high J scores. For example, in PSMs with two amino acid tags, ∼50% of target PSMs were false assignments. In PSMs with at least five amino acid tags, no decoy PSM was identified. Several prevalent hybrid methods, such as GutenTag (37) and InsPecT (38), require a minimal tag length of three amino acids in order to directly extract peptide or protein sequences from a database without filtering by precursor ion mass. By contrast, JUMP uses the list of tag sequences to query candidate peptides after filtering by precursor ion mass. In this run, JUMP identified 9.6% (630/6546) PSMs with supporting tags of less than three amino acids. Therefore, JUMP is able to use tags as short as one amino acid to improve identification sensitivity.
Fig. 4.
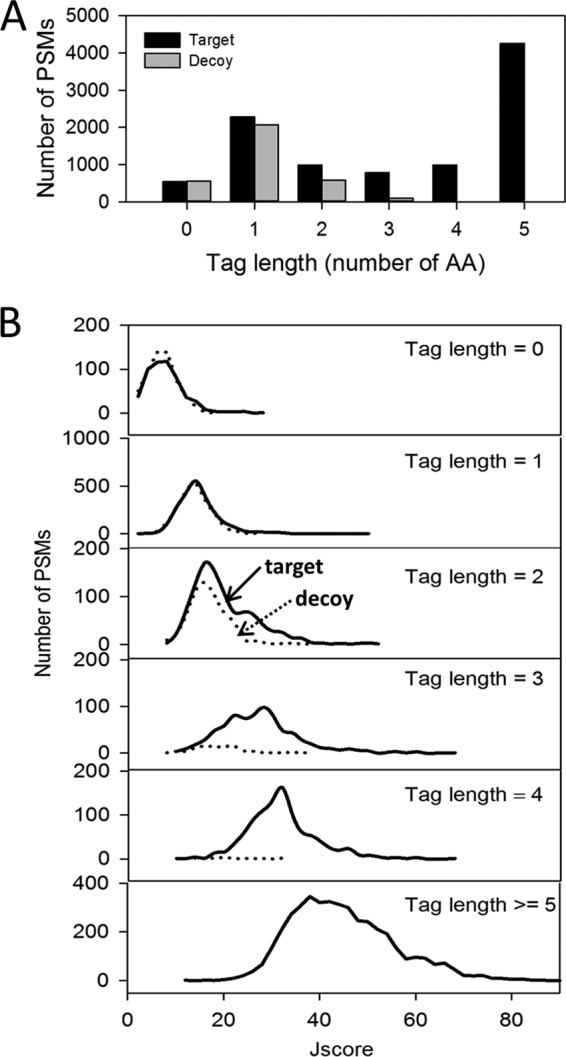
The effect of tag length on peptide identification. A, distribution of the number of identified target and decoy PSMs with different tag lengths. B, J score distributions for target and decoy PSMs with different tag lengths.
Performance Comparison of JUMP, SEQUEST, and Mascot
To fully assess the performance (i.e. sensitivity and specificity) of JUMP, we processed the same dataset with JUMP, SEQUEST, and Mascot, as the other two search engines are commercially available and widely used in the proteomics community. In a pseudo–receiver operating characteristic plot (accepted PSMs versus FDR), JUMP clearly had the best performance. At the threshold of 1% FDR, JUMP identified 6352 PSMs, 28.7% and 33.2% more than SEQUEST and Mascot, respectively (Fig. 5A). A close examination revealed that the JUMP score distinguished between targets and decoys better than the other two programs (Figs. 5B–5D). In SEQUEST, another δ correlation score (dCn) indicates the Xcorr difference between the first and second matches of the same spectrum, which reflects the specificity of peptide scoring. We computed a similar score in JUMP (dJn) to distinguish targets and decoys (Fig. 5E). A direct comparison of dCn and dJn distributions also demonstrated higher specificity of the dJn score (Fig. 5F).
Fig. 5.

Performance comparison of JUMP, SEQUEST, and Mascot. A, JUMP shows the best result on a pseudo–receiver operating characteristic plot. B–D, the distribution of target and decoy PSMs along matching scores of JUMP (J score), Mascot (E-value), and SEQUEST (Xcorr). E, the distribution of target and decoy PSMs in the J score–dJn plot. F, the distribution of δ correlation scores for JUMP (dJn) and SEQUEST (dCn) in PSMs accepted at a 1% FDR.
The improvement of specificity in JUMP may be attributed to two main features: (i) in addition to precursor ion mass filtering, the peptide database is subjected to tag filtering, which markedly reduces the search space for random matching; (ii) the J score incorporates both the pattern matching score and the tag score, emphasizing the contribution of tags, because random matched peaks are more likely to be dispersed along the full spectrum but rarely generate de novo tags. Indeed, the Yates group originally reported this rule for manual validation of PSMs, which required “some continuity to the b or y ion series” (49) that generates peptide tags.
Identification of Multiple Precursors in Mixed MS/MS Spectra
When complex peptide samples are analyzed via LC-MS/MS, a significant number of peptides with close m/z values are co-eluted, leading to co-isolation and mixed MS/MS spectra (18, 50, 51). Several search engines, such as Andromeda (18) and ProbIDtree (51), introduced a function to allow the second peptide identification. In JUMP, we implemented a similar feature to select multiple precursor ions within the isolation window for database searches. For instance, in one precursor isolation window (Fig. 6A), the strongest precursor ion was triply charged, and its percentage of precursor intensity was 77%. The second precursor ion was doubly charged, and its percentage of precursor intensity was 23%. Both ions were isolated during fragmentation to produce a mixed MS/MS spectrum. Users can define multiple precursor ions by setting a maximal precursor number for one MS/MS spectrum or the minimal percentage of precursor intensity. Relative to a search result with only one precursor ion per MS/MS spectrum, multiple precursor ion selection increased the PSMs by 18.3% at 1% FDR (Fig. 6B). The identified PSMs were primarily contributed by the first precursor ions (Figs. 6C and 6D).
Fig. 6.

Identification of multiple precursor ions in mixed tandem MS spectra. A, an example to show two co-isolated ions within the isolation window. The percentage of precursor intensity (PPI) is calculated for all precursors within the isolation window. B, inclusion of multiple precursors for the same MS/MS spectra allowed more identification than the selection of only one precursor. The cutoff was set at 1% FDR. C, D, the distribution of target and decoy PSMs along matching scores for the top precursors and for the remaining precursors.
Partial de Novo Sequencing of Novel Peptides Not Present in the Database
In some cases, tandem MS spectra lead to highly confident tags with long amino acid sequences but cannot be matched to any theoretical peptides. It is likely that the corresponding peptides are simply not present in the searched peptide database, possibly because of unconventional protease cleavage, novel modifications, unknown protein mutations, or protein contaminants that are not included in the database. In the first dataset analysis, 712 MS/MS spectra failed to identify peptides, of which 214 spectra did not generate any tag because of poor quality, and the remaining spectra generated tags of various lengths. We manually examined 13 spectra with the top tags of at least eight amino acids and performed a BLAST search against the NCBI non-redundant human database. These tags were mapped to proteins with missense mutations. For example, the top-ranked tag with 14 amino acids was derived from a high-quality spectrum (Fig. 7A), mapped to the protein PSMD13 with one mismatch (Figs. 7B and 7C). Our whole-genome sequencing and RNA-sequencing data further confirmed the missense mutation (supplemental Figs. S4A and S4B). Because JUMP implements partial de novo sequencing, it is possible to derive peptides with amino acid mutations and novel post-translational modifications for those unassigned spectra of high quality (e.g. with long tags).
Fig. 7.
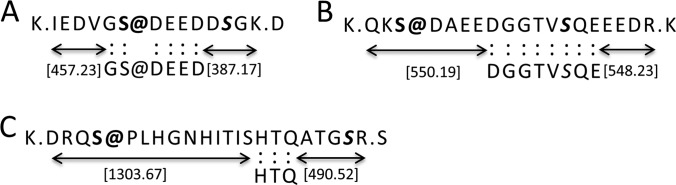
JUMP-derived tags facilitate the assignment of modification sites. A, the modification site in a tag. B, the modifiable site in a tag, but not modified. C, the modifiable site not in the tag. The flanking masses of the tag provide restrictions for site assignment.
Tag-based Assignment of Post-translational Modification Sites
To unambiguously assign the modified sites in peptide sequences, several programs have been developed to re-score local product ions to improve the determination of modification sites (52, 53). Similarly, JUMP generates partial de novo tags that provide local product ion information to assign modification sites by three rules: (i) the modified site may be directly found inside a tag (Fig. 8A); (ii) a dynamically modifiable residue may be included in a tag, but it is not modified (Fig. 8B), which excludes this possibility; and (iii) the modifiable site does not present in a tag, but the flanking masses of the tag narrow down the options for site assignment (Fig. 8C). We applied JUMP to search a phosphorylation dataset of 1836 MS/MS spectra and identified 698 PSMs and 494 peptides (1% FDR), including 673 phospho-PSMs and 487 phosphopeptides. In these phosphopeptides, 136, 172, and 179 peptides were supported by the three rules, respectively. Because JUMP yields de novo tags in assigning modifications, we believe that the program can provide reliable results to distinguish ambiguous peptides.
Fig. 8.

De novo sequencing of a novel peptide. A, a tag with 14 amino acids was derived from a high-quality spectrum, but no reliable peptide was identified during database searching. B, an amino acid change (N13S) was found in the novel tag when the tag was searched against the database. C, a nucleotide change (A → G; rs1045288) from whole-genome sequencing and RNA-sequencing confirmed the novel tag sequence.
Evaluation of JUMP and Other Programs Using Large Datasets
Advances in MS technology substantially increase scan speed, enlarge raw file size, and demand rapid processing by database search engines. For example, we routinely acquire hundreds of thousands of data-dependent MS/MS spectra in single long-gradient LC-MS/MS runs. It would be valuable for a search engine to be able to search such datasets with high sensitivity and specificity in a relatively short time. Thus, we used the third large dataset of 1,718,768 MS/MS spectra (Fig. 9) to evaluate the performance of two commonly used programs (SEQUEST and Mascot) and three hybrid programs (JUMP, InsPecT, and PEAKS DB). JUMP identified 658,392 PSMs, which was 26.9% and 32.1% more than SEQUEST and Mascot, respectively. In addition, JUMP identified 5.3% and 37.8% more PSMs than PEAKS DB and InsPecT, respectively. Similar results were obtained when we compared identified peptides.
Fig. 9.

Performance comparison of five programs using a large-scale dataset. The human dataset of LC/LC-MS/MS analysis contained ∼1.7 million MS/MS spectra. The accepted PSMs and peptides at 1% FDR are shown.
In addition, we further evaluated JUMP performance on a large-scale phosphorylation dataset of 108,654 MS/MS spectra. JUMP identified 17,937 phospho-PSMs and 6542 phosphopeptides, and these identifications included 18.2% more phospho-PSMs and 22.1% more phosphopeptides than those made by SEQUEST, as well as 25.9% more phospho-PSMs and 36.7% more phosphopeptides than identified with Mascot (supplemental Fig. S5).
CONCLUSION
We have demonstrated that JUMP is a sensitive and specific database search method for peptide identification. JUMP is able to achieve better sensitivity and specificity than other search engines such as SEQUEST, Mascot, InsPecT, and PEAKS DB. A number of features contribute to the strong performance of JUMP: (i) JUMP uses a novel algorithm for tag scoring that combines the peak intensity-based score and the peak position-based score; (ii) JUMP generates a J score that merges the local tag score and the global pattern matching score; (iii) JUMP uses tags as short as one amino acid; and (iv) JUMP is designed as a modular program for high-performance computing systems. In addition, JUMP is capable of identifying multiple candidate peptides from mixture spectra and producing de novo sequence tags. In summary, we have demonstrated that JUMP is an independent and complementary tool for use with existing software for identifying peptide/protein sequences from MS/MS raw data.
Acknowledgments
We thank all other members in the Peng Lab and in St. Jude Proteomics Facility for helpful discussions.
Footnotes
Author contributions: X.W. and J.P. designed research; X.W., Y.L., Z.W., H.W., H.T., and J.P. performed research; Z.W., H.W., H.T., and J.P. contributed new reagents or analytic tools; X.W. and Y.L. analyzed data; X.W. and J.P. wrote the paper.
* This work was partially supported by National Institutes of Health grant AG039764, NS08157, American Cancer Society grant RSG-09-181, and ALSAC (American Lebanese Syrian Associated Charities).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- FDR
- false discovery rate
- PSM
- peptide spectrum match.
REFERENCES
- 1. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 2. Sadygov R. G., Cociorva D., Yates J. R., 3rd (2004) Large-scale database searching using tandem mass spectra: looking up the answer in the back of the book. Nat. Methods 1, 195–202 [DOI] [PubMed] [Google Scholar]
- 3. Nesvizhskii A. I., Vitek O., Aebersold R. (2007) Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods 4, 787–797 [DOI] [PubMed] [Google Scholar]
- 4. Eng J. K., Searle B. C., Clauser K. R., Tabb D. L. (2011) A face in the crowd: recognizing peptides through database search. Mol. Cell. Proteomics 10, R111.009522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Noble W. S., MacCoss M. J. (2012) Computational and statistical analysis of protein mass spectrometry data. PLoS Comput. Biol. 8, e1002296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shteynberg D., Nesvizhskii A. I., Moritz R. L., Deutsch E. W. (2013) Combining results of multiple search engines in proteomics. Mol. Cell. Proteomics 12, 2383–2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mann M., Kulak N. A., Nagaraj N., Cox J. (2013) The coming age of complete, accurate, and ubiquitous proteomes. Mol. Cell 49, 583–590 [DOI] [PubMed] [Google Scholar]
- 8. Aebersold R. (2011) Editorial: from data to results. Mol. Cell. Proteomics 10, E111.014787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eng J. K., Mccormack A. L., Yates J. R. (1994) An approach to correlate tandem mass-spectral data of peptides with amino-acid-sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 10. Perkins D. N., Pappin D. J. C., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 11. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 12. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 13. Kapp E. A., Schutz F., Connolly L. M., Chakel J. A., Meza J. E., Miller C. A., Fenyo D., Eng J. K., Adkins J. N., Omenn G. S., Simpson R. J. (2005) An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics 5, 3475–3490 [DOI] [PubMed] [Google Scholar]
- 14. Chalkley R. J., Baker P. R., Huang L., Hansen K. C., Allen N. P., Rexach M., Burlingame A. L. (2005) Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting, quadrupole collision cell, time-of-flight mass spectrometer: II. New developments in Protein Prospector allow for reliable and comprehensive automatic analysis of large datasets. Mol. Cell. Proteomics 4, 1194–1204 [DOI] [PubMed] [Google Scholar]
- 15. Tabb D. L., Fernando C. G., Chambers M. C. (2007) MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 6, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park C. Y., Klammer A. A., Kall L., MacCoss M. J., Noble W. S. (2008) Rapid and accurate peptide identification from tandem mass spectra. J. Proteome Res. 7, 3022–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim S., Mischerikow N., Bandeira N., Navarro J. D., Wich L., Mohammed S., Heck A. J., Pevzner P. A. (2010) The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol. Cell. Proteomics 9, 2840–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 19. Ryu S., Goodlett D. R., Noble W. S., Minin V. N. (2012) A statistical approach to peptide identification from clustered tandem mass spectrometry data. Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine Oct. 4–7, Philadelphia, PA, pp. 643–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wenger C. D., Coon J. J. (2013) A proteomics search algorithm specifically designed for high-resolution tandem mass spectra. J. Proteome Res. 12, 1377–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lam H., Deutsch E. W., Eddes J. S., Eng J. K., King N., Stein S. E., Aebersold R. (2007) Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7, 655–667 [DOI] [PubMed] [Google Scholar]
- 22. Dasari S., Chambers M. C., Martinez M. A., Carpenter K. L., Ham A. J., Vega-Montoto L. J., Tabb D. L. (2012) Pepitome: evaluating improved spectral library search for identification complementarity and quality assessment. J. Proteome Res. 11, 1686–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peng J., Elias J. E., Thoreen C. C., Licklider L. J., Gygi S. P. (2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res. 2, 43–50 [DOI] [PubMed] [Google Scholar]
- 24. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 25. Kall L., Storey J. D., MacCoss M. J., Noble W. S. (2008) Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 7, 29–34 [DOI] [PubMed] [Google Scholar]
- 26. Zhang J., Xin L., Shan B., Chen W., Xie M., Yuen D., Zhang W., Zhang Z., Lajoie G. A., Ma B. (2012) PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics 11, M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bell A. W., Deutsch E. W., Au C. E., Kearney R. E., Beavis R., Sechi S., Nilsson T., Bergeron J. J. (2009) A HUPO test sample study reveals common problems in mass spectrometry-based proteomics. Nat. Methods 6, 423–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cooper B. (2011) The problem with peptide presumption and low Mascot scoring. J. Proteome Res. 10, 1432–1435 [DOI] [PubMed] [Google Scholar]
- 29. Taylor J. A., Johnson R. S. (1997) Sequence database searches via de novo peptide sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 11, 1067–1075 [DOI] [PubMed] [Google Scholar]
- 30. Ma B., Zhang K., Hendrie C., Liang C., Li M., Doherty-Kirby A., Lajoie G. (2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 31. Fischer B., Roth V., Roos F., Grossmann J., Baginsky S., Widmayer P., Gruissem W., Buhmann J. M. (2005) NovoHMM: a hidden Markov model for de novo peptide sequencing. Anal. Chem. 77, 7265–7273 [DOI] [PubMed] [Google Scholar]
- 32. Frank A., Pevzner P. (2005) PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 77, 964–973 [DOI] [PubMed] [Google Scholar]
- 33. Chi H., Sun R. X., Yang B., Song C. Q., Wang L. H., Liu C., Fu Y., Yuan Z. F., Wang H. P., He S. M., Dong M. Q. (2010) pNovo: de novo peptide sequencing and identification using HCD spectra. J. Proteome Res. 9, 2713–2724 [DOI] [PubMed] [Google Scholar]
- 34. Pan C., Park B. H., McDonald W. H., Carey P. A., Banfield J. F., VerBerkmoes N. C., Hettich R. L., Samatova N. F. (2010) A high-throughput de novo sequencing approach for shotgun proteomics using high-resolution tandem mass spectrometry. BMC Bioinformatics 11, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jeong K., Kim S., Pevzner P. A. (2013) UniNovo: a universal tool for de novo peptide sequencing. Bioinformatics 29, 1953–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mann M., Wilm M. (1994) Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem. 66, 4390–4399 [DOI] [PubMed] [Google Scholar]
- 37. Tabb D. L., Saraf A., Yates J. R., 3rd (2003) GutenTag: high-throughput sequence tagging via an empirically derived fragmentation model. Anal. Chem. 75, 6415–6421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanner S., Shu H., Frank A., Wang L. C., Zandi E., Mumby M., Pevzner P. A., Bafna V. (2005) InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 77, 4626–4639 [DOI] [PubMed] [Google Scholar]
- 39. Bern M., Cai Y., Goldberg D. (2007) Lookup peaks: a hybrid of de novo sequencing and database search for protein identification by tandem mass spectrometry. Anal. Chem. 79, 1393–1400 [DOI] [PubMed] [Google Scholar]
- 40. Tabb D. L., Ma Z. Q., Martin D. B., Ham A. J., Chambers M. C. (2008) DirecTag: accurate sequence tags from peptide MS/MS through statistical scoring. J. Proteome Res. 7, 3838–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang J., Xin L., Shan B., Chen W., Xie M., Yuen D., Zhang W., Zhang Z., Lajoie G. A., Ma B. (2012) PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics 11, M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 43. Sadygov R. G., Yates J. R., 3rd (2003) A hypergeometric probability model for protein identification and validation using tandem mass spectral data and protein sequence databases. Anal. Chem. 75, 3792–3798 [DOI] [PubMed] [Google Scholar]
- 44. Kall L., Canterbury J. D., Weston J., Noble W. S., MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 45. Jeong K., Kim S., Bandeira N. (2012) False discovery rates in spectral identification. BMC Bioinformatics 13 Suppl 16, S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kall L., Storey J. D., MacCoss M. J., Noble W. S. (2008) Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 7, 29–34 [DOI] [PubMed] [Google Scholar]
- 47. Xu P., Duong D. M., Peng J. (2009) Systematical optimization of reverse-phase chromatography for shotgun proteomics. J. Proteome Res. 8, 3944–3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kettenbach A. N., Gerber S. A. (2011) Rapid and reproducible single-stage phosphopeptide enrichment of complex peptide mixtures: application to general and phosphotyrosine-specific phosphoproteomics experiments. Anal. Chem. 83, 7635–7644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Link A. J., Eng J., Schieltz D. M., Carmack E., Mize G. J., Morris D. R., Garvik B. M., Yates J. R., 3rd (1999) Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 17, 676–682 [DOI] [PubMed] [Google Scholar]
- 50. Houel S., Abernathy R., Renganathan K., Meyer-Arendt K., Ahn N. G., Old W. M. (2010) Quantifying the impact of chimera MS/MS spectra on peptide identification in large-scale proteomics studies. J. Proteome Res. 9, 4152–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang N., Li X. J., Ye M., Pan S., Schwikowski B., Aebersold R. (2005) ProbIDtree: an automated software program capable of identifying multiple peptides from a single collision-induced dissociation spectrum collected by a tandem mass spectrometer. Proteomics 5, 4096–4106 [DOI] [PubMed] [Google Scholar]
- 52. Beausoleil S. A., Villen J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 53. Fermin D., Walmsley S. J., Gingras A. C., Choi H., Nesvizhskii A. I. (2013) LuciPHOr: algorithm for phosphorylation site localization with false localization rate estimation using target-decoy approach. Mol. Cell. Proteomics 12, 3409–3419 [DOI] [PMC free article] [PubMed] [Google Scholar]