

Abstract
Cell compartmentalization serves both the isolation and the specialization of cell functions. After synthesis in the cytoplasm, over a third of all proteins are targeted to other subcellular compartments. Knowing how proteins are distributed within the cell and how they interact is a prerequisite for understanding it as a whole. Surface and secreted proteins are important pathogenicity determinants. Here we present the STEP database (STEPdb) that contains a comprehensive characterization of subcellular localization and topology of the complete proteome of Escherichia coli. Two widely used E. coli proteomes (K-12 and BL21) are presented organized into thirteen subcellular classes. STEPdb exploits the wealth of genetic, proteomic, biochemical, and functional information on protein localization, secretion, and targeting in E. coli, one of the best understood model organisms. Subcellular annotations were derived from a combination of bioinformatics prediction, proteomic, biochemical, functional, topological data and extensive literature re-examination that were refined through manual curation. Strong experimental support for the location of 1553 out of 4303 proteins was based on 426 articles and some experimental indications for another 526. Annotations were provided for another 320 proteins based on firm bioinformatic predictions. STEPdb is the first database that contains an extensive set of peripheral IM proteins (PIM proteins) and includes their graphical visualization into complexes, cellular functions, and interactions. It also summarizes all currently known protein export machineries of E. coli K-12 and pairs them, where available, with the secretory proteins that use them. It catalogs the Sec- and TAT-utilizing secretomes and summarizes their topological features such as signal peptides and transmembrane regions, transmembrane topologies and orientations. It also catalogs physicochemical and structural features that influence topology such as abundance, solubility, disorder, heat resistance, and structural domain families. Finally, STEPdb incorporates prediction tools for topology (TMHMM, SignalP, and Phobius) and disorder (IUPred) and implements the BLAST2STEP that performs protein homology searches against the STEPdb.
All cells have evolved specialized compartments demarcated by biological membranes. Compartmentalization isolates biological processes, divides labor, controls molecular flows; elevates solute and macromolecular concentrations and increases reaction efficiencies.
Bacterial cells comprise a cytoplasmic aqueous volume enclosed in a single (Gram+) or double (Gram−) lipid bilayer. The Gram− cytoplasm is surrounded by a multilayered cell envelope (CE)1. The CE consists of the plasma or inner membrane (IM) phospholipid bilayer and an additional external lipid bilayer, the outer membrane (OM) that also contains anchored lipopolysaccharide molecules (1). Between the two membranes lies the periplasm, a crowded space that contains proteins, small molecules and a peptidoglycan mesh layer (1) (Fig. 1).
Fig. 1.
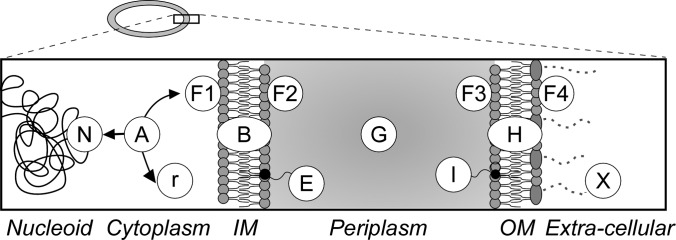
Protein subcellular location categories. Cartoon representation of E. coli cell that comprises of the cytoplasm surrounded by the inner (IM) and outer (OM) membranes. Proteins are classified in 13 categories: N: nucleoid-associated r: ribosomal A: cytoplasmic F1: peripherally associated with the IM facing with cytoplasm, B: integral IM proteins, F2: peripherally associated with the IM facing the periplasm, E: IM lipoproteins, G: periplasmic, I: OM lipoproteins F3: peripherally associated with the OM facing the periplasm, H: integral OM proteins, F4: peripherally associated with the OM facing the extra-cellular space, X: extra-cellular space. For Gene Ontology correlations see supplemental Table 3.
E. coli proteins associated with the IM and with the CE are involved in many cellular processes such as membrane biogenesis, maintenance of cell structure, transport of biomolecules, signaling and chemotaxis. These proteins are found in various cellular locations (Fig. 1) either associated with membranes, or freely diffusing between them.
Protein trafficking is experimentally tractable via in vivo tools such as monitoring of fluorescently tagged polypeptides through microscopy. Such approaches have been utilized for individual proteins but not for the complete E. coli proteome (2, 3). One disadvantage is that the fluorescent protein derivatives they employ, fused to a protein of interest, do not always get translocated in a functional form across the IM (4). In some cases, such folded proteins can be exported through the TAT pathway (5). Subcellular localization can also be studied by in vitro fractionation (6). One limitation of such approaches is the loss of cellular localization context and the possibility for cross-compartment contamination (7).
Localization of many proteins can be predicted using bioinformatics tools (8–11). Certain elements such as the signal peptide are excellent identifiers of secretory proteins (8, 9, 12, 13) that use the main, ubiquitous and essential “Sec” pathway (14) or the minor TAT pathway (15). However, this is not the case for all secreted proteins some of which have no readily identifiable “export signals” or even use “piggy-back” mechanisms or undergo the so called nonclassical secretion (16). Additional secondary structural elements such as TMs (17, 18), beta-barrels (19–21), amphiphilic alpha-helical anchors (22–24) and functional domains such as peptidoglycan (25, 26) and DNA (27, 28) binding domains can also serve as indicators of subcellular location.
Cataloging the subcellular location of proteins and their interactions is a first step towards a physicochemical understanding and in silico modeling of cells. Moreover, it is becoming increasingly obvious that several proteins undergo dynamic changes of their location, e.g. from the nucleoid to the membrane or from the cytoplasm to the extracellular space. These dynamics are regulated by different stimuli and are fundamental to the biology of the cell.
Comprehensive protein localization annotation at the proteome level is not yet available for E. coli, an exhaustively dissected model organism. General databases (e.g. Uniprot (29)) and others dedicated to E. coli (e.g. EcoWiki (30) and EcoCyc (31)), incorporate partial or mostly predicted cellular compartment annotation (EchoLOCATION (32)) for the E. coli K-12 proteome. Here we present STEPdb, a database that brings together, corrects, resolves conflicts, and re-annotates available subcellular annotation for E. coli K-12, contributes additional validated topological annotation, amalgamates this with proteome-wide biophysical information and provides visualization of proteins in a cell-context through a “cell atlas.” STEPdb organizes proteins based on their subcellular class and summarizes their features. It incorporates a plethora of information including protein complexes, proteomic, transcriptomic, biochemical data and structural, functional, and abundance annotation.
All the information collected after an exhaustive manual curation process is organized in a database easily accessible by a web interface and assisted by supporting bioinformatics tools. Specifically, STEPdb implements a multipredictor tool (SignalP, TMHMM, Phobius, and IUPred) that can be used to perform structural and topological feature predictions and BLAST2STEP that can perform similarity inquiries into the E. coli K-12 proteome. STEPdb lays the foundation towards mapping of proteome dynamics, interactions, and trafficking in the E. coli model.
In the results sections below we first detail all the steps taken to complete the annotation and manual curation of the E. coli K-12 proteome then we describe the interface through which the user can access this information.
EXPERIMENTAL PROCEDURES
The E. coli K-12 Reference Proteome and Data Sources
Two databases Uniprot (29) and EcoLOCATION (32) and the proposed IM proteome (33) were the main initial starting points for the complete subcellular categorization of K-12 described here. The E. coli K-12/MG1655 strain is one of the microbial proteomes whose comprehensive annotation is of the highest priority in Uniprot (29). This is the “reference proteome” for E. coli, contains 4303 proteins, and has been annotated here. Our annotation has been formulated in such a way that it can be easily incorporated in Uniprot.
EchoLOCATION has an easily accessible table that maps gene names to subcellular locations. However, mapping the gene names given by EchoLOCATION to the respective protein identifiers in Uniprot was not straightforward. Unfortunately, gene names cannot serve as unique identifiers of a protein sequence. In more than 100 cases the gene name of a predicted protein in EchoLOCATION when searched against Uniprot gave as a result more than one K-12 protein hits. That is because there are proteins that have common synonymous gene names with the primary gene name of others.
To retrieve updated Uniprot accession identifiers and to map Uniprot accessions identifiers to EchoLOCATION identifiers (termed: EchoBASE IDs) we used the “ID mapping” function of Uniprot. In cases where the only provided identifiers were the gene names, we used mySQL queries to compare with the primary and alternative gene names in Uniprot. In cases where multiple matches existed for the same gene name, we manually resolved the differences based on other information (e.g. protein description, mass etc.).
The annotation of pseudogenes, mobile elements, transposons, and insertion elements relied on EcoGene (34), Uniprot (29), and Ochman et al. (35). The list of E. coli K-12 complexes was retrieved from EcoCyc (31) and literature searches.
Bioinformatics Tools and Parameter Definitions
The proposed topologies in the three resources were compared between them and extensively re-evaluated with bioinformatics tools. The primary prediction tools utilized were: 1) SignalP4.0 (8) and LipoP (12) that predict signal peptides cleaved by signal peptidases I and II respectively; 2) TatP (13), which identifies the twin arginine motif present on TAT pathway signal peptides; and 3) TMHMM v2.0 (36) and Phobius (9) that detect transmembrane (TM) α-helixes of IM. For predictions with Phobius and TMHMM, LipoP and TatP the applied thresholds of the developers were used. In SignalP we selected the default d-cutoff values and selected the option for input sequences that may include TM regions. Lipoproteins that are anchored in the inner membrane were distinguished from those anchored to the outer membrane based on the amino acid at the position +2 of their mature domains (Asp or Glu in IM lipoproteins). TatP was based on the predefined regular expression best describing the form of the twin arginine motif (13). However, discrepancies in accurate prediction of known proteins made us annotate as TAT secretory proteins only those verified by biochemical studies (5) including some lacking any signal peptide (TAT-piggybacks; see export systems below).
Specific structural features that reveal subcellular locations were identified by a combination of both homology and structural domain searches and specialized tools such as Amphipaseek for the prediction of in-plane membrane anchors (supplemental Fig. S2). Other ancillary tools that were used to predict subcellular topologies were PSORT-B (11) and sosuiGramN, that is based on the physicochemical properties of the sequences (10), LocTree3, a SVM based tool (37) and ClubSub-P, that performs clusters-based homology searches for Gram− bacteria (38). With ClubSub-P we used 70% length coverage for query and hit sequences and a sequence identity threshold of 40%. These tools are based on modern machine learning algorithms that are trained by more complete and less adulterated training sets. EcoGene (34) and EcoCyc (31) were used for additional information on the proteins and their interactions.
Potential unstructured regions were examined with IUPred (39), which sums up the pair-wise stabilizing energy (interaction energy) of the constituting amino acids. It makes the assumption that IDPs are unable to stabilize their structure because of poor stabilizing contacts. The stabilizing energy of all amino acid pairs is summarized in an energy-predictor matrix (P-matrix). P-matrix values have been calculated based on the inter-residue interactions of globular protein structures.
Sequence Similarity Analysis
We run BLAST (33) queries to determine homologies between proteins. Potential outer membrane proteins carrying the autotransporter domain were determined through their homology with Ag43. The Ag43 protein sequence was run against the reference K-12 proteome, then we applied score and e-value thresholds >40 and <10−4 correspondingly.
Additionally, we identified potential PIM proteins by their similarity to well characterized peripheral membrane proteins. We used FadD13 protein of Mycobacterium, that is an ATP-binding protein membrane associated through distinctive regions rich in aromatics (40). Six proteins have been identified as “By similarity” PIM proteins based on homology to FadD13 (EntE, Acs, CaiC, MenE, FadK, and PrpE). Another set of potential PIM proteins were identified as homologs of MalK, the ATPase component of the maltose transport system. MalK is a peripheral inner membrane protein involved in maltose transport in complex with MalF and the integral inner membrane protein MalG (41). Nineteen proteins homologous to MalK have been identified in E. coli K-12. These proteins are ATPase subunits of similar ATP-binding cassette (ABC) transporters.
Evaluation of Experimental Procedures
The confidence level of the experimental evidence depends on the experimental methodology followed. For example fractionation methods coupled with proteomic analysis are known to have commonly cross-compartment contamination (7). There are also concerns about the validity of the statistical analysis following peptide identification. Early proteomic studies tend to apply less stringent criteria and use less sensitive mass spectrometers.
In the annotation of E. coli we followed some confidence criteria regarding the experimental evidence that was discovered. The order of reliability was microscopy and biochemical experiments, then proteomic studies. Therefore when a protein was identified by prefractionation MS-based proteomics, but also identified by biochemical studies the latter was considered as stronger evidence and the protein was annotated accordingly.
Comparison of E. coli Strains
Two E. coli proteomes (K-12 and BL21(DE3)) were compared with each other using the BLAST+ library (34) in order to define the “common proteome.” Each proteome was run against the other and each K-12 protein was compared against the whole proteome of BL21 and vice versa. Best hits with an identity of <40% (i.e. percent of identical residues between two sequences normalized to their total length) or an e-value <10−3 were discarded.
We defined the “core” proteome (i.e. common between all E. coli strains) using 43 sequenced E. coli strains (supplemental Table S10). Sequence alignment of all K-12 proteins against all E. coli complete proteomes was performed. The best hit for each K-12 protein in each of the 43 E. coli proteomes was selected. Next, only the best hits with identity of >40% and e-value<0.001 were selected as homologous. “Core proteins” are those with homologs that satisfy the above criteria for all the E. coli strains. In both “common” and “core” proteome analysis, protein hits were filtered using the “best hit” algorithm of the blastp routine (threshold set to 0.2). The “core proteome” comprises 2583 proteins (supplemental Table 1).
Expressed Genome
Active genes (i.e. transcribed at mRNA level) were considered to be all genes that have transcripts identified as “present” at either of four microarray datasets (42–46) or quantified in single cell analyses (44).
Database Implementation
STEPdb is a web-based database of protein subcellular locations. Data are organized and accessed through the mySQL database management system. The web interface of STEPdb was designed and generated by PHPMaker, a PHP- and Javascript-based Content Management System. The STEPdb web interface is currently available through an Apache web server (http://httpd.apache.org/). Supplementary visual interventions have been implemented with the jQuery library.
IUPred (39) source code and SignalP (8), Phobius (9) and TMHMM (36) binaries were downloaded under academic licenses and incorporated into a single multiselection tool written in PHP.
Disorder probability, solubility, and IM topology graphs are plotted using the JpGraph Object-Oriented chart library (http://jpgraph.net/). Subcellular location distributions under the cell cartoon are drawn using the “Highcharts” chart API (http://www.highcharts.com/). Complexes in the “Complexome” section are drawn in PHP with the graphics drawing library.
The BLAST2STEP web-tool was developed in PHP and currently does not support user defined identity and e-value thresholds. It uses a predefined setup and hits are filtered using the “best hit” algorithm of the blastp routine (threshold set to 0.4) and only one hit (the best) for each query is returned.
RESULTS
The Basic Proteome of E. coli K-12
E. coli strains survive in different environments and have accumulated evolutionary traits that range from those allowing adaptation to various habitats to silent genes (47). We first defined the “basic” proteome of K-12, that is devoid of proteins that are likely not synthesized and/or are encoded in genomic insertions enriched in defective prophages, transposons, pseudogenes, integrases, and mobile elements (supplemental Table S1 and S2). The “basic” K-12 proteome comprises 3897 proteins (supplemental Table S4A). The transcripts encoding 3849 of these polypeptides (42–45) as well as 3178 of these polypeptides have been detected in cells grown in LB broth (48, 49).
Subcellular Classification of the E. coli Proteome
The E. coli proteome was divided into 13 subcellular locations (10 for the CEP, 3 for the cytoplasm) based on, and extending, the formalisms of EchoLOCATION (Fig. 1). We correlated each subcellular location to a distinct GO (gene ontology) classifier (supplemental Fig. S1; supplemental Table S3 (50)). Ribosomal proteins (r) together with rRNAs constitute the large and small subunits of the ribosome; nucleoid proteins (N), include DNA/RNA binding proteins such as DNA helicases, polymerases, histone-like proteins, sigma factors, repair enzymes, and transcription factors. Proteins that have been either captured experimentally in the cytoplasm or for which there is no indication that they are located in any other location, are classified as cytoplasmic (A). In general, all nucleoid or peripheral membrane proteins will have an obvious “cytoplasmic” state so this is not explicitly identified for these classes of proteins.
Existing Annotations and Inconsistencies
First we compared the annotations provided by the three resources. Uniprot assigns subcellular locations to 48% of the K-12 reference proteome (Table I, supplemental Table S4B). EchoLOCATION organizes 4345 protein sequences into 11 subcellular locations. Bernsel and Daley, characterize 1133 IM proteins. We were able to map 4243 of the 4345 predicted proteins in EchoLOCATION and 1108 out of 1133 proposed IM proteins to the reference E. coli K-12 proteome (Table I; supplemental Tables S4D–S4E; Fig. 2). Proteins from either resource that did not correspond to entries in the E. coli reference proteome were excluded from further analysis. These were: unknown coding sequences, putative pseudogenes/transposases, duplicate entries in EchoLOCATION, proteins that have been deleted from Uniprot, or belonging to other E. coli strains (supplemental Table S4E).
Table I. Summary of previously existing subcellular annotation and comparison with STEPdb. Uniprot and EchoLOCATION databases contributed 2050 and 3979 protein annotations correspondingly and together with the theoretical IM proteome (33) gave a combined 4111 initially annotated proteins. Comparison of the three resources revealed some matches in proposed subcellular locations (“Matching annotation”) but also some differences (“Conflicting annotation”). IM proteins with matching annotations in only in two of three resources are referred as “Unique IM proteome.” All proteins with existing annotations in only one source are referred as “Unique (total).” STEPdb multicombinatorial analysis contributed 36 newly classified proteins that were of unknown location (“STEPdb de novo annotated”), 674 proteins that have been reassigned to locations other than previously proposed (“STEPdb revised”) and 601 proteins with contradicting subcellular annotations that have been unresolved (“STEPdb unresolved”).
| Uniprot | EchoLOCATION | Bernsel & Daley (2009)33 | Total | |
|---|---|---|---|---|
| Reference proteome (E. coli K-12) | 4303 | 4345a | 1133 | 4303 |
| Matching annotations | 1613 | 1646 | 850 | 1652 |
| Unique (IM proteome) | 11 | 29 | 4 | 44 |
| Unique (total) | 12 | 1998 | 4 | 2014 |
| Contradicting annotations | 425 | 599 | 254 | 601 |
| Existing annotations | 2050 | 4243 | 1108 | 4267 |
| % of reference proteome | 48% | 98% | 26% | 99% |
| Missing annotations | 2253 | 60 | – | 36 |
| Missing and unresolved annotations | 2678 | 659 | 254 | 637 |
| % of reference proteome | 62% | 15% | 6% | 18% |
| STEPdb total contribution over previous annotations | 3352 | 1333 | 560 | 1311 |
| de novo annotated | 2253 | 60 | 84 | 36 |
| Revised | 674 | 674 | 222 | 674 |
| Resolved | 425 | 599 | 254 | 601 |
| Experimental validations | 1205 | |||
| References added | 118 | |||
| % of reference proteome | 76.89% | 35.37% | 7.87% | 32.81% |
a This is the estimation of the total proteome of EchoLOCATION.
Fig. 2.

Annotation of subcellular topologies of the E. coli K-12 proteome. The E. coli K-12 reference proteome was downloaded from Uniprot (July 2014; (29)). Respective subcellular annotation from Uniprot (29) was retrieved for 2050 proteins (∼46% of the total proteome; Table I). Subcellular annotation was also downloaded from EchoLOCATION (32) that lists 4345 proteins that were matched to 3957 proteins of the E. coli reference proteome (Table I). The annotation of IM proteins we based on a proteomic analysis that contributed 1108 IM proteins (33). The subcellular terminologies of the two databases were assigned to a STEPdb subcellular class (supplemental Table S4). Amalgamation of these three resources contributed a total of 4111 proteins with an already existing proposed annotation in at least one resource leaving 195 proteins of unassigned topologies and 576 proteins with contradicting proposed subcellular location (Table I). To determine the a subcellular location for the unknown proteins and to resolve the annotation differences we sought to utilize bioinformatics tools that can predict subcellular location or other structural motifs and sequence alignment (supplemental Fig. S2). The core tools utilized were: SignalP, TatP, LipoP, Phobius (8, 9, 12, 13) used for the prediction of secretion motifs, PSORT-B (11) for the prediction of subcellular location and TMHMM, Phobius (9, 36) for the prediction of transmembrane helices. A set of additional bioinformatics tools (Prediction Tools 2) comprising Protscale for hydrophobicity (116), SOSUI, ClubSub, LocTree3 for subcellular location (10, 37, 38) AmphipaSeek for amphipathic in-plane membrane anchors (51) and BLAST (117) was employed to locate additional autotransporters. The comparison between the existing annotations and the predictions of the bioinformatics tools lead to more conflicts regarding the proposed subcellular topologies (supplemental Table S8). To resolve these annotation differences we sought experimental evidence in proteomic, genomic and biochemical studies (supplemental Table S7A).
Side-by-side comparison of the three resources (containing only annotations related to the reference proteome) revealed that they either proposed the same (“Matching”) or different (“Conflicting”), or “Unknown” locations (supplemental Fig. S3; supplemental Table S5A). ∼14% of the 4303 proteins (∼15% of the basic reference proteome) had conflicting subcellular annotations (Table I; Fig. 2; supplemental Table S8). Thirty-six proteins remained without a proposed assigned location in at least one of the three resources (Table I; supplemental Table S7). Twenty-four of the unknown proteins belong to the K-12 “basic proteome” whereas none belongs to the “core proteome.”
Some location assignments were accompanied by experimental evidence (398 for Uniprot; 506 for EchoLOCATION; only 105 common; Fig. 3A). Many other annotations are theoretical or predicted. Uniprot defines three levels of nonexperimental identifiers that we also adopted: “potential” (topologies that are predicted), “probable” (at least some experimental indication exists), and “by similarity” (indications exist in other(s) bacterial strains or species). EchoLOCATION incorporates two levels of evidence: “theoretical” for predicted proteins and “experimental.”
Fig. 3.
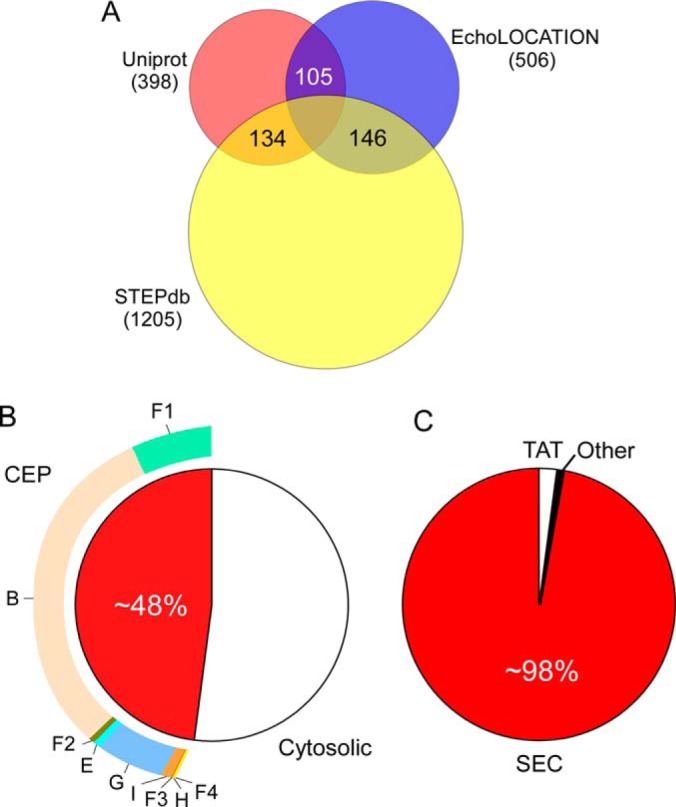
Summary of annotated topologies and experimentally verified proteins. A, Proteins with experimentally verified topologies in the two databases compared with STEPdb. B, CEP proteome and distribution of proteins within its subcellular compartments. C, Utilization of the main export systems in E. coli.
To resolve conflicts, determine the “unknown” topologies, obtain additional experimental evidence, and complete the subcellular location analysis of the E. coli proteome we re-examined the previous annotations ab initio.
De novo Comprehensive Topological Annotation of E. coli K-12
Towards the complete topological annotation of E. coli we followed a number of steps that included prediction tools of subcellular locations and structural domains, homology searches, proteomic/biochemical data, other databases and, as a central tool, manual curation through literature searches (Fig. 2). A decision tree describes the hierarchical clustering of the information from the various sources that were combined, based on the confidence we attach to the information (supplemental Fig. S3). Location assignment of high confidence was for proteins that are accompanied by experimental evidence. The robustness of the annotation for these experimentally verified proteins was generally accepted, although in some cases (described in supplementary material, “annotation conflicts”) some proposed topologies were reconsidered and overruled.
In the two following sections, we describe in more detail the process that we followed and provide some examples.
De novo Annotation of Proteins of “Unknown” Location
For the annotation of the 36 proteins of “Unknown” location we first used prediction tools and literature searches. This led to several assignments, for example, for MntS (cytoplasmic), OmpP (OM), and SgrT (PIM) (supplemental Table S7). YshB has a prediction for a potential amphipathic helix (1–10) (51) and is thus annotated as potential PIM protein. The remaining proteins were annotated as “Potential” based on the predictions of the tools (most of them as cytoplasmic; supplemental Table S7).
Elucidation of Previous Annotation Conflicts
The “Conflicting” proposals for subcellular compartments of 601 proteins were resolved in a series of steps (supplemental Fig. S3).
1. Large Scale Localization Studies
First we analyzed high throughput proteomic (supplemental Table S6C), biochemical (3, 52), genomic (27), and microscopy studies (53).
Proteins identified in either of the proteomic studies in specific locations were annotated as “experimental” unless overridden by more dedicated biochemical studies (see step 4). There were cases where two proteomic studies suggested different locations. One example is BtuB that has been identified in the outer membrane and the cytoplasm and Fis identified both in association with the inner membrane and in nucleoids collected by sucrose gradient centrifugation (supplemental Table S6A). If additional evidence was lacking, proteins were annotated as being in either of the two locations.
2. Bioinformatic Prediction Tools
Proteins with remaining conflicting annotations were further examined with the core prediction tools. When all prediction tools agreed (e.g. SignalP, LipoP, and Phobius predicted the existence of a signal peptide or both TMHMM and Phobius predicted a TM) the proteins were annotated as “potential.” When primary predictions failed to resolve issues we made additional use of recent classification tools (see Experimental Procedures).
3. Structural Elements that Correlate with Subcellular Localization
Certain structural features can also be indicative of protein localization. These include the TMs, β-barrels, and amphipathic α-helices and folds such as the “autotransporter domain.”
Some PIM proteins interact with the IM via amphipathic helices (22–24, 54–56). STEPdb identifies nine of them: DhnA (57), PbpB (22), FtsA (55), MinD (24), GlpD (54), FtsY (23), Rne (58), Rnb (59), and MinE (60). Proteins with similar features (e.g. DacA (56)) have also been identified in the periplasmic face of the IM.
The “autotransporter domain,” is located at the C terminus of OM autotransporters (AT) (61). STEPdb identifies 10 ATs mainly based on homology to the well characterized Ag43 and the presence of the at-1 InterPro family motif (62). However, three of these proteins (YcgI, YcgV, and YdeU) do not possess detectable signal peptides.
Peptidoglycan (PG)-binding domains can also be indicators of peripherally associated proteins of the OM (F3 class; Fig. 1). STEPdb identifies five L, d-transpeptidases. Three of them (YbiS, ErfK, and YcfS) cross-link Braun's lipoprotein (Lpp) to the PG and two of them form direct cross links with the PG (63). MotB is both PG-tethered through its conserved C-terminal region and IM-anchored through its N-terminal TM (64). The lipoprotein Pal is anchored in the OM and associates with the PG through its C terminus (65).
One common DNA-binding element is the helix-turn-helix (H-T-H) motif. Sigma factors (RpoS, RpoE, and RpoH) contain the H-T-H motif that mediates interaction with RpoA (RNA polymerase subunit) and with the −35 element in promoter DNA. Another set of DNA-binding proteins are the histone-like proteins (66). The signature motif of this family is a twenty residue sequence that includes three perfectly conserved positions. Four members of this family exist in K-12 (DhbA and B and IhfA and B).
The BON (bacterial OsmY and nodulation) domain is related to association with phospholipid membranes (67). Apart from OsmY, which contains two BON domains, YraP also contains two and YgaU one such domain.
4. Manual Curation
For all the remaining unresolved localizations we performed literature searches. This led to additional new experimental documentation for 1205 proteins (Fig. 3A), derived from 118 studies. For 152 proteins that remained with unassigned localizations we applied certain criteria (supplemental Table S5A) and annotated them as “Potential.” Literature searches also provided experimental evidence for proteins that had “Matching” proposed topologies by the three resources.
Summarizing the results from the conflict resolution efforts described above: for 200 proteins we have found enough evidence and propose more certain subcellular topologies. For 227 proteins the primary prediction tools agree among themselves and for 152 proteins the topology has been decided based on additional criteria (supplemental Table S5). Some detailed examples of resolved conflicts are given (supplementary Results: “Annotation conflicts” and supplemental Table S8).
The Challenge of the PIM Proteome
PIM proteins are soluble yet membrane-interacting and a particular annotation challenge (6). They mediate communication of the cytoplasm with the cell envelope through the IM for a plethora of biological processes (6). PIM proteins are under-represented in current databases, probably because of the absence of any bioinformatics tools to predict them. EchoLOCATION labels only 10 of them as “membrane-associated” whereas Uniprot lists 139 “membrane-related” proteins of which 127 are proposed to “associate with the IM from the cytoplasmic side.” PIM proteins can be identified experimentally by their physical interaction with the IM (6). STEP collects 550 PIM proteins of which 37 have multiple locations (see below). 392 are experimentally verified, 366 of them were characterized recently (6). From the remaining PIM proteins, 76 were annotated as “Probable,” 20 as “Potential,” and 26 “By similarity”.
Proteins with Multiple Subcellular Locations
Our analysis indicates that 60 of the proteins of K-12 may exist in multiple subcellular locations (Fig. 2, “MT”) making the proteome much more topologically dynamic then currently known. Proteins with multiple subcellular locations are not specifically demarcated in Uniprot or EchoLOCATION. Of these, 37 are annotated both as nucleoid and PIM proteins (N, F1); nine as both nucleoid and IM proteins (N, B). This suggests that they act as important physical connectors between the inner membrane and the genetic material. In total 53 of the 60 have at least one verified localization and the remaining were annotated as “Probable” or “Potential.” Five of these (ArcD, YgjI, ClcB, RodZ, and CadC) have been experimentally verified as being IM (3, 68, 69) and peripheral proteins.
SecM and YgfZ have been verified to localize both in the cytoplasm and the periplasm (70) (A, G). OsmY, ChiA, and HlyE have been characterized as both periplasmic and extracellular proteins. ChiA becomes secreted when the cryptic T2S system is activated (71).
Cytoplasmic peptidyl-tRNA hydrolase ArfB can also bind to ribosomes through its C-tail (72). Three OM lipoproteins (Wza, CsgG, and Lpp), normally residing in the inner leaflet of the OM, have also been demonstrated to be partially or fully surface exposed (73–75).
Two flagellar proteins, FliK and the anti-sigma factor FlgM, are cytoplasmic proteins that get secreted at the appropriate time through the flagellum. Two integral inner membrane proteins, CyoA and YiaD, have also been shown to be signal peptidase II substrates, thus both annotated as lipoproteins. Finally, enolase can be found in three states, cytoplasmic, peripherally associated to the IM and cell-surface bound (76, 77).
To denote multiple localization possibilities that have been experimentally established we introduced the comma “,” formalism whereas a slash “/” denotes two or more possible subcellular locations that have not yet been experimentally determined.
Comparing Two E. coli Proteomes
The proteome of K-12 derivative MG1655 was compared with strain BL21(DE3). BL21(DE3) is the most widely employed host for the production of recombinant proteins and is considered more robust than K12 strains because of reduced acetate production and absence of some proteases (42, 78). More than 90% of each proteome is “common,” leaving 266 and 359 proteins unique to K-12 and BL21(DE3) respectively (supplemental Table S1). The subcellular locations of the “common” proteins in BL21(DE3) was extrapolated from those of K-12. Proteins unique to BL21(DE3) were annotated de novo following the same process followed for K-12.
Protein Sorting and Secretion
Our annotation reveals that a remarkable 48% of the K-12 proteome that is synthesized in the cytoplasm, is transported to various locations in the CE (supplemental Table S4A; Fig. 3B). Before they reach their final location, CE proteins have to overcome membrane barriers that pose major energetic hurdles. E. coli cells have developed several export mechanisms to negotiate crossing into or across the IM or OM. STEPdb annotates the 12 protein export systems identified in K-12 (Fig. 4) (14, 61, 79–87). STEPdb also determines, where known, the respective exported proteins that utilize them.
Fig. 4.

E. coli K-12 main protein export pathways. Schematic representation of all currently known E. coli K-12 export systems along with their respective structural and chaperone/targeting components. Sec-dependent and non-Sec dependent export systems exist. The Sec-dependent export systems are: SEC, the essential Sec secretory pathway from which most of the proteins are secreted across or inserted into the plasma membrane; SRP-SEC, co-translational export pathway, known to mainly target and insert transmembrane proteins into the plasma membrane; LOL, Lipoprotein sorting system; BAM, the complex for the β-Barrel Membrane protein assembly; TAM, Translocation and assembly module for Autotransporters; CU, Chaperone Usher export system; T5S, the secretion pathway of autotransporters (composed of three functional domains: leader sequence, the passenger domain and the β-domain); Curli, extracellular amyloid fibers; OMV, outer membrane vesicles; T2S, Type two secretion system, which mediates secretion of proteins that are folded in the periplasm. The non-Sec dependent export systems are: Flagellum, is a motion generation organelle able to secrete some of its constituent proteins; TAT, twin arginine translocation system. Also YidC, a protein involved in membrane insertion of some proteins together with SecYEG, is also known to act independently thus defining its own pathway. Auxiliary components of the export systems presented include the periplasmic chaperone SecB, the signal recognition ribonucloprotein particle Srp, the periplasmic chaperones (Skp and DegP), the peptidyl prolyl cis-trans isomerases (SurA, FkbAB, and PpiAD) and the periplasmic disulfide oxidoreductases DsbABCD (14). The list of proteins shown is not comprehensive (see database).
A. Protein Export Systems
Protein export systems in K-12 can be classified as Sec-dependent (LOL, BAM, TAM, CU, T5SS, Curli, OMV, and T2SS) and non-Sec dependent (Flagellum and TAT)(Fig. 4). This generally reflects the protein export systems found in all bacteria although additional export systems, not present in K-12, are known to exist in other Gram− and Gram+ bacteria. The bulk of protein translocation across and protein integration into the IM is carried out by the Sec pathway (>98%; Fig. 3C; Fig. 4)(14). Sec-dependent systems generally deal with sorting proteins to the periplasm, outer membrane and beyond after the IM has been negotiated via the Sec translocase.
B. Secretory Proteins
STEPdb organizes exported proteins in two levels of secretion that are facilitated by two levels of chaperone/targeting factor pathways. The two export levels reflect the export machineries responsible for secretion through the IM (Sec, TAT, or YidC) or the OM (e.g. BAM, TAM, or CU) whereas the two levels of targeting refer to the soluble components that are responsible for guiding exported proteins to the specific membrane (e.g. SecB for the SecA/SecYEG translocase, SRP for the SecYEG translocase, Skp for the BAM etc.).
The annotation of exported proteins utilizing export systems was mainly based on tools that can predict well characterized secretion motifs such as the Sec/TAT signal peptides and TM regions (8, 12, 13) and proteomic/biochemical data (3, 5, 54, 88–90) from both in vivo (91), in vitro (92), and proteomic (93) studies. However, there are proteins that get secreted through the Sec system without having an obvious signal peptide like the Rhizobium SodA (SodM and SodF homologs in K-12) (94).
All predicted and experimentally verified Sec and TAT signal peptides were included in STEPdb along with a synopsis of their lengths and physicochemical properties. Two types of exported proteins make use of the Sec system (95). In type I secretory proteins (e.g. periplasmic, OM, or extracellular proteins) the signal peptide is excised by signal peptidase I (SPaseI), at the periplasmic face of the plasma membrane. Type II proteins have evolved signal sequences that are cleaved off by SPaseII. Type II proteins comprise of lipoproteins anchored either on the IM or the OM (81). Some lipoproteins are secreted in an unfolded form through the Sec pathway with the help of YidC (96). Lipoproteins are covalently modified with lipids at a cysteinyl amino-terminal residue (+1 position relative to the cleavage site).
IM proteins are thought to utilize mainly the SRP pathway for their targeting and are cotranslationally inserted into the IM by the Sec translocase (97). In the absence of proteome-level experimental data for all IM proteins, we adopt this proposal and categorize them all as being SRP and Sec substrates. One known exception is HyaA, an IM protein with a single TM helix. HyaA is targeted by the SRP but secreted through the TAT pathway (5). YidC-dependent IM proteins have been defined through fluorescent tagging (98). IM insertion after depletion of YidC was monitored for ∼400 IM protein using GFP tags (98). YidC appears to be responsible for the membrane integration of 77 IM proteins of K-12 and some small proteins produced by phages such as M13 and Pf3 (89, 98). Negative charges on the periplasmic segments or within the TMs have been proposed to act as possible determinants of YidC substrates (99).
In total, 33 proteins follow the TAT pathway for secretion and most of them possess characteristic signal peptides (5). Interestingly, some proteins that use this pathway lack TAT signal peptides (e.g. DmsB) and are translocated by “piggy-backing” on other TAT proteins (e.g. DmsA) that do possess bona fide TAT signal peptides (100, 101).
Finally, at least two proteins (HlyE and YebF) are known to be exported through outer membrane vesicles that bleb off the cell's surface (79, 102). Similarly, periplasmic OsmY can also be secreted into the medium.
The Web Interface of STEPdb and the Embedded Bioinformatics Tools
In this section we present the STEPdb web-interface, the included information and the auxiliary web-tools developed. STEPdb is navigated through a panel menu on the left. This is divided in two groups of buttons: 1) “Strains,” includes items giving access to the lists of proteins of the two E. coli strains as well as the list of complexes, PIM proteins, Sec/TAT secretory proteins and IM proteins, and the pairing of the two E. coli strains and 2) “Downloads and Tools” (see below), which includes a series of prediction tools, references and information on specifics of STEPdb under “About.”
The comparison of subcellular locations of the two E. coli strains is accessible through the “K-12 versus BL21” button. Below the K-12 branch lie some subclasses of the K-12 proteome along with their summarized features (PIM, IM, and the SEC/TAT secretomes). The list of proteins of each class can be viewed by the corresponding “Sequences” link and the summarized features through the “Features” link.
Beneath the IM proteome branch lies an additional button, “Topology,” where users can visualize the transmembrane orientation of ∼700 IM proteins. These are based on predictions made by Phobius and then corrected by the experimentally verified localization of the C termini of these proteins (3). A strong descriptor of IM proteins is the existence of TM regions. These can be accurately predicted by various tools (9, 36) along with the information of their orientation in the IM (i.e. which of their regions are exposed to either the cytoplasm or the periplasm). Based on these data TMHMM predicts correctly the orientation of only 78% (3) and Phobius of only ∼81% of these proteins (data not shown). Membrane proteins are classified in STEPdb depending on the location of their termini but also based on whether they are bitopic (single TMs) or polytopic (multiple TMs) (Fig. 5).
Fig. 5.
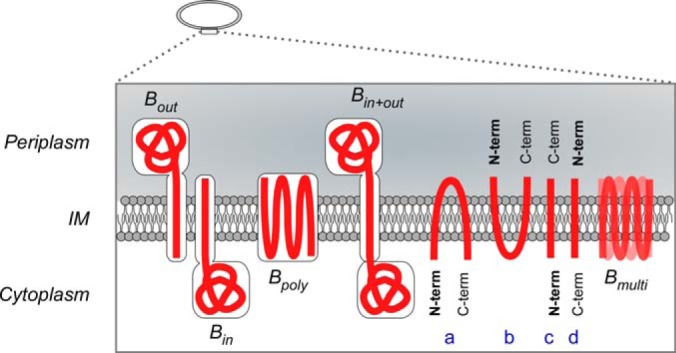
Inner Membrane protein topology. Categorization of IM proteins based on their topology and orientation in the IM. Bin, Bout bitopic IM proteins anchored in the IM with their soluble region in cytoplasm or periplasm correspondingly; Bpoly polytopic IM proteins; Bin+out single pass IM proteins with soluble regions in both cytoplasmic and periplasmic volumes; A, B, IM proteins with both their termini facing the same side cytoplasm or periplasm correspondingly; C, D, IM proteins that their N termini lay on opposite sides of the IM plane Bmulti proteins with multiple orientations.
K-12 export systems are summarized in cartoon representation in the correspondingly named menu link. In this cartoon proteins are represented as colored circles that contain active links to information for each protein. Furthermore, the complete list of the proteins participating in each export system can be selected and viewed by clicking the export system names at the bottom of the cartoon. More information regarding each system can be viewed by clicking the “References” button.
Finally, in the “Solubility” page, STEPdb summarizes the solubility features of each subcellular protein class as experimentally determined by large scale in vitro studies (103).
In the “Downloads and Tools” section of the navigation menu, the user can access: 1) the correspondence of STEPdb nomenclature to Gene Ontology terms, 2) the BLAST2STEP tool, 3) a multiprediction tool for protein motifs, 4) files for download, 5) complete list of references that have been collected after manual curation of the database entries, and 6) specific explanations on STEPdb content and the terminology used (“about”).
Bioinformatic Predictors Incorporated in STEPdb
Four widely used bioinformatics tools have been incorporated in STEPdb: TMHMM, Phobius, and SignalP, that predict secreted and membrane protein features, and IUPred that predicts protein disordered regions (39). Searches can be run via the dedicated “Predict Topology” page or accessed through “Predict Now” within the “more info” slide panel of each protein entry.
Homology searches of protein sequences against the STEPdb database can be run through BLAST2STEP. This tool can be used to predict topology for new proteins/omes based on our well-characterized subcellular topologies.
Structural/physicochemical Features of Proteins
Unstructured regions within proteins have been related to function, including trafficking (104). Proteins that include them are called intrinsically disordered (IDPs) and many of them have fundamental cellular roles (104). IDPs have regions that lack any tertiary structure under native conditions.
IDPs take part in signaling (105) and regulation pathways (106) and they often bind to DNA molecules (107), ribosomes (72), small molecules, and proteins (108).
STEPdb incorporates the IUPred tool (39) that can predict disordered areas in proteins. IUPred can be accessed either through the “more info” button where it can be executed for the selected protein or through the “Predict Location” multiprediction tool button.
The physicochemical properties of proteins (e.g. hydropathy profile and solubility), are reflected in and influence their structural attributes, their interaction with chaperones and their trafficking. Niwa et al. (103) have studied protein solubility in E. coli, in a chaperone-free reconstituted translation system using a centrifugation assay. The IM proteome consists of highly aggregation-prone proteins (solubility <30%), whereas 65% of ribosomal proteins are highly soluble (solubility >70%). Solubility features of the E. coli proteome are summarized under the “Solubility” menu button.
“More Info” Panel
Conclusions that derived from manual curation or bioinformatics predictions accompany each protein entry in STEPdb (supplemental Fig. S4). These can be found in a sliding menu panel that can be opened through a “More info” button at the left of each protein entry (supplemental Fig. S4). The “More info” panel is organized in four subsections.
The leftmost section is dedicated to manual curation information: localization, experimental evidence level, manual curation comments, and literature references. It contains additional data such as mRNA molecules/cell (44) and protein abundance as calculated by single cell screening (44) and mass spectrometry [PaxDb; (109)].
Next follows a subsection that contains information about structural motifs from the SCOP (110) database, Multifun terms, functional annotation provided by PFAM and SMART databases (111, 112). In this section a button redirects the user to protein complexes of the respective protein.
The third subsection contains physicochemical features of the protein: experimentally quantified protein solubility (103), heat stability, and protein disorder (IUPred tool; (39)). The rightmost panel brings together the results of the various classification and prediction tools. Use of the incorporated prediction tools is via the “Predict Now” button.
Protein Complexes and Visualization
STEPdb includes the first attempt to ascribe PIM protein complexes and interactions (6) and can be accessed through the “Complexome” button. The “complexome” section of STEPdb contains all known K-12 complexes, mainly drawn from EcoCyc that incorporates the only currently available curated catalog of E. coli complexes (957 complexes including subunit composition, stoichiometry, and functionality).
During manual curation we identified 61 new complexes (38 of these are heteromeric) not present in EcoCyc (supplemental Table 6B). An interesting example is Psd that gets proteolytically cleaved and forms a heteromeric complex with itself (113).
For the newly identified complexes, STEPdb retains the nomenclature, subunit composition, and Multifun (114) functional annotation formalisms of EcoCyc. Each complex can be dynamically drawn and “visualized” on demand in cartoon form upon clicking on the corresponding “draw” button (supplemental Fig. S5).
Depending on their localization complexes can be accessed through the linked cell cartoon at the top of the “Complexome” section. Protein complexes that span more than one subcellular compartment are annotated as a concatenation of all different compartments. For example the flagellum that spans both membranes and is constituted also by extracellular components is annotated as “B&H&F4.”
Subcellular annotation of protein complexes lays the foundation for future analysis and visualization of cellular functions and metabolic pathways. STEPdb colocalizes for the first time all known protein complexes of K-12 within the cell based on thorough location annotation of their corresponding subunits. This is an extension of the recently published map of the PIM proteins (6). It organizes protein complexes found in EcoCyc in nine functional categories and incorporated protein–protein interactions as registered in the Intact database (115).
The E. coli complexome map can be accessed under the “Cell Atlas” item or the “Cell Atlas” button accessible in all pages under a “cell-subsites” cartoon. It contains active links to proteins and protein complexes.
DISCUSSION
The complete elucidation of protein localization in subcellular compartments is a cornerstone for further study of any cell. The ever increasing need for proteomics analyses requires well described reference proteomes. Here we present STEPdb, an integrated database that includes protein subcellular location characterization of all proteins of E. coli K-12.
Subcellular annotation of STEPdb was based on bioinformatic prediction tools that were combined with the updated annotations of databases (29, 32) that were further corroborated by biochemical and proteomic data and a manual curation process. Manual curation contributed 1547 proteins of experimentally verified subcellular location that was based on 397 literature studies.
Collectively the multicombinatorial analysis used to derive STEPdb, revises previously existing annotation for ∼15% of the E. coli K-12 proteins (Table I; 640 of 4303 proteins), categorizes for the first time ∼4.5% of proteome (Table I; “STEPdb de novo annotated”) and resolves conflicts for 576 of 4303 proteins.
Our analysis has demonstrated that bioinformatic tools are not yet sufficient to predict comprehensively the subcellular location of all E. coli K-12 proteins. This is reflected on the contradictions between the predictions of currently available tools or by the absence of prediction tools. Another example is the prediction of TM regions by Phobius and TMHMM. In older versions of these tools the N-terminal signal peptide was frequently miss-predicted as a TM region. Even though the recent releases of SignalP and Phobius take into account the possibility of a TM or a signal peptide correspondingly, the predictions do not always agree with each other.
Towards the goal of comprehensive annotation of subcellular location, manual curation, and the inclusion of experimental data is essential. This is particularly true for protein classes such as PIM proteins that have no currently traceable bioinformatic signatures. Also, experimental knowledge of the machineries by which proteins find their final locations adds to the accuracy of the annotations. This process is ongoing and we expect it to be revisited as more experimental information becomes available. STEPdb can be used as a starting point for users looking for subcellular locations of unknown bacterial proteins. The comprehensive subcellular annotation in STEPdb will also, in the long run, provide constant feedback that will fine-tune the performance of the prediction tools and serve as a reference dataset for bacterial proteomics.
Supplementary Material
Acknowledgments
We thank W. Studier and E. Coudert (Uniprot) for insight on E. coli protein annotation, T.Minamino for providing information of flagellar protein export systems, M.Aivaliotis for help with EcoCyc complexes, and M.Papanastasiou for the PIM proteomic data.
Footnotes
Author contributions: A.E. designed research; G.O. and A.E. performed research; G.O. and A.E. analyzed data; G.O. and A.E. wrote the paper.
* This work was supported by the European Commission the Operational Programme “Competitiveness and Entrepreneuriship” (OPCE II - EPAN II) - National Strategic Reference Framework (NSRF 2007–2013) [grant codes: 09SYN-13–705 and 09SYN-13–832], an Excellence grant (#1473 to A.E.; General Secretariat of Research), and the Onassis foundation predoctoral program.
 This article contains supplemental Results, Figs S1 to S5 and Tables S1 to S10.
This article contains supplemental Results, Figs S1 to S5 and Tables S1 to S10.
The current version of STEPdb is available at http://stepdb.eu.
1 The abbreviations used are:
- CE
- cell envelope
- PIM
- Peripheral Inner Membrane
- CEP
- Cell Envelope Proteome
- IM
- Inner Membrane
- IMP
- integral inner membrane proteome
- OM
- Outer Membrane
- TM
- transmembrane.
REFERENCES
- 1. Silhavy T. J., Kahne D., Walker S. (2010) The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2:a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reyes-Lamothe R. (2012) Use of fluorescently tagged SSB proteins in in vivo localization experiments. Methods Mol. Biol. 922, 245–253 [DOI] [PubMed] [Google Scholar]
- 3. Daley D. O., Rapp M., Granseth E., Melen K., Drew D., von Heijne G. (2005) Global topology analysis of the Escherichia coli inner membrane proteome. Science 308, 1321–1323 [DOI] [PubMed] [Google Scholar]
- 4. Feilmeier B. J., Iseminger G., Schroeder D., Webber H., Phillips G. J. (2000) Green fluorescent protein functions as a reporter for protein localization in Escherichia coli. J. Bacteriol. 182, 4068–4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tullman-Ercek D., DeLisa M. P., Kawarasaki Y., Iranpour P., Ribnicky B., Palmer T., Georgiou G. (2007) Export pathway selectivity of Escherichia coli twin arginine translocation signal peptides. J. Biol. Chem. 282, 8309–8316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Papanastasiou M., Orfanoudaki G., Koukaki M., Kountourakis N., Sardis M. F., Aivaliotis M., Karamanou S., Economou A. (2013) The Escherichia coli peripheral inner membrane proteome. Mol. Cell. Proteomics 12, 599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chandramouli K., Qian P. Y. (2009) Proteomics: challenges, techniques, and possibilities to overcome biological sample complexity. Hum. Genomics Proteomics 1, 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Petersen T. N., Brunak S., von Heijne G., Nielsen H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 9. Kall L., Krogh A., Sonnhammer E. L. (2007) Advantages of combined transmembrane topology and signal peptide prediction – the Phobius web server. Nucleic Acids Res. 35, W429–W432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Imai K., Asakawa N., Tsuji T., Akazawa F., Ino A., Sonoyama M., Mitaku S. (2008) SOSUI-GramN: high performance prediction for subcellular localization of proteins in gram-negative bacteria. Bioinformation 2, 417–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gardy J. L., Spencer C., Wang K., Ester M., Tusnady G. E., Simon I., Hua S., deFays K., Lambert C., Nakai K., Brinkman F. S. (2003) PSORT-B: Improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res. 31, 3613–3617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Juncker A. S., Willenbrock H., Von Heijne G., Brunak S., Nielsen H., Krogh A. (2003) Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 12, 1652–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bendtsen J. D., Nielsen H., Widdick D., Palmer T., Brunak S. (2005) Prediction of twin-arginine signal peptides. BMC Bioinformatics 6, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chatzi K. E., Sardis M. F., Karamanou S., Economou A. (2013) Breaking on through to the other side: protein export through the bacterial Sec system. Biochem. J. 449, 25–37 [DOI] [PubMed] [Google Scholar]
- 15. Patel R., Smith S. M., Robinson C. (2014) Protein transport by the bacterial Tat pathway. Biochim. Biophys. Acta 1843, 1620–1628 [DOI] [PubMed] [Google Scholar]
- 16. Bendtsen J. D., Kiemer L., Fausboll A., Brunak S. (2005) Nonclassical protein secretion in bacteria. BMC Microbiol. 5, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wissel M. C., Wendt J. L., Mitchell C. J., Weiss D. S. (2005) The transmembrane helix of the Escherichia coli division protein FtsI localizes to the septal ring. J. Bacteriol. 187, 320–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lemmin T., Soto C. S., Clinthorne G., DeGrado W. F., Dal Peraro M. (2013) Assembly of the transmembrane domain of E. coli PhoQ histidine kinase: implications for signal transduction from molecular simulations. PLoS Comput. Biol. 9, e1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koronakis V., Sharff A., Koronakis E., Luisi B., Hughes C. (2000) Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 405, 914–919 [DOI] [PubMed] [Google Scholar]
- 20. Conlan S., Zhang Y., Cheley S., Bayley H. (2000) Biochemical and biophysical characterization of OmpG: a monomeric porin. Biochemistry 39, 11845–11854 [DOI] [PubMed] [Google Scholar]
- 21. Wimley W. C. (2003) The versatile beta-barrel membrane protein. Curr. Opin. Struct. Biol. 13, 404–411 [DOI] [PubMed] [Google Scholar]
- 22. Sung M. T., Lai Y. T., Huang C. Y., Chou L. Y., Shih H. W., Cheng W. C., Wong C. H., Ma C. (2009) Crystal structure of the membrane-bound bifunctional transglycosylase PBP1b from Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 106, 8824–8829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parlitz R., Eitan A., Stjepanovic G., Bahari L., Bange G., Bibi E., Sinning I. (2007) Escherichia coli signal recognition particle receptor FtsY contains an essential and autonomous membrane-binding amphipathic helix. J. Biol. Chem. 282, 32176–32184 [DOI] [PubMed] [Google Scholar]
- 24. King G. F., Rowland S. L., Pan B., Mackay J. P., Mullen G. P., Rothfield L. I. (1999) The dimerization and topological specificity functions of MinE reside in a structurally autonomous C-terminal domain. Mol. Microbiol. 31, 1161–1169 [DOI] [PubMed] [Google Scholar]
- 25. Hizukuri Y., Morton J. F., Yakushi T., Kojima S., Homma M. (2009) The peptidoglycan-binding (PGB) domain of the Escherichia coli pal protein can also function as the PGB domain in E. coli flagellar motor protein MotB. J. Biochem. 146, 219–229 [DOI] [PubMed] [Google Scholar]
- 26. Duncan T. R., Yahashiri A., Arends S. J., Popham D. L., Weiss D. S. (2013) Identification of SPOR domain amino acids important for septal localization, peptidoglycan binding, and a disulfide bond in the cell division protein FtsN. J. Bacteriol. 195, 5308–5315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ishihama A. (2012) Prokaryotic genome regulation: a revolutionary paradigm. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 88, 485–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rigali S., Schlicht M., Hoskisson P., Nothaft H., Merzbacher M., Joris B., Titgemeyer F. (2004) Extending the classification of bacterial transcription factors beyond the helix-turn-helix motif as an alternative approach to discover new cis/trans relationships. Nucleic Acids Res. 32, 3418–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dimmer E. C., Huntley R. P., Alam-Faruque Y., Sawford T., O'Donovan C., Martin M. J., Bely B., Browne P., Mun Chan W., Eberhardt R., Gardner M., Laiho K., Legge D., Magrane M., Pichler K., Poggioli D., Sehra H., Auchincloss A., Axelsen K., Blatter M. C., Boutet E., Braconi-Quintaje S., Breuza L., Bridge A., Coudert E., Estreicher A., Famiglietti L., Ferro-Rojas S., Feuermann M., Gos A., Gruaz-Gumowski N., Hinz U., Hulo C., James J., Jimenez S., Jungo F., Keller G., Lemercier P., Lieberherr D., Masson P., Moinat M., Pedruzzi I., Poux S., Rivoire C., Roechert B., Schneider M., Stutz A., Sundaram S., Tognolli M., Bougueleret L., Argoud-Puy G., Cusin I., Duek-Roggli P., Xenarios I., Apweiler R. (2012) The UniProt-GO Annotation database in 2011. Nucleic Acids Res. 40, D565–D570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McIntosh B. K., Renfro D. P., Knapp G. S., Lairikyengbam C. R., Liles N. M., Niu L., Supak A. M., Venkatraman A., Zweifel A. E., Siegele D. A., Hu J. C. (2012) EcoliWiki: a wiki-based community resource for Escherichia coli. Nucleic Acids Res. 40, D1270–D1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Keseler I. M., Bonavides-Martinez C., Collado-Vides J., Gama-Castro S., Gunsalus R. P., Johnson D. A., Krummenacker M., Nolan L. M., Paley S., Paulsen I. T., Peralta-Gil M., Santos-Zavaleta A., Shearer A. G., Karp P. D. (2009) EcoCyc: a comprehensive view of Escherichia coli biology. Nucleic Acids Res. 37, D464–D470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Horler R. S., Butcher A., Papangelopoulos N., Ashton P. D., Thomas G. H. (2009) EchoLOCATION: an in silico analysis of the subcellular locations of Escherichia coli proteins and comparison with experimentally derived locations. Bioinformatics 25, 163–166 [DOI] [PubMed] [Google Scholar]
- 33. Bernsel A., Daley D. O. (2009) Exploring the inner membrane proteome of Escherichia coli: which proteins are eluding detection and why? Trends Microbiol. 17, 444–449 [DOI] [PubMed] [Google Scholar]
- 34. Rudd K. E. (2000) EcoGene: a genome sequence database for Escherichia coli K-12. Nucleic Acids Res. 28, 60–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ochman H., Davalos L. M. (2006) The nature and dynamics of bacterial genomes. Science 311, 1730–1733 [DOI] [PubMed] [Google Scholar]
- 36. Krogh A., Larsson B., von Heijne G., Sonnhammer E. L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 [DOI] [PubMed] [Google Scholar]
- 37. Goldberg T., Hecht M., Hamp T., Karl T., Yachdav G., Ahmed N., Altermann U., Angerer P., Ansorge S., Balasz K., Bernhofer M., Betz A., Cizmadija L., Do K. T., Gerke J., Greil R., Joerdens V., Hastreiter M., Hembach K., Herzog M., Kalemanov M., Kluge M., Meier A., Nasir H., Neumaier U., Prade V., Reeb J., Sorokoumov A., Troshani I., Vorberg S., Waldraff S., Zierer J., Nielsen H., Rost B. (2014) LocTree3 prediction of localization. Nucleic Acids Res. 42, W350–W355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paramasivam N., Linke D. (2011) ClubSubP: cluster-based subcellular localization prediction for gram-negative bacteria and archaea. Front Microbiol. 2, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dosztanyi Z., Csizmok V., Tompa P., Simon I. (2005) IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21, 3433–3434 [DOI] [PubMed] [Google Scholar]
- 40. Andersson C. S., Lundgren C. A., Magnusdottir A., Ge C., Wieslander A., Martinez Molina D., Hogbom M. (2012) The Mycobacterium tuberculosis very-long-chain fatty acyl-CoA synthetase: structural basis for housing lipid substrates longer than the enzyme. Structure 20, 1062–1070 [DOI] [PubMed] [Google Scholar]
- 41. Ehrmann M., Ehrle R., Hofmann E., Boos W., Schlosser A. (1998) The ABC maltose transporter. Mol. Microbiol. 29, 685–694 [DOI] [PubMed] [Google Scholar]
- 42. Yoon S. H., Han M. J., Jeong H., Lee C. H., Xia X. X., Lee D. H., Shim J. H., Lee S. Y., Oh T. K., Kim J. F. (2012) Comparative multi-omics systems analysis of Escherichia coli strains B and K-12. Genome Biol. 13, R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang L., Li J., March J. C., Valdes J. J., Bentley W. E. (2005) luxS-dependent gene regulation in Escherichia coli K-12 revealed by genomic expression profiling. J. Bacteriol. 187, 8350–8360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taniguchi Y., Choi P. J., Li G. W., Chen H., Babu M., Hearn J., Emili A., Xie X. S. (2010) Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329, 533–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Patten C. L., Kirchhof M. G., Schertzberg M. R., Morton R. A., Schellhorn H. E. (2004) Microarray analysis of RpoS-mediated gene expression in Escherichia coli K-12. Mol. Genet. Genomics 272, 580–591 [DOI] [PubMed] [Google Scholar]
- 46. Oberto J., Nabti S., Jooste V., Mignot H., Rouviere-Yaniv J. (2009) The HU regulon is composed of genes responding to anaerobiosis, acid stress, high osmolarity, and SOS induction. PLoS One 4, e4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Choudhury R., Tsai Y. S., Dominguez D., Wang Y., Wang Z. (2012) Engineering RNA endonucleases with customized sequence specificities. Nat. Commun. 3, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pan J.-Y., Li H., Ma Y., Chen P., Zhao P., Wang S.-Y., Peng X.-X. (2010) Complexome of Escherichia coli Envelope Proteins under Normal Physiological Conditions. J. Proteome Res. 9, 3730–3740 [DOI] [PubMed] [Google Scholar]
- 49. Iwasaki M., Miwa S., Ikegami T., Tomita M., Tanaka N., Ishihama Y. (2010) One-dimensional capillary liquid chromatographic separation coupled with tandem mass spectrometry unveils the Escherichia coli proteome on a microarray scale. Anal. Chem. 82, 2616–2620 [DOI] [PubMed] [Google Scholar]
- 50. Consortium T. G. O. (2013) Gene ontology annotations and resources. Nucleic Acids Research 41, D530–D535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sapay N., Guermeur Y., Deleage G. (2006) Prediction of amphipathic in-plane membrane anchors in monotopic proteins using a SVM classifier. BMC Bioinformatics 7, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gonnet P., Rudd K. E., Lisacek F. (2004) Fine-tuning the prediction of sequences cleaved by signal peptidase II: a curated set of proven and predicted lipoproteins of Escherichia coli K-12. Proteomics 4, 1597–1613 [DOI] [PubMed] [Google Scholar]
- 53. Watt R. M., Wang J., Leong M., Kung H. F., Cheah K. S., Liu D., Danchin A., Huang J. D. (2007) Visualizing the proteome of Escherichia coli: an efficient and versatile method for labeling chromosomal coding DNA sequences (CDSs) with fluorescent protein genes. Nucleic Acids Res. 35, e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Walz A. C., Demel R. A., de Kruijff B., Mutzel R. (2002) Aerobic sn-glycerol-3-phosphate dehydrogenase from Escherichia coli binds to the cytoplasmic membrane through an amphipathic alpha-helix. Biochem. J. 365, 471–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shiomi D., Margolin W. (2008) Compensation for the loss of the conserved membrane targeting sequence of FtsA provides new insights into its function. Mol. Microbiol. 67, 558–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Phoenix D. A., Pratt J. M. (1990) pH-induced insertion of the amphiphilic alpha-helical anchor of Escherichia coli penicillin-binding protein 5. Eur. J. Biochem. 190, 365–369 [DOI] [PubMed] [Google Scholar]
- 57. Villegas J. M., Volentini S. I., Rintoul M. R., Rapisarda V. A. (2011) Amphipathic C-terminal region of Escherichia coli NADH dehydrogenase-2 mediates membrane localization. Arch. Biochem. Biophys. 505, 155–159 [DOI] [PubMed] [Google Scholar]
- 58. Murashko O. N., Kaberdin V. R., Lin-Chao S. (2012) Membrane binding of Escherichia coli RNase E catalytic domain stabilizes protein structure and increases RNA substrate affinity. Proc. Natl. Acad. Sci. U.S.A. 109, 7019–7024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lu F., Taghbalout A. (2013) Membrane association via an amino-terminal amphipathic helix is required for the cellular organization and function of RNase II. J. Biol. Chem. 288, 7241–7251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shih Y. L., Huang K. F., Lai H. M., Liao J. H., Lee C. S., Chang C. M., Mak H. M., Hsieh C. W., Lin C. C. (2011) The N-terminal amphipathic helix of the topological specificity factor MinE is associated with shaping membrane curvature. PLoS One 6, e21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Henderson I. R., Cappello R., Nataro J. P. (2000) Autotransporter proteins, evolution and redefining protein secretion. Trends Microbiol. 8, 529–532 [DOI] [PubMed] [Google Scholar]
- 62. Hunter S., Apweiler R., Attwood T. K., Bairoch A., Bateman A., Binns D., Bork P., Das U., Daugherty L., Duquenne L., Finn R. D., Gough J., Haft D., Hulo N., Kahn D., Kelly E., Laugraud A., Letunic I., Lonsdale D., Lopez R., Madera M., Maslen J., McAnulla C., McDowall J., Mistry J., Mitchell A., Mulder N., Natale D., Orengo C., Quinn A. F., Selengut J. D., Sigrist C. J., Thimma M., Thomas P. D., Valentin F., Wilson D., Wu C. H., Yeats C. (2009) InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sanders A. N., Pavelka M. S. (2013) Phenotypic analysis of Eschericia coli mutants lacking L,D-transpeptidases. Microbiology 159, 1842–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. O'Neill J., Xie M., Hijnen M., Roujeinikova A. (2011) Role of the MotB linker in the assembly and activation of the bacterial flagellar motor. Acta Crystallogr. D Biol. Crystallogr. 67, 1009–1016 [DOI] [PubMed] [Google Scholar]
- 65. Godlewska R., Wisniewska K., Pietras Z., Jagusztyn-Krynicka E. K. (2009) Peptidoglycan-associated lipoprotein (Pal) of Gram-negative bacteria: function, structure, role in pathogenesis, and potential application in immunoprophylaxis. FEMS Microbiol. Lett. 298, 1–11 [DOI] [PubMed] [Google Scholar]
- 66. Pettijohn D. E. (1988) Histone-like proteins and bacterial chromosome structure. J. Biol. Chem. 263, 12793–12796 [PubMed] [Google Scholar]
- 67. Yeats C., Bateman A. (2003) The BON domain: a putative membrane-binding domain. Trends Biochem. Sci 28, 352–355 [DOI] [PubMed] [Google Scholar]
- 68. Bendezu F. O., Hale C. A., Bernhardt T. G., de Boer P. A. J. (2009) RodZ (YfgA) is required for proper assembly of the MreB actin cytoskeleton and cell shape in E-coli. Embo Journal 28, 193–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rauschmeier M., Schuppel V., Tetsch L., Jung K. (2014) New Insights into the Interplay Between the Lysine Transporter LysP and the pH Sensor CadC in Escherichia coli. Journal of Molecular Biology 426, 215–229 [DOI] [PubMed] [Google Scholar]
- 70. Rajapandi T., Dolan K. M., Oliver D. B. (1991) The first gene in the Escherichia coli secA operon, gene X, encodes a nonessential secretory protein. J. Bacteriol. 173, 7092–7097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Francetic O., Belin D., Badaut C., Pugsley A. P. (2000) Expression of the endogenous type II secretion pathway in Escherichia coli leads to chitinase secretion. EMBO J. 19, 6697–6703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Handa Y., Inaho N., Nameki N. (2011) YaeJ is a novel ribosome-associated protein in Escherichia coli that can hydrolyze peptidyl-tRNA on stalled ribosomes. Nucleic Acids Res. 39, 1739–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Robinson L. S., Ashman E. M., Hultgren S. J., Chapman M. R. (2006) Secretion of curli fibre subunits is mediated by the outer membrane-localized CsgG protein. Mol. Microbiol. 59, 870–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Drummelsmith J., Whitfield C. (2000) Translocation of group 1 capsular polysaccharide to the surface of Escherichia coli requires a multimeric complex in the outer membrane. EMBO J. 19, 57–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cowles C. E., Li Y., Semmelhack M. F., Cristea I. M., Silhavy T. J. (2011) The free and bound forms of Lpp occupy distinct subcellular locations in Escherichia coli. Mol. Microbiol. 79, 1168–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Boel G., Pichereau V., Mijakovic I., Maze A., Poncet S., Gillet S., Giard J. C., Hartke A., Auffray Y., Deutscher J. (2004) Is 2-phosphoglycerate-dependent automodification of bacterial enolases implicated in their export? J. Mol. Biol. 337, 485–496 [DOI] [PubMed] [Google Scholar]
- 77. Taghbalout A., Rothfield L. (2007) RNaseE and the other constituents of the RNA degradosome are components of the bacterial cytoskeleton. Proc. Natl. Acad. Sci. U.S.A. 104, 1667–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Veit A., Polen T., Wendisch V. F. (2007) Global gene expression analysis of glucose overflow metabolism in Escherichia coli and reduction of aerobic acetate formation. Appl. Microbiol. Biotechnol. 74, 406–421 [DOI] [PubMed] [Google Scholar]
- 79. Prehna G., Zhang G., Gong X., Duszyk M., Okon M., McIntosh L. P., Weiner J. H., Strynadka N. C. (2012) A protein export pathway involving Escherichia coli porins. Structure 20, 1154–1166 [DOI] [PubMed] [Google Scholar]
- 80. Busch A., Waksman G. (2012) Chaperone-usher pathways: diversity and pilus assembly mechanism. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 1112–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Miyamoto S., Tokuda H. (2007) Diverse effects of phospholipids on lipoprotein sorting and ATP hydrolysis by the ABC transporter LolCDE complex. Biochim. Biophys. Acta 1768, 1848–1854 [DOI] [PubMed] [Google Scholar]
- 82. Solov'eva T. F., Novikova O. D., Portnyagina O. Y. (2012) Biogenesis of beta-barrel integral proteins of bacterial outer membrane. Biochemistry 77, 1221–1236 [DOI] [PubMed] [Google Scholar]
- 83. Selkrig J., Mosbahi K., Webb C. T., Belousoff M. J., Perry A. J., Wells T. J., Morris F., Leyton D. L., Totsika M., Phan M. D., Celik N., Kelly M., Oates C., Hartland E. L., Robins-Browne R. M., Ramarathinam S. H., Purcell A. W., Schembri M. A., Strugnell R. A., Henderson I. R., Walker D., Lithgow T. (2012) Discovery of an archetypal protein transport system in bacterial outer membranes. Nat. Struct. Mol. Biol. 19, 506–510, S501 [DOI] [PubMed] [Google Scholar]
- 84. Barnhart M. M., Chapman M. R. (2006) Curli biogenesis and function. Annu. Rev. Microbiol. 60, 131–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Douzi B., Filloux A., Voulhoux R. (2012) On the path to uncover the bacterial type II secretion system. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 1059–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lee P. A., Tullman-Ercek D., Georgiou G. (2006) The bacterial twin-arginine translocation pathway. Annu. Rev. Microbiol. 60, 373–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Van Gerven N., Waksman G., Remaut H. (2011) Pili and flagella biology, structure, and biotechnological applications. Prog. Mol. Biol. Transl. Sci. 103, 21–72 [DOI] [PubMed] [Google Scholar]
- 88. Hemm M. R., Paul B. J., Miranda-Rios J., Zhang A., Soltanzad N., Storz G. (2010) Small stress response proteins in Escherichia coli: proteins missed by classical proteomic studies. J. Bacteriol. 192, 46–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fontaine F., Fuchs R. T., Storz G. (2011) Membrane localization of small proteins in Escherichia coli. J. Biol. Chem. 286, 32464–32474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Molloy M. P., Herbert B. R., Slade M. B., Rabilloud T., Nouwens A. S., Williams K. L., Gooley A. A. (2000) Proteomic analysis of the Escherichia coli outer membrane. Eur. J. Biochem. 267, 2871–2881 [DOI] [PubMed] [Google Scholar]
- 91. Hiniker A., Bardwell J. C. (2004) In vivo substrate specificity of periplasmic disulfide oxidoreductases. J. Biol. Chem. 279, 12967–12973 [DOI] [PubMed] [Google Scholar]
- 92. Martinez-Hackert E., Hendrickson W. A. (2009) Promiscuous substrate recognition in folding and assembly activities of the trigger factor chaperone. Cell 138, 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Baars L., Ytterberg A. J., Drew D., Wagner S., Thilo C., van Wijk K. J., de Gier J. W. (2006) Defining the role of the Escherichia coli chaperone SecB using comparative proteomics. J. Biol. Chem. 281, 10024–10034 [DOI] [PubMed] [Google Scholar]
- 94. Krehenbrink M., Edwards A., Downie J. A. (2011) The superoxide dismutase SodA is targeted to the periplasm in a SecA-dependent manner by a novel mechanism. Mol. Microbiol. 82, 164–179 [DOI] [PubMed] [Google Scholar]
- 95. Dalbey R. E., Kuhn A. (2012) Protein traffic in Gram-negative bacteria - how exported and secreted proteins find their way. Fems. Microbiol. Rev. 36, 1023–1045 [DOI] [PubMed] [Google Scholar]
- 96. Froderberg L., Houben E. N., Baars L., Luirink J., de Gier J. W. (2004) Targeting and translocation of two lipoproteins in Escherichia coli via the SRP/Sec/YidC pathway. J. Biol. Chem. 279, 31026–31032 [DOI] [PubMed] [Google Scholar]
- 97. Tian P., Bernstein H. D. (2009) Identification of a post-targeting step required for efficient cotranslational translocation of proteins across the Escherichia coli inner membrane. J. Biol. Chem. 284, 11396–11404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gray A. N., Henderson-Frost J. M., Boyd D., Sharafi S., Niki H., Goldberg M. B. (2011) Unbalanced charge distribution as a determinant for dependence of a subset of Escherichia coli membrane proteins on the membrane insertase YidC. Mbio 2, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dalbey R. E., Kuhn A., Zhu L., Kiefer D. (2014) The membrane insertase YidC. Biochim. Biophys. Acta 1843, 1489–1496 [DOI] [PubMed] [Google Scholar]
- 100. Bilous P. T., Cole S. T., Anderson W. F., Weiner J. H. (1988) Nucleotide sequence of the dmsABC operon encoding the anaerobic dimethylsulphoxide reductase of Escherichia coli. Mol. Microbiol. 2, 785–795 [DOI] [PubMed] [Google Scholar]
- 101. Neumann M., Mittelstadt G., Iobbi-Nivol C., Saggu M., Lendzian F., Hildebrandt P., Leimkuhler S. (2009) A periplasmic aldehyde oxidoreductase represents the first molybdopterin cytosine dinucleotide cofactor containing molybdo-flavoenzyme from Escherichia coli. FEBS J. 276, 2762–2774 [DOI] [PubMed] [Google Scholar]
- 102. Lee E. Y., Bang J. Y., Park G. W., Choi D. S., Kang J. S., Kim H. J., Park K. S., Lee J. O., Kim Y. K., Kwon K. H., Kim K. P., Gho Y. S. (2007) Global proteomic profiling of native outer membrane vesicles derived from Escherichia coli. Proteomics 7, 3143–3153 [DOI] [PubMed] [Google Scholar]
- 103. Niwa T., Ying B. W., Saito K., Jin W., Takada S., Ueda T., Taguchi H. (2009) Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins. Proc. Natl. Acad. Sci. U.S.A. 106, 4201–4206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tompa P. (2002) Intrinsically unstructured proteins. Trends Biochem. Sci. 27, 527–533 [DOI] [PubMed] [Google Scholar]
- 105. Kishii R., Falzon L., Yoshida T., Kobayashi H., Inouye M. (2007) Structural and functional studies of the HAMP domain of EnvZ, an osmosensing transmembrane histidine kinase in Escherichia coli. J. Biol. Chem. 282, 26401–26408 [DOI] [PubMed] [Google Scholar]
- 106. Dunker A. K., Silman I., Uversky V. N., Sussman J. L. (2008) Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 18, 756–764 [DOI] [PubMed] [Google Scholar]
- 107. Chang Y. C., Oas T. G. (2010) Osmolyte-induced folding of an intrinsically disordered protein: folding mechanism in the absence of ligand. Biochemistry 49, 5086–5096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gajiwala K. S., Burley S. K. (2000) HDEA, a periplasmic protein that supports acid resistance in pathogenic enteric bacteria. J. Mol. Biol. 295, 605–612 [DOI] [PubMed] [Google Scholar]
- 109. Wang M., Weiss M., Simonovic M., Haertinger G., Schrimpf S. P., Hengartner M. O., von Mering C. (2012) PaxDb, a database of protein abundance averages across all three domains of life. Mol. Cell. Proteomics 11, 492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Andreeva A., Howorth D., Chandonia J. M., Brenner S. E., Hubbard T. J., Chothia C., Murzin A. G. (2008) Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 36, D419–D425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Finn R. D., Bateman A., Clements J., Coggill P., Eberhardt R. Y., Eddy S. R., Heger A., Hetherington K., Holm L., Mistry J., Sonnhammer E. L., Tate J., Punta M. (2014) Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Letunic I., Doerks T., Bork P. (2012) SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–D305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Tyhach R. J., Hawrot E., Satre M., Kennedy E. P. (1979) Increased synthesis of phosphatidylserine decarboxylase in a strain of Escherichia coli bearing a hybrid plasmid. Altered association of enzyme with the membrane. J. Biol. Chem. 254, 627–633 [PubMed] [Google Scholar]
- 114. Serres M. H., Riley M. (2000) MultiFun, a multifunctional classification scheme for Escherichia coli K-12 gene products. Microb. Comp. Genomics 5, 205–222 [DOI] [PubMed] [Google Scholar]
- 115. Aranda B., Achuthan P., Alam-Faruque Y., Armean I., Bridge A., Derow C., Feuermann M., Ghanbarian A. T., Kerrien S., Khadake J., Kerssemakers J., Leroy C., Menden M., Michaut M., Montecchi-Palazzi L., Neuhauser S. N., Orchard S., Perreau V., Roechert B., van Eijk K., Hermjakob H. (2010) The IntAct molecular interaction database in 2010. Nucleic Acids Res. 38, D525–D531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wilkins M. R., Gasteiger E., Bairoch A., Sanchez J. C., Williams K. L., Appel R. D., Hochstrasser D. F. (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 [DOI] [PubMed] [Google Scholar]
- 117. Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., Madden T. L. (2008) NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.