

Abstract
Human radiolabel studies are traditionally conducted to provide a definitive understanding of the human absorption, distribution, metabolism and excretion (ADME) properties of a drug. However, advances in technology over the past decade have allowed alternative methods to be employed to obtain both clinical ADME and pharmacokinetic (PK) information. These include microdose and microtracer approaches using accelerator mass spectrometry, and the identification and quantification of metabolites in samples from classical human PK studies using technologies suitable for non-radiolabelled drug molecules, namely liquid chromatography-mass spectrometry and nuclear magnetic resonance spectroscopy. These recently developed approaches are described here together with relevant examples primarily from experiences gained in support of drug development projects at GlaxoSmithKline. The advantages of these study designs together with their limitations are described. We also discuss special considerations which should be made for a successful outcome to these new approaches and also to the more traditional human radiolabel study in order to maximize knowledge around the human ADME properties of drug molecules.
Keywords: investigational drugs, metabolism, pharmacokinetics, radioactive tracers
Introduction
In the development of potential new medicines, it is important to understand the human absorption, distribution, metabolism and excretion (ADME) properties of the drug in order to comprehend fully the impact of the drug molecule and any metabolites on its pharmacology, safety and ultimately, drug labelling recommendations. It is also a requirement of regulatory agencies worldwide 1–3. It is essential to ascertain the pharmacokinetics (PK) of the unchanged drug to evaluate drug safety and efficacy and to aid drug design, including decisions on salt form, drug formulation, dose level, administration route and dosing regimen. The disposition of the unchanged drug must also be adequately defined to understand potential inter-patient variability through different disease states such as hepatic or renal impairment and in specific populations such as the elderly, paediatrics and either gender. These factors are generally determined by measuring the concentration of unchanged parent drug in blood, plasma or serum collected during the multitude of clinical studies in both healthy subjects and patients conducted during phases 1–4 of drug development. Liquid chromatography-tandem mass spectrometry (LC/MS/MS) methods are most commonly employed.
It is also important to determine the nature of any drug metabolites to ascertain whether the pharmacology of the dosed drug is attributed to unchanged drug only or whether there are active metabolites contributing to efficacy, side effects or drug–drug interactions (DDIs). Historically, the standard approach for understanding these additional ADME properties involved dosing 14C-radiolabelled material and collecting excreta and plasma 4,5. PK profiles for any metabolites of concern were subsequently determined using LC/MS/MS methods to discharge any safety risks.
In recent years, a number of novel approaches for obtaining clinical drug disposition information have been adopted including, microdose and microtracer approaches 6,7 and the identification and quantification of metabolites in samples from classical human PK studies using technologies suitable for non-radiolabelled drug molecules 8–11. Additional information on biliary excretion has also been made routine through non-invasive bile collection with the Entero-Test® string device 12. At GlaxoSmithKline (GSK), these approaches have been successfully used instead of, or in addition to, conventional PK or human radiolabel studies (HRS) across a variety of drug candidates at different stages of the drug development program to address specific ADME issues.
This paper outlines these alternative approaches with a focus on small molecules, using recent examples primarily from drugs in development within our organization. It also discusses decisions which should be made around healthy volunteer vs. patient populations and dose level, route and frequency for a successful outcome to both these studies and the more traditional HRS to maximize knowledge around the disposition of drug molecules in humans.
Alternative approaches to understanding PK in humans
Knowledge of the human PK profile of a drug candidate is important early in the development process to establish whether the drug is likely to have suitable properties to become a viable medicine. Traditionally, these data are first obtained in the first-in-human (FIH) single ascending dose (SAD) study using LC/MS/MS methodology. Predictive methods can also be applied to estimate human PK and the utility of various methods have been reviewed 13–18. However there are instances where in vitro and in vivo differences, or cross species differences, can affect the confidence in these predictions and alternative approaches as described in the following sections, have been successfully employed.
Microdose studies
Microdosing is defined as administration of ≤100 μg of drug molecule, ≤1/100th the non-clinical no observed adverse effect level (NOAEL) and ≤1/100th pharmacologically active dose to humans 2. It is regarded within the pharmaceutical industry as a valuable technique that can be used to establish the clinical PK profile early in development, thereby allowing earlier risk evaluation and drug development decisions to be made 6,19. As a result of the low doses involved, microdose studies use highly sensitive analytical methods, either LC/MS/MS or LC in conjunction with accelerator mass spectrometry (LC+AMS) to extrapolate the PK profile of drug candidates to higher clinically relevant doses assuming linearity of exposure with dose. The arguments around the choice of approach have been reviewed 20. LC/MS/MS is generally preferable to AMS as it avoids the additional resources required to synthesize radiolabelled drug and allows for a more rapid turnaround of data. However, it may not be possible to develop an adequately sensitive LC/MS/MS method, necessitating the need for a microdose study with radiolabelled drug. One of the advantages of using radiolabelled drug is the availability of additional information on, for example, possible routes of excretion. An outline of the microdose approach is summarized schematically in Figure 1.
Figure 1.
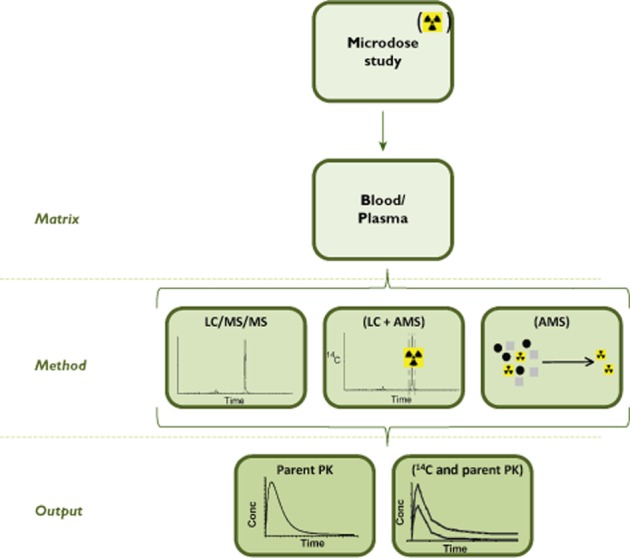
Schematic of the microdose approach (information in parenthesis indicates use of 14C drug)
In GSK, the microdosing approach with a non-labelled drug molecule has been used to address the human PK profile for a novel EP1 receptor antagonist, previously in development for the treatment of inflammatory pain 21. Inconsistencies between preclinical in vitro metabolic stability and in vivo PK data, and uncertainties with respect to allometric predictions of the human PK profile were a critical development liability for the molecule. Before commitment of resources to support a formal phase 1 study, a microdose study was conducted in two groups of healthy human volunteers by administering either a single oral or intravenous (i.v.) microdose. Drug concentrations in plasma were measured for up to 24 h post-dose using LC/MS/MS. The data revealed a favourable PK profile, consistent with a clinically acceptable dosing regimen and thereby permitted continued development of the drug candidate. Disappointingly, the drug molecule could not be developed further as additional human PK predictions established that it could not be administered to humans at therapeutically relevant dose levels due to unexpected toxicology in subsequent safety studies in the dog. The decision to terminate development could be made with the added confidence provided by the human PK from the microdose study.
A contrasting microdose approach using radiolabelled drug was conducted with an allosteric antagonist of human CCR4 in development as an oral therapy for the treatment of allergic bronchopulmonary aspergillosis 22. The drug molecule showed markedly different systemic PK profiles in rat and dog, leading to variable predictions of the human PK profile. Before commitment of resources to support a formal phase 1 study, a microdose study was conducted in healthy volunteers by administering a single i.v. microdose with an aim to inform on i.v. PK parameters only (bioavailability from oral administration was not considered to be a major concern at the time of the microdose study). Plasma and urine were collected for 48 and 24 h post-dose, respectively, and total radioactivity was measured directly by AMS. Drug concentrations in plasma were determined using an LC+AMS method. This study indicated low to moderate plasma clearance and unchanged drug accounted for about 50% of the total plasma radioactivity with the remaining circulating drug-related material assumed to be comprised of one or more metabolites. Approximately 20% of the dose was eliminated in the urine. The data provided confidence that the molecule would have sufficiently slow systemic clearance and high volume of distribution to provide an adequate opportunity for demonstration of efficacy, allowing continued development of the drug candidate. Development of the drug candidate continued. However, unfortunately, oral bioavailability proved to be an issue in a subsequent clinical study which led to termination of the development of this project.
In summary, employing a microdose strategy enables liability surrounding PK predictions to be discharged at an earlier stage of development, with potential reduced cost and resource allocation (minimal drug and animal requirements) than may otherwise have been required to initiate a conventional FIH study. It is the potential for early termination of a drug candidate which is the main attraction of this approach, thereby allowing subsequent selection of an alternative more promising drug molecule. However, if a drug is found to have an acceptable PK profile for further progression then the overall development timeframe will be lengthened. This may be seen as an unacceptable diversion, and a more acceptable use of the microdose approach is the comparison of PK parameters across several potential drug candidates in order to select the most favourable for development. A common concern with this approach may be a lack of agreement between the PK profiles following microdose administration vs. those at higher therapeutic doses, which may reflect non-linearity in absorption, distribution, metabolism and/or excretion over the dose range. Studies to date have not generally corroborated these latter concerns 23,24.
In addition to providing early information on human PK, the microdose technique can be used to understand the DDI potential for a candidate drug. A proof of principle study was conducted using a CCR5 inhibitor previously in development for rheumatoid arthritis, the development of which had been terminated in part, due its DDI liability. A microdose was administered to healthy volunteers, both alone and in the presence of the CYP3A4 inhibitor, ketoconazole, which was dosed repeatedly to achieve steady-state systemic exposure 25. Concentrations of the drug candidate in plasma were quantified by LC+AMS and showed approximately 10-fold increases in both Cmax and AUC in the presence of ketoconazole. These results were found to be comparable with the interaction previously observed at a therapeutic dose of the CCR5 inhibitor drug (50 mg) with a fixed dose combination product of lopinavir and ritonavir, a potent CYP3A4 and CYP2D6 inhibitor. The microdosing DDI approach is of particular value when there is limited in vivo information on the metabolic clearance of a drug in humans. It allows in vitro inhibition data to be put into clinical context and thereby give increased confidence in the DDI predictions.
Microtracer studies
The emergence of the microtracer approach in combination with predictive tools to replace animal models of PK and metabolism has been previously described 26. Intravenous microtracer technology generally utilizes a 14C-i.v. microdose (as described above) co-administered with a therapeutic non-radiolabelled oral dose 23,27. Stable isotopically labelled drug may also be used as the microdose although our organization has no clinical experience of this approach 28. The i.v. microdose is generally between a hundredth and a thousandth of the extravascular oral dose and is administered at the time of maximum systemic concentration (tmax) from the extravascular dose. This ensures that the body handles the tracer i.v. dose simultaneously and in the same manner as the extravascular therapeutic dose 29–31. Administration of the i.v. microdose can be conducted without additional preclinical testing to support i.v. or radiolabel dosing due to the extremely low chemical and radioactive doses given 2. A key advantage of the microtracer approach is the ability to add an iv microdose onto an existing traditional PK study design, thereby reducing the number of clinical studies in the development programme. An outline of the microtracer approach is shown schematically in Figure 2.
Figure 2.

Schematic of the microtracer approach
A microtracer approach was used in conjunction with a regional absorption study design in the development of a 5-lipoxygenase-activating protein inhibitor for the treatment of asthma and chronic obstructive pulmonary disease to understand issues around bioavailability and PK variability 32. The microtracer dose was given as a 15 min infusion at the approximate tmax of one of four oral non-labelled therapeutic doses in healthy volunteers. Results showed similar elimination in plasma from the i.v. tracer and oral therapeutic doses and allowed determination of absolute bioavailability for each of the oral doses with reference to the i.v. tracer. An advantage of administering a low 14C-labelled dose (or even 13C-labelled where analytical sensitivity allows) means that simpler formulations can be administered for example, a saline solution for the very low doses involved, and without the additional toxicology requirements of a high i.v. dose. This study allowed characterization of different oral formulations and delivered regional absorption data without lengthy preclinical and formulation pre-work which informed on a potential strategy for future formulation investigations.
The microtracer design has also been utilized to predict dosage regimens in highly compromised patient populations for an administration route not previously studied. A motilin agonist that accelerates gastric emptying is under development for several indications including that of the intensive care setting. Despite knowledge of its PK following oral administration, information on PK from i.v. dosing was required to inform dosage regimens in this patient population. A radiolabelled i.v. microtracer dose was given as a 15 min infusion at the approximate tmax of an oral non-labelled therapeutic dose to healthy subjects 33. Blood was collected for 120 h post-dose and plasma analysed for both total radioactivity and parent drug by AMS and LC+AMS, respectively. Unchanged parent drug from the non-labelled oral dose was quantified in plasma using LC/MS/MS. Despite the 500-fold difference in dose between the two different dose routes, the profiles for the terminal phase following both i.v. and oral administration were parallel. The compound exhibited low to moderate clearance and high volume of distribution. The data from this microtracer study allowed PK simulations to be performed which were targeted at regimens in the difficult intensive care unit setting with the advantage that this vulnerable patient population did not need to be exposed to ineffective doses.
For drug molecules where a traditional HRS is planned, maximized study efficiency may be gained using a double-tracer approach whereby a 13C-i.v. microdose is administered concomitant with a conventional 14C-labelled oral dose in an attempt to assess efficiently absolute bioavailability in the same study as the definitive human ADME data. While our organization has no experience with this study design to date, the determination of PK parameters for 13C-labelled i.v. tofogliflozin have been obtained together with the quantification of circulating drug related material, excretion balance, full metabolic profile and metabolite identification from the oral 14C-tofogliflozin dose 34.
The key advantage for implementing a microtracer approach is that oral and i.v. PK parameters are available from the same subject in the same temporal frame, due to co-administration of the two doses, leading to improved data integrity and reduced subject numbers. In the authors' experience, the regulators have demonstrated a keen awareness and acceptance of this approach and have even provided valuable input into the design of a recently conducted GSK study 35. Additional ADME endpoints, including information on metabolism and excretion routes are also available which can be used in conjunction with the PK information to refine further clinical plans. This will be discussed in detail later in this article.
Alternative approaches to facilitate understanding of metabolism and excretion in humans
There is a regulatory expectation that information on the human metabolites of a drug molecule should be available as early as possible in the drug development programme before exposing large numbers of human subjects to the drug or treating patients for a lengthy period 1,2. HRS are the accepted gold standard to facilitate understanding of the human ADME properties of a drug molecule because the use of radiolabelled material helps to ensure that all drug-related material can be accounted for.
The conduct of these studies has been well described and extensively discussed 4. An outline of the approach is shown schematically in Figure 3. In brief, typically four to six healthy adult subjects receive a single clinically relevant dose of drug containing 2–4 MBq (approximately 50–100 μCi) of 14C-labelled drug. Excreta and blood are collected for typically 7–10 days until approximately 90% of the administered radioactivity is recovered or until <1% of the radioactivity is collected on 2 consecutive days. Samples are then analysed for unchanged parent drug and metabolites.
Figure 3.

Schematic of the HRS approach
It is generally accepted within the pharmaceutical industry, that these studies are integral to the drug development process by generating a comprehensive picture of the overall disposition of the drug and that they are conducted to provide information in four key areas: (1) the presence and, where possible, identities of drug-related material in excreta to provide knowledge of the proposed clearance mechanisms which may impact on inter-patient variability or DDI, (2) the identities of circulating metabolites which may contribute to pharmacology or safety, (3) indicators for covalent binding namely, low extraction recoveries in plasma or metabolites in excreta formed from potentially reactive moieties and (4) ensuring that there is preclinical safety coverage for any notable metabolites and metabolic routes 5. Recently, alternative approaches have been established which provide valuable metabolism information without the complexities and some of the ethical considerations of an HRS. Examples of these approaches are described below.
Conventional PK studies with additional ADME investigations
A preliminary understanding of the metabolic profile of a drug in humans can be obtained using residual or additional samples collected from clinical PK studies which may be analysed for metabolites. A variety of analytical techniques are used, typically liquid chromatography-mass spectrometry (LC/MS) and/or semi-preparative LC followed by nuclear magnetic resonance (NMR) spectroscopy of the resulting fractions, if larger doses and sample volumes are available 8,36. The maximum dose administered clinically during drug development often occurs in the phase 1 ascending dose study and these studies are a good source of samples for definitive metabolite identification. Blood or plasma is analysed for parent drug to generate PK information and any remaining samples can then be used for metabolite evaluation using samples pooled across subjects and time points to generate one sample with concentrations proportional to the PK area under the curve (AUC) 37. Since the publication of the FDA Guidance for Industry on Safety Testing of Drug Metabolites in 2008 1 and the EMEA ICH M3 guidance document in 2009 [2, 3], there has been considerable focus on the identification and quantification of metabolites from early clinical studies and the various analytical techniques and the strategies employed have been well reviewed 8–11.
The preferred approach within our organization is to use quantitative NMR, if dose level permits, in conjunction with LC/MS 36 because unique to NMR, quantification can be performed without the need for metabolite standards. Typically, 20–50 ml of a single plasma pool representing AUC(0,24 h) for all subjects, is extracted and fractionated by preparative LC prior to quantitative NMR of the individual fractions. In general, this method has been found to be successful in the identification and quantification of circulating metabolites in line with regulatory guidelines when the plasma pool contains at least 5 μg of unchanged parent drug. This means that individual metabolites equivalent to 10% drug-related material (DRM) can be detected with a lower limit of quantification of 0.5 μg. For example, this technique was used to investigate the circulating metabolites following oral administration of an 8-aminoquinoline analogue in development for the treatment of visceral leishmaniasis 10. Circulating components included unchanged parent drug and three metabolites at levels exceeding 10% DRM. All were detected at levels adequate for human exposure coverage in one or more of the default toxicological species. Importantly, however, NMR revealed the presence of a unique human circulating metabolite, an N-nitrosamine present at approximately 5% DRM. This metabolite was subsequently synthesized for further safety studies to investigate the potential genotoxic risk.
If there is not sufficient drug-related material for analysis by NMR, metabolites in plasma may be identified and quantified by LC/MS/MS using standards of known concentration for comparison. Ideally, synthetic standards of the individual metabolites are used. However typically these are not available at the time of the early clinical studies and an alternative source for these standards needs to be determined. Metabolite reference standards may be isolated from a biological matrix, for example, human urine from the same clinical study or in vitro incubations with the drug. The concentrations of these metabolite standards can then be estimated by NMR prior to their use in an LC/MS/MS assay 38. Alternatively, samples obtained from an in vitro or non-clinical metabolism study with radiolabelled drug can be used 39. For example, preliminary information was obtained by LC/MS and NMR on human circulating metabolites following single oral administration to healthy human subjects of a prolyl hydroxylase inhibitor under development for the treatment of anaemia in chronic kidney disease (unpublished data on file within DMPK Department, GSK). The levels of metabolites in pooled plasma [representative of AUC(0,24 h)] were estimated using LC/MS by comparing metabolite concentrations with those in human hepatocytes incubated with radiolabelled drug. The absolute levels of unchanged parent drug and all observed metabolites in the plasma pool were subsequently determined and their total was used to estimate the percentage levels of individual metabolites. Six metabolites were found to be present at levels approaching or exceeding 10% observed DRM. These metabolites were not present in the default toxicological species. Therefore additional investigations were initiated to identify an appropriate toxicological species that generated the metabolites. Cynomolgus monkeys were shown to produce the major human metabolites and definitive toxicological studies were conducted in which the metabolites were quantified using LC/MS/MS methods with chemically synthesized metabolite standards. Adequate exposure coverage was demonstrated, allowing the contribution of the metabolites to the overall toxicity assessment to be evaluated.
Urine collected in clinical studies can be used not only as a source of additional material for definitive identification of metabolite structures and quantification of circulating metabolites but also, to determine the elimination pathways of the drug molecule 36,40. As with plasma, pooled urine can be fractionated by preparative LC prior to quantitative NMR of the individual fractions. Typically, 600 ml of urine (0–24 h), pooled proportionally across total volume excreted per subject, is fractionated for NMR. In combination with LC/MS, unchanged drug and metabolites may then be identified and quantified without the need for synthetic standards of individual metabolites. Information on individual metabolite concentrations, total renal clearance and minimum dose absorbed can be estimated from the quantitative NMR data alone. For example, following 1H NMR analysis of semi-preparative LC fractions of urine from the visceral leishmaniasis patients in the aforementioned study, it was determined that approximately 17% of the oral dose was eliminated through renal excretion over 24 h, with 30% (approximately 5 mg) of the eliminated material formed via the N-nitrosation pathway 10.
Typically, 1H is the preferred nuclei for NMR analysis. However for fluorinated drug molecules 19F NMR can be advantageous. There is no fluorine background in biological matrices and therefore any 19F NMR signals observed in the biological sample are likely to be from drug-related material. This leads to less complex NMR spectra for each preparative LC fraction thus permitting easier quantification of DRM than if 1H NMR were used. The techniques' specificity may also permit the direct analysis of urine without prior separation of the individual components allowing fluorine containing metabolites to be simultaneously detected and quantified, although not identified in a single run 40.
Biliary secretion can be a major route of elimination for drugs and their metabolites. Therefore information on drug-related material in bile may prove useful in understanding the full PK and ADME properties of a drug. Characterization of hepatobiliary elimination permits a more thorough understanding of the routes of metabolism and their relationship to the overall clearance of a drug which ultimately assists with the assessment of the risks of DDIs with co-administered medications. Biliary disposition information is rarely available because historically, the collection of bile was complex and invasive 41,42. Examination of human faeces from a conventional HRS is typically used as a surrogate matrix for bile, to quantify and identify drug-related material excreted via non-renal routes. Unfortunately, faeces may provide misleading information for some drugs, particularly drugs with poor oral absorption. Furthermore, material secreted in bile is susceptible to metabolism by gut microflora, for example, glucuronide conjugates which may not be subsequently detected in faeces. This may result in a reduced understanding of the overall clearance mechanisms of a drug. Faecal analysis is also not recommended for non-labelled studies due to potential issues with extraction and recovery of DRM which would not be fully understood without a radiolabel marker.
More recently, Guiney et al. have reported the use of the Entero-Test® as a non-invasive and simple technique for the collection of duodenal bile 12. In initial studies to evaluate this technique, duodenal bile was successfully collected from healthy volunteers dosed orally with a tool compound, simvastatin and the major metabolites identified were consistent with those reported previously for intubated hypercholesterolaemic patients dosed with 14C-simvastatin 43,44.
In a later study, duodenal bile was collected from healthy elderly volunteers dosed orally with a CXCR2 antagonist under development for the treatment of chronic obstructive pulmonary disease, to gain an understanding of the enzymes involved in its clearance. This permitted an assessment of the potential risk of the drug candidate being the victim of drug interactions with co-administered CYP inhibitors, prior to further clinical trials 45. The drug was shown to be cleared predominantly by glucuronidation with an O-glucuronidated metabolite eliminated in both urine and bile. This reduced the concern of the drug being victim to drug interactions with CYP3A4 inhibitors which are commonly administered in this therapeutic area and thereby enabled the progression of clinical trials in the target patient population.
As demonstrated, valuable metabolite information can be obtained using the Entero-Test® for duodenal bile collection which can help guide further work. It is a simple, safe and cost-effective technique which has been used in clinical studies with both non- and radiolabelled drug candidates in healthy subjects. However, it could equally be used for patients. Limited quantitative information is available using the technique and in general, only a single time point has been examined. Other limitations of the technique have been described previously and include complications when differentiating between unabsorbed and biliary secreted drug after oral dosing 12. Nevertheless, use of the Entero-Test® for high dose clinical studies where biliary secretion of drug and/or metabolites is predicted may complement metabolite information from other matrices, such as plasma and urine, and is increasingly being employed within our organization.
As summarized in Figure 4, supplemental ADME investigations added onto a PK study can provide a wealth of information. Within GSK, the analysis of samples from SAD and multiple ascending dose (MAD) phase 1 studies using non-labelled drug is now routine. The resulting data have allowed earlier decisions to be made on the non-clinical safety testing of the metabolites of our drug candidates. By identifying the nature of drug-related material in excreta through collection of urine and bile from a planned clinical PK study, it also provides a preliminary understanding of the clearance mechanisms which may impact on inter-patient variability or DDIs. Similarly, the identities and quantities of circulating metabolites may be determined by analysis of residual plasma or blood from this study allowing non-clinical safety coverage for any notable metabolites and routes of metabolism to be established. A key advantage of this approach is that a preliminary understanding of the metabolism and excretion pathways of a drug molecule can be achieved without additional dosing of subjects or the need for a conventional HRS. Notwithstanding the use of a microtracer approach as described in the following section, subjects are therefore not exposed to higher levels of radioactivity for drugs that fail to progress successfully from early phase clinical trials whilst preliminary information is available to guide future clinical and non-clinical safety studies. A limitation of this approach compared with one utilizing radiolabelled drug is that there is no information on blood/plasma extraction efficiencies, a potential measure of covalent binding. Metabolites formed via reactive pathways provide some information on the potential for covalent binding but without a radiolabel, the true extent cannot be established. This issue may be overcome by employing a microtracer approach (see below) which may be designed with the additional advantage of providing a full excretion balance and quantification of metabolites in all matrices.
Figure 4.
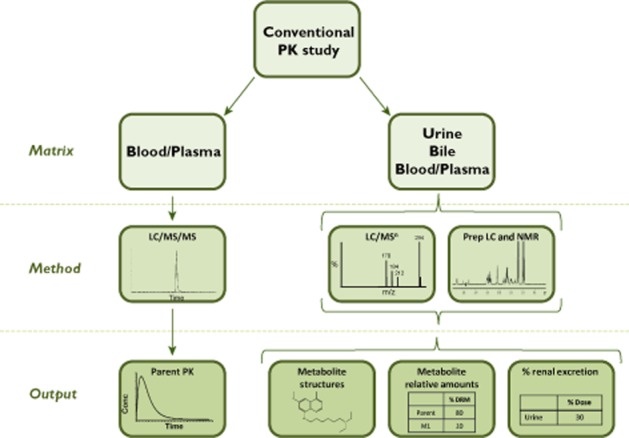
Schematic of the approach for a conventional PK study with additional ADME investigations
Microtracer studies
In addition to oral and i.v. PK parameters, a key advantage to the microtracer approach described previously, is the ability to add a 14C-i.v. microtracer dose onto an existing oral study design to provide additional ADME information. The methodology described above to explore the metabolism and excretion of a drug candidate in non-labelled studies can be equally applied to a microtracer study to gain an understanding of the ADME properties of the non-labelled oral dose. In addition, AMS can be used to determine the excretion balance and metabolic profile of the 14C-i.v. dose as well as provide information on blood/plasma extraction efficiencies.
For example, in the motilin agonist microtracer study described above, plasma, duodenal bile, urine and faeces were collected in an attempt to understand fully the metabolism and elimination of the drug 33. The metabolic profiles were shown by AMS to be relatively simple. Two major components were identified by LC/MS in urine and bile, an oxidized sulfate conjugate and unchanged parent drug. The desulfated product of the former was observed in faeces after i.v. dosing and was postulated to be formed by the gut flora mediated deconjugation of the sulfate moiety excreted in bile. This study exemplifies the information available from a well-designed microtracer study providing value to the project whilst using minimal resources. In the authors' opinion, if a microtracer approach is to be employed early in the development of a drug molecule for absolute bioavailability, then with minimal additional resource, it would seem prudent to determine metabolism and excretion profiles in this study. The additional ADME information may help guide future decisions and ultimately in the future, may mean that no additional HRS is required for drug registration.
Special considerations
Route of administration
Traditionally, the metabolism and distribution of a molecule is determined after administration of the radiolabelled drug candidate by the intended clinical route. However, for drugs administered by the ocular, inhaled or intranasal routes, i.v. and/or oral administration may prove to be more feasible. For example, for respiratory drugs it is difficult to produce the radioactive drug in the inhalation device to be used commercially and also to quantify reliably the exhaled dose. Any quantitative data from the HRS would therefore be incomplete and misrepresentative of the clinical situation. A low dose oral/i.v. approach is preferred such that the i.v. dose is considered a surrogate for the inhaled portion of the dose (the venous supply delivers the dose directly to the heart and lung via the pulmonary artery and vein) and the oral dose represents the swallowed portion. For example, the metabolism and distribution of fluticasone furoate, a glucocorticoid receptor agonist in development as an intranasal treatment for patients with symptoms of rhinitis, was investigated in humans 46. 14C-fluticasone furoate was administered orally (2 mg) to healthy male volunteers and i.v. (0.25 mg) to the same subjects 4 weeks later. The major routes of metabolism of fluticasone furoate in humans were identified for both dosing routes (thioester hydrolysis and oxidative defluorination) with the majority of radioactivity excreted in the faeces after both routes of administration. In a later study, vilanterol, an inhaled novel long acting β2-adrenoceptor agonist used in combination therapy with fluticasone furoate for chronic obstructive pulmonary disease and asthma, was investigated after oral administration only 47. In this case, radiolysis of the 14C-isotope led to instability over the period required to manufacture and release an i.v. or inhaled formulation therefore it was impossible to administer 14C-vilanterol by either of these routes. Fortunately, vilanterol demonstrates oral absorption which is not necessarily the case for all inhaled drug candidates and in this instance, an oral study design could be employed as a compromise. The primary routes of metabolism and excretion were therefore identified for an oral dose only together with the major circulating metabolites. These were subsequently synthesized and measured in non-clinical and clinical studies following inhalation administration to eliminate potential safety concerns.
Understanding the ADME properties of dermally applied drugs is a challenge due to low systemic exposures combined with inadequate analytical sensitivity of methods of detection. Investigations are currently on-going within our organization to evaluate the absorption of drug through the skin in a clinical study where the radioactive dose is administered by the dermal route, thereby allowing assessment of systemic drug accumulation upon repeat administration. This approach involves administration of a relatively conventional amount of 14C-labelled drug with detection of both total radioactivity and parent drug concentrations in plasma using AMS technology.
Dosing frequency
Human metabolism studies are generally conducted following a single administration of the radiolabelled drug and the exposures of any circulating metabolites can be subsequently simulated at steady-state 48. Any metabolites deemed worthy of additional evaluation are synthesized and monitored at steady-state in later non-clinical and clinical studies using LC/MS/MS assays. Human exposures are then compared with the exposures observed in the non-clinical toxicity studies to assess their safety. It is the metabolite profile at steady-state which is important as this typically reflects the treatment paradigm for most drugs however, an evaluation may be based on data from a single dose HRS assuming that there is neither unexpected accumulation of metabolites upon repeat dose nor the appearance of notable metabolites following repeat administration only 1,2. In the case of darapladib, an inhibitor of lipoprotein-associated phospholipase A2 under development as a potential anti-atherosclerotic agent, the accumulation of unchanged drug after repeat dosing was notably lower than had been predicted from the single dose PK data. In order to gain an insight into the possible mechanisms behind this disparity, the relative disposition and metabolism of radiolabelled drug was compared at steady-state and after single oral and i.v. doses of 14C-drug 49. The design of the steady-state arm of the study was such that a single radiolabelled dose was administered on day 11 during the 14 day repeat oral dosing of non-labelled drug. No additional metabolites were detected after repeat dosing nor were there any appreciable differences between relative amounts of each metabolite after the two dosing regimens. Nonetheless, systemic exposure to radioactive material was reduced within the repeat dose regimen, consistent with time-dependent pharmacokinetics resulting from enhanced clearance or reduced absorption through potential induction of either CYP3A and/or P-glycoprotein, darapladib being a substrate for both. This study design did not allow the absolute amounts of individual metabolites to be determined at steady-state which did limit any definitive conclusions on potential mechanisms, including enzyme induction. Repeat administration of 14C-drug would have provided additional information to help substantiate any hypothesis.
Advances in LC/MS and NMR technologies mean that an assessment of human metabolism can now be conducted (as described above) for any single or repeat dose clinical PK study, if sufficient dose is given and as stated previously, this is now routinely performed within GSK. For example, quantitative NMR was used to evaluate metabolite exposures for a novel CCR3 antagonist at steady-state to gain an early understanding of the metabolism of the drug molecule in humans 36. Plasma samples from the repeat dose phase 1 clinical studies were analysed by LC/MS and quantitative NMR and two circulating metabolites were identified at levels exceeding parent drug. This was subsequently confirmed using LC/MS/MS with chemically synthesized metabolite standards. It should be noted, that the major metabolite was poorly ionized by MS and may have been undetected if the NMR approach had not been used. In addition, even if a definitive HRS had been performed, this metabolite may have remained elusive as it was the result of an amide hydrolysis with the loss of the radionuclide from the synthesized 14C-labelled moiety. For this drug candidate, a dual 14C-label or two versions of the drug candidate with 14C-labels on opposite ends of the molecule would have been required for a successful outcome to the definitive HRS.
Radioactive dose
Traditionally, the HRS often involves administration of a radioactive dose in the region of 3.7 MBq (100 μCi) but there are several scenarios in which this level of radiation burden is not acceptable or achievable. These include dosimetry based limitations where data from animal studies dictate a lower than traditional 14C-dose be administered to keep the human exposure within a specific ionizing radiation risk category, limited chemical mass due to drug potency thereby limiting the radioactive dose that can be administered, protracted elimination of drug-related material in humans, or indeed prohibitive costs of production of sufficient quantities of 14C-labelled drug to allow traditional doses. The resulting low specific activity leads to additional analytical challenges and may necessitate the use of AMS to facilitate determination of mass balance, PK of total radioactivity and metabolite profiling. For example, a dopamine receptor antagonist under development for schizophrenia was predicted to have protracted elimination based upon animal data. Due to radiation dosimetry concerns, the HRS was therefore conducted with a dose of low specific activity drug (only 18.5 kBq in 16 mg) to minimize the exposure of the subjects to ionizing radiation 50. Excreta and blood were collected for 15 days post-dosing followed by periodic collections of excreta for 55 days. All samples were analysed by AMS for excretion balance and total radioactivity in plasma. A cumulative mean recovery of >85% was obtained by extrapolations based on plots of amount excreted per day vs. time. The metabolites in urine and plasma were also successfully profiled by AMS allowing identification of the major metabolites. This study demonstrated that with highly sensitive analytical technology and extrapolation of data from limited but protracted sample collections, a successful HRS may be achieved despite the low specific activity of the dosed drug candidate.
Patient populations
HRS are typically conducted using healthy male adult subjects although females are included should the target indication be predominantly in females, for example, breast or ovarian cancer, contraception or migraine. However for some drugs, in particular those used in oncology, the cytotoxic nature of these drugs or clinical safety considerations necessitate enrolling patients instead of healthy subjects for an HRS 51. Recruitment for oncology subjects can be slow and complicated by the limited number of approved clinical HRS sites which are often not located near oncology centres 52. In order to balance the slow recruitment whilst meeting study objectives, a rolling assessment of the study results may therefore be used to allow the study to be assessed continuously and stopped at fewer than the target number of patients once the results meet an acceptable criteria. Recruitment is often opened to females. Unique to oncology and other life-threatening indications, it may also be unethical to discontinue treatment once the HRS is complete due to the potential benefits of treatment with the drug. Cancer patients may therefore be offered the opportunity to continue treatment with non-labelled drug under a roll-over protocol 53.
For example, a single oral dose of 14C-trametinib (2 mg, 2.9 MBq) was administered to patients with advanced solid tumours to determine the absorption, metabolism and excretion of trametinib, a MEK inhibitor approved for the treatment of unresectable or metastatic melanoma with BRAF V600E or V600K mutation 54. The low chemical dose and long half-life (circa 10 days) posed technical challenges for metabolite analysis, study design and data interpretation, exacerbated by the need to enrol cancer patients rather than healthy subjects. Assessment of the emerging results indicated a higher than anticipated radiation exposure due to protracted elimination of radioactivity (<50% recovered over 10 days) and revised dosimetry estimates indicated that the risk associated with dosing additional patients was considered ethically unacceptable. The study was therefore terminated after only two patients had been dosed. However in combination with metabolism information from the FIH study, it was accepted that the metabolism and disposition of trametinib had been adequately assessed.
It may be considered inappropriate for ethical reasons to dose radiolabelled material to patients and non-labelled drug to healthy subjects. Equally, conducting an HRS in healthy subjects may not adequately describe the ADME properties of a drug in the intended patient population. In these situations it may be prudent to collect blood, urine and even bile from a routine clinical PK study using patients dosed with non-labelled drug in order to investigate the metabolic and excretion profiles of the drug using cold technologies. For example, the toxicity of the 8-aminoquinoline analogue discussed previously prevented dosing to healthy subjects. However, it was also deemed inappropriate to dose radiolabel to patients with visceral leishmaniasis. Human metabolism was therefore established through MS and quantitative NMR analyses of plasma and urine from the non-radiolabelled study in patients. Importantly, the regulators accepted that the overall metabolism (both quantitative and qualitative) of the drug was adequately defined even without radiolabelled drug and there was no requirement for an additional HRS 10.
Advances in LC/MS and NMR technologies offer a mechanism by which ADME can be studied in special patient groups such as those with renal or hepatic impairment. The metabolic and excretion profiles of a drug molecule can be established using cold technologies and subsequently compared with data from healthy subjects. Equally, a microtracer approach or low radioactive dose by the primary therapeutic route may be employed if a full excretion profile is required.
Conclusions
HRS are the accepted gold standard for providing an understanding of the human ADME properties of a drug molecule. They are complex and resource intensive. However we have demonstrated how recent advances in technology have enabled additional and in general, less complex approaches to be conducted. As summarized in Figure 5, the advantages and limitations emphasize the importance of deciding upon the optimum approach on a case-by-case basis dependent on the specific ADME (and PK) information required together with the clinical dose, target therapeutic area and portfolio requirements. The increasing availability of these alternative approaches may ultimately present opportunities to obtain the necessary information earlier in the drug development process thereby allowing more informed decisions to be made on drug progression and further clinical and toxicological safety studies. Ultimately, bespoke study designs within a bespoke development plan are foreseen for all new drug candidates. Equally, these alternative approaches may extend ADME investigations into additional disease-state or pediatric populations and eventually, may negate the need to conduct a traditional HRS, assigning it to the history books.
Figure 5.

Summary of alternative approaches for understanding the PK and ADME properties of a drug in humans
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare that they are employees of GlaxoSmithKline and hold shares in the company.
All GSK-sponsored studies referred to were conducted in accordance with the guiding principles of the Declaration of Helsinki and in compliance with good clinical practices and local regulatory guidelines. The protocols and informed consent forms were approved by an institutional review board or independent ethics committee at each study site before any subject was enrolled or study procedure performed. All human biological samples were sourced ethically and, where applicable, patient consent was obtained for their use for research.
All GSK-sponsored animal studies were conducted in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals and were reviewed by the Institutional Animal Care and Use Committee either at GSK or by the ethical review process at the institution where the work was performed.
The authors acknowledge Dr Stephanie North, Dr Gordon Dear, Mr Andy Roberts and Mr Andy Harrell for their scientific contributions to the strategies presented and for their review of this manuscript. They also acknowledge the contributions made by past and present colleagues in the design, conduct and interpretation of the studies discussed here.
References
- FDA Guidance for Industry. 2008. Safety testing of drug metabolites Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079266.pdf2008 (last accessed 24 June 2014)
- 2009. ICH Topic M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf (last accessed 24 June 2014)
- 2012. ICH guideline M3 (R2) – questions and answers Available at http://www.ema.europa.eu/docs/en_GB/document_library/Other/2011/07/WC500109298.pdf (last accessed 24 June 2014)
- Penner N, Klunk LJ, Prakash C. Human radiolabeled mass balance studies: objectives, utilities and limitations. Biopharm Drug Dispos. 2009;30:185–203. doi: 10.1002/bdd.661. [DOI] [PubMed] [Google Scholar]
- Roffey SJ, Obach RS, Gedge JI, Smith DA. What is the objective of the mass balance study? A retrospective analysis of data in animal and human excretion studies employing radiolabeled drugs. Drug Metab Rev. 2007;39:17–43. doi: 10.1080/03602530600952172. [DOI] [PubMed] [Google Scholar]
- Rowland M. Microdosing: a critical assessment of human data. J Pharm Sci. 2012;101:4067–4074. doi: 10.1002/jps.23290. [DOI] [PubMed] [Google Scholar]
- Lappin G, Stevens L. Biomedical accelerator mass spectrometry: recent applications in metabolism and pharmacokinetics. Expert Opin Drug Metab Toxicol. 2008;4:1021–1033. doi: 10.1517/17425255.4.8.1021. [DOI] [PubMed] [Google Scholar]
- Dear GJ, Beaumont C, Roberts A, Squillaci B, Thomas S, Nash M, Fraser D. Approaches for the rapid identification of drug metabolites in early clinical studies. Bioanal. 2011;3:197–213. doi: 10.4155/bio.10.186. [DOI] [PubMed] [Google Scholar]
- Gao H, Obach RS. Addressing MIST (Metabolites in Safety Testing): bioanalytical approaches to address metabolite exposures in humans and animals. Curr Drug Metab. 2011;12:578–586. doi: 10.2174/138920011795713661. [DOI] [PubMed] [Google Scholar]
- Nedderman ANR, Dear GJ, North S, Obach RS, Higton D. From definition to implementation: a cross-industry perspective of past, current and future MIST strategies. Xenobiotica. 2011;41:605–622. doi: 10.3109/00498254.2011.562330. [DOI] [PubMed] [Google Scholar]
- Ramanathan R, Josephs JL, Jemal M, Arnold M, Humphreys WG. Novel MS solutions inspired by MIST. Bioanalysis. 2010;2:1291–1313. doi: 10.4155/bio.10.83. [DOI] [PubMed] [Google Scholar]
- Guiney WJ, Beaumont C, Thomas SR, Robertson DC, McHugh SM, Koch A, Richards D. Use of Entero-Test, a simple approach for non-invasive clinical evaluation of the biliary disposition of drugs. Br J Clin Pharmacol. 2011;72:133–142. doi: 10.1111/j.1365-2125.2011.03956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin P, Jones HM, Jones RD, Yates JWT, Gibson CR, Chien JY, Ring BJ, Adkison KK, He H, Vuppugalla R, Marathe P, Fischer V, Dutta S, Sinha VK, Björnsson T, Lavé T, Ku MS. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 1: goals, properties of the PhRMA dataset, and comparison with literature datasets. J Pharm Sci. 2011;100:4050–4073. doi: 10.1002/jps.22554. [DOI] [PubMed] [Google Scholar]
- Jones RD, Jones HM, Rowland M, Gibson CR, Yates JWT, Chien JY, Ring BJ, Adkison KK, Ku MS, He H, Vuppugalla R, Marathe P, Fischer V, Dutta S, Sinha VK, Björnsson T, Lavé T, Poulin P. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 2: comparative assessment of prediction methods of human volume of distribution. J Pharm Sci. 2011;100:4074–4089. doi: 10.1002/jps.22553. [DOI] [PubMed] [Google Scholar]
- Ring BJ, Chien JY, Adkison KK, Jones HM, Rowland M, Jones RD, Yates JWT, Ku MS, Gibson CR, He H, Vuppugalla R, Marathe P, Fischer V, Dutta S, Sinha VK, Björnsson T, Lavé T, Poulin P. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 3: comparative assessement of prediction methods of human clearance. J Pharm Sci. 2011;100:4090–4110. doi: 10.1002/jps.22552. [DOI] [PubMed] [Google Scholar]
- Vuppugalla R, Marathe P, He H, Jones RDO, Yates JWT, Jones HM, Gibson CR, Chien JY, Ring BJ, Adkison KK, Ku MS, Fischer V, Dutta S, Sinha VK, Björnsson T, Lavé T, Poulin P. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 4: prediction of plasma concentration–time profiles in human from in vivo preclinical data by using the Wajima approach. J Pharm Sci. 2011;100:4111–4126. doi: 10.1002/jps.22551. [DOI] [PubMed] [Google Scholar]
- Poulin P, Jones RDO, Jones HM, Gibson CR, Rowland M, Chien JY, Ring BJ, Adkison KK, Ku MS, He H, Vuppugalla R, Marathe P, Fischer V, Dutta S, Sinha VK, Björnsson T, Lavé T, Yates JWT. PHRMA CPCDC initiative on predictive models of human pharmacokinetics, part 5: prediction of plasma concentration–time profiles in human by using the physiologically-based pharmacokinetic modeling approach. J Pharm Sci. 2011;100:4127–4157. doi: 10.1002/jps.22550. [DOI] [PubMed] [Google Scholar]
- Kostewicz ES, Aarons L, Bergstrand M, Bolger MB, Galetin A, Hatley O, Jamei M, Lloyd R, Pepin X, Rostami-Hodjegan A, Sjögren E, Tannergren C, Turner DB, Wagner C, Weitschies W, Dressman J. PBPK models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci. 2014;57:300–321. doi: 10.1016/j.ejps.2013.09.008. [DOI] [PubMed] [Google Scholar]
- Lappin G, Noveck R, Burt T. Microdosing and drug development: past, present and future. Expert Opin Drug Metab Toxicol. 2013;9:817–834. doi: 10.1517/17425255.2013.786042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Jiang H, Christopher LJ, Shen JX, Zeng J, Arnold ME. Sensitivity based analytical approaches to support human absolute bioavailability studies. Bioanalysis. 2014;6:497–504. doi: 10.4155/bio.13.318. [DOI] [PubMed] [Google Scholar]
- Ostenfeld T, Beaumont C, Bullman J, Beaumont M, Jeffrey P. Human microdose evaluation of the novel EP1 receptor antagonist GSK269984A. Br J Clin Pharmacol. 2012;74:1033–1044. doi: 10.1111/j.1365-2125.2012.04296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahn A, Hodgson S, Wilson R, Robertson J, Watson J, Beerahee M, Hughes SC, Young G, Graves R, Hall D, van Marle S, Solari R. Safety, tolerability, pharmacokinetics and pharmacodynamics of GSK2239633, a CC-chemokine receptor 4 antagonist, in healthy male subjects: results from an open-label and from a randomised study. BMC Pharmacol Toxicol. 2013;14:14. doi: 10.1186/2050-6511-14-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappin G, Kuhnz W, Jochemsen R, Kneer J, Chaudhary A, Oosterhuis B, Drijfhout WJ, Rowland M, Garner RC. Use of microdosing to predict pharmacokinetics at the therapeutic dose: experience with 5 drugs. Clin Pharmacol Ther. 2006;80:203–215. doi: 10.1016/j.clpt.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Lappin G, Shishikura Y, Jochemsen R, Weaver RJ, Gesson C, Houston JB, Oosterhuis B, Bjerrum OJ, Grynkiewicz G, Alder J, Rowland M, Garner C. Comparative pharmacokinetics between a microdose and therapeutic dose for clarithromycin, sumatriptan, propafenone, paracetamol (acetaminophen) and phenobarbital in human volunteers. Eur J Pharm Sci. 2011;43:141–150. doi: 10.1016/j.ejps.2011.04.009. [DOI] [PubMed] [Google Scholar]
- Mahar KM, Sehon C, Tai G, Haws T, Howe D, Young G, Danoff T. Comparison of a GSK706769 microdose CYP3A4 drug–drug interaction study with a previous pharmacological dose study. 2012. American College of Clinical Pharmacology, Annual Meeting September 23rd-25th, San Diego, California abstract #1393294.
- Harrison A, Gardner I, Hay T, Dickins M, Beaumont K, Phipps A, Purkins L, Allan G, Christian R, Duckworth J, Gurrell I, Kempshall S, Savage M, Seymour M, Simpson M, Taylor L, Turnpenny P. Case studies addressing human pharmacokinetic uncertainty using a combination of pharmacokinetic simulation and alternative first in human paradigms. Xenobiotica. 2012;42:57–74. doi: 10.3109/00498254.2011.622418. [DOI] [PubMed] [Google Scholar]
- Vogel JS, Turteltaub KW. Accelerator mass spectrometry as a bioanalytical tool for nutritional research. Adv Exp Med Biol. 1998;445:397–410. doi: 10.1007/978-1-4899-1959-5_25. [DOI] [PubMed] [Google Scholar]
- Jiang H, Zeng J, Li W, Bifano M, Gu H, Titsch C, Easter J, Burrell R, Kandoussi H, Aubry AF, Arnold M. Practical and efficient strategy for evaluating oral absolute bioavailability with an intravenous microdose of a stable isotopically-labeled drug using a selected reaction monitoring mass spectrometry assay. Anal Chem. 2012;84:10031–10037. doi: 10.1021/ac3024558. [DOI] [PubMed] [Google Scholar]
- Sarapa N, Hsyu PH, Lappin G, Garner RC. The application of accelerator mass spectrometry to absolute bioavailability studies in humans: simultaneous administration of an intravenous microdose of 14C-nelfinavir mesylate solution and oral nelfinavir to healthy volunteers. J Clin Pharmacol. 2005;45:1198–1205. doi: 10.1177/0091270005280051. [DOI] [PubMed] [Google Scholar]
- Stevens LA, Evans P, Dueker SR, Lohstroh PN, Giacomo J, Tu D, Shen Z, Yeh L, Ong V, Densel M, Rowlings C, Quart B. Microdose and microtracer intravenous pharmacokinetics of RDEA806 in healthy subjects. Clin Pharmacol Ther. 2009;85:S24–25. [Google Scholar]
- Wu A, Stevens LA, Savant I, Wang X, Laskin OL. Absolute bioavailability of CC-11050, a low water soluble NCE using an i.v. microdose of [14C]-CC-11050 solution concomitantly with an oral unlabelled dose. Clin Pharmacol Ther. 2010;87:S88. [Google Scholar]
- Daley-Yates P, Norris V, Ambery C, Preece A. Early clinical evaluation of a novel 5-lipoxygenase activating protein (FLAP) inhibitor (GSK2190915A). Pharmacokinetics, bioavailability and dose form selection: influence of age, food, drug interactions and regional absorption (LPA112071, LPA112362, LPA114604) 2012. Proceedings of the British Pharmacological Society, BPS Winter Meeting Available at http://www.pa2online.org/abstract/abstract.jsp?abid=30851&author=preece&cat=-1&period=-1 (last accessed 30 November 2013)
- Vasist Johnson LS, Young MA, Stevens LA, Cozens SJ, Collier J, Robertson DC, Dukes GE. A microtracer study of GSK962040, a motilin receptor agonist, to support dosing regimens in the critical care setting. Clin Pharmacol Ther. 2012;91:S80. [Google Scholar]
- Schwab D, Portron A, Backholer Z, Lausecker B, Kawashima K. A novel double-tracer technique to characterize absorption, distribution, metabolism and excretion (ADME) of [14C]tofogliflozin after oral administration and concomitant intraveous microdose administration of [13C]tofogliflozin in humans. Clin Pharmacokinet. 2013;52:463–473. doi: 10.1007/s40262-013-0051-z. [DOI] [PubMed] [Google Scholar]
- Leonowens C, Pendry C, Bauman J, Young GC, Ho M, Henriquez F, Fang L, Morrison RA, Orford K, Ouellet D. Concomitant oral and intravenous pharmacokinetics of trametinib, a MEK inhibitor, in subjects with solid tumours. Br J Clin Pharmacol. 78:524–532. doi: 10.1111/bcp.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dear GJ, Roberts AD, Beaumont C, North SE. Evaluation of preparative high performance liquid chromatography and cryoprobe-nuclear magnetic resonance spectroscopy for the early quantitative estimation of drug metabolites in human plasma. J Chromatogr B. 2014;876:182–190. doi: 10.1016/j.jchromb.2008.10.040. 2008. [DOI] [PubMed] [Google Scholar]
- Hop CECA, Wang Z, Chen Q, Kwei G. Plasma-pooling methods to increase throughput for in vivo pharmacokinetic screening. J Pharm Sci. 1998;87:901–903. doi: 10.1021/js970486q. [DOI] [PubMed] [Google Scholar]
- Vishwanathan K, Babalola K, Wang J, Espina R, Yu L, Adedoyin A, Talaat R, Mutlib A, Scatina J. Obtaining exposures of metabolites in preclinical species through plasma pooling and quantitative NMR: addressing metabolites in safety testing (MIST) guidance without using radiolabeled compounds and chemically synthesized metabolite standards. Chem Res Toxicol. 2009;22:311–322. doi: 10.1021/tx8003328. [DOI] [PubMed] [Google Scholar]
- Yu C, Chen CL, Gorycki FL, Neiss TG. A rapid method for quantitatively estimating metabolites in human plasma in the absence of synthetic standards using a combination of liquid chromatography/mass spectrometry and radiometric detection. Rapid Commun Mass Spectrom. 2007;21:497–502. doi: 10.1002/rcm.2863. [DOI] [PubMed] [Google Scholar]
- Ismail IM, Dear GJ, Roberts AD, Plumb RS, Ayrton J, Sweatman BC, Bowers GD. N-O glucuronidation: a major human metabolic pathway in the elimination of two novel anticonvulsant drug candidates. Xenobiotica. 2002;32:29–43. doi: 10.1080/00498250110081623. [DOI] [PubMed] [Google Scholar]
- Ghibellini G, Johnson BM, Kowalsky RJ, Heizer WD, Brouwer KLR. A novel method for the determination of biliary clearance in humans. AAPS J. 2004;6:45–52. doi: 10.1208/aapsj060433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghibellini G, Leslie EM, Brouwer KLR. Methods to evaluate biliary excretion of drug in human: an updated review. Mol Pharm. 2006;3:198–211. doi: 10.1021/mp060011k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Schwartz MS, Vickers S, Gilbert JD, Amin RD, Depuy B, Liu L, Rogers JD, Pond SM, Duncan CA, Olah TV, Bayne WF. Metabolic disposition of simvastatin in patients with T-tube drainage. Drug Metab Dispos. 1994;22:139–142. [PubMed] [Google Scholar]
- Vickers S, Duncan CA, Vyas KP, Kari PH, Arison B, Prakash SR, Ramjit HG, Pitzenberger SM, Stokker G, Duggan DE. In vitro and in vivo biotransformation of simvastatin, an inhibitor of HMG CoA reductase. Drug Metab Dispos. 1990;18:476–483. [PubMed] [Google Scholar]
- Bloomer JC, Nash M, Webb A, Miller BE, Lazaar AL, Beaumont C, Guiney WJ. Assessment of potential drug interactions by characterization of human drug metabolism pathways using non-invasive bile sampling. Br J Clin Pharmacol. 2013;75:488–496. doi: 10.1111/j.1365-2125.2012.04352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SC, Shardlow PC, Hollis FJ, Scott RJ, Motivaras DS, Allen A, Rousell VM. Metabolism and disposition of fluticasone furoate, an enhanced-affinity glucocorticoid, in humans. Drug Metab Dispos. 2008;36:2337–2344. doi: 10.1124/dmd.108.022137. [DOI] [PubMed] [Google Scholar]
- Harrell AW, Siederer SK, Bal J, Patel NH, Young GC, Felgate CC, Pearce SJ, Roberts AD, Beaumont C, Emmons AJ, Pereira AI, Kempsford RD. Metabolism and disposition of vilanterol, a long-acting β2-adrenoreceptor agonist for inhalation use in humans. Drug Metab Dispos. 2013;41:89–100. doi: 10.1124/dmd.112.048603. [DOI] [PubMed] [Google Scholar]
- Prakash C, Li Z, Orlandi C, Klunk L. Assessment of Exposure of Metabolites in preclinical species and humans at steady state from the singl-dose radiolabelled absorption, distribution, metabolism, and excretion studies: a case study. Drug Metab Dispos. 2012;40:1308–1320. doi: 10.1124/dmd.112.044933. [DOI] [PubMed] [Google Scholar]
- Dave M, Nash M, Young GC, Ellens H, Magee M, Roberts AD, Taylor MA, Greenhill RW, Boyle GW. Disposition and metabolism of darapladib, a lipoprotein-associated phospholipase A2 inhibitor, in humans. Drug Metab Dispos. 2014;42:415–430. doi: 10.1124/dmd.113.054486. [DOI] [PubMed] [Google Scholar]
- Young GC, Ellis WJ. AMS in drug development in GSK. Nucl Instrum Methods Phys Res B. 2007;259:752–757. [Google Scholar]
- Beumer JH, Beijnen JH, Schellens JHM. Mass balance studies, with a focus on anticancer drugs. Clin Pharmacokinet. 2006;45:33–58. doi: 10.2165/00003088-200645010-00003. [DOI] [PubMed] [Google Scholar]
- Deng Y, Sychterz C, Suttle AB, Dar MM, Bershas D, Negash K, Qian Y, Chen EP, Gorycki PD, Ho MYK. Bioavailability, metabolism and disposition of oral pazopanib in patients with advanced cancer. Xenobiotica. 2013;43:443–453. doi: 10.3109/00498254.2012.734642. [DOI] [PubMed] [Google Scholar]
- Bershas DA, Ouellet D, Mamaril-Fishman DB, Nebot N, Carson SW, Blackman SC, Morrison RA, Adams JL, Jurusik KE, Knecht DM, Gorycki PD, Richards-Peterson LE. Metabolism and disposition of oral dabrafenib in cancer patients: proposed participation of aryl nitrogen in carbon-carbon bond cleavage via decarboxylation following enzymatic oxidation. Drug Metab Dispos. 2013;41:2215–2224. doi: 10.1124/dmd.113.053785. [DOI] [PubMed] [Google Scholar]
- Ho MYK, Morris MJ, Pirhalla JL, Bauman JW, Pendry B, Orford KW, Morrison RA, Cox DS. Trametinib, a first-in-class oral MEK inhibitor mass balance study with limited enrolment of two male subjects with advanced cancers. Xenobiotica. 2014;44:352–368. doi: 10.3109/00498254.2013.831143. [DOI] [PubMed] [Google Scholar]